
Abstract
Bile acids (BAs) not only play critical roles in liver-gut immune homeostasis but also participate in regulating lipid, glucose, and energy metabolism. BAs transporter defect or signaling pathways abnormal activation are linked to cholestasis, inflammation, fibrosis, carcinogenesis, and metabolic disorders. BAs and related signaling pathways have become attractive therapeutic targets for inflammation, fibrosis, and metabolic diseases, including type 2 diabetes mellitus and non-alcoholic fatty liver disease (NAFLD). Hydrophilic BAs, including ursodeoxycholic acid (UDCA), tauroursodeoxycholic (TUDCA), and 24-norursodeoxycholic (nor-UDCA), have hepatoprotective properties and are widely used in cholestatic liver diseases. Here, we provide an overview of the mechanism and recent clinical application of UDCA in hepatobiliary diseases, as well as BAs cross-talk with the gut microbiota in health and diseases. Targeting bile-acid signaling for liver cirrhosis is a promising and effective strategy. Evidences from clinical trials suggest that UDCA treatment has beneficial effects on cirrhosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
6.1 Bile Acids and History of Ursodeoxycholic Acid
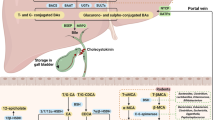
Bile is an important secretion necessary for the digestion and absorption of lipids in the gut. About 500 mg of cholesterol is converted into bile acids (BAs) in the adult liver each day. Newly synthesized BAs are transported into the lumen of the small intestine via the biliary duct, where they act as emulsifiers to help the digestion and absorption of dietary lipids, cholesterol, and fat-soluble nutrients [1]. The solubilized substances are incorporated into lipoproteins, which are delivered to the liver and metabolized. The enterohepatic circulation is a complex pathway in order to maintain the homeostasis of BAs. Generally, BAs move from the hepatocyte into canalicular bile, flow through the biliary tract and into the duodenum. Most BAs are actively recycled in the distal ileum, with a small fraction passively absorbed in the large intestine. Then, they are transported to the liver through portal vein, and efficiently taken up by the hepatocyte [2]. The majority of BAs (>95%) are effectively reabsorbed in the gut via the enterohepatic circulation, and the remaining 5% are newly synthesized in the liver [1].
Ursodeoxycholic acid (UDCA; 3α,7β-dihydroxy5β-cholanoic acid) is a primary component of human bile, physiologically. It is a type of hydrophilic BAs produced by intestinal bacteria and accounts for 1–3% of human BAs [3]. The earliest use of UDCA to cure diseases can be traced back more than 1000 years ago, when traditional Chinese medicine practitioners in the Tang Dynasty discovered the efficacy of bear bile in treating chronic liver diseases [4]. Until 1902, Hammarsten first found the presence of an unknown BA in the bile of the polar bear that he called “ursocholeinic acid.” In 1927, the chemical form of UDCA was identified by Shoda firstly. In 1936, the characterization of the chemical structure of UDCA was done by Iwasaki, which promoted its sufficient synthesis for use in clinical practice [5]. Then, in the 1950s, it was proposed that the therapeutic effects of the bear bile were likely related to high concentrations of the taurine-conjugated form of UDCA and tauroursodeoxycholic (TUDCA) [6]. Subsequently, the therapeutical effect of UDCA in hepatobiliary diseases, such as gallbladder stones [7, 8] and primary biliary cirrhosis (PBC) [9], had been reported in succession.
Nowadays, UDCA has a defined role in preventing and treating patients with cholestatic liver diseases. Of note, UDCA also showed beneficial effects in some other diseases, including treating chronic heart failure [10], shrinking tumors [11], and improving vision [12]. This chapter will provide an overview of the mechanism and clinical application of hydrophilic BAs in hepatobiliary diseases, as well as BAs cross-talk with the gut microbiota in health and disease.
6.2 The Mechanisms of Bile Acids in Hepatobiliary Diseases
6.2.1 Bile Acid Transport, Bile Acid-Induced Toxicity, and Hepatocellular Adaptive Responses in Cholestasis
6.2.1.1 Bile Acid Transport
The transport of BAs is critical for maintaining the enterohepatic BAs circulation, and the regulation of BAs transporters is required for the maintenance of BAs homeostasis [13]. The transporters of BAs include a variety of transport proteins and enzymes located in hepatocytes, as follows: the sinusoidal transporter sodium taurocholate co-transporting polypeptide (NTCP/SLC10A1), members of the anion transporting polypeptide (OATPs/SLCO) family, conjugation enzymes, and the ATP-dependent efflux pump bile salt export pump (BSEP, also known as ABCB) [11, 13, 14]. These transporters are important for a rapid transition of BAs from blood to bile and maintain a low intracellular BA concentration [15, 16]. In the gut, apart from a few passive uptakes of BAs in the proximal small intestine and colon, they are actively absorbed mainly in the terminal ileum via an apical sodium-dependent BA transporter (ASBT) [17]. Then, BAs are bound to the ileal bile acid-binding protein (IBABP, also known as ileal lipid-binding protein ILBP and fatty acid-binding protein 6, FABP6) and exported into the portal blood via organic solute transporter alpha/beta (OSTα/OSTβ) [17]. Furthermore, the BAs in the enterocytes can induce the production of the intestinal peptide hormone fibroblast growth factor 15 (FGF15) in mice (a homolog of human FGF 19), which inhibits the BAs synthesis in hepatocytes in an endocrine manner [18], facilitates gallbladder refilling [19], and downregulates the expression of ASBT expression in a paracrine manner [20], altogether causing a reduction of circulating BAs.
6.2.1.2 Bile Acid-Induced Toxicity
The hydrophobicity of BAs depends on the number, position, and orientation of the hydroxyl groups, which are key factors in determining their degree of toxicity. Regarding the order of hydrophobicity of BAs, it is generally considered that UDCA < cholic acid (CA) < chenodeoxycholic acid (CDCA) < deoxycholic acid (DCA) < lithocholic acid (LCA) [21]. The accumulation of hydrophobic BAs in hepatocytes, like CDCA and DCA, has been considered as the main cause of liver injury in cholestatic liver disease. Hydrophobic BAs are known to directly injure isolated hepatocytes [22], cultured hepatocytes [23], and whole liver [24], but the mechanisms of their toxicity need to be further studied. Here are several hypotheses that may account for the cytotoxicity associated with the most hydrophobic BAs [25]. BAs can cause cell damage by their detergent effects on lipid components [26]. Moreover, it can also enhance the reactive oxygen species (ROS) generation that, in turn, oxidatively modify lipids, proteins, and nucleic acids, and eventually resulting in an increase in hepatocyte apoptosis [27]. Additionally, they can activate Kupffer cells to generate ROS, further aggravating the hepatocyte injury [28].
There are two main pathways of cell death caused by the accumulation of BAs within the hepatocyte; lower concentrations of BAs induce hepatocellular apoptosis [29,30,31,32], whereas higher concentrations induce necrosis [23, 33]. However, the contribution of these two types of cell death in promoting cholestatic liver injury is still in dispute. A brief introduction of them is as follows. Apoptosis is characterized by the maintenance of cellular ATP content. Hydrophobic BAs can induce apoptosis through the extrinsic death receptor-mediated pathway or the intrinsic mitochondria-mediated pathway according to the early evidence [34, 35]. It is confirmed recently that the changes of calcium signaling caused by ER stress can induce apoptosis as well [34, 35]. In contrast to BA-induced cell apoptosis, cellular necrosis is often induced by a high concentration of BAs with the character of cell swelling and intracellular and plasma membranes disruption. The mechanisms for BA-induced hepatocellular necrosis include direct membrane damage due to the detergent-like properties of hydrophobic BAs [26], depletion of ATP, ion dysregulation, mitochondrial and cellular swelling, plasma membrane failure, and cell lysis, releasing intracellular contents [22].
Conversely, as a hydrophilic BA, UDCA can treat cholestatic liver diseases by modulating hydrophobic BAs induced injury in hepatocytes. The hepatoprotective effects of hydrophilic BA have been found in different animal models, such as cholestatic liver diseases and metabolic diseases [36, 37]. And their potential mechanisms [38] like protection against liver inflammation and fibrosis will be discussed in the following paragraphs.
6.2.1.3 Hepatocellular Adaptive Responses in Cholestasis
Cholestasis is a blockage in bile flow caused by mechanical obstruction of biliary ducts or by hepatic transporter defects. During cholestasis, hepatic BAs synthesis and transport will be disturbed, the levels of intrahepatic BAs and plasma BAs will increase, and only small quantities of BAs will reach the colon to participate in enterohepatic circulation, which leads to the BAs profiles, localization and signal transduction alteration [35].
In order to avoid the damage from cholestasis, compensatory changes in the expression of hepatic BAs transporters occur [39]. These changes mainly include downregulation of BAs uptake and synthesis, and upregulation of BAs excretion through increased BSEP or transporters that are able to facilitate the BAs excretion [40, 41]. Several nuclear receptors will be involved in the responses above, such as farnesoid X receptor (FXR), pregnane X receptor (PXR), Constitutive Androstane Receptor (CAR), and the small heterodimer partner (SHP), as well as FGF19 [13, 40]. FXR is a BA-activated nuclear receptor, which influences a myriad of pathways in hepatocytes and other hepatic nonparenchymal cells, including Kupffer cells, endothelial cells, and hepatic stellate cells [13]. FXR/SHP in hepatocytes represses BAs synthesis by mediating a downregulation of NTCP and cholesterol 7α-hydroxylase (CYP7A1) to repress. FXR can also promote BA excretion through directly upregulating BSEP [13]. In humans, hepatic production of FGF-19 may also induce the downregulation of CYP7A1 [42]. Furthermore, a variety of alternative excretory transporters are upregulated during cholestasis, such as the heteromeric transporter OSTα/β and the ABC transporters MRP3 and MRP4, which are often located on the basolateral membrane of hepatocytes, and their expression levels are low under normal physiological conditions [13]. Therefore, if BA secretion is impaired, adaptive responses reduce the accumulation of BAs in the liver and protect hepatocytes against damage to a certain extent. If these responses are insufficient, apoptosis or necrosis of liver cells may occur inevitably [16].
6.2.2 Bile Acids and Cholangiocytes in Cholestasis
Cholangiocytes are polarized epithelial cells lining the intra- and extrahepatic bile ducts, which play a key role in bile composition and flow by solute transport processes [43]. Despite comprising ~5% of the cells in the liver, cholangiocytes account for up to 30% of total bile flow in humans, with the other 70% originating from hepatocyte canalicular secretion [44]. Cholangiocytes contain a large number of transporters that can secret large amounts of bicarbonate, water, and chloride. Specifically, secretin stimulates the apical insertion of intracellular vesicles containing anion exchange protein 2 (AE2), cystic fibrosis transmembrane conductance regulator (CFTR), and aquaporin 1 (AQP1), resulting in chloride secretion through CFTR that is exchanged with bicarbonate via AE2. This bicarbonate generates osmotic force and facilitates the movement of water through AQP1. An alkaline barrier, also called “biliary bicarbonate umbrella”, was formed by the biliary secretion of bicarbonate which can render the BAs polar, de-protonated, and membrane impermeable [45]. Moreover, biliary bicarbonate neutralizes gastric acid contained in food and facilitates the absorption of nutrients [46].
Cholangiocytes also express BAs transporters (like ASBT) that contribute to the absorption of conjugated BAs. Also, passive absorption of protonated unconjugated BA can occur. Cholangiocytes reuptake BAs and then secret them into the peribiliary plexuses blood. This process is called as “cholehepatic shunt pathway,” which is an alternative mechanism to the enterohepatic circulation of BAs, and leads to BAs return to hepatocytes for re-secretion into bile, enhancing its choleretic effect. Furthermore, several experiments indicated that the concentration and composition of BAs may activate different signaling pathways (i.e., calcium protein kinase C, phosphoinositide 3-kinase, mitogen-activated protein kinase, and extracellular signal-regulated protein kinase) to regulate the function of cholangiocytes.
Cholangiocyte damage is a major manifestation in certain cholestatic diseases, thus, the responses of cholangiocytes to injury are also important for understanding the pathophysiology and treatment of cholestatic diseases [43, 47]. Once cholangiocytes are injured, they transform into a neuroendocrine phenotype and cause bile duct hyperplasia, a common histological manifestation of cholestatic liver diseases [48]. Injury of biliary cells can either be immune mediated or non-immune mediated, such as drug-induced liver injury, mechanical biliary obstruction, and so on. Whatever the cause, the accumulation of toxic BAs in the bile ducts will damage cholangiocytes through cholangiocyte membrane disruption, induction of autophagy, and mediation of the secretion of pro-inflammatory and pro-fibrotic factors [48]. In addition, “bicarbonate umbrella” is formed by secreted bicarbonate and cholangiocyte glycocalyx, which can protect the apical membrane of cholangiocytes against BAs induced damage [45, 49].
6.2.3 Hepatoprotective Properties of Hydrophilic Bile Acids (UDCA, TUDCA, nor-UDCA)
Hydrophilic BAs are usually used as therapeutic approaches for cholestasis, including UDCA, TUDCA, and nor-UDCA. TUDCA is the taurine conjugate of UDCA. In cholestasis, UDCA and TUDCA can counteract many of the cellular changes induced by hydrophobic BAs. Their hepatoprotective properties [4] are summarized as follows. (1) Hydrophilic BAs, such as UDCA, can stabilize cell membrane structure and prevent hydrophobic BAs from damaging the cell membrane. (2) Hydrophilic BAs can also inhibit cell apoptosis mainly through blocking mitochondrial damage. (3) Treatment with hydrophilic BAs can promote bicarbonate secretion by several mechanisms including an increase in the anion exchanger 2 expressions. The detergent effects of hydrophobic BAs will be antagonized by bicarbonate. (4) Hydrophilic BAs also have various functions, such as preventing oxidative stress, regulating immunity, and alleviating the damage caused by cholestasis together with the above mechanisms.
24-norursodeoxycholic (nor-UDCA) is a non-amidated, side chain-shortened C23 derivative of UDCA. Instead of undergoing a full enterohepatic circulation, like other conjugated BAs, nor-UDCA undergoes cholehepatic shunting. Since a nor-UDCA anion is secreted into canalicular bile in the unconjugated form, it is protonated by a hydrogen ion derived from carbonic acid that was generated by the hydration of luminal CO2, a process catalyzed by cholangiocellular carbonic anhydrase [50, 51]. The protonated BA is absorbed, thus generating a bicarbonate anion. Nor-UDCA passes through the cholangiocyte, returns to the sinusoids via the periductular capillary plexus, and is re-secreted into bile. This process is termed “cholehepatic shunting”, which generates bicarbonate anion, reinforcing the “biliary bicarbonate umbrella”. Cholehepatic shunting also enables “ductular targeting” to injured bile ducts, which plays a critical role of direct anti-inflammatory, anti-fibrosis, and anti-proliferation [52, 53].
Recently, hydrophilic tetrahydroxylated bile acids (THBA) have attracted the attention of researchers. THBA is more hydrophilic and less cytotoxic than the di- or tri-hydroxylated BAs, which can suppress BA-induced liver damage in mice [54]. Scientists found that feeding THBA to Mdr2−/− mice led to lower levels of toxic secondary BA, LCA, compared with the mice fed the base diet, while feeding of UDCA at equivalent doses led to an average increase in LCA of more than one thousand-fold in the feces and 300-fold in plasma. While the significance of such an increase in LCA was not explored [3], it does find possible adverse consequences of raising LCA during UDCA treatment. For example, treatment with UDCA has been reported to increase the incidence of colon cancer in primary sclerosing cholangitis (PSC) patients with inflammatory bowel disease, where most colon carcinomas develop in the early years after UDCA treatment. Thus, the finding that THBA feeding leads to lower or unchanged LCA production in comparison to UDCA and other BA derivatives may have special implications in terms of the therapeutic potential of THBA for reducing the toxicity of the BA pool.
6.2.3.1 Hydrophilic Bile Acids and Liver Inflammation
The BAs-induced inflammation plays an important role in the process of liver injury [55]. Thus, the modulation of the inflammatory responses via hydrophilic BAs is a potential target in treating cholestasis. UDCA was approved in 1997 for treatment in PBC at a dose of 13–15 mg/kg/day. Many clinical studies showed that UDCA improved liver biochemical indexes, delayed the progress of diseases, and increased survival free of liver transplantation [56,57,58]. A study evaluated the efficacy of TUDCA by analyzing 199 Chinese PBC patients who received TUDCA or UDCA for 24 weeks. A similar proportion of patients in both groups achieved a 25% or 40% reduction in ALP compared to baseline values. In addition, a phase II study of 159 patients with PSC treated with placebo vs. 500, 1000, or 1500 mg of nor-UDCA showed that nor-UDCA reduced ALP levels in a dose-dependent manner. Of note, the anti-inflammatory effect of nor-UDCA is more obvious when compared to UDCA in S. mansoni induced liver injury, and nor-UDCA can directly repress antigen presentation of antigen-presenting cells and subsequent T-cell activation in vitro [59].
Except for PBC and PSC, hydrophilic BAs have also achieved a good result in other chronic liver diseases. In a mouse model of hepatic ischemia reperfusion (HIR), TUDCA attenuated HIR injury by improving liver function in vivo and decreasing hepatocyte apoptosis in vitro. Moreover, TUDCA diminishes the expression and secretion of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α by suppressing ER stress in Kupffer cells via the IRE1α/TRAF2/NF-κB pathway [60]. Likewise, in a non-alcoholic fatty liver disease (NAFLD) model, TUDCA alleviates gut inflammatory responses via downregulation of pro-inflammatory cytokines, such as IL-1β, CCL2, CCL4, and Icam1, and improves intestinal barrier function by increasing levels of tight junction molecules and the solid chemical barrier [61].
6.2.3.2 Hydrophilic Bile Acids and Liver Fibrosis
Chronic liver inflammation will cause liver fibrosis, cirrhosis and, even hepatocellular carcinoma. Hepatic fibrosis is a pathological process that results from the excessive accumulation of extracellular matrix (ECM) proteins, which replace the damaged normal liver tissue. There are two main causes of chronic liver injuries: hepatoxic injury (caused by hepatitis B or hepatitis C virus) and cholesterol injury (like PBC and PSC). Upon removal of the etiological source of the chronic injury, liver fibrosis can be reversed [62]. The transcription of proteins, such as BSEP and CYP7A1, participating in numerous signaling pathways, such as BAs synthesis, detoxification, and fibrogenesis, play a key role in the pathogenesis of cholestatic liver fibrosis [41, 63, 64]. A recent study found that NTCP expression is linearly associated with the severity of liver fibrosis, and antagonizing BAs uptake may be a therapeutic target for preventing disease progression [65].
Many experiments confirmed that hydrophilic BAs could inhibit liver fibrosis in different disease models [66,67,68,69,70], but its detailed mechanism remains to be investigated. The latest study revealed that UDCA displayed antifibrotic role by protecting HSC against the production of collagen and inhibiting cellular viability involving autophagy inhibition [71]. Notably, in the Mdr2−/− mice, a model of sclerosing cholangitis, nor-UDCA strongly reversed biliary fibrosis and injury, which was superior to UDCA treatment [72]. Similarly, in a rat model of thioacetamide-induced liver fibrosis, although nor-UDCA and UDCA exhibited therapeutic effects on fibrosis, nor-UDCA was more effective than UDCA, especially in the experiment with liver fibrosis regression. A similar role has also been reported for TUDCA. A study confirmed that TUDCA could inhibit carbon tetrachloride-induced liver fibrosis in rats [73], and its beneficial effects may be attributed to decreased hepatic unfolded protein response signaling and apoptotic cell death [74].
6.2.3.3 Hydrophilic Bile Acids and Liver Lipid Metabolism
FXR, a dedicated BA receptor, plays a critical role in lipid homeostasis. Prior studies revealed that FXR agonists can reduce circulating triglycerides (TGs) [75] and hepatic steatosis [76]. This beneficial remodeling of lipid metabolism is regulated by the FXR-SHP axis, which represses sterol regulatory element-binding protein 1c (SREBP1c), a master regulator of hepatic de novo lipogenesis [77], and by FXR-dependent interference of ChREBP binding to the liver pyruvate kinase (LPK) promoter [78]. Similarly, whole-body FXR−/− mice display an increase in serum TGs and cholesterol levels, together with an accumulation of hepatic lipid deposits and enhanced levels of lipogenic genes in the liver [79,80,81].
However, whether hydrophilic BAs, like UDCA, can lower the lipid levels is now uncertain and needs to be further studied. In basic research, UDCA is usually recognized as an agent with lipid-lowering activity. For instance, UDCA-treated mice showed higher expression levels of ABCG8, ABCB11, and CYP27A1, and lower expression levels of LXR and PPAR-α, which suggested that UDCA can improve lipid metabolism [82]. UDCA significantly inhibited lipid accumulation in a NAFLD cell model, which may repress the activation of AKT/mTOR/SREBP-1 signaling pathway [83]. But in clinical trials, it remains difficult to draw a firm conclusion. Some studies observed a significant decrease in total cholesterol levels after UDCA treatment [84,85,86,87]; however, other studies found no beneficial effect on lipid metabolism [88,89,90,91]. A meta-analysis [92] pooled the data from 15 randomized placebo-controlled trials and summarized the impact of UDCA on circulating lipid concentrations. Total cholesterol was reduced after UDCA treatment, while LDL-C, HDL-C, and TG were not significantly altered by UDCA administration. Moreover, UDCA reduced the levels of total cholesterol and LDL-C without affecting TG and HDL-C in PBC patients.
6.2.3.4 Hydrophilic Bile Acids and Gut Microbiome
There is a close and bidirectional interplay between BAs and the gut microbiota: the gut microbiome shapes the BAs pool, and cholestasis may alter intestinal microbial communities. Few studies have focused on the gut microbiota in cholestatic liver diseases [93, 94]. Notably, a recent study found that the diversity of gut microbiota reduced significantly in PBC patients, which is partially relieved by UDCA administration [95]. Similarly, reduced intraindividual bacterial diversity has been found in stool samples from PSC patients [96], but it remains unknown if they are primary or secondary to the bile secretory failure present in cholestatic disorders. Moreover, loss of gut microbiota in mdr2−/− mice, a mouse model of PSC deficient of canalicular transporter of phospholipid that can induce biliary injury, can also lead to increased liver damage [97]. Furthermore, the germ-free mdr2−/− mice exhibited significantly more severe liver chemistry and histological injuries compared to the control group [36]. These findings suggested the importance of commensal microbiota and its metabolites in protecting against injuries to bile duct.
Recent studies explored the effects of UDCA on gut microbiota composition in human and mice models [98,99,100]. Interestingly, UDCA influenced bacterial populations inducing a marked decrease in abundance of Bifidobacterium, Lactobacillus, and Lactobacillaceae [98]. UDCA could also improve colitogenic dysbiosis. A recent study indicated that UDCA, TUDCA, or glycoursodeoxycholic (GUDCA) equally lowered the severity of dextran sodium sulfate-induced colitis in mice and ameliorated colitis-associated fecal dysbiosis at the phylum level [101]. In a human study, the UDCA treatment can increase the abundances of F. prausnitzii, but reduce Ruminococcus gnavus, and this finding was associated with the lower risk of colorectal adenoma in men than in women [99]. In general, hydrophilic BAs seem to be a protective substance in both health and disease, but it remains to be determined if these effects are relevant to the therapeutic action of hydrophilic BAs.
6.3 Clinical Applications of Hydrophilic Bile Acids (UDCA, TUDCA, nor-UDCA) in Cirrhosis
6.3.1 Primary Biliary Cirrhosis
6.3.1.1 UDCA
PBC is characterized by progressive immune-mediated destruction of the small-to-medium-sized bile ducts, resulting in chronic cholestasis, portal inflammation, and fibrosis that can develop to cirrhosis, and even liver failure [102]. The diagnosis is based on anti-mitochondrial antibody (AMA) or anti-nuclear antibody (ANA) positive in the presence of a cholestatic biochemical profile, histologic confirmation being mandatory only in seronegative cases [103]. These patients usually have fatigue and pruritus, both of which occur independently of disease severity. It is prevalent among women, white patients, and patients 60–70 years old [104].
UDCA is the only drug approved by the US Food and Drug Administration and the European Association for the Study of the Liver for the treatment of PBC [105]. UDCA is the 7-β-epimer of the primary bile acid chenodeoxycholic acid, a naturally-occurring hydrophilic BA. The inflammation state created by BA accumulation in hepatocytes resulted in cell necrosis and apoptosis. UDCA increases the elimination of toxic substances from hepatocytes by inhibiting intestinal absorption of BAs and increasing biliary BAs secretion. The secretion stimulation depends on a dual MAPK- and integrin-dependent mechanism and activating hepatocytes and cholangiocytes vesicular exocytosis as well as carrier insertion into their apical membranes [106, 107] Meanwhile, it stimulates the secretion of a bicarbonate-rich fluid from cholangiocytes, which decreases cholestasis. Finally, UDCA augments micelle formation to prevent the toxic effect of BAs to cell membranes [108]. It is also reported that the gut microbial profile in PBC patients is altered and partially restored after UDCA therapy [95].
UDCA has been shown to improve serum hepatic biochemistries and prevent histological progression [109, 110], but it could not relieve the symptoms of fatigue and pruritus [111]. A retrospective review including 550 patients with PBC who accepted UDCA treatment or placebo control revealed that UDCA improved the survival free of liver transplantation [58]. The survival rate of patients with stage 1 or 2 disease was similar to that of a healthy control population when given long-term UDCA [112]. A meta-analysis of 4845 patients enrolled in long-term cohort studies revealed that UDCA treatment improved the transplant-free survival of 90% at 5 years, 78% at 10 years, and 66% at 15 years, compared with 79% at 5 years, 59% at 10 years, and 32% at 15 years in untreated group [56]. The latest research in 2021 showed that in a cohort of predominantly male patients with cirrhosis, UDCA response contributes to a reduction in decompensation, all-cause, and liver-related death or transplantation, with the highest benefit in patients with portal hypertension [113]. African American and Asian American/American Indian/Pacific Island (ASINPI) patients who did not receive UDCA had significantly higher mortality than white patients [114].
The recommended dose of UDCA is 13–15 mg/kg per day for all patients with PBC, usually for life, unless intolerance occurs. Loose stool, headache, and mild weight gain are the most frequently reported adverse effects of UDCA [115]. High doses of UDCA (28–30 mg/kg/day) is not recommended especially for patients with varices or liver transplantation, because UDCA has slight side effects which may be ineffective and harmful [115]. UDCA should be given to all PBC patients lifelong, including during pregnancy and breastfeeding [105]. What is more, preventive UDCA after liver transplantation for PBC reduces the risk of disease recurrence, graft loss, and death [116].
About 40% of patients will not have an adequate biochemical response to UDCA, who have relative risk of 5.51 (95% CI 1.70–15.99) of death or liver transplantation compared with those with a response [117]. Women presenting at younger than age 50 has the lowest response rates and highest levels of symptoms [118]. Besides, serum vitamin D level is also associated with disease severity and response to UDCA in PBC [119].
Stratification to recognize those high-risk patients with shorter survival using serum liver tests has been evaluated extensively across different cohorts worldwide, which is suggested for all patients following 1 year of UDCA therapy. This stratification is fundamental to recognizing those patients that should be considered for new disease-modifying therapy.
There are several classifications to define incomplete response to UDCA [120] (Table 6.1).
However, large-scale follow-up data have recently shown that even an incomplete response to UDCA in PBC is associated with better survival [127], which strongly suggests that UDCA therapy in PBC must be continued for life, regardless of biochemical response.
PBC patient outcomes can be predicted by biochemical indexes. The GLOBE and UK-PBC risk scoring systems were proven to be good predictors for future cirrhosis-related complications [120, 128] (Table 6.2).
6.3.1.2 Combined with Obeticholic Acid, Fibrates, Corticosteroids, and Other Drugs
No unified treatment was recommended to PBC patients who have an incomplete response to UDCA. UDCA combination with obeticholic acid (OCA), fibrates, and budesonide may be effective, but long-term efficacy is still a needed step to study.
6.3.1.3 Combined with Obeticholic Acid
For those adult PBC patients who are incompletely responsive to UDCA for at least 1 year or cannot tolerate UDCA as monotherapy, OCA was firstly recommended by EMA and FDA as a combined drug of UDCA [120, 131]. OCA can regulate BA synthesis, absorption, transport, secretion, and metabolism as an FXR agonist [132, 133]. A randomized control study assessed the effect of OCA on BA hepatobiliary excretion in PBC patients with an inadequate response to UDCA [134]. This study showed that, compared to placebo, OCA increased the transport of the conjugated BA tracer 11C-CSar and accelerated the transportation of endogenous conjugated BAs from hepatocytes into biliary canaliculi, which revealed that OCA can reduce the time hepatocytes are exposed to potentially cytotoxic BAs. A research revealed that OCA demonstrated choleretic and antifibrotic effects by regulating FXR as well as immune response and inflammation [135]. Several other researches suggested that OCA reduced ALP levels compared with placebo along with or without UDCA [136,137,138]. OCA 5 mg once daily is recommended for adult PBC patients who are in inadequate biochemical response to the UDCA treatment with adequate doses for at least 1 year or who are intolerant to UDCA. If ALP or total bilirubin level has not gained any adequate reduction after 6 month-treatment at this dose, the OCA dosage can be increased to the maximum recommended dose of 10 mg/day once daily. The side effects of OCA are itch and dyslipidemia. As the benefit is not well determined in decompensated PBC patients, OCA is not recommended for these patients [131].
6.3.1.4 Combined with Fibrates
As agonists of peroxisome proliferator-activated receptors (PPARs), fibrates have anti-inflammatory, anticholestatic, and antifibrotic functions [139, 140]. Several placebo-controlled trials showed that patients treated with bezafibrate in combination with UDCA had a higher biochemical response and lower predicted mortality or need for liver transplantation than those treated with placebo plus UDCA [116, 141, 142]. Another study revealed that bezafibrate combined with UDCA significantly decreased the predicted risk of mortality [143]. Besides, bezafibrate also has a function of improving pruritus, fibrosis, and inflammatory histological scores [141, 144]. Overall, bezafibrate is the only drug currently available to improve symptoms, serological indicators, and prognosis in PBC patients. In spite of this, there are still several PBC patients who had a low response to bezafibrate combined with UDCA. Similarly, fenofibrate combined with UDCA for those PBC patients who have an inadequate response to UDCA can also improve serological indicators [145, 146] as well as fibrosis and ductular injury [147], and enhance transplant-free and decompensation-free survival [148]. But there were also some side effects of using fibrates, including myalgias, elevation in serum bilirubin levels/creatinine levels/aminotransaminase levels. At the same time, fibrates are also not recommended to treat PBC patients with decompensated liver cirrhosis.
6.3.1.5 Combined with Corticosteroids
The role of glucocorticoids in treating PBC inflammation is controversial [140], especially when UDCA is combined use of budesonide. Budesonide, as an agonist of PXR/glucocorticoid receptor (GR), is also involved in the synthesis, transport, and metabolism of BA, with high receptor affinity and high primary metabolism [149]. Several studies illustrated that budesonide improved the level of ALP and liver histology compared to placebo when combined with UDCA [150, 151] In addition, budesonide has severe osteoporosis complications and minor action of improving biochemical parameters as well as liver histology [152]. Besides, a recent placebo-controlled randomized trial disclosed that the addition of budesonide improved liver-related serological parameters, but had little effect on liver histology [153]. The effect of budesonide is closely related to the disease stage of PBC. Steroid-related side effects are the main adverse effects of budesonide, and also include portal vein thrombosis as well as osteoporosis [154]. Therefore, budesonide is not suitable for the treatment of advanced stage of PBC.
6.3.1.6 Combined with Other Drugs
PBC is a type of disease associated with an autoimmune state, and the role of several immunosuppressants and immunomodulators has been evaluated over the past few decades, such as methotrexate [155, 156], colchicine [157], azathioprine [158] and so on. However, the effects of these drugs were largely unsatisfactory, with patients showing no significant improvement in serological indicators, liver pathology, and overall survival, and/or reporting unacceptable risk of adverse events [159,160,161,162]. These demonstrated that autoimmune characteristics only partly reflected the nature of PBC.
6.3.2 Primary Sclerosing Cholangitis (PSC)
PSC is a rare disease with unknown etiology. It is mainly manifested as chronic progressive cholestasis, which eventually leads to end-stage liver disease. Multifocal intrahepatic or extrahepatic bile duct inflammation and fibrotic stenosis are the main characteristics [163]. Inflammatory bowel disease (IBD), which occurs most frequently in men aged 30–40, may be an important risk factor for 60%–80% of patients [164]. In addition, the risk of developing hepatobiliary or colorectal cancer is very high. About 40% of PSC patients die of cancer, with a mortality rate four times that of the general population [165]. Currently, the treatment of PSC has not been determined.
A number of studies since 1992 found that low-dose (13-15 mg/kg) and medium-dose (17–23 mg/L) UDCA had significant effects on improving liver biochemical indexes of PSC patients [166, 167]. However, there was no statistically significant improvement in mortality, liver transplantation, and cholangiocarcinoma [168,169,170]. In addition, high doses (28-32 mg/kg) of UDCA can lead to PSC progression to cirrhosis, esophageal varices, cholangiocellular carcinoma (CCA), colorectal dysplasia, liver transplantation, or death [115]. There are currently conflicting treatment guidelines for PSC. In 2019, the British Gastroenterological Association recommended that UDCA should not be routinely treated in newly diagnosed PSC patients [171]. As recommended by the British Gastroenterology Association, the American Association for the Study of Liver Diseases (AASLD) clinical practice guidelines do not recommend UDCA for patients with PSC [172]. However, the European Association for the Study of the Liver (EASL) has no specific recommendation on whether UDCA can be used for PSC [173]. For patients already using UDCA, discontinuation of UDCA leads to deterioration of liver symptoms, biochemical indices, and Mayo risk scores [174]. Therefore, patients already treated with UDCA need to decide whether to continue UDCA treatment after 6 months of use based on biochemical reactions and itching relief [175]. At present, the optimal dose of UDCA is 17–23 mg/kg, which has the most significant improvement on liver biochemical indexes [176], and the usual dosage for most doctors is 20 mg/kg.
TUDCA is a hydrophilic BA that is a taurine conjugate of UDCA. The role of UDCA in the liver is mostly generated by the non-conjugated form and its taurine conjugated TUDCA, and there is little difference between the two [4]. Eight patients with pancreatic cancer-induced biliary tract obstruction, but no liver or intestinal disease, were randomly treated with TUDCA and UDCA, and their absorption and BA secretion were similar [177]. Toxicity of BAs is inversely proportional to hydrophilicity, and coupling with taurine makes UDCA more polar, which indicates that TUDCA has a higher therapeutic effect [178,179,180]. In patients with cholestatic liver disease treated with UDCA or TUDCA, 85% of the PBC cholestase decreased, but not in the PSC group [181]. At present, the efficacy of TUDCA on PSC is still lacking more evidence, and further exploration is needed.
Nor-UDCA and UDCA have similar physiological structure, with one methylene less side chain than UDCA, relatively resistant to amidation, hepatobiliary shunting and the ability to directly stimulate bile duct cells to secrete bicarbonate. It has a strong ability to resist biliary tract injury caused by BAs [45] and has a bright prospect for the treatment of cholestatic liver and bile duct diseases. In typical PSC models of multidrug resistance gene 2 knockout mice (Mdr2−/−), nor-UDCA significantly improved sclerosing cholangitis in mice [72]. Nor-UDCA also reduced liver damage in selective bile duct ligation (SBDL) mice, while UDCA was significantly more toxic to common bile duct ligation (CBDL) mice [182]. A multicenter randomized controlled trial of 161 patients with PSC found significant reductions in ALP levels after 12 weeks of nor-UDCA500 mg/day, 1000 mg/day, and 1500 mg/day, showing a good safety profile similar to placebo. There was no difference between itch reports and comfort groups [183]. Nor-UDCA is currently being evaluated in a phase III clinical study in patients with PSC (ClinicalTrials.gov, NCT01755507).
6.3.3 NAFLD-Related Cirrhosis
NAFLD has become one of the most common chronic liver diseases in the world, which is associated with obesity, hyperlipidemia, hyperlipidemia, type 2 diabetes, and metabolic syndrome [184, 185]. Non-alcoholic steatohepatitis (NASH), a subtype of NAFLD, has hepatocellular necrosis, inflammation, and fibrosis, and can lead to cirrhosis and even liver cancer in some patients [185]. Currently, there are no FDA-approved therapeutic drugs for this disease, and lifestyle changes, such as diet modification and exercise, are effective treatment methods [185]. BAs not only promote intestinal fat digestion and absorption but also act as ligands to bind BA receptors and regulate lipid metabolism and glucose metabolism through various signaling pathways [186,187,188]. Meanwhile, there is evidence that BA homeostasis is imbalanced in NASH patients [189], so BA analogs and their compounds affecting BA signaling pathways are expected to be effective drugs for the treatment of NAFLD/NASH.
UDCA, a hydrophilic BA with several hepatoprotective properties, has also been tested in NAFLD/NASH, but the results appear to be less than satisfactory. Treatment studies of UDCA in NAFLD reported a decade ago showed improvement in NAFLD transaminases with both low [190] and high dose [191] therapies, but this was not confirmed in large cohort studies that also used lower [192] and higher [193] dose treatments for more than 1 year. Therefore, there has been controversy about the efficacy of UDCA in NAFLD, and enthusiasm for its research in the disease has waned. The results of two recently conducted trials were again opposite, with one conventional dose (20 mg/kg/day), short-term (3 weeks) therapy demonstrating increased liver steatosis, disease activity and fibrosis in patients treated with UDCA [91, 194], while the other 6-month UDCA treatment at a dose of 15 mg/kg/day produced significant normalization of liver enzymes and improvement in lipids and liver steatosis [69]. Overall, the question of whether UDCA plays a role in NAFLD that is more beneficial or more detrimental has not been confirmed at this experimental stage, so it is not recommended in the current guidelines as a treatment for NAFLD [195]. However, some trials have shown beneficial effects of UDCA in combination with other drugs (e.g., vitamin E, curcumin) [190, 196, 197], and this may be considered in the future. The UDCA derivative nor-UDCA has been shown to improve steatohepatitis in a mouse model of NASH [198], and a recent phase II trial in patients with the disease also significantly reduced transaminase levels [199], indicating a potential therapeutic role for nor-UDCA in NAFLD disease.
OCA is a semi-synthetic analog of CDCA, a highly selective receptor agonist for FXR [200]. FXR expression in the terminal ileum and liver plays a role in the treatment of NAFLD [201]. When intestinal BA levels are elevated, the reabsorbed BAs enter the enterocytes to activate FXR and release human FGF19, which reaches the liver and binds to FGF receptor 4 (FGFR4), inhibiting BA synthesis by directly inhibiting the expression of CYP7A1 [202]. Hepatic FXR activation also inhibits CYP7A1, which manifests to promote bile excretion. In addition, in animal models of liver disease, FXR activation inhibits adipogenesis to reduce steatosis and exerts anti-inflammatory and antifibrotic effects [203, 204]. Thus, the results of the OCA Phase II and Phase III clinical trials in NASH showed considerable beneficial effects of OCA - improvement of fibrosis [204, 205]. However, OCA treatment also decreased HDL and increased LDL cholesterol [204], increasing cardiovascular risk, while its side effect of pruritus was surprising and disappointing, so the development of an alternative to OCA without pruritic side effects is an urgent priority.
6.3.4 Drug-Induced Cholestasis
Drug-induced cholestasis is common and accounts for approximately 17% of all hepatic adverse drug reactions (ADRs) [206]. Some drugs only cause simple cholestasis, such as estrogens, anabolic steroids. Some drugs can induce cholestatic hepatitis, drug-induced sclerosing cholangitis, and the vanishing bile duct syndrome (VBDS), some cases even progress to cholestatic cirrhosis. Chlorpromazine, ketoconazole, and amoxycillin-clavulanate are typical drugs. There is no pretreatment for drug-induced cholestasis, but early recognition and prompt drug withdrawal are the more important [206]. According to the published individual case reports and open cohort studies, UDCA is effective to relieve jaundice, pruritus, fatigue, and liver biochemical abnormalities in approximately two-thirds of treated cases [206,207,208,209,210]. Considering the important methodological limitations, it is difficult to preclude a generalization of the results on some retrospective and prospective cohort studies [211,212,213]. High-quality controlled studies are required to explore the effect of UDCA in drug-induced cholestasis. However, it is difficult to conduct these experiments, given that a wide variety of drugs have been involved and the nature of these cases has been isolated [214].
6.3.5 Intrahepatic Cholestasis of Pregnancy
Intrahepatic cholestasis of pregnancy (ICP) is one of the most common pregnancy-specific liver diseases, which often occurs in the second and third trimester of pregnancy. Clinical syndromes of ICP include generalized pruritus and elevated BAs, with normal or abnormal liver function. ICP is associated with multiple adverse pregnancy outcomes, including preterm birth (iatrogenic and spontaneous), amniotic fluid staining, neonatal depression, respiratory distress syndrome, and increased risk of stillbirth [215]. In normal pregnancies, BAs are transported from the fetus to the mother, whereas in ICP pregnancies, placental transport occurs in the opposite direction. As a result, both maternal and fecal BA levels increased. Increased levels of total bile acid (TBA) are associated with the induction of oxidative stress and apoptosis, resulting in damage to liver cells and other tissues, and an increased risk of harmful effects on the fetus with increased levels of TBA in maternal blood [216].
UDCA is the therapeutic choice for ICP. The mechanism includes the replacement of hydrophobic BAs to ensure the protection of hepatocyte membranes and to stimulate the expulsion of BAs from the fetus through the placenta [216]. Since 1991, after the publication of the first article showing that UDCA can improve serum bile salt levels and pruritus symptoms, many articles further confirmed that UDCA is effective on pruritus and in decreasing liver transaminase and bilirubin in ICP patients [217, 218]. A meta-analysis found that UDCA also can effectively improve fetal prognosis [219]. The incidence of fetal distress/asphyxia was lower in the UDCA group than in the placebo group, but the difference was not statistically significant [219]. However, an RCT trial involving 605 women found that UDCA treatment, although harmless, did not reduce maternal BA concentrations, nor can it reduce the adverse perinatal outcomes in women with ICP [220]. Meanwhile, a large meta-analysis of 5557 ICP cases and 165,136 controls showed that BAs are important for fetal prognosis, however, UDCA treatment did not significantly affect the relationship between BA levels and fetal prognosis [221]. Therefore, the relevance of UDCA for the treatment of ICP should be reconsidered. UDCA is still the first-line treatment for ICP and is recommended in six national guidelines for the management of ICP.
Although the usefulness of UDCA is now in doubt, no studies have been published to date to report any adverse effects of UDCA on mothers or fetuses. But Chappell recommended that the lack of evidence of in vivo benefits should prevent further routine clinical use of UDCA, which does no harm but avoids unproven treatment for women [221]. In conclusion, the findings of the latest study undermine the role of UDCA as a first-line treatment for ICP, and more research is needed to further explore the implications of UDCA for pregnant women and fetuses.
6.3.6 Total Parenteral Nutrition-Associated Cholestasis
Long-term total parenteral nutrition treatment is a risk factor to cause transient or persistent liver damage, manifested as cholestasis with increased serum ALP and bilirubin levels [222]. Clinical studies indicated that orally administered UDCA in doses of 10–30 mg/kg body weight per day is effective to improve cholestatic abnormalities caused by parenteral nutrition-associated cholestasis in neonates [223,224,225,226]. Recently, a retrospective research in neonates demonstrated that UDCA therapy was associated with a faster decline of conjugated bilirubin and greater weight gain, but not associated with the duration of parenteral nutrition-associated liver disease [227]. UDCA is recommended to treat parenteral nutrition-associated cholestasis by the American Society for Parenteral and Enteral Nutrition Clinical Guidelines (2014). However, this suggestion is lacking of high-quality evidence, and more relevant studies are required to verify its effect [228]. Evidence of a benefit of the application of UDCA in adults with parenteral nutrition-associated liver disease is more limited, with a single study showing that treatment with an average of 11.2 mg oral UDCA/kg body weight per day is related to a decline in GGT and ALT levels, but not ALP, AST, or bilirubin levels [229].
6.3.7 Chronic Graft-Versus-Host Disease Involving the Liver
Graft-versus-host disease (GVHD) is a common complication following allogeneic bone marrow transplantation with cholestasis and veno-occlusive disease. Up to now, preliminary studies indicated that short-term treatment with UDCA improves the cholestasis in GVHD [230]. A prospective, single-center study showed that the long-term treatment of UDCA results in clinical and biochemical beneficial effects in individuals with limited GVHD of the liver. The data suggests that long-term therapy is safe and tolerable [231]. Another randomized, open-label multicenter research indicated that in addition to short-term benefits, UDCA prophylaxis improves long-term survival and reduces non-relapse mortality without causing any adverse effects [232].
6.3.8 Liver Disease in Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal recessive disease that occurs more often in Caucasians. The odds of the disease are about 1/2000–3000 [233]. The mutations of the CFTR gene lead to dysfunction of chloride channels in the apical epithelial cells of the gut, pancreas, and bile duct systems, and cause dehydration of secretions and mucus hyperplasia, while affecting bile production, leading to multisystem disease [234,235,236].
While CF predominantly causes damage in the lung, now people are shedding more light on how to deal with the extrapulmonary manifestations of CF for advances in patient care have altered the course of CF and led to a significant increase in life expectancy. The clinical manifestations of CFLD may include elevated liver enzymes, cholangitis, and hepatic steatosis, as well as focal fibrosis and focal cirrhosis [236]. CF is now considered the third leading cause of death following respiratory and transplant complications.
UDCA is currently the primary treatment for primary liver disease and can improve the flow of BAs by inducing the flow of hydrogen carbonate bile [237], but its use as a CFID treatment remains controversial. A population-based longitudinal cohort study from the UK has found that the prevalence of CFID is slowly increasing. After stratifying patients for cirrhosis of the liver, the use of UDCA was found to be associated with longer survival, especially in patients without cirrhosis, but not in patients with cirrhosis [238]. Another study reported that UDCA can reduce cirrhosis in patients with mild liver disease, thereby preventing the development of cirrhosis, which is consistent with earlier observational studies in CF patients with mild liver disease [239]. We, therefore, suspect that UDCA might have a beneficial effect in patients with early or mild CF disease. However, most studies prove it of no obvious effect to use UDCA for the long-term survival of CFID. A review based on four RCTs concluded that UDCA treatment had no significant effect on CF patients, except for a slight effect on liver enzyme reduction, but given that these studies were short-term trials, there is no enough evidence to support the UDCA’s role in improving survival [237]. A multicenter cohort study found that using UDCA did not reduce the incidence of portal hypertension [240].
In short, more evidences are needed to confirm the effect of UDCA in CTID. In the future, RCTs which involve a larger sample size and longer observation time are required. Due to the absence of additional useful medicine for CFID, it is still recommended to start UDCA treatment once diagnosed with CFID.
6.3.9 Progressive Familial Intrahepatic Cholestasis
Progressive familial intrahepatic cholestasis (PFIC) is an autosomal recessive disorder that results from defect in bile secretion and is characterized by intrahepatic cholestasis. PFIC is usually onsets in infancy and childhood and can lead to liver cirrhosis. According to the gene mutation location, PFIC is divided into three types. Type1 (PFIC1) is defective in ATP8B1 gene which encodes the FIC1 protein. Type2 (PFIC2) has mutations in ABCB11 gene encoding BSEP. Type 3 (PFIC3) is associated with mutations in ABCB4 gene encoding the canalicular translocator of phosphatidylcholine MDR3 [241,242,243]. The severity of liver disease and the response to pharmacological therapy vary among PFIC children. Cholestatic jaundice and pruritus are the main clinical presentations. PFIC1/2 usually manifests with normal serum gamma-glutamyl transpeptidase (GGT) while that is raised in patients with PFIC3. Although the optimal strategy for the treatment of PFIC has not been fully defined, liver transplantation has been considered to be a definitive therapy, with a survival rate of approximately 92% at 5 years at present [244]. The combination of UDCA and standard nutritional support, including adequate calories, supplementation of fat-soluble vitamins, and medium-chain triglycerides, is the essential treatment for PFIC. UDCA 20–30 mg/kg/day for 2–4 years is safe and decreases the ALT and GGT levels and improves the nutritional condition, hepatosplenomegaly, and pruritus [245, 246]. Currently, UDCA is the first-line therapy for patients with ABCB4 deficiency (PFIC III). Its efficacy is associated with the type of ABCB4 variant and the changes in MDR3 expression/function which result from the former factors. For patients with normal or reduced MDR3 activity, UDCA can be used as an effective treatment method, and it improves the liver function, even restores it to be normal. However, patients with nearly complete or complete loss of MDR3 function are ineffective with the treatment of UDCA [247, 248]. In addition, studies have shown that UDCA can induce the insertion of Bsep into the microtubule membrane of hepatocytes, thereby increasing the microtubule expression of Bsep, which can be used for the treatment of patients with ABCB11 deficiency (PFIC II) [249, 250]. In patients with ATP8B1 deficiency (PFIC I), the efficacy of UDCA is not ideal. For these patients, partial biliary diversion surgery is an option worth considering [251].
6.3.10 Other Pediatric Cholestatic Disorders
Pediatric cholestatic disorders include biliary atresia, Alagille syndrome, BA synthesis defects, ductal plate abnormalities, including Caroli syndrome and congenital hepatic fibrosis, and certain metabolic diseases [252]. In addition to liver transplantation in childhood, UDCA is an adjunctive therapy for pediatric cholestatic diseases, especially for biliary atresia [253, 254]. Despite the compelling evidence lacking to verify its exact effect, given the low side effect risk profile of standard-dose UDCA (10–20 mg/kg/day), it is often used in those children who suffer from pediatric chronic cholestasis [252].
References
Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–74.
Hofmann AF. The enterohepatic circulation of bile acids in mammals: form and functions. Frontiers in bioscience (Landmark edition). 2009;14:2584–98.
Hofmann AF. Pharmacology of ursodeoxycholic acid, an enterohepatic drug. Scand J Gastroenterol Suppl. 1994;204:1–15.
Cabrera D, Arab JP, Arrese M. UDCA, NorUDCA, and TUDCA in liver diseases: a review of their mechanisms of action and clinical applications. Handb Exp Pharmacol. 2019;256:237–64.
Lazaridis KN, Gores GJ, Lindor KD. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J Hepatol. 2001;35:134–46.
Li S, Tan HY, Wang N, et al. Substitutes for bear bile for the treatment of liver diseases: research Progress and future perspective. Evid Based Complement Alternat Med. 2016;2016:4305074.
Leuschner U, Leuschner M, Sieratzki J, et al. Gallstone dissolution with ursodeoxycholic acid in patients with chronic active hepatitis and two years follow-up. A pilot study. Dig Dis Sci. 1985;30:642–9.
Nakagawa S, Makino I, Ishizaki T, et al. Dissolution of cholesterol gallstones by ursodeoxycholic acid. Lancet. 1977;2:367–9.
Poupon R, Chrétien Y, Poupon RE, et al. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis? Lancet. 1987;1:834–6.
von Haehling S, Schefold JC, Jankowska EA, et al. Ursodeoxycholic acid in patients with chronic heart failure: a double-blind, randomized, placebo-controlled, crossover trial. J Am Coll Cardiol. 2012;59:585–92.
Goossens JF, Bailly C. Ursodeoxycholic acid and cancer: from chemoprevention to chemotherapy. Pharmacol Ther. 2019;203:107396.
Pardue MT, Allen RS. Neuroprotective strategies for retinal disease. Prog Retin Eye Res. 2018;65:50–76.
Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol. 2013;58:155–68.
Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. 2003;83:633–71.
Jansen PL, Ghallab A, Vartak N, et al. The ascending pathophysiology of cholestatic liver disease. Hepatology. 2017;65:722–38.
Woolbright BL, Jaeschke H. Therapeutic targets for cholestatic liver injury. Expert Opin Ther Targets. 2016;20:463–75.
Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50:2340–57.
Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–25.
Choi M, Moschetta A, Bookout AL, et al. Identification of a hormonal basis for gallbladder filling. Nat Med. 2006;12:1253–5.
Sinha J, Chen F, Miloh T, et al. Beta-klotho and FGF-15/19 inhibit the apical sodium-dependent bile acid transporter in enterocytes and cholangiocytes. Am J Physiol Gastrointest Liver Physiol. 2008;295:G996–G1003.
Thomas C, Pellicciari R, Pruzanski M, et al. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–93.
Spivey JR, Bronk SF, Gores GJ. Glycochenodeoxycholate-induced lethal hepatocellular injury in rat hepatocytes. Role of ATP depletion and cytosolic free calcium. J Clin Invest. 1993;92:17–24.
Galle PR, Theilmann L, Raedsch R, et al. Ursodeoxycholate reduces hepatotoxicity of bile salts in primary human hepatocytes. Hepatology. 1990;12:486–91.
Sokol RJ, McKim JM Jr, Goff MC, et al. Vitamin E reduces oxidant injury to mitochondria and the hepatotoxicity of taurochenodeoxycholic acid in the rat. Gastroenterology. 1998;114:164–74.
Attili AF, Angelico M, Cantafora A, et al. Bile acid-induced liver toxicity: relation to the hydrophobic-hydrophilic balance of bile acids. Med Hypotheses. 1986;19:57–69.
Billington D, Evans CE, Godfrey PP, et al. Effects of bile salts on the plasma membranes of isolated rat hepatocytes. Biochem J. 1980;188:321–7.
Sokol RJ, Straka MS, Dahl R, et al. Role of oxidant stress in the permeability transition induced in rat hepatic mitochondria by hydrophobic bile acids. Pediatr Res. 2001;49:519–31.
Ljubuncic P, Fuhrman B, Oiknine J, et al. Effect of deoxycholic acid and ursodeoxycholic acid on lipid peroxidation in cultured macrophages. Gut. 1996;39:475–8.
Rodrigues CM, Fan G, Ma X, et al. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest. 1998;101:2790–9.
Rodrigues CM, Ma X, Linehan-Stieers C, et al. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 1999;6:842–54.
Yerushalmi B, Dahl R, Devereaux MW, et al. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology. 2001;33:616–26.
Patel T, Bronk SF, Gores GJ. Increases of intracellular magnesium promote glycodeoxycholate-induced apoptosis in rat hepatocytes. J Clin Invest. 1994;94:2183–92.
Sokol RJ, Winklhofer-Roob BM, Devereaux MW, et al. Generation of hydroperoxides in isolated rat hepatocytes and hepatic mitochondria exposed to hydrophobic bile acids. Gastroenterology. 1995;109:1249–56.
Faubion WA, Guicciardi ME, Miyoshi H, et al. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest. 1999;103:137–45.
Higuchi H, Bronk SF, Takikawa Y, et al. The bile acid glycochenodeoxycholate induces trail-receptor 2/DR5 expression and apoptosis. J Biol Chem. 2001;276:38610–8.
Mariotti V, Cadamuro M, Spirli C, et al. Animal models of cholestasis: an update on inflammatory cholangiopathies. Biochim Biophys Acta Mol basis Dis. 2019;1865:954–64.
Mariotti V, Strazzabosco M, Fabris L, et al. Animal models of biliary injury and altered bile acid metabolism. Biochim Biophys Acta Mol basis Dis. 2018;1864:1254–61.
Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677–89.
Arrese M, Trauner M. Molecular aspects of bile formation and cholestasis. Trends Mol Med. 2003;9:558–64.
Arrese M, Karpen SJ. Nuclear receptors, inflammation, and liver disease: insights for cholestatic and fatty liver diseases. Clin Pharmacol Ther. 2010;87:473–8.
Wagner M, Zollner G, Trauner M. Nuclear receptor regulation of the adaptive response of bile acid transporters in cholestasis. Semin Liver Dis. 2010;30:160–77.
Jansen PL, Schaap FG, Beuers UH. Fibroblast growth factor 19, an anticholestatic drug produced by human liver. Gastroenterology. 2012;142:e29–30.
Banales JM, Huebert RC, Karlsen T, et al. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol. 2019;16:269–81.
Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol. 2006;12:3496–511.
Hohenester S, Wenniger LM, Paulusma CC, et al. A biliary HCO3- umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology. 2012;55:173–83.
Tabibian JH, Masyuk AI, Masyuk TV, et al. Physiology of cholangiocytes. Comprehensive. Physiology. 2013;3:541–65.
Sato K, Meng F, Giang T, et al. Mechanisms of cholangiocyte responses to injury. Biochim Biophys Acta Mol basis Dis. 2018;1864:1262–9.
Cheung AC, Lorenzo Pisarello MJ, LaRusso NF. Pathobiology of biliary epithelia. Biochim Biophys Acta Mol basis Dis. 2018;1864:1220–31.
Beuers U, Trauner M, Jansen P, et al. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol. 2015;62:S25–37.
Hofmann AF, Zakko SF, Lira M, et al. Novel biotransformation and physiological properties of norursodeoxycholic acid in humans. Hepatology. 2005;42:1391–8.
Yoon YB, Hagey LR, Hofmann AF, et al. Effect of side-chain shortening on the physiologic properties of bile acids: hepatic transport and effect on biliary secretion of 23-nor-ursodeoxycholate in rodents. Gastroenterology. 1986;90:837–52.
Lazarević S, Đanić M, Goločorbin-Kon S, et al. Semisynthetic bile acids: a new therapeutic option for metabolic syndrome. Pharmacol Res. 2019;146:104333.
Trauner M, Halilbasic E, Claudel T, et al. Potential of nor-Ursodeoxycholic acid in Cholestatic and metabolic disorders. Digestive Diseases (Basel, Switzerland). 2015;33:433–9.
Sheps JA, Wang R, Wang J, et al. The protective role of hydrophilic tetrahydroxylated bile acids (THBA). Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866:158925.
Li M, Cai SY, Boyer JL. Mechanisms of bile acid mediated inflammation in the liver. Mol Asp Med. 2017;56:45–53.
Lammers WJ, van Buuren HR, Hirschfield GM, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology. 2014;147:1338–49.e5. quiz e15
Poupon RE, Bonnand AM, Chrétien Y, et al. The UDCA-PBC Study Group. Ten-year survival in ursodeoxycholic acid-treated patients with primary biliary cirrhosis. Hepatology. 1999;29:1668–71.
Poupon RE, Lindor KD, Cauch-Dudek K, et al. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology. 1997;113:884–90.
Sombetzki M, Fuchs CD, Fickert P, et al. 24-nor-ursodeoxycholic acid ameliorates inflammatory response and liver fibrosis in a murine model of hepatic schistosomiasis. J Hepatol. 2015;62:871–8.
Xu X, Wang M, Li JZ, et al. Tauroursodeoxycholic acid alleviates hepatic ischemia reperfusion injury by suppressing the function of Kupffer cells in mice. Biomedicine & Pharmacotherapy = Biomedecine & pharmacotherapie. 2018;106:1271–81.
Wang W, Zhao J, Gui W, et al. Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non-alcoholic fatty liver disease. Br J Pharmacol. 2018;175:469–84.
Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18:151–66.
Cai SY, Boyer JL. The role of bile acids in cholestatic liver injury. Anna Translational Med. 2021;9:737.
Fickert P, Fuchsbichler A, Moustafa T, et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol. 2009;175:2392–405.
Salhab A, Amer J, Lu Y, et al. Sodium(+)/taurocholate cotransporting polypeptide as target therapy for liver fibrosis. Gut. 2021; https://doi.org/10.1136/gutjnl-2020-323345. Online ahead of print.
Buko VU, Lukivskaya OY, Naruta EE, et al. Protective effects of Norursodeoxycholic acid versus Ursodeoxycholic acid on Thioacetamide-induced rat liver fibrosis. J Clin Exp Hepatol. 2014;4:293–301.
Corpechot C, Carrat F, Bonnand AM, et al. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology. 2000;32:1196–9.
Mas N, Tasci I, Comert B, et al. Ursodeoxycholic acid treatment improves hepatocyte ultrastructure in rat liver fibrosis. World J Gastroenterol. 2008;14:1108–11.
Nadinskaia M, Maevskaya M, Ivashkin V, et al. Ursodeoxycholic acid as a means of preventing atherosclerosis, steatosis and liver fibrosis in patients with nonalcoholic fatty liver disease. World J Gastroenterol. 2021;27:959–75.
Pan XL, Zhao L, Li L, et al. Efficacy and safety of tauroursodeoxycholic acid in the treatment of liver cirrhosis: a double-blind randomized controlled trial. Journal of Huazhong University of Science and Technology Medical sciences. 2013;33:189–94.
Ye HL, Zhang JW, Chen XZ, et al. Ursodeoxycholic acid alleviates experimental liver fibrosis involving inhibition of autophagy. Life Sci. 2020;242:117175.
Fickert P, Wagner M, Marschall HU, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465–81.
Wang D, Yang L, Huang J-M, et al. Tauroursodeoxycholic acid inhibits carbon tetrachloride-induced liver fibrosis in rats. World Chinese J Digestol 2010:1979–84.
Paridaens A, Raevens S, Devisscher L, et al. Modulation of the unfolded protein response by tauroursodeoxycholic acid counteracts apoptotic cell death and fibrosis in a mouse model for secondary biliary liver fibrosis. Int J Mol Sci. 2017;18.
de Groot P, Scheithauer T, Bakker GJ, et al. Donor metabolic characteristics drive effects of faecal microbiota transplantation on recipient insulin sensitivity, energy expenditure and intestinal transit time. Gut. 2020;69:502–12.
Pathak P, Liu H, Boehme S, et al. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem. 2017;292:11055–69.
Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–18.
Caron S, Huaman Samanez C, Dehondt H, et al. Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Mol Cell Biol. 2013;33:2202–11.
Duran-Sandoval D, Cariou B, Percevault F, et al. The farnesoid X receptor modulates hepatic carbohydrate metabolism during the fasting-refeeding transition. J Biol Chem. 2005;280:29971–9.
Kong B, Luyendyk JP, Tawfik O, et al. Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther. 2009;328:116–22.
Sinal CJ, Tohkin M, Miyata M, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–44.
Fan N, Meng K, Zhang Y, et al. The effect of ursodeoxycholic acid on the relative expression of the lipid metabolism genes in mouse cholesterol gallstone models. Lipids Health Dis. 2020;19:158.
Hu J, Hong W, Yao KN, et al. Ursodeoxycholic acid ameliorates hepatic lipid metabolism in LO2 cells by regulating the AKT/mTOR/SREBP-1 signaling pathway. World J Gastroenterol. 2019;25:1492–501.
Balan V, Dickson ER, Jorgensen RA, et al. Effect of ursodeoxycholic acid on serum lipids of patients with primary biliary cirrhosis. Mayo Clin Proc. 1994;69:923–9.
Méndez-Sánchez N, González V, Chávez-Tapia N, et al. Weight reduction and ursodeoxycholic acid in subjects with nonalcoholic fatty liver disease. A double-blind, placebo-controlled trial. Ann Hepatol. 2004;3:108–12.
Miettinen TA, Färkkilä M, Vuoristo M, et al. Serum cholestanol, cholesterol precursors, and plant sterols during placebo-controlled treatment of primary biliary cirrhosis with ursodeoxycholic acid or colchicine. Hepatology. 1995;21:1261–8.
Mouillot T, Beylot M, Drai J, et al. Effect of bile acid supplementation on endogenous lipid synthesis in patients with short bowel syndrome: a pilot study. Clinical Nutrition (Edinburgh, Scotland). 2020;39:928–34.
Battezzati PM, Podda M, Bianchi FB, et al. Italian multicenter group for the study of UDCA in PBC. Ursodeoxycholic acid for symptomatic primary biliary cirrhosis. Preliminary analysis of a double-blind multicenter trial. J Hepatol. 1993;17:332–8.
Braga MF, Grace MG, Lenis J, et al. Efficacy and safety of ursodeoxycholic acid in primary, type IIa or IIb hypercholesterolemia: a multicenter, randomized, double-blind clinical trial. Atherosclerosis. 2009;203:479–82.
Fromm H, Roat JW, Gonzalez V, et al. Comparative efficacy and side effects of ursodeoxycholic and chenodeoxycholic acids in dissolving gallstones. A double-blind controlled study. Gastroenterology. 1983;85:1257–64.
Mueller M, Thorell A, Claudel T, et al. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol. 2015;62:1398–404.
Simental-Mendía LE, Simental-Mendía M, Sánchez-García A, et al. Impact of ursodeoxycholic acid on circulating lipid concentrations: a systematic review and meta-analysis of randomized placebo-controlled trials. Lipids Health Dis. 2019;18:88.
Li Y, Tang R, Leung PSC, et al. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun Rev. 2017;16:885–96.
Quigley EM. Primary biliary cirrhosis and the microbiome. Semin Liver Dis. 2016;36:349–53.
Tang R, Wei Y, Li Y, et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut. 2018;67:534–41.
Kummen M, Holm K, Anmarkrud JA, et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut. 2017;66:611–9.
Tabibian JH, O’Hara SP, Trussoni CE, et al. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology. 2016;63:185–96.
Kim DJ, Yoon S, Ji SC, et al. Ursodeoxycholic acid improves liver function via phenylalanine/tyrosine pathway and microbiome remodelling in patients with liver dysfunction. Sci Rep. 2018;8:11874.
Pearson T, Caporaso JG, Yellowhair M, et al. Effects of ursodeoxycholic acid on the gut microbiome and colorectal adenoma development. Cancer Med. 2019;8:617–28.
Winston JA, Rivera A, Cai J, et al. Secondary bile acid ursodeoxycholic acid alters weight, the gut microbiota, and the bile acid pool in conventional mice. PLoS One. 2021;16:e0246161.
Van den Bossche L, Hindryckx P, Devisscher L, et al. Ursodeoxycholic acid and its taurine- or glycine-conjugated species reduce Colitogenic Dysbiosis and equally suppress experimental colitis in mice. Appl Environ Microbiol 2017; 83:e02766–16.
Selmi C, Bowlus CL, Gershwin ME, et al. Primary biliary cirrhosis. Lancet. 2011;377:1600–9.
Terziroli Beretta-Piccoli B, Mieli-Vergani G, Vergani D, et al. The challenges of primary biliary cholangitis: what is new and what needs to be done. J Autoimmun. 2019;105:102328.
Lu M, Li J, Haller IV, et al. Factors associated with prevalence and treatment of primary biliary cholangitis in United States health systems. Clin Gastroenterol Hepatol. 2018;16:e6.
Easl E. EASL clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67:145–72.
Haussinger D, Kurz AK, Wettstein M, et al. Involvement of integrins and Src in tauroursodeoxycholate-induced and swelling-induced choleresis. Gastroenterology. 2003;124:1476–87.
Kurz AK, Graf D, Schmitt M, et al. Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology. 2001;121:407–19.
Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an overview of their mechanisms of action. Clin Res Hepatol Gastroenterol. 2012;36:S3–12.
Angulo P, Batts KP, Therneau TM, et al. Long-term ursodeoxycholic acid delays histological progression in primary biliary cirrhosis. Hepatology. 1999;29:644–7.
Combes B, Carithers RL Jr, Maddrey WC, et al. A randomized, double-blind, placebo-controlled trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology. 1995;22:759–66.
Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet (London, England). 2015;386:1565–75.
Corpechot C, Carrat F, Bahr A, et al. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology. 2005;128:297–303.
John BV, Khakoo NS, Schwartz KB, et al. Ursodeoxycholic acid response is associated with reduced mortality in primary biliary cholangitis with compensated cirrhosis. Am J Gastroenterol. 2021;116:1913–23.
Gordon SC, Wu KH, Lindor K, et al. Ursodeoxycholic acid treatment preferentially improves overall survival among African Americans with primary biliary cholangitis. Am J Gastroenterol. 2020;115:262–70.
Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. 2009;50:808–14.
Corpechot C, Rousseau A, Chazouillères O. Switching vs. add-on strategy in PBC treatment: lessons from UDCA and bezafibrate experience. J Hepatol. 2020;72:1210–1.
Pares A, Caballeria L, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology. 2006;130:715–20.
Carbone M, Mells GF, Pells G, et al. Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology. 2013;144:560–9 e7. quiz e13-4
Guo GY, Shi YQ, Wang L, et al. Serum vitamin D level is associated with disease severity and response to ursodeoxycholic acid in primary biliary cirrhosis. Aliment Pharmacol Ther. 2015;42:221–30.
Montano-Loza AJ, Corpechot C. Definition and Management of Patients with Primary Biliary Cholangitis and an incomplete response to therapy. Clin Gastroenterol Hepatol. 2021;19:2241–51.e1.
Corpechot C, Abenavoli L, Rabahi N, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48:871–7.
Corpechot C, Chazouilleres O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long-term outcome. J Hepatol. 2011;55:1361–7.
Kuiper EM, Hansen BE, de Vries RA, et al. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology. 2009;136:1281–7.
Kumagi T, Guindi M, Fischer SE, et al. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis. Am J Gastroenterol. 2010;105:2186–94.
Momah N, Silveira MG, Jorgensen R, et al. Optimizing biochemical markers as endpoints for clinical trials in primary biliary cirrhosis. Liver Int. 2012;32:790–5.
Azemoto N, Abe M, Murata Y, et al. Early biochemical response to ursodeoxycholic acid predicts symptom development in patients with asymptomatic primary biliary cirrhosis. J Gastroenterol. 2009;44:630–4.
Harms MH, van Buuren HR, Corpechot C, et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J Hepatol. 2019;71:357–65.
Efe C, Tascilar K, Henriksson I, et al. Validation of risk scoring Systems in Ursodeoxycholic Acid-Treated Patients with Primary Biliary Cholangitis. Am J Gastroenterol. 2019;114:1101–8.
Carbone M, Sharp SJ, Flack S, et al. The UK-PBC risk scores: derivation and validation of a scoring system for long-term prediction of end-stage liver disease in primary biliary cholangitis. Hepatology. 2016;63:930–50.
Lammers WJ, Hirschfield GM, Corpechot C, et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving Ursodeoxycholic acid therapy. Gastroenterology. 2015;149:e4.
Lindor KD, Bowlus CL, Boyer J, et al. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69:394–419.
Chávez-Talavera O, Tailleux A, Lefebvre P, et al. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. 2017;152:1679–94.e3.
Jia W, Wei M, Rajani C, et al. Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein Cell. 2021;12:411–25.
Kjærgaard K, Frisch K, Sørensen M, et al. Obeticholic acid improves hepatic bile acid excretion in patients with primary biliary cholangitis. J Hepatol. 2021;74:58–65.
Gomez E, Garcia Buey L, Molina E, et al. Effectiveness and safety of obeticholic acid in a southern European multicentre cohort of patients with primary biliary cholangitis and suboptimal response to ursodeoxycholic acid. Aliment Pharmacol Ther. 2021;53:519–30.
Hirschfield GM, Mason A, Luketic V, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148:751–61.e8.
Kowdley KV, Luketic V, Chapman R, et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology. 2018;67:1890–902.
Nevens F, Andreone P, Mazzella G, et al. A placebo-controlled trial of Obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375:631–43.
Bolier R, de Vries ES, Parés A, et al. Fibrates for the treatment of cholestatic itch (FITCH): study protocol for a randomized controlled trial. Trials. 2017;18:230.
Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol Hepatol. 2020;17:93–110.
Reig A, Sesé P, Parés A. Effects of Bezafibrate on outcome and pruritus in primary biliary cholangitis with suboptimal Ursodeoxycholic acid response. Am J Gastroenterol. 2018;113:49–55.
Tanaka A, Hirohara J, Nakano T, et al. Association of bezafibrate with transplant-free survival in patients with primary biliary cholangitis. J Hepatol. 2021;75:565–71.
Honda A, Tanaka A, Kaneko T, et al. Bezafibrate improves GLOBE and UK-PBC scores and long-term outcomes in patients with primary biliary cholangitis. Hepatology. 2019;70:2035–46.
Sorda JA, González Ballerga E, Barreyro FJ, et al. Bezafibrate therapy in primary biliary cholangitis refractory to ursodeoxycholic acid: a longitudinal study of paired liver biopsies at 5 years of follow up. Aliment Pharmacol Ther. 2021;54:1202–12.
Grigorian AY, Mardini HE, Corpechot C, et al. Fenofibrate is effective adjunctive therapy in the treatment of primary biliary cirrhosis: a meta-analysis. Clin Res Hepatol Gastroenterol. 2015;39:296–306.
Zhang Y, Li S, He L, et al. Combination therapy of fenofibrate and ursodeoxycholic acid in patients with primary biliary cirrhosis who respond incompletely to UDCA monotherapy: a meta-analysis. Drug Des Devel Ther. 2015;9:2757–66.
Wang L, Sun K, Tian A, et al. Fenofibrate improves GLOBE and UK-PBC scores and histological features in primary biliary cholangitis. Minerva Med 2021. https://doi.org/10.23736/S0026-4806.21.07316-X. Online ahead of print.
Cheung AC, Lapointe-Shaw L, Kowgier M, et al. Combined ursodeoxycholic acid (UDCA) and fenofibrate in primary biliary cholangitis patients with incomplete UDCA response may improve outcomes. Aliment Pharmacol Ther. 2016;43:283–93.
Jung D, Fantin AC, Scheurer U, et al. Human ileal bile acid transporter gene ASBT (SLC10A2) is transactivated by the glucocorticoid receptor. Gut. 2004;53:78–84.
Leuschner M, Maier KP, Schlichting J, et al. Oral budesonide and ursodeoxycholic acid for treatment of primary biliary cirrhosis: results of a prospective double-blind trial. Gastroenterology. 1999;117:918–25.
Rautiainen H, Kärkkäinen P, Karvonen AL, et al. Budesonide combined with UDCA to improve liver histology in primary biliary cirrhosis: a three-year randomized trial. Hepatology. 2005;41:747–52.
Angulo P, Jorgensen RA, Keach JC, et al. Oral budesonide in the treatment of patients with primary biliary cirrhosis with a suboptimal response to ursodeoxycholic acid. Hepatology. 2000;31:318–23.
Hirschfield GM, Beuers U, Kupcinskas L, et al. A placebo-controlled randomised trial of budesonide for PBC following an insufficient response to UDCA. J Hepatol. 2021;74:321–9.
Hempfling W, Grunhage F, Dilger K, et al. Pharmacokinetics and pharmacodynamic action of budesonide in early- and late-stage primary biliary cirrhosis. Hepatology. 2003;38:196–202.
Combes B, Emerson SS, Flye NL, et al. Methotrexate (MTX) plus ursodeoxycholic acid (UDCA) in the treatment of primary biliary cirrhosis. Hepatology. 2005;42:1184–93.
Hendrickse MT, Rigney E, Giaffer MH, et al. Low-dose methotrexate is ineffective in primary biliary cirrhosis: long-term results of a placebo-controlled trial. Gastroenterology. 1999;117:400–7.
Gong Y, Gluud C. Colchicine for primary biliary cirrhosis: a Cochrane Hepato-biliary group systematic review of randomized clinical trials. Am J Gastroenterol. 2005;100:1876–85.
Gong Y, Christensen E, Gluud C. Azathioprine for primary biliary cirrhosis. Cochrane Database Syst Rev 2007:Cd006000.
Giljaca V, Poropat G, Stimac D, et al. Methotrexate for primary biliary cirrhosis. Cochrane Database Syst Rev 2010:Cd004385.
Harms MH, van Buuren HR, van der Meer AJ. Improving prognosis in primary biliary cholangitis–therapeutic options and strategy. Best Pract Res Clin Gastroenterol. 2018;34-35:85–94.
Kaplan MM, Cheng S, Price LL, et al. A randomized controlled trial of colchicine plus ursodiol versus methotrexate plus ursodiol in primary biliary cirrhosis: ten-year results. Hepatology. 2004;39:915–23.
Saffioti F, Gurusamy KS, Eusebi LH, et al. Pharmacological interventions for primary biliary cholangitis: an attempted network meta-analysis. Cochrane Database Syst Rev 2017; 3:Cd011648.
Karlsen TH, Folseraas T, Thorburn D, et al. Primary sclerosing cholangitis - a comprehensive review. J Hepatol. 2017;67:1298–323.
Rawla P, Sunkara T, Raj JP. Role of biologics and biosimilars in inflammatory bowel disease: current trends and future perspectives. J Inflamm Res. 2018;11:215–26.
Boonstra K, Weersma RK, van Erpecum KJ, et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology. 2013;58:2045–55.
Beuers U, Spengler U, Kruis W, et al. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo-controlled trial. Hepatology. 1992;16:707–14.
Olsson R, Boberg KM, de Muckadell OS, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology. 2005;129:1464–72.
Shi J, Li Z, Zeng X, et al. Ursodeoxycholic acid in primary sclerosing cholangitis: meta-analysis of randomized controlled trials. Hepatol Res. 2009;39:865–73.
Poropat G, Giljaca V, Stimac D, et al. Bile acids for primary sclerosing cholangitis. Cochrane Database Syst Rev. 2011;2011:Cd003626.
Triantos CK, Koukias NM, Nikolopoulou VN, et al. Meta-analysis: ursodeoxycholic acid for primary sclerosing cholangitis. Aliment Pharmacol Ther. 2011;34:901–10.
Chapman MH, Thorburn D, Hirschfield GM, et al. British Society of Gastroenterology and UK-PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut. 2019;68:1356–78.
Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51:660–78.
EASL. EASL clinical practice guidelines: management of cholestatic liver diseases. J Hepatol, 2009:237–67.
Wunsch E, Trottier J, Milkiewicz M, et al. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology. 2014;60:931–40.
Lazaridis KN, LaRusso NF. Primary Sclerosing cholangitis. N Engl J Med. 2016;375:1161–70.
Lindor KD, Kowdley KV, Harrison ME. ACG clinical guideline: primary Sclerosing cholangitis. Am J Gastroenterol. 2015;110:646–59. quiz 60
Rudolph G, Kloeters-Plachky P, Sauer P, et al. Intestinal absorption and biliary secretion of ursodeoxycholic acid and its taurine conjugate. Eur J Clin Investig. 2002;32:575–80.
Heuman DM, Bajaj R. Ursodeoxycholate conjugates protect against disruption of cholesterol-rich membranes by bile salts. Gastroenterology. 1994;106:1333–41.
Heuman DM, Mills AS, McCall J, et al. Conjugates of ursodeoxycholate protect against cholestasis and hepatocellular necrosis caused by more hydrophobic bile salts. In vivo studies in the rat. Gastroenterology. 1991;100:203–11.
Tsukahara K, Kanai S, Ohta M, et al. Taurine conjugate of ursodeoxycholate plays a major role in the hepatoprotective effect against cholestasis induced by taurochenodeoxycholate in rats. Liver. 1993;13:262–9.
Reggiani A, Dizioli P, Quattrocchi D, et al. Intrahepatic cholestasis: clinical and nosological classification. Critical survey of personal experience. Minerva Gastroenterol Dietol. 1997;43:71–81.
Fickert P, Pollheimer MJ, Silbert D, et al. Differential effects of norUDCA and UDCA in obstructive cholestasis in mice. J Hepatol. 2013;58:1201–8.
Fickert P, Hirschfield GM, Denk G, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol. 2017;67:549–58.
Sanyal AJ. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2019;16:377–86.
Sheka AC, Adeyi O, Thompson J, et al. Nonalcoholic Steatohepatitis: A Review. JAMA. 2020;323:1175–83.
Arab JP, Karpen SJ, Dawson PA, et al. Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology. 2017;65:350–62.
Lefebvre P, Cariou B, Lien F, et al. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–91.
Li Y, Jadhav K, Zhang Y. Bile acid receptors in non-alcoholic fatty liver disease. Biochem Pharmacol. 2013;86:1517–24.
Segovia-Miranda F, Morales-Navarrete H, Kücken M, et al. Three-dimensional spatially resolved geometrical and functional models of human liver tissue reveal new aspects of NAFLD progression. Nat Med. 2019;25:1885–93.
Dufour JF, Oneta CM, Gonvers JJ, et al. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin e in nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2006;4:1537–43.
Ratziu V, de Ledinghen V, Oberti F, et al. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011;54:1011–9.
Lindor KD, Kowdley KV, Heathcote EJ, et al. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology. 2004;39:770–8.
Leuschner UF, Lindenthal B, Herrmann G, et al. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52:472–9.
Mueller M, Castro RE, Thorell A, et al. Ursodeoxycholic acid: effects on hepatic unfolded protein response, apoptosis and oxidative stress in morbidly obese patients. Liver Int. 2018;38:523–31.
Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67:328–57.
Gheibi S, Gouvarchin Ghaleh HE, Motlagh BM, et al. Therapeutic effects of curcumin and ursodexycholic acid on non-alcoholic fatty liver disease. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie. 2019;115:108938.
Pietu F, Guillaud O, Walter T, et al. Ursodeoxycholic acid with vitamin E in patients with nonalcoholic steatohepatitis: long-term results. Clin Res Hepatol Gastroenterol. 2012;36:146–55.
Beraza N, Ofner-Ziegenfuss L, Ehedego H, et al. Nor-ursodeoxycholic acid reverses hepatocyte-specific nemo-dependent steatohepatitis. Gut. 2011;60:387–96.
Traussnigg S, Schattenberg JM, Demir M, et al. Norursodeoxycholic acid versus placebo in the treatment of non-alcoholic fatty liver disease: a double-blind, randomised, placebo-controlled, phase 2 dose-finding trial. Lancet Gastroenterol Hepatol. 2019;4:781–93.
Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574–82.e1.
Yuan L, Bambha K. Bile acid receptors and nonalcoholic fatty liver disease. World J Hepatol. 2015;7:2811–8.
Jahn D, Rau M, Hermanns HM, et al. Mechanisms of enterohepatic fibroblast growth factor 15/19 signaling in health and disease. Cytokine Growth Factor Rev. 2015;26:625–35.
Verbeke L, Mannaerts I, Schierwagen R, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep. 2016;6:33453.
Younossi ZM, Ratziu V, Loomba R, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (London, England). 2019;394:2184–96.
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet (London, England). 2015;385:956–65.
Velayudham LS, Farrell GC. Drug-induced cholestasis. Expert Opin Drug Saf. 2003;2:287–304.
Piotrowicz A, Polkey M, Wilkinson M. Ursodeoxycholic acid for the treatment of flucloxacillin-associated cholestasis. J Hepatol. 1995;22:119–20.
Agca E, Akcay A, Simsek H. Ursodeoxycholic acid for terbinafine-induced toxic hepatitis. Ann Pharmacother. 2004;38:1088–9.
Katsinelos P, Vasiliadis T, Xiarchos P, et al. Ursodeoxycholic acid (UDCA) for the treatment of amoxycillin-clavulanate potassium (Augmentin)-induced intra-hepatic cholestasis: report of two cases. Eur J Gastroenterol Hepatol. 2000;12:365–8.
Studniarz M, Czubkowski P, Cielecka-Kuszyk J, et al. Amoxicillin/clavulanic acid-induced cholestatic liver injury after pediatric liver transplantation. Ann Transplant. 2012;17:128–31.
Asgarshirazi M, Shariat M, Dalili H, et al. Ursodeoxycholic acid can improve liver transaminase quantities in children with anticonvulsant drugs hepatotoxicity: a pilot study. Acta Med Iran. 2015;53:351–5.
Lang SM, Ortmann J, Rostig S, et al. Ursodeoxycholic acid attenuates hepatotoxicity of multidrug treatment of mycobacterial infections: a prospective pilot study. International journal of mycobacteriology. 2019;8:89–92.
Wree A, Dechêne A, Herzer K, et al. Steroid and ursodesoxycholic acid combination therapy in severe drug-induced liver injury. Digestion. 2011;84:54–9.
Yu YC, Mao YM, Chen CW, et al. CSH guidelines for the diagnosis and treatment of drug-induced liver injury. Hepatol Int. 2017;11:221–41.
Wood AM, Livingston EG, Hughes BL, et al. Intrahepatic cholestasis of pregnancy: a review of diagnosis and management. Obstet Gynecol Surv. 2018;73:103–9.
Piechota J, Jelski W. Intrahepatic cholestasis in pregnancy: review of the literature. J Clin Med 2020; 9.
Grand’Maison S, Durand M, Mahone M. The effects of ursodeoxycholic acid treatment for intrahepatic cholestasis of pregnancy on maternal and fetal outcomes: a meta-analysis including non-randomized studies. J Obstet Gynaecol Can. 2014;36:632–41.
Joutsiniemi T, Timonen S, Leino R, et al. Ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy: a randomized controlled trial. Arch Gynecol Obstet. 2014;289:541–7.
Bacq Y, Sentilhes L, Reyes HB, et al. Efficacy of ursodeoxycholic acid in treating intrahepatic cholestasis of pregnancy: a meta-analysis. Gastroenterology. 2012;143:1492–501.
Chappell LC, Bell JL, Smith A, et al. Ursodeoxycholic acid versus placebo in women with intrahepatic cholestasis of pregnancy (PITCHES): a randomised controlled trial. Lancet (London, England). 2019;394:849–60.
Kumar P, Kulkarni A. UDCA therapy in intrahepatic cholestasis of pregnancy? J Hepatol. 2020;72:586–7.
Calmus Y, Gane P, Rouger P, et al. Hepatic expression of class I and class II major histocompatibility complex molecules in primary biliary cirrhosis: effect of ursodeoxycholic acid. Hepatology. 1990;11:12–5.
Chen CY, Tsao PN, Chen HL, et al. Ursodeoxycholic acid (UDCA) therapy in very-low-birth-weight infants with parenteral nutrition-associated cholestasis. J Pediatr. 2004;145:317–21.
De Marco G, Sordino D, Bruzzese E, et al. Early treatment with ursodeoxycholic acid for cholestasis in children on parenteral nutrition because of primary intestinal failure. Aliment Pharmacol Ther. 2006;24:387–94.
Levine A, Maayan A, Shamir R, et al. Parenteral nutrition-associated cholestasis in preterm neonates: evaluation of ursodeoxycholic acid treatment. Journal of Pediatric Endocrinology & Metabolism: JPEM. 1999;12:549–53.
Spagnuolo MI, Iorio R, Vegnente A, et al. Ursodeoxycholic acid for treatment of cholestasis in children on long-term total parenteral nutrition: a pilot study. Gastroenterology. 1996;111:716–9.
Thibault M, McMahon J, Faubert G, et al. Parenteral nutrition-associated liver disease: a retrospective study of ursodeoxycholic acid use in neonates. The Journal of Pediatric Pharmacology and Therapeutics. 2014;19:42–8.
Wales PW, Allen N, Worthington P, et al. A.S.P.E.N. clinical guidelines: support of pediatric patients with intestinal failure at risk of parenteral nutrition-associated liver disease. JPEN J Parenter Enteral Nutr. 2014;38:538–57.
Beau P, Labat-Labourdette J, Ingrand P, et al. Is ursodeoxycholic acid an effective therapy for total parenteral nutrition-related liver disease? J Hepatol. 1994;20:240–4.
Fried RH, Murakami CS, Fisher LD, et al. Ursodeoxycholic acid treatment of refractory chronic graft-versus-host disease of the liver. Ann Intern Med. 1992;116:624–9.
Arat M, Idilman R, Soydan EA, et al. Ursodeoxycholic acid treatment in isolated chronic graft-vs.-host disease of the liver. Clin Transpl. 2005;19:798–803.
Ruutu T, Juvonen E, Remberger M, et al. Improved survival with ursodeoxycholic acid prophylaxis in allogeneic stem cell transplantation: long-term follow-up of a randomized study. Biol Blood Marrow Transplant. 2014;20:135–8.
Sakiani S, Kleiner DE, Heller T, et al. Hepatic manifestations of cystic fibrosis. Clin Liver Dis. 2019;23:263–77.
Al Sinani S, Al-Mulaabed S, Al Naamani K, et al. Cystic fibrosis liver disease: know more. Oman Med J. 2019;34:482–9.
Rafeeq MM, Murad HAS. Cystic fibrosis: current therapeutic targets and future approaches. J Transl Med. 2017;15:84.
Staufer K. Current treatment options for cystic fibrosis-related liver disease. Int J Mol Sci 2020;21:8586.
Cheng K, Ashby D, Smyth RL. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database Syst Rev. 2017;9:Cd000222.
Toledano MB, Mukherjee SK, Howell J, et al. The emerging burden of liver disease in cystic fibrosis patients: a UK nationwide study. PLoS One. 2019;14:e0212779.
van der Feen C, van der Doef HP, van der Ent CK, et al. Ursodeoxycholic acid treatment is associated with improvement of liver stiffness in cystic fibrosis patients. J Cyst Fibros. 2016;15:834–8.
Colombo C, Alicandro G, Oliver M, et al. Ursodeoxycholic acid and liver disease associated with cystic fibrosis: a multicenter cohort study. J Cyst Fibros. 2022;21:220–6.
Davit-Spraul A, Fabre M, Branchereau S, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010;51:1645–55.
Jacquemin E, De Vree JM, Cresteil D, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120:1448–58.
van Mil SW, Houwen RH, Klomp LW. Genetics of familial intrahepatic cholestasis syndromes. J Med Genet. 2005;42:449–63.
Wanty C, Joomye R, Van Hoorebeek N, et al. Fifteen years single center experience in the management of progressive familial intrahepatic cholestasis of infancy. Acta Gastro-Enterol Belg. 2004;67:313–9.
Jacquemin E, Hermans D, Myara A, et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology. 1997;25:519–23.
Agarwal S, Lal BB, Rawat D, et al. Progressive familial intrahepatic cholestasis (PFIC) in Indian children: clinical Spectrum and outcome. J Clin Exp Hepatol. 2016;6:203–8.
Gordo-Gilart R, Andueza S, Hierro L, et al. Functional analysis of ABCB4 mutations relates clinical outcomes of progressive familial intrahepatic cholestasis type 3 to the degree of MDR3 floppase activity. Gut. 2015;64:147–55.
Khabou B, Mahjoub B, Barbu V, et al. Phenotypic variability in Tunisian PFIC3 patients harboring a complex genotype with a differential clinical outcome of UDCA treatment. Clinica Chimica acta. 2018;486:122–8.
Gonzales E, Grosse B, Schuller B, et al. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology. 2015;62:558–66.
Kagawa T, Orii R, Hirose S, et al. Ursodeoxycholic acid stabilizes the bile salt export pump in the apical membrane in MDCK II cells. J Gastroenterol. 2014;49:890–9.
van der Woerd WL, Houwen RH, van de Graaf SF. Current and future therapies for inherited cholestatic liver diseases. World J Gastroenterol. 2017;23:763–75.
Kriegermeier A, Green R. Pediatric Cholestatic liver disease: review of bile acid metabolism and discussion of current and emerging therapies. Front Med. 2020;7:149.
Verkade HJ, Bezerra JA, Davenport M, et al. Biliary atresia and other cholestatic childhood diseases: advances and future challenges. J Hepatol. 2016;65:631–42.
Burns J, Davenport M. Adjuvant treatments for biliary atresia. Translational Pediatrics. 2020;9:253–65.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Gao, W. et al. (2022). Ursodeoxycholic Acid in Liver Cirrhosis: A Chinese Perspective. In: Qi, X., Yang, Y. (eds) Pharmacotherapy for Liver Cirrhosis and Its Complications. Springer, Singapore. https://doi.org/10.1007/978-981-19-2615-0_6
Download citation
DOI: https://doi.org/10.1007/978-981-19-2615-0_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-2614-3
Online ISBN: 978-981-19-2615-0
eBook Packages: MedicineMedicine (R0)