
Abstract
Recently, several genes that predispose to type 2 diabetes have been discovered. There is ample evidence to indicate a genetic predisposition to the microvascular complication of nephropathy in people with both type 1 and type 2 diabetes, in addition to the well-known and potent effects of environmental variables. In populations all over the world, familial aggregation of phenotypes such as end-stage renal disease, albuminuria, and chronic kidney disease has frequently been recorded. Heritability estimations for albuminuria and glomerular filtration rate also show considerable influences from inherited variables. Recent genome-wide linkage analyses have examined positional candidate genes under numerous chromosomal areas that are more likely to contain genes that increase the risk of developing diabetic nephropathy. The hereditary elements of diabetic kidney disease are reviewed in this book chapter, with a focus on recently identified genes and pathways. It appears likely that inheriting risk alleles at numerous susceptibility loci, in the presence of hyperglycemia, increases the risk for diabetes-associated kidney damage. In contrast to the molecular genetic studies, which have already been fully reviewed elsewhere, this book chapter focuses on the gathered data on hereditary factors from family studies in order to assess the role of genetic vulnerability in diabetic nephropathy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Diabetes mellitus (DM) is recognised to have a wide range of consequences, including microvascular and macrovascular issues as well as other metabolic issues. These have been chosen as a group to increase their global influence. The frequency of diabetes is rapidly increasing, having significant negative socioeconomic impacts in economically developed and underdeveloped countries (Tandon et al., 2018). Additionally, the rise in DM-related issues, specifically diabetic diabetes-related retinopathy, diabetic neuropathy, diabetic nephropathy, and other related cardiovascular conditions are placing enormous demands on healthcare. According to the International Diabetes Federation (IDF), around 463 million persons worldwide had diabetes as of 2019, and by 2030, the population could reach 578 million worldwide (Saeedi et al., 2019). According to estimates, 30 to 40 percent of people with diabetes mellitus go on to develop the disease, later developing nephropathy into end-stage renal disease (ESRD) (Wei et al., 2018). There are fundamental mechanisms underlying type 1 (T1DM) and type 2 diabetic kidney disease. Despite the overall clinical appearance of both diseases is remarkably similar, and T2DM histology findings. The primary cause of the increase in hyperglycemic levels is acknowledged to be the development of renal lesions that precisely detect DN progression.
Diabetic nephropathy is rarely seen in type 2 diabetes in the first 10 years after diagnosis; however, roughly about 3 percent of patients with type 2 diabetes, recently diagnosed with diabetes, will be already have overt nephropathy. The variance in this part reveals the role of comorbidities and the link between age-related effects of hypertension as well as coexisting atherosclerosis. Although the major reason for DN is hemodynamic and metabolic factors, it has been indicated that DN is an inflammatory process including immune cells (Duran-Salgado & Rubio-Guerra, 2014). The key pathology variations of diabetic nephropathy are factors such as hypertension, proteinuria, and aggregation of advanced glycation end products (AGEs) as well growth factors along with a perpetual worsening in the function of the kidney. The etiology of DN is multifactorial, accompanied by genetic and ecological factors (Magee et al., 2017). Albumin/creatinine ratios of more than 300 mg/g creatinine or abnormally high levels of albuminuria, which are found in at least two out of three samples and show an obvious result, as well as the occurrence of diabetic retinopathy condition and the non-appearance of other renal diseases, are characteristics of diabetic nephropathy (Forbes & Cooper, 2013). According to studies by Parving et al. (2011), Alter et al. (2012), and Bonventre (2012), it is also a clinical disease characterised by extreme extracellular matrix protein deposition, reduced glomerular filtration rate (GFR), lower creatinine clearance, reduced excretion of albumin, tubulointerstitial fibrosis, increased thickness of glomerular basement membrane, and glomerular hypertrophy. Through repeated measurement and rising values of albuminuria within the usual and unusual ranges are linked with an increased threat in the progression of cardiovascular and renal disease, the normal level of albuminuria is defined as being 30 mg per gram or 30 mg in 24 h, whereas the unusual range is beyond 30 mg per gram.
The most frequent cause of ESRD progression is diabetic nephropathy. Although T2DM is a preventable and treatable foundation for DN to ESRD, it is known that T2DM-related ESRD cases account for more than 50% of all cases globally (American Diabetes Association, 2017). High morbidity and mortality in cardiovascular disease are also associated with DN, most often in the initial stages of the disease. Although the pathophysiology of DN is very well recognised, several causes of the disease still need to be found. A number of previously published studies have shown that some non-modifiable risk variables, such as family history, higher GFR following the diagnosis of type 2 diabetes mellitus, and ethnicity, are associated with DN. Obesity, hypertension, smoking, excessive blood sugar, and dyslipidemia are the modifiable causal factors for the development of DN (Shaza et al., 2005) (Fig. 1).

Pathophysiology of diabetic nephropathy
Family clustering and the fact that not all diabetes patients proceed to obvious DN despite long-standing poor glycaemic management reveal the genetic component of DN vulnerability. The family clustering studies provide more evidence for the major role that inherited variables play in ESRD and diabetic nephropathy. A strategy to distinguish between the mechanisms of DN initiation and advancement is the investigation of predisposed genetic loci or genes. Due to this, multiple determinations have been undertaken to recognise the genetic relationships between DN and various traits using an assortment of techniques, together with genome-wide association studies (GWAS), and candidate gene linkage analysis (Liu et al., 2015).
2 Epidemiology
Diabetes renal disease is well documented to have a substantial part in the progression of ESRD worldwide, particularly in nations like the USA, Japan, and Europe. Numerous population-based studies have found that Blacks and Asians with diabetes are more likely than White people to have the end-stage renal disease (ESRD) (Karter et al., 2002). When compared to White people, Hispanic people with and without diabetes have a higher incidence of renal illness (Young et al., 2003). Despite having low body mass indices, diabetic patients in India frequently have high levels of inflammatory markers, low levels of adiponectin, and insulin resistance (Pugh et al., 1988). While overt diabetes is very common among older Indian patients in other places as well, intolerance is well-established within the Indian people. However, the dangerously high prevalence of prediabetes and obesity in young individuals raises serious concerns in the current environment (Mohan et al., 2003, 2007; Ravikumar et al., 2011).
Along with the rise in diabetes, there was also a marked rise in the frequency of DN, which has appeared as the leading cause of ESRD. Currently, chronic renal disease in elderly persons accounts for no less than 46% of cases of diabetic nephropathy. Overt nephropathy and microalbuminuria were found to be prevalent in urban residents with diabetes, with respective incidences of 2.1% and 26.9%, according to the Urban-Rural Epidemiology Study (Goyal et al., 2010). It is quite concerning that Southeast Asians have a considerably higher risk of developing impaired glucose tolerance and low fasting glucose levels than the local inhabitants of European ancestry (Unnikrishnan et al., 2007). Additionally, it was noted in the UK and Canada that people of Asian descent had a higher prevalence of chronic kidney-related issues, notably DN, which was reportedly caused by differing lifestyles and genetics (Gray et al., 2010).
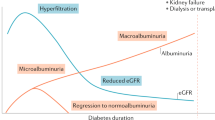
When present in a diabetic patient, proteinuria of more than 300 mg/day was a clear indicator of diabetic nephropathy (Barbour et al., 2010). Studies have shown that DN patients exhibit a variety of clinical manifestations as ESRD progresses, despite the fact that the hallmark of DN is a steady progression of micro- to macroalbuminuria, and hyperfiltration in the initial stage leads to the impairment of kidney function. There are several phases involved in the progression of DN in diabetic patients, which leads to end-stage renal disease.
3 Stage I: Early Hypertrophy-Hyperfiltration
Past investigations have mostly revealed that several renal alterations are involved from the clinical onset of diabetes mellitus. As well as the early stage of hypertrophy and hyperfiltration, these modifications mostly depend on the function, biochemistry, and structure providing the definition of a new entity (American Diabetes Association, 2004). The majority of individuals with newly diagnosed diabetes experience this stage, which is characterised by an upsurge in glomerular filtration (GF) rate and a rise in capillary permeability that causes microalbuminuria (MAU) and, ultimately, renal hypertrophy. Glomerular filtration and microalbuminuria may worsen as a result of insulin therapy (Satchell & Tooke, 2008).
4 Stage II: Latent Diabetic Nephropathy
This is a stage that advances slowly and manifests structural lesions in biopsy after many years without any obvious clinical indications of renal disease. While an obvious glomerular impairment will be present, the glomerular filtration rate either stays high or rebounds to normal. Some diabetics will remain in stage II for the rest of their lives. In this stage as well, improved glucose management can result in a reduction in glomerular filtration rate (MacIsaac et al., 2017).
5 Stage III: Incipient Diabetic Nephropathy
The glomerular basement membrane (GBM) dilatation, increased albuminuria of between 30–300 mg/day (20–200 g/min), and a practically unchanged glomerular filtration rate are the hallmarks of this stage. The progression of albuminuria in the setting of declining renal function is significantly influenced by blood pressure. Another symptom is microalbuminuria; thus screening for it once a year in all diabetic patients can help identify the potential renal disease and cardiovascular risk (Satchell & Tooke, 2008; Clarke et al., 2003).
6 Stage IV: Overt Nephropathy
Irreversible proteinuria, ongoing hypertension, and GFR under 60 ml/min/1.73 m2 are the hallmarks of this stage. It has been determined that between 20 and 40 percent of T2DM patients who have microalbuminuria are more likely to develop overt nephropathy (Clarke et al., 2003). When albumin levels in the urine exceed 300 mg per day or 200 g/min after 10 to 15 years, hypertension is also known to worsen in almost all patients. On the other hand, when glomerular damage worsens, the kidneys’ ability to filter out albumin is increasingly diminished, causing a rise in urine albumin excretion of roughly 10–20 percent annually (Molitch et al., 2004). This stage typically leads to renal insufficiency and nephrotic syndrome with concurrent advancement of additional diabetic-related complications like diabetic nephropathy, diabetic neuropathy, retinopathy, or diabetic foot as well as increased hypertension if blood sugar and blood pressure are not strictly controlled (Drummond & Mauer, 2002).
7 Stage V: End-Stage Renal Disease
The glomerular filtration rate (2–20 ml/min/year), which is known to be extremely varied in individuals, steadily decreases with the occurrence of overt nephropathy, necessitating kidney replacement therapy (peritoneal dialysis, hemodialysis, and kidney transplantation). The variability of diseases, such as glomerular filtration barrier malfunction and loss in kidney function among individuals at this stage, points to a variety of disease pathways associated with a variety of hereditary determinants (Kramer et al., 2003; Ellis et al., 2012). The research of relative risk alleles and low-frequency alleles of the diabetic nephropathy phenotype provided experimental study support for this interpretation (Placha et al., 2005).
Variations in structural and functional characteristics identify diabetic nephropathy. Glomerular hyperfiltration, glomerular basement membrane dilation, mesangial cell growth, and nodular glomerulosclerosis are typical symptoms in the early stages of DN. Diabetes induces significant histopathological changes in the kidney structure, according to Kimmelstiel-Wilson (Chan et al., 2014). It is well established that DN’s pathophysiological mechanisms are complex. In the early stages of DN, tubular hypertrophy is seen to exist, and this gradually gives rise to interstitial fibrosis, tubular atrophy, and arteriolar hyalinosis (Kimmelstiel & Wilson, 1936). Hyperglycemia is the initiating cause that leads to structural and functional alterations, which progress to mesangial matrix buildup, overt proteinuria, glomerular hyperfiltration, tubular epithelial hypertrophy, and ultimately end-stage renal disease (Weil et al., 2012).
The development and progression of DN, which affect a variety of cell types including podocytes, inflammatory cells, endothelial, mesangial kidney cells, as well as renal tubular and accumulation duct systems, are known to be significantly influenced by high glucose levels. These procedures involve a number of different paths. Transforming growth factor (TGF) cellular expression and strong reactive oxygen species (ROS) generation in the mitochondria are important effects of hyperglycemia (Vallon & Komers, 2011). In addition to increasing the expression of growth factors, cytokines, extracellular matrix proteins (ECM), and leucocyte adhesion molecules in the kidneys, reactive oxygen species also stimulate the signalling and transcription factors (Fig. 2).

Schematic representation of the consequences of diabetes mellitus
TGF stimulates the production of the ECM, which results in cellular hypertrophy and better collagen synthesis (Weil et al., 2012). The diffuse mesangial enlargement is a recognisable sign of diabetic nephropathy (Forbes & Cooper, 2013). Wilson nodules, or areas of strong mesangial deposition, are visible in 40 to 50 percent of patients who later develop proteinuria (Chan et al., 2014). The mesangial matrix expansion also has a relationship to proteinuria and declining kidney function. The accumulation of matrix in the mesangial area, which lowers the capillary surface area accessible for the filtration process, causes advanced loss of renal function (Bilous, 1997). The glomerular basement membrane dilatation caused by a change in the structure and composition of the membrane is another distinct characteristic of diabetic glomerulopathy; however, it is not strictly related because it occurs regardless of nephropathy (Fioretto & Mauer, 2007). Although it is more permeable to proteins, the thick GBM contributes as an efficient barrier in the filtering of proteins (Arif & Nihalani, 2013). The lack of charge selectivity by the GBM, which permits the passage of positively charged proteins like albumin, is shown by a decrease in negatively charged proteoglycans. The excretion of proteinuria helps to explain this effect in part. The glomerular visceral epithelial cell is the primary barrier separating plasma proteins from the vasculature (slit diaphragm of podocytes). The podocytes’ variants are adaptable. When microalbuminuria first develops, there is a noticeable drop in the number of podocytes as well as a broadening of the pedicels, but these changes are unrelated to the degree of proteinuria. The majority of the podocytes will be lost as DN progresses, and the remaining podocytes should compensate for this loss (Lin & Susztak, 2016).
The fall in proteinuria and GFR is a result of all the aforementioned alterations in the glomerular filtration barrier, which is made up of glomerular endothelium, podocytes, and GBM. The tubules that have interstitial fibrosis and nephron atrophy are observed to be negatively impacted by proteinuria (Williams, 2005). As a result, tubular transport will be impaired by ongoing hyperglycemia, resulting in hemodynamic imbalance and a decline in renal function. Hyaline arteriosclerosis, which also causes glomerular dysfunction, is another early sign of DN. This syndrome is made worse by hyperlipidemia and hypertension, which constrict the arteriole lumen and increase renal vascular hypertension (Nishi et al., 2000).
In the initial phases of diabetic nephropathy, the glomerulus experiences hemodynamic alterations, including hyperfiltration and hyperperfusion damage. This stage is thus multifaceted. A rise in glomerular capillary pressure, which in turn enhances the transcapillary hydraulic pressure gradient and intensifies the glomerular plasma flow, results from a reduction in the arteriolar resistance (Hostetter, 2003). There is a vast range of probable processes and intermediates. There are sufficiently clear indications that diabetic hyperfiltration is caused by an imbalance among the vasoactive factors that regulate the afferent and efferent arteriolar tonus, in addition to the fact that metabolic variables are linked to hyperglycemia (Vallon & Komers, 2011). Hyperperfusion and hyperfiltration are produced as a result of variables including growth hormone, prostanoids, insulin, atrial natriuretic factor, nitric oxide, glucagon, and angiotensin-II (ANG-II) (Chawla et al., 2010). On the other hand, glomerular basement membrane thickening increased mesangial cell matrix synthesis, and elevated intraglomerular pressure are recognised to be the causes of glomerulosclerosis. Hyperglycemia causes effects on the renal cells that are hemodynamic, inflammatory, and profibrogenic, which enhance the production of angiotensin-II. Cytokines like TGF and vascular endothelial growth factors (VEGF) are the main mediators of hyperfiltration (Wolf et al., 2003).
It is well-recognised that beta plays a significant role in diabetic vascular instability. Thus, this process increases arginine re-synthesise and endothelial NOS (nitric oxide synthase) mRNA expression, both of which lead to an increase in nitric oxide production (Sharma et al., 2003). The enhanced glomerular filtration will be a result of the elevated nitric oxide levels in the kidneys. According to recent research, endothelial nitric oxide (NOS3, eNOS) is a causal enzyme that contributes to the excess nitric oxide generation in diabetic kidneys. Additionally, this leads to an increase in the amount of ROS in the diabetic kidneys (Veelken et al., 2000; Sugimoto et al., 1998). Other research has also indicated that neuronal NOS plays a significant role in hyperfiltration, modulating the negative tubuloglomerular feedback loop (Ito et al., 2001).
Additional variables that are connected to the pathophysiology of diabetic nephropathy are also regulated by nitric oxide. Growth factors and cytokines are released via the autocrine and paracrine pathways when glomerular hemodynamics are altered as a result of mechanical strain and shear stress (Chawla et al., 2010). Further structural changes to DN are caused by this hemodynamic stress and are initiated locally by growth factors and cytokines. Because of the macula densa mechanism and tubular hypertrophy, the increase in sodium chloride reabsorption in the loops of Henle increases the frequency of the glomerular filtration rate. As a result, it facilitates sodium chloride’s accelerated reabsorption, which may be crucial in this phase and once again links structural alterations to hemodynamic variation in diabetic nephropathy (Thomson et al., 2004).
Hyperglycemia, elevated glucagon levels, and growth factors that characterise diabetes mellitus are considered metabolic variables. It is well-established that changes in insulin levels have a clear effect on microvascular problems (Vithian & Hurel, 2010). Even after many histological defects in their kidneys, the majority of diabetes people will not advance to renal failure. Thus, it is clear from this that hyperglycemia is a necessary but insufficient component to trigger renal damage that ultimately results in kidney failure. The glucose transport activity is a known important regulator in the extracellular growth of mesangial cells. The key regulator that facilitates the entry of glucose into kidney cells is the glucose transporter 1 (GLUT1). Additional metabolic pathways are stimulated by glucose, which results in the growth and formation of mesangial cell matrix (Mishra et al., 2005). Even though the glucose levels are normal, changes in the renal cells will result from GLUT1 overexpression (Heilig et al., 1995). The mesangial cells’ presence of GLUT1, a brain-type glucose transporter, and GLUT4, an insulin-sensitive extracellular glucose transporter, makes it simple for a high level of glucose to enter the cell in an insulin-independent fashion (Haneda et al., 2003; Heilig et al., 1995).
Numerous experimental investigations show that diabetes significantly increases the rate of matrix and glomerular basement membrane components, which improves collagen synthesis. As a result of higher glycaemic levels, GLUT1 is typically overexpressed; however, the pathophysiological role of glucose transporter 1 in DN is not yet known (Heilig et al., 1995). It is also known that glucose interacts with structural proteins, causing a process known as non-enzymatic glycosylation that leads to the formation of advanced glycosylation end products (Nishikawa et al., 2000). The end products of this reaction distress the glomerular basement membrane components, leading to the formation of cross-links that are prominent to diminished sensitivity, which results in the buildup and deposit of proteases (Brownlee et al., 1986). AGEs alter the macrophage removal system by impeding the mechanisms of mesangial clearance, which causes an expansion of the mesangial cell and eventually results in glomerular blockage (Forbes et al., 2001). The final result of a series of as-yet-unknown metabolic processes is advanced glycation end products (AGEs). Although there is a clear difference between regulated glycaemic levels as expressed by HbA1c (glycated haemoglobin), AGE formation is high in diabetes induced by persistent hyperglycemia (Cohen et al., 2003). There is also known to be a strong association between the decrease in kidney function and the accumulation of AGE in kidney disorders because filtration of the kidney plays a critical role in the clearance of circulating AGEs (Nitta et al., 2013).
Due to hemodynamic changes brought on by non-enzymatic glycosylation, protein kinase C (PKC) is also activated, accelerating the polyol pathway and stimulating the production of TGF, VEGF, interleukin-6, interleukin-1, interleukin-18, and tumour necrosis factor-alpha (TNF). Oxidative stress and the formation of ROS both destroy DNA and proteins and act as signalling amplifiers for stress pathways like mitogen-activated protein kinase (MAPK), PKC, and nuclear factor kappa B (NF-KB) (Ha & Lee, 2000). Aldose reductase converts extra glucose into sorbitol, activating the polyol pathways. Sorbitol dehydrogenase then converts fructose, causing oxidative stress due to an increase in the nicotinamide adenine dinucleotide phosphate (NADPH) ratio (Srivastava et al., 2005). Protein structural and functional variation, the expression of inflammatory cytokines and growth factors, and oxidative stress are all consequences of AGE formation, which is caused by the non-enzymatic binding of lipids, glucose, and nucleic acids to proteins as well as nucleic acids (Sheetz & King, 2002). All of these lead to increased proteinuria, glomerulosclerosis, and undoubtedly tubulointerstitial fibrosis by generating the glomerular basement membrane’s high albumin permeability and extracellular matrix buildup.
The generation of ROS by oxidative stress is believed to be a causative factor in the development of diabetes complications (Brownlee, 2001). The cytotoxic nature of ROS is known to promote fibrosis and inflammation. In addition, molecular disruption is caused by the oxidation of macromolecules such as carbohydrates, proteins, lipids, and DNA (Nishikawa et al., 2000). According to studies, oxidative stress is significantly influenced by insulin resistance because free glucose and low NADPH ratios activate the polyol and aldose reductase pathways. Through the de novo synthesis of diacylglycerol (DAG), the elevated intracellular glucose level activates the PKC (Geraldes & King, 2010). The transition of metal-catalysed Fenton reactions, peroxidases, deficits in the mitochondrial respiratory chain, xanthine oxidase activity, NADPH oxidase coupled with NO synthase, and autooxidation of glucose is known to contribute to the formation of ROS in diabetes (Fakhruddin et al., 2017). The production of ROS in diabetes is significantly influenced by the activity and expression of NADPH oxidase (Wautier et al., 2001). The membranous p22phox and gp91phos, also known as nox-1 and nox-4 homologues, the cytosolic p47phox and p67phox subunits, and the regulatory G protein rac-1 make up the five subunits that make up vascular NADPH (Harrison et al., 2003). Due to the stimulation, the cytosolic and membranous subunits will come together to create a functional oxidase that will use NADPH as an electron donor, resulting in a finely defined oxidative burst. In experimental diabetes, there has been a rise in the cytosolic NADPH oxidase subunit, p47phox, in the kidney and vascular system (Christ et al., 2002).
It is well established that a rise in blood sugar levels can boost the mitochondria’s production of ROS. The increased glucose absorption also stimulates the citric acid cycle and glycolysis, which results in an excess synthesis of electron donors such as NADH and flavin adenine dinucleotide (FADH2) (Brownlee, 2001). The inner membrane of the mitochondria contains a localised electron transport chain, and when the number of electron donors increases, this pumping of protons across the inner membrane raises the membrane potential. As a result, the transit of electrons at complex III is restricted by lengthening the half-life of the coenzyme Q’s free radical intermediates, which further reduces O2 to superoxide. Furthermore, although type 2 diabetic people can also develop renal lesions without diabetes, the distinguishing features of DN are recognised to be brought on by diabetes. All of these judgments reach the conclusion that detecting the accumulation of AGEs significantly indicates hyperglycemia and also advances knowledge of other metabolic variables. The excessive production of superoxides and cellular pseudohypoxia also contributes to inflammatory processes and oxidative stress. In diabetic individuals, the accumulation of AGE is well recognised as an early indicator of renal problems (Amore et al., 2004). Numerous familial studies, as well as epidemiological studies, have shown that the main factors that show an important role in the progression of nephropathy as well hyperglycemia, obesity, insulin resistance, dyslipidemia, and genetic susceptibility along with other environmental factors and dietary habits (Adler et al., 2003). Age, race, ethnicity, and family history are non-modifiable characteristics. Modifiable factors typically have to do with lifestyle choices and environment-related elements that can be changed.
Individual differences in the risk of developing DN in diabetes patients bring the genetic predisposition relationship to a close. Long-term effects of DN include increased blood pressure, declining renal function, and ESRD, according to knowledge. A combination of metabolic and hemodynamic disturbances increased expression of inflammatory cytokines, growth hormones, and genetic susceptibility works together to activate these causes (Jacobsen, 2005). It is yet unknown how nephropathy is genetically transmitted. Its multifactorial character makes polygenic transmission more likely than other modes of transmission. Several familial investigations have produced ample data which confirms the abovementioned information (Adler et al., 2003).
It is well-recognised that ethnicity can also affect the likelihood of developing diabetic nephropathy. Numerous studies have shown that Asians with diabetes are more likely than Caucasians to develop ESRD, which raises their likelihood of receiving kidney replacement therapy by 5.8 times (Burden et al., 1992). According to a cross-sectional study done in the UK, South Asians are further probable than Europeans to have microalbuminuria. Another study that supported this finding showed that South Asians had a proteinuria prevalence rate of 21%, while White people in the UK only had a prevalence rate of 14% (Dreyer et al., 2009). In a follow-up study, it was shown that South Asians had a 1.45 times larger drop in eGFR and were four times more likely to develop albuminuria than Europeans (Shaw et al., 2006). Hence, all these findings emphasise the notion that an increased understanding of the risk factors which lead to the pathogenesis of DN specifically in the South Asian population is crucial. Learning more about the pathogenesis of DN in South Asians will be essential for preventing the disease’s start and delaying its progression.
One of the main complications of diabetes is diabetic nephropathy, which is characterised by elevated levels of albuminuria that accelerate the loss of renal function. Because elevated levels of albuminuria do not manifest until there has been severe kidney damage, clinical identification of nephropathy is difficult (Parving et al., 2011). Heavy albuminuria rate is currently the gold standard for DN detection. Typically, after 15 to 20 years of uncontrolled diabetes, DN incidence is noted (Nazar, 2014). Another fact that not all diabetes patients with microalbuminuria proceed to DN is still not fully understood. This emphasises the necessity of developing genetic indicators for the prognosis of DN at the onset of the disease. The fact that not all patients with diabetes progress to diabetic nephropathy (DN) can shed light on the role of hereditary factors in the development of diabetes and its progression to DN. According to this assertion, inherited genetic characteristics in the family may have an impact on a person who has both diabetes and diabetic nephropathy. However, each person experiences renal functional impairment at the same rate. When compared to patients with a normal amount of albumin excretion, studies have shown that those with nephropathy had a higher incidence of diabetes risk factors (Brennan et al., 2013). Comparing first-degree relatives of diabetes patients with and without DN, familial investigations have shown that these features are more common in DN patients (Fogarty et al., 2000). As a result, the development of diabetic nephropathy may have been influenced by genetic factors, which further suggest a vulnerability to cardiovascular illnesses. The impact of genetic factors, in addition to the link to single-gene changes, can likely be complicated and multivariate. Consequently, it is also known that gene-gene interactions and gene-environment interactions show a substantial role in the development of disease (Liu et al., 2015).
Numerous genetic investigations have already been conducted to categorise potential candidate genes in large sample size studies, which also adds to the study of diabetic nephropathy pathogenesis. The identification of DN-related genes will also aid in identifying those people who are more likely to experience this renal problem. Additionally, it increases understanding of the illness progression mechanisms. Due to their small sample sizes and weak statistical power, the majority of earlier studies about the genes of DN are often constrained. Recent observations on diversity in genotype expression brought on by intraglomerular pressure and high glucose levels make it more difficult to make sense of the results. The prediction of diabetic people who are likely to develop a high risk of DN will benefit from the discovery of vulnerable genes and causal variations. It will also help to identify potential prognostic indicators for early nephropathy detection and inhibition, as well as for delaying the progression to the subsequent advanced stages. The major goal is to raise the patients with DN’s survival rate. Single-nucleotide polymorphisms (SNPs), extensive, structural chromosomal disruptions, and other variations of genetic deviation can all be found in the human genome. The SNPs linked to gene expression may also be involved in a number of disorders brought on by disruptions in the homeostasis of the genes. This could include transcriptional mechanisms such as transcription factor binding, atypical gene splicing, and messenger RNA degradation (Cheung & Spielman, 2009). The human genome has been sequenced, and it shows that 99.9% of each person’s DNA is the same. Only 0.1 percent of people differ from one another. This distinction is what determines how each individual’s phenotype is unique. Single base deviations in the human genome are known as single-nucleotide polymorphisms, which are small genetic changes. Despite the relatively low danger that the normal genetic variants display, bigger populations indicate increased risk due to their larger frequencies. A genetic locus is often considered polymorphic if the frequency of the sporadic allele exceeds 1% and results in a heterozygotic frequency of at least 2%. It is generally recognised that a combination of hereditary genetic polymorphisms and numerous environmental circumstances can predispose someone to specific diseases or change how a disease develops. Additionally, recent research has demonstrated that genetic polymorphisms might influence a person’s susceptibility to conditions like diabetes mellitus, osteoporosis, hypertension, and Alzheimer’s disease. Comparatively to the detection of single gene mutations or other significant chromosomal aberrations, the discovery of such vulnerable genes is difficult because a single polymorphism seems to display only a minimal effect on the phenotype. Since the occurrence of many sensitive alleles at a multiplex locus may not always result in overt clinical symptoms, it is hypothesised that the change in environmental factors will be the cause (Holtzman & Marteau, 2000). With the completion of the Human Genome Project (HGP), opportunities for identifying the genes linked to a variety of multifaceted diseases have also quickly advanced. This has improved our understanding of the haplotype structure of the genome along with recent developments in epidemiological studies relating to DN. Genome-wide association studies, genetic linkage studies, association analysis studies/case-control studies, and candidate gene approach studies are a basic list of genetic techniques (Smith et al., 2004). The application of GWAS in the realm of medical genetics is well established. Investigating the relationship between particular genetic variations and particular diseases is done using this method. The described method further analyses the genomes of numerous people to find genetic indicators that can be used for disease prognosis. Once these genetic markers have been discovered, they may be utilised to research the role of genes in the development of a disease and the creation of superior preventive and therapeutic approaches. One benefit of GWAS is that it can find the genes regardless of whether their function has previously been established. However, problems including low statistical power, high costs, ambiguous genetic effects, and unreliable phenotypic definitions are some of the causes of partial success (Cooke et al., 2008). However, SNP-based genome-wide association studies are utilised in humans to significantly identify the regions that are important for the onset of some diseases, including diabetes, Crohn’s disease, and even other hereditary problems (Barrett et al., 2008; Sladek et al., 2007). Additionally, this approach has been utilised to identify specific genetic loci, namely, for diabetic kidney disease (DKD), eGFR, and albuminuria in people with diabetes mellitus. This method pinpoints the chromosomal location of genes that are naturally associated with the disease. It simply works by keeping an eye on the genes that remain close to one another on the same chromosome and are recognised as being linked throughout the meiotic stage (Altshuler et al., 2008). The description of a specific phenotype associated with each gene is essential. The two main categories of linkage analysis investigations include parametric and nonparametric test methods. In the conventional linkage analysis, the method of inheritance, phenocopies, as well as gene frequencies must be identified (Lin et al., 2008). For linkage analysis research, choosing suitable genetic markers is also essential. Microsatellite markers were used in the initial linkage studies; however, SNPs are now more frequently used due to their equivalent importance and low cost (Table 1).
The comprehensive information from the entire genome is known to be delivered through genome-wide linkage analysis. The downside of this strategy, though, is that it is less sensitive than association studies. However, it also has the capacity to identify genomic regions that include a variety of genes and gene variations. Because it is typically undertaken on a large number of instructional families, it is more difficult, expensive, and time-consuming. Pima Indians’ entire genome linkage investigation resulted in the identification of DN-susceptible loci on chromosomes 3, 7, and 20 (Imperatore et al., 1998). This method entails the identification of genetic variants in particular candidate genes that may alter or alter the structure and function of proteins, causing the advancement of specific diseases. In case-control studies, participant selection is the key element. Case-control association studies are used to investigate candidate genetic polymorphisms by comparing the frequency of polymorphisms in certain genes between patients and control participants. To avoid unfavourable outcomes, it is necessary to properly recruit a sizable number of carefully characterised cases and control individuals. It is necessary to enroll a high-enough percentage of qualified patients to guarantee that there is no chance of developing a significant association.
To achieve this, uniform diagnostic tests for the illness should be investigated, especially if subgroup analysis is being done with varied disease stages and severity. Given that occupational characteristics and changes in lifestyle have a significant impact on susceptibility to various illnesses, it is imperative to develop a relevant questionnaire that accounts for all of these variables and to ensure that all participants complete it (Daly & Day, 2001). The family-based association analysis is another common technique used in genetic association investigations. The transmission disequilibrium test (TDT) is a different approach to investigating the frequency of diseased alleles that are transmitted or not to the affected offspring from the diseased heterozygous parents (Feng et al., 2007). There is no need for a control group because the genotypes of the patient’s parents are already known. It is thought that the illness allele is connected with the trait if allele transmission occurs in more than half of the cases. It is well-known that looking into the relationships between alleles in these groups can provide light on the population association and assist localise the vulnerable locus within a 2 Mb-wide area. These chromosomal regions may be incredibly small, yet they will be big enough to allow for physical mapping. Further discovery of causative polymorphisms and thorough genetic alterations will also be possible. In case-control studies, the candidate gene method is essential for identifying the genes that are predisposed to diabetic nephropathy. The candidate gene study approach can be well understood in comparison to genome-wide studies because DN-associated genes will be selectively chosen for the study. Around the world, candidate gene studies for DN are being carried out in an effort to find genetic indicators that may help predict the likelihood that a diabetic patient would eventually develop diabetic nephropathy. Single-nucleotide polymorphisms in one or more genes that suggest a potential role in the pathology of a certain disease are studied while examining the candidate genes. Therefore, even when a gene’s influence on a disease is minimal, this approach appears to be helpful. It is thought to be an appropriate investigation for complicated genetic transmission. Additionally, this can be particularly helpful in situations when the genetic influence is very low but the prevalence of disease-associated alleles is high (Patnala et al., 2013). Although candidate gene studies are known to have strong statistical power, no novel genes will be found. Linkage analyses, case-control association studies, and genome-wide association studies, among others, have demonstrated the role of genetic vulnerability in the development of DN in people with type 2 diabetes mellitus.
However, as is the case with the majority of complicated diseases, it has been demonstrated that it is difficult to pinpoint the precise genetic variants that cause nephropathy. Therefore, a deeper comprehension of the genetic heritability underpinning DN may even aid in the development of novel biomarkers to identify illness vulnerability and support novel therapy modalities. The combination of multiple information sources tends to be more beneficial to improve the understanding of the pathophysiology of illnesses with diverse phenotypes, such as diabetic nephropathy (Brennan et al., 2013). When compared to external environmental influences, genes have a significant impact on the development of nephritis, which is already a well-known and established fact. Single-nucleotide polymorphism analysis is used to study the impact of these polymorphisms on parameters such as illness susceptibility, healthcare to change treatment trajectories, and drug resistance.
In light of this, a wide variety of genes have been examined to determine their relationship to DN and the contribution of multiple SNPs found in the DN-associated genes. The fact that different ethnic groups exhibit different susceptibility patterns is another obsession. Positive associations of DN with several genes that have already undergone investigation are evident. Profibrotic growth factors genes like TGF-1, connective tissue growth factor (CTGF), VEGF, intercellular adhesion molecule 1 (ICAM1), the receptor for advanced glycation end products (RAGE), monocyte chemoattractant protein-1 (MCP-1) and antagonists, as well as genes linked to dyslipidemia, are a few of the genes. Predisposed maps for diseases will benefit from the Indian Genome Variation Consortium (IGVC) data as well as additional epidemiology and related phenotypic information (Narang et al., 2010).
This will raise awareness for those who are more susceptible to developing DN. Genetic association studies are widely employed and sensitive when contrasted to the scant information on genetic linkage research. Fatty acid-binding protein-2 (FABP2), angiotensin-converting enzyme (ACE), GLUT1, and ectonucleotide pyrophosphatase (ENPP1) are a few other DN-associated genes that have been discovered using the candidate genetic association technique (Tang et al., 2015). It has been established that the genetic variants chosen for the current study play a substantial role in the many signs and symptoms of diabetic nephropathy, including renal hypertrophy, mesangial cell growth, and endothelial dysfunction.
TGF-1 is a multifunctional cytokine that is linked to the pathogenesis of many renal issues, including diabetic nephropathy, by advancing renal hypertrophy and increasing extracellular matrix buildup (Reeves & Andreoli, 2000). The TGF-1 gene, which is at the chromosomal location 19q13.1–q13.3, is known to contain seven exons and six big introns. Currently, information on ten polymorphic loci that are visible between introns and exons as well as in various flanking regions is being acknowledged (Watanabe et al., 2002). The transforming growth factor is known to promote renal hypertrophy and fibrosis, while other cytokines, such as TNF and monocyte chemoattractant protein-1 (MCP-1), interfere with the CCR2 and CCR5 chemokine receptors to further regulate the influx of macrophages into the kidney (Prasad et al., 2007). High glycaemic levels seen in people with type 2 diabetes mellitus are linked to the activation of glucose transport 1, which prompts the renal and mesangial tubular cells to overexpress TGF. Additionally, an increase in intraglomerular pressure, activation of the renin-angiotensin system, reactive oxygen species, and advanced glycation end products cause the renal system to produce TGF (Qian et al., 2008). Numerous studies have looked at significant individual differences and shown that genetic variables may regulate the amounts of TGF-1 in the serum and mRNA (Ahluwalia et al., 2009). The two genetic polymorphisms, 915G > C and 869 T > C, were examined in the German population, and the results revealed a substantial connection between 869 T > C with DN but not with the other SNP, 915 G > C. (Babel et al., 2006). In a study conducted by Valladares-Salgado et al. (2010), it was important to explore the relationship between cholesterol levels and the TGF-1 gene’s T869C polymorphism. Results showed a significant correlation between total cholesterol levels and the CC + CT genotype. Although many different variations of this gene are expected to exist and be deposited in the dbSNP database, there is very little information regarding the function and significance of these variants in relation to renal problems. Numerous investigations in various populations revealed an enhanced correlation between the 915G > C polymorphism and a high risk of ESRD and chronic renal failure. However, it is still unclear how exactly this polymorphism affects diabetes patients whose condition worsens and eventually leads to DN. There hasn’t been any research on the association between the TGF-1915G > C polymorphism and diabetic nephropathy in the South Indian population. The genetic polymorphism was included in the proposed study since it was found to play a significant role in the pathophysiology of DN and was associated with greater plasma levels in diabetic patients in other groups. Translocation of a variant plasmacytoma one gene is well-known for being involved in frequent translocations between the area of chromosome 2 that codes for it and the region of chromosome 22 (Graham & Adams, 1986). Chromosome 8q24 is where it is situated (Millis et al., 2007). Although it has long been known that the kidney contains significant quantities of PVT1, its function is not yet understood. Through the use of a genome-wide SNP association research among American Indians with diabetes mellitus, the gene for plasmacytoma variant translocation 1 is identified as a candidate gene for end-stage renal disease (ESRD) (Nazar, 2014). PVT1 has been found to have an impact on unchecked cell proliferation, especially mesangial cell expansion, which is a well-known key characteristic of diabetic kidney disorders. PVT1 has been found to affect the formation and progression of DN by a variety of processes, including the aggregation of extracellular matrix proteins in the glomerular mesangium (Alvarez & DiStefano, 2011). It has been demonstrated that PVT1 increases TGFB1 and plasminogen activator inhibitor 1 in human mesangial cells, which are two of the main causes of extracellular matrix buildup in the glomeruli under high glucose circumstances (Alvarez et al., 2013). The PVT1 gene cluster was found to contain the markers that were found to be strongly linked with ESRD. The expression of the PVT1 gene results in a number of transcript variations (Nelson et al., 1988). PVT1 is a 1.9 kb long non-coding RNA that further encodes a number of transcripts but is unable to synthesise proteins. According to Alvarez et al. (2013), it is the first and first non-coding RNA to have demonstrated a relationship with renal disorders. Breast and ovarian cancers are also known to be linked to the overexpression of PVT1 (Guan et al., 2007). A genome-wide single-nucleotide polymorphism association study among Pima Indians demonstrated a connection between the rs2648875 gene polymorphism of the PVT1 gene and ESRD. It is well-known that a rise in glucose and TGF-1 levels causes PVT1 to be increased (Hanson et al., 2007). The genetic variation rs2648875 has been linked to diabetic mellitus, which further contributes to DN.
To learn more about the association between PVT1 and DN and to research the precise role of the gene causing ESRD, additional studies in various groups are needed. It is known that pentraxin 3 belongs to the long pentraxin group and shares structural similarities with short pentraxins like C-reactive protein and serum amyloid P component (SAP) (CRP). But when it comes to cell formation and stimulus activation, it shows a variety of variances. While IL-6 is known to trigger the formation of CRP and SAP in liver cells, PTX3 is known to be produced by a variety of cell types as a result of activation brought on by specific stimuli. Additionally, it has been discovered that PTX3 is stimulated by inflammatory signals and changed lipoproteins in cells such vascular endothelium and smooth muscle cells. Additionally, epithelial cells, neurons, fibroblasts, glial cells, and adipocytes have all been discovered to contribute to its formation (Bottazzi et al., 2016). According to certain research, acute ischemia-reperfusion renal injuries and chronic kidney disorders both cause higher-than-normal plasma levels of PTX3. PTX3 is a proinflammatory cytokine with a documented propensity for having a pathogenic effect (Yilmaz et al., 2009). A biomarker of immune-inflammatory response and a proven conservative defence mechanism, high plasma levels of PTX3 are likely to protect the kidneys as well (Speeckaert et al., 2013).
As established by prior investigations, DN is an inflammatory disease with changed plasma levels that may also play a role in the disease’s progression. Similar findings demonstrated that plasma levels of PTX3 were negatively correlated with eGFR before positively correlating with the levels of proteinuria in DN patients. This led to the further suggestion of PTX3’s involvement in the development of DN (Uzun et al., 2016). Additionally, it has been discovered that the SNPs in this gene affect the amounts of circulating PTX3, which is once again connected to several additional illnesses, including hepatocellular carcinoma, primary graft malfunction, and pulmonary tuberculosis (Carmo et al., 2016; Diamond et al., 2012; Olesen et al., 2007), while only one study, conducted in the Chinese population, found a link between DN and genetic variations that increase susceptibility to diabetes (Zhu et al., 2017). According to linkage studies, the chromosome 3q is linked to diabetes and nephropathy in a number of populations. Additionally, the gene that codes for PTX3 is situated in the region of 3q25.32 that is too close to the linkage region (Vionnet et al., 2006). This strengthens the connection between PTX3 and DN that may exist. Further research on the genetic variation of PTX3 will pave the path for the creation of a potential biomarker for the early diagnosis of DN in diabetic patients. Endothelial dysfunction is another defining feature of DN. Nitric oxide synthase activity in endothelial cells is decreased as a result of endothelial dysfunction, a risk factor. Additionally, this action is critical to the pathophysiology of DN (Nakagawa et al., 2007). The nitric oxide synthase 3 gene is located on chromosome 7 in the 7q35–7q36 region. With 26 exons and 25 introns, it has a total length of 21 kilobases (Zanchi et al., 2000).
Endothelial nitric oxide synthase, an enzyme, identifies nitric oxide as a crucial endothelial cell by product that is from L-arginine (eNOS). It has been demonstrated that the NO plays a variety of physiological and regulatory activities as well as taking part in processes like blocking platelet aggregation, neurotransmission, controlling blood pressure, and relaxing smooth muscle (Syed et al., 2011). Inducible NOS (iNOS), endothelial NOS (eNOS), and neuronal NOS (nNOS) are the three isoforms of nitric oxide synthase (Noiri et al., 2002). The likelihood of developing issues connected to diabetes increases when ROS generation increases, particularly superoxide anion (O2-) (Mclennan et al., 2000). Peroxynitrite (ONOO-), which is known as a weak agonist necessary to activate the cyclic guanosine monophosphate, is created when the superoxide anion combines with nitric oxide (cGMP). O2- effectively deactivates the NO as a result. Nitric oxide activity may be reduced by the NADPH oxidase induction seen in diabetes mellitus. By reducing the expression of matrix metalloproteinases and boosting the expression of tissue inhibitor of metalloproteinases in the kidney, this further accelerates the mRNA expression required for the fibronectin and transforming growth factor-1 (TGF-1) present in the glomerulus (Chiarelli et al., 2009).
It has been established that the eNOS gene variations both reduce nitric oxide production and contribute to endothelial dysfunction. The 786 T > C alteration found in the promoter region of the eNOS gene is one of the most significant single-nucleotide polymorphisms (b). Glu is changed to Asp at codon 298 by the 894 G > T alteration found in exon 7 (c). Intron 4 contains a 27-bp variable tandem repeat (VNTR) polymorphism that has been shown to exhibit an insertion and deletion with two alleles (Asakimori et al., 2002). The 894G > T polymorphism among these has been found to continue to be significantly related with DN. Studies have shown that the eNOS 894G > T polymorphism T allele and TT genotype significantly increase the chance of developing DN. Diabetic individuals from Tunisia and Japan have a higher prevalence of this polymorphism, which has further contributed to renal failure (Santos et al., 2011).
However, other studies have also claimed to have found no appreciable effects in the specific population. This variation can be explained by ethnic and geographic factors that may affect the connection of polymorphisms and hence affect whether DN will proceed or not. Other illnesses such acute myocardial infarction, coronary artery disease, hypertension, and atherosclerosis are associated with an increased frequency of this polymorphism (Colombo et al., 2002; Cilingir et al., 2019). The polymorphism can be proposed as a primary factor for the progression of diabetic nephropathy specifically in individuals with diabetes, based on the results of previous studies, it can be concluded; furthermore, the majority of the Caucasian and Asian populations were associated with the 894G > T polymorphism’s significant risk of DN among diabetic patients as opposed to in participants who were not diabetic (Chalasova et al., 2014).
8 Conclusion
DN and ESRD continue to be major issues despite greatest attempts to reduce the disease’s effect on such end-organ damage. A multifactorial strategy is still the most rational one in the extremely complicated environment of diabetes, where no one medication can stop the progression of DN. This should include single RAS inhibition for hypertension or albuminuria, as well as appropriate glycaemic management. The general health around the world is seriously threatened by diabetes mellitus (DM). One of the most frequent side effects of diabetes and the main contributor to end-stage renal disease is diabetic nephropathy (DN) (ESRD). The progression of DM patients to ESRD is estimated to be between 30 and 40 percent worldwide, highlighting the influence of hereditary variables on DN. Family clustering further supports the crucial part that inherited variables play in DN and ESRD. In order to find susceptibility genes in various diabetic cohorts, numerous genetic investigations have been conducted. It has only recently been discovered that DN and ESRD have extensive susceptibility genes.
References
Adler, A. I., Stevens, R. J., Manley, S. E., Bilous, R. W., Cull, C. A., Holman, R. R., & UKPDS Group. (2003). Development and progression of nephropathy in type 2 diabetes: The United Kingdom prospective diabetes study (UKPDS 64). Kidney International, 63(1), 225–232.
Ahluwalia, T. S., Khullar, M., Ahuja, M., Kohli, H. S., Bhansali, A., Mohan, V., Venkatesan, R., Rai, T. S., Sud, K., & Singal, P. K. (2009). Common variants of inflammatory cytokine genes are associated with risk of nephropathy in type 2 diabetes among Asian Indians. PLoS One, 4(4), e5168.
Alter, M. L., Ott, I. M., Von Websky, K., Tsuprykov, O., Sharkovska, Y., Krause-Relle, K., Raila, J., Henze, A., Klein, T., & Hocher, B. (2012). DPP-4 inhibition on top of angiotensin receptor blockade offers a new therapeutic approach for diabetic nephropathy. Kidney and Blood Pressure Research, 36(1), 119–130.
Altshuler, D., Daly, M. J., & Lander, E. S. (2008). Genetic mapping in human disease. Science, 322(5903), 881–888.
Alvarez, M. L., & DiStefano, J. K. (2011). Functional characterization of the plasmacytoma variant translocation 1 gene (PVT1) in diabetic nephropathy. PLoS One, 6(4), e18671.
Alvarez, M. L., Khosroheidari, M., Eddy, E., & Kiefer, J. (2013). Role of microRNA 1207-5P and its host gene, the long non-coding RNA Pvt1, as mediators of extracellular matrix accumulation in the kidney: Implications for diabetic nephropathy. PLoS One, 8(10), e77468.
American Diabetes Association. (2004). Nephropathy in diabetes (position statement). Diabetes Care, 27, 79–83.
American Diabetes Association. (2017). 10. Microvascular complications and foot care. Diabetes Care, 40, S88–S98.
Amore, A., Cirina, P., Conti, G., Cerutti, F., Bagheri, N., Emancipator, S. N., & Coppo, R. (2004). Amadori-configurated albumin induces nitric oxide-dependent apoptosis of endothelial cells: A possible mechanism of diabetic vasculopathy. Nephrology Dialysis Transplantation, 19(1), 53–60.
Arif, E., & Nihalani, D. (2013). Glomerular filtration barrier assembly: An insight. Postdoc Journal: A Journal of Postdoctoral Research and Postdoctoral Affairs, 1(4), 33.
Asakimori, Y., Yorioka, N., Taniguchi, Y., Ito, T., Ogata, S., Kyuden, Y., & Kohno, N. (2002). T-786→C polymorphism of the endothelial nitric oxide synthase gene influences the progression of renal disease. Nephron, 91(4), 747–751.
Babel, N., Gabdrakhmanova, L., Hammer, M. H., et al. (2006). Predictive value of cytokine gene polymorphisms for the development of end-stage renal disease. Journal of Nephrology, 19(6), 802–807.
Barbour, S. J., Er, L., Djurdjev, O., Karim, M., & Levin, A. (2010). Differences in progression of CKD and mortality amongst Caucasian, oriental Asian and South Asian CKD patients. Nephrology Dialysis Transplantation, 25(11), 3663–3672.
Barrett, J. C., Hansoul, S., Nicolae, D. L., Cho, J. H., Duerr, R. H., Rioux, J. D., Brant, S. R., Silverberg, M. S., Taylor, K. D., Barmada, M. M., & Bitton, A. (2008). Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nature Genetics, 40(8), 955–962.
Bilous, R. W. (1997). The pathology of diabetic nephropathy. In K. Alberti, P. Zimmet, R. A. DeFronzo, & H. Keen (Eds.), International textbook of diabetes mellitus (pp. 1349–1362). Wiley.
Bonventre, J. V. (2012). Can we target tubular damage to prevent renal function decline in diabetes. Seminars in Nephrology, 32(5), 452–462.
Boright, A. P., Paterson, A. D., Mirea, L., Bull, S. B., Mowjoodi, A., Scherer, S. W., Zinman, B., & DCCT/EDIC Research Group. (2005). Genetic variation at the ACE gene is associated with persistent microalbuminuria and severe nephropathy in type 1 diabetes: The DCCT/EDIC Genetics Study. Diabetes, 54(4), 1238–1244.
Bottazzi, B., Inforzato, A., Messa, M., Barbagallo, M., Magrini, E., Garlanda, C., & Mantovani, A. (2016). The pentraxins PTX3 and SAP in innate immunity, regulation of inflammation and tissue remodelling. Journal of Hepatology, 64(6), 1416–1427.
Brennan, E., McEvoy, C., Sadlier, D., Godson, C., & Martin, F. (2013). The genetics of diabetic nephropathy. Genes, 4(4), 596–619.
Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature, 414(6865), 813–820.
Brownlee, M., Vlassara, H., Kooney, A., Ulrich, P., & Cerami, A. (1986). Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science, 232(4758), 1629–1632.
Buraczynska, M., Zukowski, P., Ksiazek, P., Kuczmaszewska, A., Janicka, J., & Zaluska, W. (2014). Transcription factor 7-like 2 (TCF7L2) gene polymorphism and clinical phenotype in end-stage renal disease patients. Molecular Biology Reports, 41(6), 4063–4068.
Burden, A. C., McNally, P. C., Feehally, J., & Walls, J. (1992). Increased incidence of end-stage renal failure secondary to diabetes mellitus in Asian ethnic groups in the United Kingdom. Diabetic Medicine, 9(7), 641–645.
Carmo, R. F., Aroucha, D., Vasconcelos, L. R., Pereira, L. M., Moura, P., & Cavalcanti, M. S. (2016). Genetic variation in PTX 3 and plasma levels associated with hepatocellular carcinoma in patients with HCV. Journal of Viral Hepatitis, 23(2), 116–122.
Chalasova, K., Dvorakova, V., Pacal, L., Bartakova, V., Brozova, L., Jarkovsky, J., & Kankova, K. (2014). NOS3 894G>T polymorphism is associated with progression of kidney disease and cardiovascular morbidity in type 2 diabetic patients: NOS3 as a modifier gene for diabetic nephropathy? Kidney and Blood Pressure Research, 38(1), 92–98.
Chan, Y., Lim, E. T., Sandholm, N., Wang, S. R., AJ, M. K., Ripke, S., Daly, M. J., Neale, B. M., Salem, R. M., Hirschhorn, J. N., & DIAGRAM Consortium. (2014). An excess of risk increasing low-frequency variants can be a signal of polygenic inheritance in complex diseases. The American Journal of Human Genetics, 94(3), 437–452.
Chawla, T., Sharma, D., & Singh, A. (2010). Role of the renin angiotensin system in diabetic nephropathy. World Journal of Diabetes, 1(5), 141.
Cheung, V. G., & Spielman, R. S. (2009). Genetics of human gene expression: Mapping DNA variants that influence gene expression. Nature Reviews Genetics, 10(9), 595–604.
Chiarelli, F., Gaspari, S., & Marcovecchio, M. L. (2009). Role of growth factors in diabetic kidney disease. Hormone and Metabolic Research, 41(08), 585–593.
Choe, E. Y., Wang, H. J., Kwon, O., Kim, K. J., Kim, B. S., Lee, B. W., Ahn, C. W., Cha, B. S., Lee, H. C., Kang, E. S., & Mantzoros, C. S. (2013). Variants of the adiponectin gene and diabetic microvascular complications in patients with type 2 diabetes. Metabolism, 62(5), 677–685.
Christ, M., Bauersachs, J., Liebetrau, C., Heck, M., Günther, A., & Wehling, M. (2002). Glucose increases endothelial-dependent superoxide formation in coronary arteries by NAD (P) H oxidase activation: Attenuation by the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor atorvastatin. Diabetes, 51(8), 2648–2652.
Chung, H. F., Long, K. Z., Hsu, C. C., Al Mamun, A., Chiu, Y. F., Tu, H. P., Chen, P. S., Jhang, H. R., Hwang, S. J., & Huang, M. C. (2014). Adiponectin gene (ADIPOQ) polymorphisms correlate with the progression of nephropathy in Taiwanese male patients with type 2 diabetes. Diabetes Research and Clinical Practice, 105(2), 261–270.
Cilingir, V., Donder, A., Milanlioğlu, A., Yilgör, A., & Tombul, T. (2019). Association between endothelial nitric oxide synthase polymorphisms T786C and G894T and ischaemic stroke. Eastern Journal of Medicine, 24(4), 472–477.
Clarke, P., Gray, A., Legood, R., Briggs, A., & Holman, R. (2003). The impact of diabetes-related complications on healthcare costs: Results from the United Kingdom prospective diabetes study (UKPDS Study No. 65). Diabetic Medicine, 20(6), 442–450.
Cohen, R. M., Holmes, Y. R., Chenier, T. C., & Joiner, C. H. (2003). Discordance between HbA1c and fructosamine: Evidence for a glycosylation gap and its relation to diabetic nephropathy. Diabetes Care, 26(1), 163–167.
Colombo, M. G., Andreassi, M. G., Paradossi, U., Botto, N., Manfredi, S., Masetti, S., Rossi, G., Clerico, A., & Biagini, A. (2002). Evidence for association of a common variant of the endothelial nitric oxide synthase gene (Glu298→Asp polymorphism) to the presence, extent, and severity of coronary artery disease. Heart, 87(6), 525–528.
Cooke, G. S., Campbell, S. J., Bennett, S., Lienhardt, C., McAdam, K. P., Sirugo, G., Sow, O., Gustafson, P., Mwangulu, F., van Helden, P., & Fine, P. (2008). Mapping of a novel susceptibility locus suggests a role for MC3R and CTSZ in human tuberculosis. American Journal of Respiratory and Critical Care Medicine, 178(2), 203–207.
Daly, A. K., & Day, C. P. (2001). Candidate gene case-control association studies: Advantages and potential pitfalls. British Journal of Clinical Pharmacology, 52(5), 489–499.
Deshmukh, H. A., Palmer, C. N., Morris, A. D., & Colhoun, H. M. (2013). Investigation of known estimated glomerular filtration rate loci in patients with type 2 diabetes. Diabetic Medicine, 30(10), 1230–1235.
Diamond, J. M., Meyer, N. J., Feng, R., Rushefski, M., Lederer, D. J., Kawut, S. M., Lee, J. C., Cantu, E., Shah, R. J., Lama, V. N., & Bhorade, S. (2012). Variation in PTX3 is associated with primary graft dysfunction after lung transplantation. American Journal of Respiratory and Critical Care Medicine, 186(6), 546–552.
Dreyer, G., Hull, S., Aitken, Z., Chesser, A., & Yaqoob, M. M. (2009). The effect of ethnicity on the prevalence of diabetes and associated chronic kidney disease. QJM: An International Journal of Medicine, 102(4), 261–269.
Drummond, K., & Mauer, M. (2002). The early natural history of nephropathy in type 1 diabetes: II. Early renal structural changes in type 1 diabetes. Diabetes, 51(5), 1580–1587.
Duran-Salgado, M. B., & Rubio-Guerra, A. F. (2014). Diabetic nephropathy and inflammation. World Journal of Diabetes, 5(3), 393.
Ellis, J. W., Chen, M. H., Foster, M. C., Liu, C. T., Larson, M. G., de Boer, I., Köttgen, A., Parsa, A., Bochud, M., Böger, C. A., & Kao, L. (2012). Validated SNPs for eGFR and their associations with albuminuria. Human Molecular Genetics, 21(14), 3293–3298.
Ewens, K. G., George, R. A., Sharma, K., Ziyadeh, F. N., & Spielman, R. S. (2005). Assessment of 115 candidate genes for diabetic nephropathy by transmission/disequilibrium test. Diabetes, 54(11), 3305–3318.
Fakhruddin, S., Alanazi, W., & Jackson, K. E. (2017). Diabetes-induced reactive oxygen species: Mechanism of their generation and role in renal injury. Journal of Diabetes Research.
Feng, B. J., Goldgar, D. E., & Corbex, M. (2007). Trend-TDT – A transmission/disequilibrium based association test on functional mini/microsatellites. BMC Genetics, 8(1), 1–8.
Fioretto, P., & Mauer, M. (2007). Histopathology of diabetic nephropathy. Seminars in Nephrology, 195–207.
Fogarty, D. G., Rich, S. S., Hanna, L., Warram, J. H., & Krolewski, A. S. (2000). Urinary albumin excretion in families with type 2 diabetes is heritable and genetically correlated to blood pressure. Kidney International, 57(1), 250–257.
Forbes, J. M., & Cooper, M. E. (2013). Mechanisms of diabetic complications. Physiological Reviews, 93(1), 137–188.
Forbes, J. M., Soulis, T., Thallas, V., Panagiotopoulos, S., Long, D. M., Vasan, S., Wagle, D., Jerums, G., & Cooper, M. E. (2001). Renoprotective effects of a novel inhibitor of advanced glycation. Diabetologia, 44(1), 108–114.
Fu, L. L., Lin, Y., Yang, Z. L., & Yin, Y. B. (2012). Association analysis of genetic polymorphisms of TCF7L2, CDKAL1, SLC30A8, HHEX genes and microvascular complications of type 2 diabetes mellitus. Chinese Journal of Medical Genetics., 29(2), 194–199.
Geraldes, P., & King, G. L. (2010). Activation of protein kinase C isoforms and its impact on diabetic complications. Circulation Research, 106(8), 1319–1331.
Goyal, R. K., Shah, V. N., Saboo, B. D., Phatak, S. R., Shah, N. N., Gohel, M. C., Raval, P. B., & Patel, S. S. (2010). Prevalence of overweight and obesity in Indian adolescent school going children: Its relationship with socioeconomic status and associated lifestyle factors. The Journal of the Association of Physicians of India, 58, 151–158.
Graham, M., & Adams, J. M. (1986). Chromosome 8 breakpoint far 3′ of the c-myc oncogene in a Burkitt's lymphoma 2; 8 variant translocation is equivalent to the murine pvt-1 locus. The EMBO Journal, 5(11), 2845–2851.
Gray, L. J., Tringham, J. R., Davies, M. J., Webb, D. R., Jarvis, J., Skinner, T. C., Farooqi, A. M., & Khunti, K. (2010). Screening for type 2 diabetes in a multiethnic setting using known risk factors to identify those at high risk: A cross-sectional study. Vascular Health and Risk Management, 6, 837.
Guan, Y., Kuo, W. L., Stilwell, J. L., Takano, H., Lapuk, A. V., Fridlyand, J., Mao, J. H., Yu, M., Miller, M. A., Santos, J. L., & Kalloger, S. E. (2007). Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clinical Cancer Research, 13(19), 5745–5755.
Ha, H., & Lee, H. B. (2000). Reactive oxygen species as glucose signalling molecules in mesangial cells cultured under high glucose. Kidney International, 58, 19–25.
Haneda, M., Koya, D., Isono, M., & Kikkawa, R. (2003). Overview of glucose signaling in mesangial cells in diabetic nephropathy. Journal of the American Society of Nephrology, 14(5), 1374–1382.
Hanson, R. L., Craig, D. W., Millis, M. P., Yeatts, K. A., Kobes, S., Pearson, J. V., Lee, A. M., Knowler, W. C., Nelson, R. G., & Wolford, J. K. (2007). Identification of PVT1 as a candidate gene for end-stage renal disease in type 2 diabetes using a pooling-based genome-wide single nucleotide polymorphism association study. Diabetes, 56(4), 975–983.
Harrison, D. G., Cai, H., Landmesser, U., & Griendling, K. K. (2003). Interactions of angiotensin II with NAD (P) H oxidase, oxidant stress and cardiovascular disease. Journal of the Renin-Angiotensin-Aldosterone System: JRAAS, 4(2), 51–61.
Heilig, C. W., Concepcion, L. A., Riser, B. L., Freytag, S. O., Zhu, M., & Cortes, P. (1995). Overexpression of glucose transporters in rat mesangial cells cultured in a normal glucose milieu mimics the diabetic phenotype. The Journal of Clinical Investigation, 96(4), 1802–1814.
Holtzman, N. A., & Marteau, T. M. (2000). Will genetics revolutionize medicine. New England Journal of Medicine, 343(2), 141–144.
Hostetter, T. H. (2003). Hyperfiltration and glomerulosclerosis. Seminars in Nephrology, 23(2), 194–199.
Imperatore, G., Hanson, R. L., Pettitt, D. J., Kobes, S., Bennett, P. H., & Knowler, W. C. (1998). Sib-pair linkage analysis for susceptibility genes for microvascular complications among Pima Indians with type 2 diabetes. Pima Diabetes Genes Group. Diabetes, 47(5), 821–830.
Ito, A., Uriu, K., Inada, Y., Qie, Y. L., Takagi, I., Ikeda, M., Hashimoto, O., Suzuka, K., Eto, S., Tanaka, Y., & Kaizu, K. (2001). Inhibition of neuronal nitric oxide synthase ameliorates renal hyper filtration in streptozotocin-induced diabetic rat. Journal of Laboratory and Clinical Medicine, 138(3), 177–185.
Jacobsen, P. K. (2005). Preventing end stage renal disease in diabetic patients—Genetic aspect (part I). Journal of the Renin-Angiotensin-Aldosterone System, 6(1), 1–14.
Karter, A. J., Ferrara, A., Liu, J. Y., Moffet, H. H., Ackerson, L. M., & Selby, J. V. (2002). Ethnic disparities in diabetic complications in an insured population. Journal of the American Medical Association, 287(19), 2519–2527.
Kim, J. H., Shin, H. D., Park, B. L., Moon, M. K., Cho, Y. M., Hwang, Y. H., Oh, K. W., Kim, S. Y., Lee, H. K., Ahn, C., & Park, K. S. (2006). SLC12A3 (solute carrier family 12 member [sodium/chloride] 3) polymorphisms are associated with end-stage renal disease in diabetic nephropathy. Diabetes, 55(3), 843–848.
Kimmelstiel, P., & Wilson, C. (1936). Intercapillary lesions in the glomeruli of the kidney. The American Journal of Pathology, 12(1), 83.
Kramer, H. J., Nguyen, Q. D., Curhan, G., & Hsu, C. Y. (2003). Renal insufficiency in the absence of albuminuria and retinopathy among adults with type 2 diabetes mellitus. Journal of the American Medical Association, 289(24), 3273–3277.
Lin, J. S., & Susztak, K. (2016). Podocytes: The weakest link in diabetic kidney disease. Current Diabetes Reports, 16(5), 1–9.
Lin, G., Wang, Z., Wang, L., Lau, Y. L., & Yang, W. (2008). Identification of linked regions using high-density SNP genotype data in linkage analysis. Bioinformatics, 24(1), 86–93.
Liu, R., Lee, K., & He, J. C. (2015). Genetics and epigenetics of diabetic nephropathy. Kidney Diseases, 1(1), 42–51.
MacIsaac, R. J., Jerums, G., & Ekinci, E. I. (2017). Effects of glycaemic management on diabetic kidney disease. World Journal of Diabetes, 8(5), 172.
Magee, C., Grieve, D. J., Watson, C. J., & Brazil, D. P. (2017). Diabetic nephropathy: A tangled web to unweave. Cardiovascular Drugs and Therapy, 31(5–6), 579–592.
Mclennan, S. V., Fisher, E., Martell, S. Y., Death, A. K., Williams, P. F., Lyons, J. G., & Yue, D. K. (2000). Effects of glucose on matrix metalloproteinase and plasmin activities in mesangial cells: Possible role in diabetic nephropathy. Kidney International, 58, 81–87.
Millis, M. P., Bowen, D., Kingsley, C., Watanabe, R. M., & Wolford, J. K. (2007). Variants in the plasmacytoma variant translocation gene (PVT1) are associated with end-stage renal disease attributed to type 1 diabetes. Diabetes, 56(12), 3027–3032.
Mishra, R., Emancipator, S. N., Kern, T., & Simonson, M. S. (2005). High glucose evokes an intrinsic proapoptotic signaling pathway in mesangial cells. Kidney International, 67(1), 82–93.
Mohan, V., Shanthirani, C. S., & Deepa, R. (2003). Glucose intolerance (diabetes and IGT) in a selected South Indian population with special reference to family history, obesity and lifestyle factors: The Chennai Urban Population Study (CUPS 14). The Journal of the Association of Physicians of India., 51, 771–777.
Mohan, V., Sandeep, S., Deepa, R., Shah, B., & Varghese, C. (2007). Epidemiology of type 2 diabetes: Indian scenario. The Indian Journal of Medical Research, 125(3), 217–230.
Molitch, M. E., DeFronzo, R. A., Franz, M. J., & Keane, W. F. (2004). Nephropathy in diabetes. Diabetes Care, 27, 79.
Möllsten, A., Vionnet, N., Forsblom, C., Parkkonen, M., Tarnow, L., Hadjadj, S., Marre, M., Parving, H. H., & Groop, P. H. (2011). A polymorphism in the angiotensin II type 1 receptor gene has different effects on the risk of diabetic nephropathy in men and women. Molecular Genetics and Metabolism, 103(1), 66–70.
Mooyaart, A. L., Valk, E. J., van Es, L. A., Bruijn, J. A., de Heer, E., Freedman, B. I., Dekkers, O. M., & Baelde, H. J. (2011). Genetic associations in diabetic nephropathy: A meta-analysis. Diabetologia, 54(3), 544–553.
Nakagawa, T., Sato, W., Glushakova, O., Heinig, M., Clarke, T., Campbell-Thompson, M., Yuzawa, Y., Atkinson, M. A., Johnson, R. J., & Croker, B. (2007). Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. Journal of the American Society of Nephrology, 18(2), 539–550.
Narang, A., Roy, R. D., Chaurasia, A., Mukhopadhyay, A., Mukerji, M., Dash, D., & Indian Genome Variation Consortium. (2010). IGVBrowser–A genomic variation resource from diverse Indian populations. Database.
Nazar, C. M. (2014). Diabetic nephropathy; principles of diagnosis and treatment of diabetic kidney disease. Journal of Nephropharmacology, 3(1), 15.
Nelson, R. G., Newman, J. M., Knowler, W. C., Sievers, M. L., Kunzelman, C. L., Pettitt, D. J., Moffett, C. D., Teutsch, S. M., & Bennett, P. H. (1988). Incidence of end-stage renal disease in type 2 (non-insulin-dependent) diabetes mellitus in Pima Indians. Diabetologia, 31(10), 730–736.
Nishi, S., Ueno, M., Hisaki, S., et al. (2000). Ultrastructural characteristics of diabetic nephropathy. Medical Electron Microscopy, 33, 65–73.
Nishikawa, T., Edelstein, D., & Brownlee, M. (2000). The missing link: A single unifying mechanism for diabetic complications. Kidney International, 58, 26–30.
Nitta, K., Okada, K., Yanai, M., & Takahashi, S. (2013). Aging and chronic kidney disease. Kidney and Blood Pressure Research, 38(1), 109–120.
Noiri, E., Satoh, H., Taguchi, J. I., Brodsky, S. V., Nakao, A., Ogawa, Y., Nishijima, S., Yokomizo, T., Tokunaga, K., & Fujita, T. (2002). Association of eNOS Glu298Asp polymorphism with end-stage renal disease. Hypertension, 40(4), 535–540.
Nomiyama, T., Tanaka, Y., Piao, L., Nagasaka, K., Sakai, K., Ogihara, T., Nakajima, K., Watada, H., & Kawamori, R. (2003). The polymorphism of manganese superoxide dismutase is associated with diabetic nephropathy in Japanese type 2 diabetic patients. Journal of Human Genetics, 48(3), 138–141.
Olesen, R., Wejse, C., Velez, D. R., Bisseye, C., Sodemann, M., Aaby, P., Rabna, P., Worwui, A., Chapman, H., Diatta, M., & Adegbola, R. A. (2007). DC-SIGN (CD209), pentraxin 3 and vitamin D receptor gene variants associate with pulmonary tuberculosis risk in West Africans. Genes & Immunity, 6, 456–467.
Parving, H. H., Mauer, M., Fioretto, P., Rossing, P., & Ritz, E. (2011). Diabetic nephropathy. In Brenner and Rector’s the Kidney. WB Saunders Company.
Patnala, R., Clements, J., & Batra, J. (2013). Candidate gene association studies: A comprehensive guide to useful in silico tools. BMC Genetics, 14(1), 1–1.
Pezzolesi, M. G., Poznik, G. D., Mychaleckyj, J. C., Paterson, A. D., Barati, M. T., Klein, J. B., Ng, D. P., Placha, G., Canani, L. H., Bochenski, J., & Waggott, D. (2009). Genome-wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes, 58(6), 1403–1410.
Placha, G., Canani, L. H., Warram, J. H., & Krolewski, A. S. (2005). Evidence for different susceptibility genes for proteinuria and ESRD in type 2 diabetes. Advances in Chronic Kidney Disease, 12(2), 155–169.
Prasad, P., Tiwari, A. K., Kumar, K. P., Ammini, A. C., Gupta, A., Gupta, R., & Thelma, B. K. (2007). Association of TGFβ1, TNFα, CCR2 and CCR5 gene polymorphisms in type-2 diabetes and renal insufficiency among Asian Indians. BMC Medical Genetics, 8(1), 20.
Pugh, J. A., Stern, M. P., Haffner, S. M., Eifler, C. W., & Zapata, M. (1988). Excess incidence of treatment of end-stage renal disease in Mexican Americans. American Journal of Epidemiology, 127(1), 135–144.
Qian, Y., Feldman, E., Pennathur, S., Kretzler, M., & Brosius, F. C. (2008). From fibrosis to sclerosis: Mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes, 57(6), 1439–1445.
Ramadan, R. A., Zaki, A. M., Magour, G. M., Zaki, M. A., Aglan, S. A., Madkour, M. A., & Shamseya, M. M. (2016). Association of XbaI GLUT1 polymorphism with susceptibility to type 2 diabetes mellitus and diabetic nephropathy. American Journal of Molecular Biology, 6, 71–78.
Ravikumar, P., Bhansali, A., Walia, R., Shanmugasundar, G., & Ravikiran, M. (2011). Alterations in HbA1c with advancing age in subjects with normal glucose tolerance: Chandigarh Urban Diabetes Study (CUDS). Diabetic Medicine, 28(5), 590–594.
Reeves, W. B., & Andreoli, T. E. (2000). Transforming growth factor β contributes to progressive diabetic nephropathy. Proceedings of the National Academy of Sciences, 97(14), 7667–7669.
Saeedi, P., Petersohn, I., Salpea, P., Malanda, B., Karuranga, S., Unwin, N., Colagiuri, S., Guariguata, L., Motala, A. A., Ogurtsova, K., & Shaw, J. E. (2019). Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the international diabetes federation diabetes atlas. Diabetes Research and Clinical Practice, 157, 107843.
Santos, K. G., Crispim, D., Canani, L. H., Ferrugem, P. T., Gross, J. L., & Roisenberg, I. (2011). Association of eNOS gene polymorphisms with renal disease in Caucasians with type 2 diabetes. Diabetes Research and Clinical Practice, 91(3), 353–362.
Satchell, S. C., & Tooke, J. E. (2008). What is the mechanism of microalbuminuria in diabetes: A role for the glomerular endothelium. Diabetologia, 51(5), 714–725.
Satirapoj, B., Tasanavipas, P., & Supasyndh, O. (2019). Role of TCF7L2 and PPARG2 gene polymorphisms in renal and cardiovascular complications among patients with type 2 diabetes: A cohort study. Kidney Diseases, 5(4), 220–227.
Seman, N. A., He, B., Ojala, J. R., Mohamud, W. N., Östenson, C. G., Brismar, K., & Gu, H. F. (2014). Genetic and biological effects of sodium-chloride cotransporter (SLC12A3) in diabetic nephropathy. American Journal of Nephrology, 40(5), 408–416.
Sharma, K., Deelman, L., Madesh, M., Kurz, B., Ciccone, E., Siva, S., Hu, T., Zhu, Y., Wang, L., Henning, R., & Ma, X. (2003). Involvement of transforming growth factor-β in regulation of calcium transients in diabetic vascular smooth muscle cells. American Journal of Physiology-Renal Physiology, 285(6), 258–270.
Shaw, P. K., Baboe, F., van Es, L. A., van der Vijver, J. C., van de Ree, M. A., de Jonge, N., & Rabelink, T. J. (2006). South-Asian type 2 diabetic patients have higher incidence and faster progression of renal disease compared with Dutch-European diabetic patients. Diabetes Care, 29(6), 1383–1385.
Shaza, A. M., Rozina, G., Izham, M. M., & Azhar, S. S. (2005). Dialysis for end stage renal disease: A descriptive study in Penang Hospital. Medical Journal of Malaysia, 60(3), 320.
Sheetz, M. J., & King, G. L. (2002). Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. Journal of the American Medical Association, 288(20), 2579–2588.
Sladek, R., Rocheleau, G., Rung, J., Dina, C., Shen, L., Serre, D., Boutin, P., Vincent, D., Belisle, A., Hadjadj, S., & Balkau, B. (2007). A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature, 445(7130), 881–885.
Smith, M. W., Patterson, N., Lautenberger, J. A., Truelove, A. L., McDonald, G. J., Waliszewska, A., Kessing, B. D., Malasky, M. J., Scafe, C., Le, E., & De Jager, P. L. (2004). A high density admixture map for disease gene discovery in African Americans. The American Journal of Human Genetics, 74(5), 1001–1013.
Speeckaert, M. M., Speeckaert, R., Carrero, J. J., Vanholder, R., & Delanghe, J. R. (2013). Biology of human pentraxin 3 (PTX3) in acute and chronic kidney disease. Journal of Clinical Immunology, 33(5), 881–890.
Srivastava, S. K., Ramana, K. V., & Bhatnagar, A. (2005). Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocrine Reviews, 26(3), 380–392.
Sugimoto, H., Shikata, K., Matsuda, M., Kushiro, M., Hayashi, Y., Hiragushi, K., Wada, J., & Makino, H. (1998). Increased expression of endothelial cell nitric oxide synthase (ecNOS) in afferent and glomerular endothelial cells is involved in glomerular hyper filtration of diabetic nephropathy. Diabetologia, 41(12), 1426–1434.
Syed, R., Biyabani, M. U., Prasad, S., Deeba, F., & Jamil, K. (2011). Evidence of association of a common variant of the endothelial nitric oxide synthase gene (Glu298 Asp polymorphism) to coronary artery disease in South Indian population. Journal of Medical Genetics and Genomics, 3(1), 13–18.
Tandon, N., Anjana, R. M., Mohan, V., Kaur, T., Afshin, A., Ong, K., Mukhopadhyay, S., Thomas, N., Bhatia, E., Krishnan, A., & Mathur, P. (2018). The increasing burden of diabetes and variations among the states of India: The Global Burden of Disease Study 1990–2016. The Lancet Global Health, 6(12), 1352–1362.
Tang, Z. H., Zeng, F., & Zhang, X. Z. (2015). Human genetics of diabetic nephropathy. Renal Failure, 37(3), 363–371.
Teumer, A., Tin, A., Sorice, R., Gorski, M., Yeo, N. C., Chu, A. Y., Li, M., Li, Y., Mijatovic, V., Ko, Y. A., & Taliun, D. (2016). Genome-wide association studies identify genetic loci associated with albuminuria in diabetes. Diabetes, 65(3), 803–817.
Thomson, S. C., Vallon, V., & Blantz, R. C. (2004). Kidney function in early diabetes: The tubular hypothesis of glomerular filtration. American Journal of Physiology-Renal Physiology, 286(1), F8–F15.
Tiongco, R. E., Aguas, I. S., Cabrera, F. J., Catacata, M., Flake, C. C., Manao, M. A., & Policarpio, A. (2020). The role of the TNF-α gene -308 G/A polymorphism in the development of diabetic nephropathy: An updated meta-analysis. Diabetes and Metabolic Syndrome: Clinical Research and Reviews, 14(6), 2123–2129. https://doi.org/10.1016/j.dsx.2020.10.032
Unnikrishnan, R., Rema, M., Pradeepa, R., Deepa, M., Shanthirani, C. S., Deepa, R., & Mohan, V. (2007). Prevalence and risk factors of diabetic nephropathy in an urban South Indian population: The Chennai Urban Rural Epidemiology Study (CURES 45). Diabetes Care, 30(8), 2019–2024.
Uzun, S., Ozari, M., Gursu, M., Karadag, S., Behlul, A., Sari, S., Koldas, M., Demir, S., Karaali, Z., & Ozturk, S. (2016). Changes in the inflammatory markers with advancing stages of diabetic nephropathy and the role of pentraxin-3. Renal Failure, 38(8), 1193–1198.
Valladares-Salgado, A. D., Angeles-Martínez, J. A., Rosas, M., García-Mena, J. A., Utrera-Barillas, D. O., Gómez-Díaz, R. I., Escobedo-De La Peña, J. O., Parra, E. J., & Cruz, M. (2010). Association of polymorphisms within the transforming growth factor-β1 gene with diabetic nephropathy and serum cholesterol and triglyceride concentrations. Nephrology, 15(6), 644–648.
Vallon, V., & Komers, R. (2011). Pathophysiology of the diabetic kidney. Comprehensive Physiology, 1(3), 1175–1232.
Varghese, S., & Kumar, S. G. (2022). Role of eNOS and TGFβ1 gene polymorphisms in the development of diabetic nephropathy in type 2 diabetic patients in South Indian population. Egyptian Journal of Medical Human Genetics, 23(1), 10.
Veelken, R., Hilgers, K. F., Hartner, A., Haas, A., & BÖHMER KP, Sterzel RB. (2000). Nitric oxide synthase isoforms and glomerular hyper filtration in early diabetic nephropathy. Journal of the American Society of Nephrology, 11(1), 71–79.
Vionnet, N., Tregouët, D., Kazeem, G., Gut, I., Groop, P. H., Tarnow, L., Parving, H. H., Hadjadj, S., Forsblom, C., Farrall, M., & Gauguier, D. (2006). Analysis of 14 candidate genes for diabetic nephropathy on chromosome 3q in European populations: Strongest evidence for association with a variant in the promoter region of the adiponectin gene. Diabetes, 55(11), 3166–3174.
Vithian, K., & Hurel, S. (2010). Microvascular complications: Pathophysiology and management. Clinical Medicine, 10(5), 505.
Watanabe, Y., Kinoshita, A., Yamada, T., Ohta, T., Kishino, T., Matsumoto, N., Ishikawa, M., Niikawa, N., & Yoshiura, K. I. (2002). A catalog of 106 single-nucleotide polymorphisms (SNPs) and 11 other types of variations in genes for transforming growth factor-β1 (TGF-β1) and its signaling pathway. Journal of Human Genetics, 47(9), 478–483.
Wautier, M. P., Chappey, O., Corda, S., Stern, D. M., Schmidt, A. M., & Wautier, J. L. (2001). Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. American Journal of Physiology-Endocrinology and Metabolism, 280(5), 685–694.
Wei, L., Xiao, Y., Li, L., Xiong, X., Han, Y., Zhu, X., & Sun, L. (2018). The susceptibility genes in diabetic nephropathy. Kidney Diseases, 4(4), 226–237.
Weil, E. J., Lemley, K. V., Mason, C. C., Yee, B., Jones, L. I., Blouch, K., Lovato, T., Richardson, M., Myers, B. D., & Nelson, R. G. (2012). Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney International, 82(9), 1010–1017.
Williams, M. E. (2005). Diabetic nephropathy: The proteinuria hypothesis. American Journal of Nephrology, 25(2), 77–94.
Wolf, G., Butzmann, U., & Wenzel, U. O. (2003). The renin-angiotensin system and progression of renal disease: From hemodynamics to cell biology. Nephron Physiology, 93(1), 3–13.
Xu, M., Chen, X., Yan, L., Cheng, H., & Chen, W. (2008). Association between (AC) n dinucleotide repeat polymorphism at the 50-end of the aldose reductase gene and diabetic nephropathy: A meta-analysis. Journal of Molecular Endocrinology, 40, 243–251.
Yilmaz, M. I., Axelsson, J., Sonmez, A., Carrero, J. J., Saglam, M., Eyileten, T., Caglar, K., Kirkpantur, A., Celik, T., Oguz, Y., & Vural, A. (2009). Effect of renin angiotensin system blockade on pentraxin 3 levels in type-2 diabetic patients with proteinuria. Clinical Journal of the American Society of Nephrology, 4(3), 535–541.
Young, B. A., Maynard, C., & Boyko, E. J. (2003). Racial differences in diabetic nephropathy, cardiovascular disease, and mortality in a national population of veterans. Diabetes Care, 26(8), 2392–2399.
Zanchi, A., Moczulski, D. K., Hanna, L. S., Wantman, M., Warram, J. H., & Krolewski, A. S. (2000). Risk of advanced diabetic nephropathy in type 1 diabetes is associated with endothelial nitric oxide synthase gene polymorphism. Kidney International, 57(2), 405–413.
Zhang, R., Zhuang, L., Li, M., Zhao, W., Ge, X., Chen, Y., Wang, F., Wang, N., Bao, Y., Liu, L., & Liu, Y. (2018). Arg913Gln of SLC12A3 gene promotes development and progression of end-stage renal disease in Chinese type 2 diabetes mellitus. Molecular and Cellular Biochemistry, 437(1), 203–210.
Zhu, H., Yu, W., Xie, Y., Zhang, H., Bi, Y., & Zhu, D. (2017). Association of pentraxin 3 gene polymorphisms with susceptibility to diabetic nephropathy. Medical Science Monitor: International Medical Journal of Experimental and Clinical Research, 23, 428.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kulanthaivel, L., Ganesan, G., Kirubhanand, C., Subbaraj, G.K. (2023). Diabetic and Nephropathy. In: Noor, R. (eds) Advances in Diabetes Research and Management. Springer, Singapore. https://doi.org/10.1007/978-981-19-0027-3_5
Download citation
DOI: https://doi.org/10.1007/978-981-19-0027-3_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-0026-6
Online ISBN: 978-981-19-0027-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)