
Abstract
The discovery of the G-protein coupled-receptor (GPCR) CXCR4 as a major coreceptor of HIV-1 entry about three decades ago explained why the chemokine SDF-1/CXCL12 inhibits specific viral strains. The knowledge that RANTES, MlP-1α, and MlP-1β specifically inhibit other primary HIV-1 strains allowed the rapid discovery of CCR5 as second major viral coreceptor and explained why individuals with deletions in CCR5 are protected against sexual HIV-1 transmission. Here, we provide an update on endogenous ligands of GPCRs that act as endogenous inhibitors of HIV-1, HIV-2, and simian immunodeficiency virus (SIV) entry. In addition, we summarize the development of optimized derivatives of endogenous GPCR ligands and their perspectives as antiviral agents and beyond. Finally, we provide examples for other endogenous peptides that may contribute to our innate immune defense against HIV-1 and other viral pathogens and offer prospects for preventive or therapeutic development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
5.1 Introduction
Our innate immune response represents the first line of defense against viral pathogens. Detection of foreign viral invaders by immune sensors induces the interferon system and consequently increases expression of numerous antiviral factors as well as activation of immune-associated proteases. Antimicrobial peptides (AMPs) are an important component of the innate immune response (Ahmed et al. 2019; Sørensen et al. 2008). While many intracellular antiviral factors restrict viral replication in cells that are already infected, AMPs usually act outside of the cell and may destroy viral particles directly and/or protect uninfected cells against viral entry. Many human AMPs, such as defensins and cathelicidin LL-37, are positively charged and helical and best known for their broad antibacterial activity (Diamond et al. 2009; Wang et al. 2014). It has become clear, however, that AMPs are structurally more versatile than initially thought and also display antiviral and immunomodulatory activities (Vilas Boas et al. 2019; Pahar et al. 2020).
In the case of HIV-1 and related nonhuman primate lentiviruses, endogenous inhibitors may not only play a role in controlling viral replication but were also instrumental in elucidating the viral entry process. Since depletion of CD4+ T cells is a hallmark of HIV-1 infection in vivo, CD4 was identified as primary receptor soon after the discovery of this virus as causative agent of AIDS (Sattentau and Weiss 1988). While it also rapidly became clear that additional factors are required for viral entry, their identification took more than a decade (Alkhatib and Berger 2007). The breakthrough came with the discovery of CXCR4 as entry cofactor of HIV-1 strains causing strong cytopathic effects in immortalized T cell lines. CXCL12 (initially named SDF-1), the chemokine ligand of CXCR4, inhibited infection of some HIV-1 strains (Ahmed et al. 2019). Many other HIV-1 strains, however, were inhibited by CCL5 (initially named RANTES) and to a lesser extent by CCL3 (MlP-1α) and CCL4 (MlP-1β), which were all already known to interact with CCR5 (Lusso 2006). This knowledge allowed the rapid discovery of CCR5 as major coreceptor of primary HIV-1 strains (reviewed in: Alkhatib and Berger 2007). Subsequent studies showed that CCR5 plays the key role in virus transmission and during chronic infection, while CXCR4-tropic HIV-1 strains emerge in about half of all AIDS patients and are associated with rapid disease progression in the absence of combined antiretroviral therapy (cART) (Connor et al. 1997; Schuitemaker et al. 2011).
Since the initial discovery of CCR5 and CXCR4 as major coreceptors of HIV-1, several other GPCRs have been reported to mediate HIV-1 entry albeit with lower efficiency (Pollakis and Paxton 2012; Wetzel et al. 2018). It has also been established that HIV-2 and related simian immunodeficiency viruses (SIVs) are more promiscuous in coreceptor usage. A variety of chemokines and peptidic GPCR ligands have been reported to specifically inhibit HIV-2 and SIV entry (Fig. 5.1). Most but not all of them also regulate the physiological signaling function of the respective GPCRs and may thus play important roles in the trafficking and function of immune cells. In this review, we summarize key findings on endogenous peptide inhibitors of HIV-1 and related primate lentiviruses, with a focus on entry inhibitors. We also discuss their mechanism of action, the optimization of endogenous agents, and their potential prospects for preventive or therapeutic development and application.

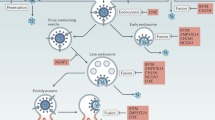
Schematic presentation of HIV and SIV entry and endogenous (poly)peptides inhibiting this process. CD4 binding of the viral glycoprotein (e.g., HIV-1 GP120) and subsequent interactions with GPCRs result in the liberation and membrane insertion of the GP41 fusion peptide (FP). Subsequent six-helix bundle formation triggers the fusion of viral and cellular membranes and ultimately leads to the release of the viral capsid into the cytosol of the host cell. Virus infection can be blocked at several stages by the indicated endogenous antiviral chemokines and peptides
5.2 Antiviral Host Defense Peptides
5.2.1 Defensins
Peptides exhibiting antimicrobial, antifungal, or antiviral activity are referred to as Host Defense Peptides (HDPs). These are commonly constitutively expressed in mucous tissue and induced early during immune activation as a first line of defense against human pathogens (Schröder and Harder 1999). Defensins form a diverse group of 30–40 amino acid long peptides sharing similar charge, morphology and antimicrobial properties (Shafee et al. 2016). While the α-defensin precursors DEFAs (Defensin, alpha) proteins, cluster on chromosome 8, β-defensin are encoded on a variety of genes and grouped based on their structure and function. All defensins are characterized by three intramolecular cysteine-disulfide bonds, β-sheet structure and their cationic and amphipathic properties (Bulet et al. 2004). The human α-defensins 1–3, and human β-defensin-2 and 3 inhibit replication of CCR5- and CXCR4-tropic strains of HIV-1, including several primary isolates in the low μM range (Hu et al. 2019; Quiñones-Mateu et al. 2003; Wu et al. 2005). Structural integrity of the three intramolecular cysteine-disulphide bonds, as well as side chain hydrophobicity, were shown to be critical for antiviral activity. Human α-defensins 1–3 and 5 are lectin-like and bind the glycosylation sites of the HIV-1 gp120, thereby inhibiting virion attachment. However, the antiviral activity of human α-defensin 5 remains controversial (Ding et al. 2013; Furci et al. 2012). α-defensin concentrations in human saliva vary between 1 and 10 μg/ml, being in the range of antimicrobial and antiviral active concentration (Gardner et al. 2009).
Of three known classes of defensins (α-, β-, and θ-defensin) only α- and β-defensins are transcribed in humans. Human θ-defensin is a pseudogene harboring a premature stop codon (Daher et al. 1986; Zhao et al. 2013). θ-defensins, which are expressed in monkeys, or synthesized based on the human pseudogene sequence, are named retrocyclins (Cole et al. 2002; Nguyen et al. 2003). Unlike α- and β-defensin, retrocyclins are circular peptides but contain the disulphide bonds that are characteristic for defensins. Retrocyclins were found to bind and restrict the HIV-1 glycoproteins gp120, gp41 and its cellular receptor CD4 due to its lectin-like ability to bind N-and O-linked carbohydrates, while having little to no effect on cell fusion of HIV-2 and SIV (Münk et al. 2003; Owen et al. 2004; Wang et al. 2003). Additionally, retrocyclins may bind the HIV-1 heptad repeats, thereby inhibiting 6-helix bundle formation of gp120 and virus-cell fusion (Cole et al. 2006; Gallo et al. 2006). The retrocyclin congener RC-101 was confirmed safe during in vivo application in pigtailed macaques. RC-101 was applied in the cervix and vagina of the primates as a quick-dissolving film, where it remained for several days and kept its activity against HIV-1 and SIV (Cole et al. 2010). Recent studies characterize the role of defensins in HIV response in human macrophage systems and connect the antiviral effect of defensins to GPCR mediated signaling pathways (Bharucha et al. 2021). It remains to be determined, whether defensins play a role in the innate defense against HIV-1 in vivo. However, their broad antiviral and antimicrobial effects make defensins interesting for future therapeutic applications (Park et al. 2018).
5.2.2 LL-37
Most HDPs are expressed as precursor proteins and proteolytic processing leads to the release of the bioactive peptides. A well-studied member of the cathelicidin protein family is hCAP-18. hCAP-18 can be processed in 16 different fragments some of which show antimicrobial and antiviral activity (Agerberth et al. 1995; Steinstraesser et al. 2005). The latter, in particular, has been described for LL-37, a 37-aa cationic peptide generated by proteinase 3 mediated cleavage of the C-terminal end of hCAP18. LL-37 has an α-helical structure and forms aggregates in solution (Shahmiri et al. 2016; Wang et al. 2014). Its amphipathic nature allows LL-37 to insert into lipid bilayers. Thus, unlike other HDPs, LL-37 is protected from proteolytic degradation (Oren et al. 1999). Further optimization of LL-37 led to the identification of FK-17 as minimal active antiviral peptide (Wang et al. 2008). The antibacterial activity of LL-37 is mediated by permeabilization of bacterial membranes. Cholesterol, which is absent in bacterial cell membranes, protects mammalian cells from LL-37 mediated pore formation (Brender et al. 2012; Sancho-Vaello et al. 2020). The antiviral mechanism of LL-37 is under debate. An inhibitory effect of LL-37 on the HIV-1 protease has been observed (Wong et al. 2011) but effects on membrane integrity of enveloped viruses have also been proposed (Wang et al. 2014). Current studies focus on the antimicrobial activity of LL-37 and its application in multivalent systems (Lakshmaiah Narayana et al. 2021; Mori et al. 2021). Notably, LL-37 was also found to directly interact with the HIV-1 cofactor CXCR4 (Pan et al. 2018; Podaza et al. 2020), but it is currently unclear whether LL-37 inhibits CXCR4-tropic HIV-1 infection.
5.3 Endogenous Ligands Targeting CXCR4-Mediated HIV Infection
C-X-C chemokine receptor type 4 (CXCR4) is a typical G-protein coupled receptor composed of seven transmembrane domains (Wu et al. 2010). It plays important roles in immunity, tissue regeneration and hematopoietic stem cell homeostasis (Pozzobon et al. 2016). CXCR4 dysfunction is associated with several malignancies, such as inflammatory diseases and cancer, making CXCR4 an important drug target (Pozzobon et al. 2016). Besides this, CXCR4 is also a major coreceptor for HIV-1 entry (Moore et al. 2004) (Fig. 5.1). HIV-1 infection is typically initiated by unspecific attachment of the virion to a host cell by its external gp120 envelope glycoprotein (Env). Subsequent binding to the primary CD4 receptor induces structural rearrangements in gp120 resulting in the interaction with viral coreceptors, mainly CXCR4 and CCR5, triggering conformational changes in gp41 that finally drive membrane fusion (reviewed in Chen 2019). CCR5-tropic HIV-1 variants dominate during acute and chronic infection. During or after AIDS progression a coreceptor switch or expansion is observed in some patients. CXCR4-tropic viruses are associated with a more rapid deterioration of the immune system leading to faster disease progression (Mosier 2008) in the absence of cART. The gp120 V3 loop determines coreceptor interaction and only a few amino acid changes are required to switch from CCR5 to CXCR4 coreceptor usage (De Jong et al. 1992). So far, it is largely unknown, which factors trigger the coreceptor switch (Connell et al. 2020; Regoes and Bonhoeffer 2005).
5.3.1 CXCL12
C-X-C motif chemokine 12 (CXCL12, formerly named SDF-1) is so far the only classical chemokine known to interact with CXCR4 and to block CXCR4-tropic HIV-1 infection (Bleul et al. 1996; Janssens et al. 2018). It is constitutively expressed in many tissues, especially the bone marrow and lymph nodes, where it acts as chemoattractant for lymphocytes (Nagasawa 2007). CXCL12 is encoded by a single gene and several splice variants are described with molecular weights between 8 and 14 kDa (Gleichmann et al. 2000; Yu et al. 2006). CXCL12 shares the common structure of chemokines: a disordered N-terminus followed by a globular core domain. Binding of CXCL12 to CXCR4 is initiated by interactions with the receptor N-terminus and then the binding pocket, which is shaped by the receptor transmembrane domains and extracellular loops (Wu et al. 2010; Xu et al. 2013). This so called “two-site”-binding model has also been implicated for gp120 interaction with CXCR4, which interacts with the N-terminus of CXCR4 and subsequently with the second and third extracellular loops of the receptor (Kalinina et al. 2013; Lin et al. 2003; Wu et al. 2010). The basic V3 loop may also penetrate the positively charged receptor binding pocket (Wu et al. 2010). Considering the similar binding modes, it is plausible that CXCL12 inhibits CXCR4-tropic HIV-1 by blocking access of the viral gp120 to the GPCR. However, CXCL12 also induces CXCR4 downmodulation and this effect correlates with the antiviral activity of CXCL12 isoforms, suggesting that receptor downmodulation contributes to its antiviral activity (Altenburg et al. 2010; Amara et al. 1997). CXCL12α and CXCL12β are the two most abundant isoforms in humans, both inhibiting CXCR4-tropic HIV-1 with IC50 values in the nanomolar range (Altenburg et al. 2007, 2010). CXCL12γ has been shown to have even more potent anti-HIV-1 activity due to increased affinity to CXCR4 and more efficient receptor internalization. However, this isoform is mainly expressed in the adult human heart and hardly detected in other tissues (Yu et al. 2006).
It is tempting to speculate that CXCL12 might be involved in HIV-1 transmission and pathogenesis. HIV-1 is mainly sexually transmitted and in most cases single, so called “transmitted/founder” (T/F) viruses establish infection (Parrish et al. 2013; Joseph et al. 2015). T/F viruses are almost exclusively CCR5-tropic, indicating a selective strong transmission barrier for CXCR4-tropic viruses (Grivel et al. 2010). The reason(s) for specific restriction of CXCR4-utilizing viruses are currently unclear. One plausible explanation is the presence of inhibitory CXCR4 ligands at sites of initial virus infection. CXCL12 is constitutively produced by epithelial vaginal cells and might contribute to selective inhibition of CXCR4-tropic viruses (Francis et al. 2016). However, CXCL12 levels in vaginal fluids vary and are frequently low (17.4–2071.5 pg/ml) (Francis et al. 2016), arguing against a major role of CXCL12 in preventing sexual transmission of CXCR4-tropic HIV-1 strains.
In infected individuals, CXCL12 plasma levels reach up to 10 ng/ml (Ikegawa et al. 2001). Concentrations may even be higher in tissues that are relevant for HIV-1 pathogenesis, such as lymph nodes and the gut (González et al. 2010; Müller et al. 2001). In addition, genetic CXCL12-polymorphisms affect disease progression (Modi et al. 2005; van Rij et al. 1998; Winkler 1998) suggesting that CXCL12 may restrict CXCR4-tropic HIV-1 in vivo. However, other studies did not confirm a role of CXCL12 in viral pathogenesis (Brambilla et al. 2000; Ioannidis 2001; Mehlotra et al. 2015; Petersen et al. 2005; Watanabe et al. 2003; Wei et al. 2018). A more recent study identified CXCR4-tropic HIV-1 variants that are resistant to inhibition by CXCL12 (Armani-Tourret et al. 2021). These variants emerged in late stage AIDS patients with low CD4 T cell counts and may show an enhanced ability to infect naive CD4 T cells surrounded by CXCL12 (Armani-Tourret et al. 2021). Altogether, the relevance of CXCL12 in viral transmission, propagation, and pathogenesis is far from clear and warrants further studies.
The identification of CXCL12 as potent inhibitor of CXCR4-tropic HIV-1 infection stimulated research to develop CXCL12-based antiviral agents for therapeutic approaches. N-terminal truncations and sequence modifications allowed to design CXCL12 analogs lacking agonistic and thus pro-inflammatory activity (Crump 1997; Heveker et al. 1998). Findings showing that not only the CXCL12 N-terminus but also residues in the loop region (Crump 1997) and the C-terminal α-helix (Luo et al. 1999a, b) contribute to receptor interaction led to the design of more sophisticated CXCL12-derivatives (Tudan et al. 2002). The lead compound, CTCE0021 is composed of CXCL12(5–14) linked to CXCL12(55–67) by a four-glycine linker mimicking the distance between the N- and C-terminal regions of CXCL12. In the optimized CTCE0214 derivative, the α-helical structure was stabilized by lactamization leading to enhanced receptor binding and the peptide was cyclized to improve plasma stability (Zhong et al. 2004). Furthermore, modifications and dimerization of the N-terminus allowed to convert derivatives into CXCR4 antagonists (Faber et al. 2007; Loetscher et al. 1998). To our best knowledge, none of these CXCL12-derived peptides has been evaluated as therapeutic agents against CXCR4-tropic HIV-1 in advanced clinical trials. Among other reasons, lack of oral bioavailability of CXCL12-derived peptides may have accounted for the termination of further development as antiviral drugs. However, some CXCL12 derivatives showed potent anti-inflammatory properties or mobilized stem cells in vivo and may be further developed for other CXCR4-linked diseases (Fan et al. 2012; Zhong et al. 2004).
5.3.2 EPI-X4
A second CXCR4 ligand with antiviral activity is EPI-X4. This peptide was identified in a fraction of a peptide library derived from human hemofiltrate that selectively inhibited CXCR4-tropic HIV-1 infection (Zirafi et al. 2015). The active compound turned out to be an 1832 Da and 16 amino acid long peptide derived from serum albumin (position 408–423), which was termed EPI-X4 (Endogenous Peptide Inhibitor of CXCR4). EPI-X4 is generated from human serum albumin under acidic conditions by aspartic proteases (e.g., Cathepsin D and E) (Buske et al. 2015; Gilg et al. 2021, Mohr et al. 2015; Zirafi et al. 2016). The peptide is evolutionary conserved and interacts with the CXCR4 binding pocket thereby antagonizing CXCL12-induced signaling and cell migration (Sokkar et al. 2021; Zirafi et al. 2015). In addition, EPI-X4 suppresses basal CXCR4 signaling, and thus also acts as inverse agonist of the receptor (Zirafi et al. 2015). Notably, EPI-X4 only interacts with CXCR4 but no other GPCRs including CXCR7. Thus, this peptide is a highly selective inhibitor of CXCR4 function. However, its physiological role remains to be clarified.
EPI-X4 not only antagonizes CXCR4 but also prevents CXCR4-tropic HIV-1 infection in cell culture with an IC50 value of ~10–20 μg/ml, while having no effect on CCR5-tropic HIV-1 infection (Harms et al. 2020a, b; Zirafi et al. 2015). EPI-X4 interacts with the CXCR4 binding pocket via its seven N-terminal amino acids, presumably blocking access of the viral glycoprotein to the coreceptor (Sokkar et al. 2021). The CXCR4 antagonizing peptide is not detectable at relevant concentrations in plasma or serum of healthy individuals or HIV-1 patients (Mohr et al. 2015; Zirafi et al. 2015), suggesting that EPI-X4 does not play a major role in controlling CXCR4-tropic HIV-1 infection in vivo (Mohr et al. 2015; Zirafi et al. 2015). However, it is currently not known whether EPI-X4 may also be locally produced in lymphoid tissues, the major sites of viral replication, and affect CXCR4-tropic HIV-1. Notably, high amounts of EPI-X4 sufficient to block CXCR4-tropic HIV-1 can be easily generated upon acidification of plasma, which activates proteolytic digestion of the abundant precursor albumin (Mohr et al. 2015; Müller et al. 2016; Zirafi et al. 2015). Acidic pH values are also characteristic for vaginal fluid (Boskey et al. 1999). Thus, it is conceivable that EPI-X4 might be locally generated from albumin-rich semen in the acidic environment of the vaginal tract, where the peptide might selectively restrict CXCR4-tropic HIV-1 upon sexual intercourse.
EPI-X4 is a promising candidate for further development as CXCR4 antagonist to treat CXCR4-tropic HIV-1 infection or other CXCR4-linked diseases (Buske et al. 2015; Zirafi et al. 2015). EPI-X4 is not cytotoxic, can be easily modified, acts as antagonist and inverse agonist of CXCR4, and was shown to reduce airway inflammation in a mouse asthma model without having side effects (Zirafi et al. 2015). Based on computational modeling and empiric approaches, EPI-X4 derivatives with increased plasma stability and reduced size (<1000 Da) were developed, that antagonize CXCR4 and inhibit CXCR4-tropic HIV-1 infection in the low nanomolar range (Harms et al. 2020a, b; Sokkar et al. 2021; Zirafi et al. 2015). The optimized EPI-X4 derivatives WSC02 and JM#21 were successfully tested in preclinical mouse models of Waldenström’s macroglobulinemia (a CXCR4-linked disease with constitutive overactivation of the receptor) and acute myeloid leukemia (Kaiser et al. 2021), as well as atopic dermatitis, and allergic asthma (Harms et al. 2020a, b). Currently, they are evaluated as antiviral agents against CXCR4-tropic HIV-1 in humanized mice. The small size of some improved EPI-X4 with molecular weights below 1000 Da might also pave the way for future oral administration (Sokkar et al. 2021).
5.3.3 Other CXCR4 Ligands
CXCL12 was long-thought to be the only chemokine ligand of CXCR4. In 2007, however, the lymphokine macrophage migration inhibitory factor (MIF) was reported as novel interaction partner for CXCR4 and CD74 (Bernhagen et al. 2007). However, in contrast to CXCL12, addition of MIF to HIV-1 infected cell cultures promoted viral replication independently of coreceptor usage (Regis et al. 2010), demonstrating that MIF does not inhibit CXCR4-tropic HIV-1 strains.
Ubiquitin is a small regulatory protein found in most human tissues (Mayor and Peng 2012). Extracellular ubiquitin functions as an immune modulator with anti-inflammatory properties (Majetschak 2011) and was shown to bind and agonize CXCR4 (Saini et al. 2010a, b). Similar to CXCL12, ubiquitin evoked signal transduction via CXCR4, and CXCR4-expressing cells migrated along a ubiquitin gradient (Saini et al. 2010a, b). However, ubiquitin did not inhibit CXCR4-tropic HIV-1 at concentrations up to 10 μM (Saini et al. 2011). Since extracellular ubiquitin levels usually do not exceed concentrations of 10 nM (Majetschak 2011), it does most likely not contribute to the control of HIV-1 in vivo.
As discussed in Sect. 5.2.2, LL-37 is an antimicrobial peptide which may also inhibit viral infections (Pahar et al. 2020). It has been reported that LL-37 affects CXCR4 distribution on the cell surface and its incorporation into lipid rafts (Wu et al. 2012). Interestingly, LL-37 induces CXCR4 signaling and internalization via interaction with an alternative binding site on the receptor, revealing it as a novel agonist for CXCR4 (Pan et al. 2018; Podaza et al. 2020). However, whether LL-37 binding to CXCR4 inhibits CXCR4-tropic HIV-1 infection is unclear.
Another host defense factor interacting with CXCR4 is the human β-defensin-3 (hBD-3) (Feng et al. 2006, 2013). hBD-3 acts as CXCR4 antagonist and inhibits CXCL12-induced receptor signaling and chemotaxis toward CXCL12. It has been reported that hBD-3 also reduces the infection by CXCR4-tropic HIV-1 in vitro, albeit only at high concentrations (20 μg/ml) that may not be reached in the human body (Feng et al. 2013; Sun et al. 2005).
More recently, the chemokine CXCL17 was described as novel ligand of CXCR4 (White et al. 2019, 2021). CXCL17 is expressed by mucosal tissues where it is presumably involved in innate immune response and angiogenesis (Burkhardt et al. 2012). CXCL17 inhibits CXCR4-mediated signaling and ligand binding via a glycosaminoglycan-containing accessory protein. If CXCL17 also has an impact on CXCR4-tropic HIV-1 and contributes to HIV-1 pathogenesis remains to be determined.
5.4 Chemokine Ligands of CCR5 Inhibit CCR5-Tropic HIV-1
C–C chemokine receptor type 5 (CCR5) is a GPCR possessing the typical seven transmembrane helical structure of all GPCRs. It is expressed on cells of the immune system including helper and effector T lymphocytes and antigen-resenting cells. CCR5 and its chemokine ligands are involved in immune regulation and inflammatory processes and have been associated with the pathogenesis of several inflammatory diseases (Vangelista and Vento 2018). CCR5-targeting strategies gained significant interest and the small molecule CCR5 antagonist Maraviroc has been approved by the FDA for HIV-1 treatment (Lieberman-Blum et al. 2008).
CCR5 interacts with several chemokines and most of them have been reported to inhibit infection by CCR5-tropic HIV-1. The first chemokines that were described to block HIV-1 infection were the CD8 T cell derived inflammatory proteins CCL3 (MIP-1β, i.e., macrophage inflammatory protein 1 beta), CCL4 (MIP-1α), the respective isoforms CCL3L1 and CCL4L1, and CCL5 (RANTES, i.e., regulated on activation, normal T expressed and secreted) (McBrien et al. 2018). It was later discovered that also CCL2, CCL7, CCL8, CCL11, CCL13, CCL14, and CCL16 are ligands for CCR5, of which CCL7, and the latter two were the only ones not reported to reduce CCR5-tropic HIV-1 infection (Blain et al. 2007; Blanpain et al. 1999; Detheux et al. 2000). Among all CCR5 chemokine ligands, CCL3, CCL4, truncated versions of CCL14 (see below) and in particular CCL5 most efficiently inhibit CCR5-tropic HIV-1 (Blanpain et al. 1999; Detheux et al. 2000; Münch et al. 2002). As discussed above for CXCR4/CXCL12, inhibition of CCR5-tropic HIV-1 by chemokines involves two distinct mechanisms: (1) downmodulation of the CCR5 receptor from the cell surface, and (2) sterically hindrance of CCR5 interaction with the viral glycoprotein gp120 (Alkhatib et al. 1997; Blanpain et al. 1999; Cocchi et al. 1995; Oberlin et al. 1996). Although native CCR5-chemokines are described to bind to G-protein coupled CCR5 with high affinity, they appear to have low affinity to the uncoupled receptor. In contrast, CCR5-tropic HIV-1 interacts with its coreceptor independently of coupled G-protein. This difference might limit antiviral activity of native chemokines and explain why CCR5-tropic HIV-1 persist despite high chemokine production at inflammatory sites (Brelot and Chakrabarti 2018).
Another CCR5 ligand, CCL14, is converted into an active chemokine by proteolytic processing (Detheux et al. 2000). Full-length CCL14 is a weak ligand for CCR1 and lacks potent chemotactic activity (Tsou et al. 1998). It is a 74 amino acid protein that shares ~46% sequence identity with CCL3 and CCL4 (Detheux et al. 2000). A truncated form of CCL14, termed CCL14[9–74], was isolated from human hemofiltrate and shown to be a potent CCR1 and CCR5 agonist that blocks CCR5-tropic HIV-1 (Detheux et al. 2000). CCL14[9–74] targets the second extracellular loop (ECL-2) of CCR5, induces CCR5 internalization, and inhibits CCR5-tropic HIV-1 strains in primary T cells and macrophages (Münch et al. 2002). CCL14[9–74] promotes calcium flux and migration of T lymphocytes, eosinophils, and monocytes (Münch et al. 2002).
β-chemokine mRNA expression is markedly upregulated in HIV-1 infected patients (Trumpfheller et al. 1998) and genetic variations in CCR5 ligands or copy numbers might be linked to progression to AIDS (Brelot and Chakrabarti 2018). Also, it has been suggested that decreased CCL5-sensitivity of CCR5-tropic primary HIV-1 isolates correlates with CD4+ T cell decline and disease progression in infected patients (Karlsson et al. 2004; Kwa et al. 2003). However, findings are debated and it is presently unclear, if the direct inhibition of CCR5-tropic HIV-1 by respective chemokines influences the course of HIV-1 infection (Brelot and Chakrabarti 2018).
Based on the anti-HIV-1 activity of natural chemokine ligands, several groups developed analogues of CCL5 to treat or prevent HIV-1 infection. To avoid receptor mediated signaling that could lead to adverse effects, initial studies focused on the design of CCR5 antagonists. It has been shown that the N-terminus of CCL5 is critical for receptor interaction and signaling (Choi et al. 2012). One of the first CCL5-derived therapeutic peptides was CCL5(9–68). Deletion of the first eight N-terminal amino acids abrogated the signaling function but also reduced binding efficiency to CCR5 (Arenzana-Seisdedos et al. 1996). A second strategy for eliminating agonistic functions of CCL5 was the extension of the N-terminus by a methionine residue. Since [Met]CCL5 showed no agonistic activity, CCL5-derivatives were generated with N-terminal modifications mimicking the hydrophobic nature of methionine (Gaertner et al. 2008; Kawamura et al. 2004; Lederman 2004; Mack et al. 1998; Wilken et al. 1999). One of those derivatives was [5P12]CCL5, which revealed no calcium signaling activity and did not induce receptor internalization (Gaertner et al. 2008; Nedellec et al. 2011). [5P12]CCL5 protected rhesus macaques against vaginal challenge with simian/human immunodeficiency virus (SHIV) (Veazey et al. 2009) and successfully surpassed pharmacokinetic studies in a sheep model following vaginal administration (McBride et al. 2017). In addition, [5P12]CCL5, demonstrated unusually high thermal and biological stability and could be produced at low-cost (Cerini et al. 2016, 2017; McBride et al. 2017). Therefore, the construct is currently further developed as vaginal and rectal microbicide for prevention of sexual HIV-1 transmission (McBride et al. 2017, 2019a, b).
Other studies focused on new CCL5-derived therapeutic molecules, including other N-terminally modified variants (Hartley et al. 2003; Nardese et al. 2001), polymer-conjugated derivatives (Shao et al. 2005), or other derivatives of the endogenous sequence (Nishiyama et al. 1999; Secchi et al. 2012; Vangelista et al. 2006; Vyroubalova et al. 2006). One of those peptides [5P7]CCL5 has been used to determine a crystal structure of CCR5 in complex with the modified chemokine analogue showing occupation of all gp120 binding sites within the receptor (Zheng et al. 2017).
One concern of using chemokine-based therapeutic agents is their potential pro-inflammatory activity, which could lead to detrimental chronic activation of the immune system (Baggiolini 2001). In addition, immune activation might lead to increased susceptibility of activated T cells to HIV-1 infection (Kinter et al. 1998). Another point to consider is the broad specificity for different chemokine receptors for most of the CCR5 ligands. CCL5 additionally interacts with CCR1, CCR3, and GPR75, what might lead to unwanted therapy-related effects. Due to the lack of CCR5 signaling, high stability and tolerability in preclinical studies, [5P12]CCL5 might be a good candidate for further development as microbicide for the prevention of HIV-1 infection. However, despite 25 years of research, none of the chemokine derivatives successfully passed through phase II/III trials or were approved for the therapy of HIV-1/AIDS.
5.5 CysC Fragments Inhibit GPR15-Mediated HIV-2 and SIV Infection
The basic entry mechanism of HIV-1, HIV-2, and SIVs are highly similar but the latter show broader coreceptor usage. As described above, HIV-1 almost exclusively utilizes CCR5 and/or CXCR4. In comparison, HIV-2 frequently also uses the CXC-chemokine receptor 6 (CXCR6) and G protein-coupled receptor 15 (GPR15) in addition to CCR5 and CXCR4 for viral entry (Gilbert et al. 2003; Mörner et al. 1999; Popper et al. 1999; Reeves et al. 1999).
GPR15 has been proposed to be a key player in mucosal immunity as it is supposedly involved in the homing and trafficking of T cells to the inflamed mucosa of the gut (Fischer et al. 2016; Nguyen et al. 2015; Suply et al. 2017). It is a GPCR with a molecular weight of ~40.8 kDa expressed by e.g. lymphocytes, endothelial cells and synovial macrophages (Cartwright et al. 2014; Clayton et al. 2001; Pan et al. 2017). In addition to its physiological functions, GPR15 is an entry cofactor for HIV-2 and SIVs (Kiene et al. 2012; Unutmaz et al. 1998). In a recent study, C-terminal fragments of the abundant plasma protein Cystatin C, e.g. CysC95-146 (5914 Da), were identified as specific, neutral ligands of GPR15 that prevent lentiviral infections via this GPCR (Hayn et al. 2021). Cystatin C is small, basic protein expressed by all nucleated cells in the human body at constant rates (Grubb et al. 1985; Onopiuk et al. 2015; Zi and Xu 2018). In healthy individuals, Cystatin C plasma levels are ~0.1 μM, however, they can reach up to 0.5–0.7 μM under conditions of uremia and inflammation (Abrahamson et al. 1986). Notably, the plasma levels of CysC are increased in HIV infected individuals and decrease with the initiation of cART (Longenecker et al. 2015).
Antiviral Cystatin C fragments can be generated by proteolytic digestion of CysC with the proteases Cathepsin D, chymase, and Napsin A (Hayn et al. 2021). In vivo, these proteases are either secreted by specialized granules or lysosomal exocytosis during immune responses and activated under acidic conditions (Okajima 2013; Rodríguez et al. 1997; Yamamoto et al. 2012). The generation of these GPR15-specific peptides shows parallels to the generation of the CXCR4 antagonist EPI-X4 (Sect. 5.3.2) from serum albumin by cathepsin D and E under acidic conditions (Zirafi et al. 2015), as well as the CCR5 agonist (CCL14[9–74]) from CCL14 (Detheux et al. 2000b; Münch et al. 2002). It is therefore tempting to speculate that these GPCR-targeting peptides are locally generated and cooperate to inhibit GPR15-, CXCR4-, and CCR5-mediated lentiviral infection. SIVs are most likely infecting primate species for millions of years (Compton et al. 2013; Gifford et al. 2008) and the presence of such endogenous peptides might have been a driving force for promiscuous coreceptor usage.
In 2017, Suply et al. reported the discovery of a chemokine ligand of GPR15, a peptide they termed GPR15L (Suply et al. 2017). GPR15L is a polypeptide consisting of 57 amino acids which is expressed in the colon, stomach, tonsils, skin, and the cervix in humans (Suply et al. 2017). GPR15L affects downstream pathways upon binding to GPR15 (Ocón et al. 2017; Suply et al. 2017) but fails to inhibit GPR15-dependent infection by lentiviral pathogens (Hayn et al. 2021). This was unexpected since the agonistic chemokine ligands of CCR5 and CXCR4, e.g. CCL5 and CXCL12, inhibit CCR5- or CXCR4-tropic HIV infection, respectively (Bleul et al. 1996; Mosier et al. 1999; Oberlin et al. 1996). Conversely, antivirally active, C-terminal CysC peptides do not induce GPR15 signaling, making them neutral ligands of this GPCR (Hayn et al. 2021). These findings show that endogenous peptide ligands may prevent a detrimental activity of a GPCR (e.g., virus entry) without compromising its physiological signaling function.
5.6 VIRIP Blocks Fusion Peptide Insertion into the Cell Membrane
Many viral glycoproteins utilize hydrophobic fusion peptides as membrane anchors (Albertini et al. 2012; Söllner 2004; White et al. 2009). Upon exposure, those 20–30 amino acid, nonpolar domains perturbate the proximal layer of its target cell membrane (Agirre et al. 2000; McMahon and Gallop 2005). FPs are enriched in hydrophobic and aromatic amino acids. In almost all FP sequences repetitive patterns of two to four hydrophobic amino acids connected by glycine are found (Epand 2003). Fusion peptides mediate an essential step during host cell entry, which makes them useful targets for therapeutic approaches (Badani et al. 2014; Fumakia et al. 2016; Vigant et al. 2015).
VIRIP (VIRus-Inhibitory Peptide) is a 20 amino acid fragment of α1-AT (α1-antitrypsin). It blocks HIV-1 entry by binding to the gp41 FP, preventing the insertion of the FP into the cellular membrane and consequently the viral anchoring and fusion process. The antiviral peptide was identified by screening of a human hemofiltrate library and found to inhibit a wide variety of HIV-1 strains (Münch et al. 2007). Full-length α1-AT can reach plasma concentrations of up to 250 μM during infection or inflammation (Brantly et al. 1988). VIRIP is produced by the proteolytic digest of α1-AT by matrix metalloproteases and enriched in the plasma of HIV-1 patients during acute viremia (Kramer et al. 2010). Notably, a HIV-1-infected patient with severe α1-AT deficiency showed very rapid progression to AIDS (Potthoff et al. 2007) suggesting that α1-AT or VIRIP may contribute in suppressing HIV-1 replication.
The unique mode of action and high barrier to resistance made VIRIP an interesting candidate for further development. A structure activity study resulted in the generation of optimized VIRIP derivatives, such as VIR-576, with IC50 values of 10–50 nM, which is about two orders of magnitude more potent than endogenous VIRIP (Münch et al. 2007). The increased antiretroviral efficacy is due to additional hydrophobic residues enhancing its interaction with the gp41 FP and a cysteine bridge stabilizing the active conformation (Münch et al. 2007; Venken et al. 2011) VIR-576 was safe and efficient in a phase I/II trial and reduced the mean plasma viral loads by 1.23 log10 copies per ml without causing severe adverse effects (Forssmann et al. 2010). The genetic barrier for HIV-1 to overcome VIRIP mediated restriction is very high. During long-term passage of HIV-1 for more than 1 year with increasing concentrations of VIR-353 on MT-4 cells, resistance was achieved but associated with strongly reduced viral fitness (Gonzalez et al. 2011; Müller et al. 2018). Despite high efficacy in vitro, treatment with VIR-576 required infusion of large amounts of the peptide in vivo. Proteolytic degradation, tissue distribution, and/or absorption to the extracellular matrix or serum components may reduce the efficacy of VIR-576 in patients (Forssmann et al. 2010). However, this study provided evidence that endogenous peptides can be optimized and suppress viral loads and established the HIV-1 FP as therapeutic target. Studies to generate VIRIP derivates with further increased antiviral activity and improved pharmacokinetic properties are ongoing.
5.7 Conclusions and Perspectives
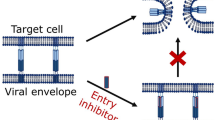
HIV-1 entry into target cells is a complex multistep process involving attachment, coreceptor binding, anchoring, and fusion. Essentially each step can be inhibited by (poly)peptides naturally existing in the human body. Defensins, retrocyclin and VIRIP directly target the virion and may prevent CD4 binding and viral fusion with the host cell. In comparison, chemokines and other GPCR ligands (EPI-X4, CysC fragments) inhibit entry through occupation and/or downregulation of the viral coreceptors. These endogenous antiviral factors may play important roles in the innate immune response against HIV-1. However, their contribution in controlling viral transmission and replication remains to be clarified. Notably, some of these peptides are generated by proteolytic processing of abundant precursor proteins with non-related function and inhibit HIV-1 entry by novel and unexpected mechanisms: Degradation of the protease inhibitor Cystatin C results in fragments that specifically bind GPR15 without agonizing or antagonizing the receptor but preventing GPR15-mediated infection. VIRIP is released from the acute phase protein α1 antitrypsin and blocks HIV-1 fusion by preventing the insertion of the viral fusion peptide into the cellular membrane. EPI-X4, a highly specific CXCR4 antagonist, is produced by processing of human serum albumin, the most abundant protein in the circulation and extravascular space. The common principle underlying the generation of these peptides is a low pH trigger, which activates acidic proteases that release the effector peptide. These endogenous viral entry inhibitors are particularly promising for development as drugs for HIV-1/AIDS or GPCR-linked diseases, because they are already evolutionarily optimized to perform their respective function(s) in humans. Furthermore, improved derivatives thereof should be better tolerated and are less immunogenic than those based on foreign antigens. Moreover, EPI-X4 and VIRIP are relatively small peptides and hence easy to modify by means of peptide synthesis and/or chemical modifications allowing to develop analogs with improved bioavailability and pharmacokinetic properties.
References
Abrahamson M, Barrett AJ, Salvesen G, Grubb A (1986) Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J Biol Chem 261(24):11282. https://doi.org/10.1016/s0021-9258(18)67380-6
Agerberth B, Gunne H, Odeberg J, Kogner P, Boman HG, Gudmundsson GH (1995) FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc Natl Acad Sci U S A 92(1):195–199. https://doi.org/10.1073/pnas.92.1.195
Agirre A, Flach C, Goñi FM, Mendelsohn R, Valpuesta JM, Wu F, Nieva JL (2000) Interactions of the HIV-1 fusion peptide with large unilamellar vesicles and monolayers. A cryo-TEM and spectroscopic study. Biochim Biophys Acta Biomembr 1467(1):153–164. https://doi.org/10.1016/S0005-2736(00)00214-5
Ahmed A, Siman-Tov G, Hall G, Bhalla N, Narayanan A (2019) Human antimicrobial peptides as therapeutics for viral infections. Viruses 11(8):704. https://doi.org/10.3390/v11080704
Albertini AAV, Baquero E, Ferlin A, Gaudin Y (2012) Molecular and cellular aspects of rhabdovirus entry. Viruses 4(1):117–139. https://doi.org/10.3390/v4010117
Alkhatib G, Berger EA (2007) HIV coreceptors: from discovery and designation to new paradigms and promise. Eur J Med Res 12(9):375–384
Alkhatib G, Locati M, Kennedy PE, Murphy PM, Berger EA (1997) HIV-1 coreceptor activity of CCR5 and its inhibition by chemokines: independence from G protein signaling and importance of coreceptor downmodulation. Virology 234(2):340–348. https://doi.org/10.1006/viro.1997.8673
Altenburg JD, Broxmeyer HE, Jin Q, Cooper S, Basu S, Alkhatib G (2007) A naturally occurring splice variant of CXCL12/stromal cell-derived factor 1 is a potent human immunodeficiency virus type 1 inhibitor with weak chemotaxis and cell survival activities. J Virol 81(15):8140–8148. https://doi.org/10.1128/JVI.00268-07
Altenburg JD, Jin Q, Alkhatib B, Alkhatib G (2010) The potent anti-HIV activity of CXCL12γ correlates with efficient CXCR4 binding and internalization. J Virol 84(5):2563–2572. https://doi.org/10.1128/JVI.00342-09
Amara A, Le Gall S, Schwartz O, Salamero J, Montes M, Loetscher P, Baggiolini M, Virelizier J-L, Arenzana-Seisdedos F (1997) HIV coreceptor downregulation as antiviral principle: SDF-1α–dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J Exp Med 186(1):139–146. https://doi.org/10.1084/jem.186.1.139
Arenzana-Seisdedos F, Virelizier J-L, Rousset D, Clark-Lewis I, Loetscher P, Moser B, Baggiolini M (1996) HIV blocked by chemokine antagonist. Nature 383(6599):400–400. https://doi.org/10.1038/383400a0
Armani-Tourret M, Zhou Z, Gasser R, Staropoli I, Cantaloube-Ferrieu V, Benureau Y, Garcia-Perez J, Pérez-Olmeda M, Lorin V, Puissant-Lubrano B, Assoumou L, Delaugerre C, Lelièvre J-D, Lévy Y, Mouquet H, Martin-Blondel G, Alcami J, Arenzana-Seisdedos F, Izopet J et al (2021) Mechanisms of HIV-1 evasion to the antiviral activity of chemokine CXCL12 indicate potential links with pathogenesis. PLoS Pathog 17(4):e1009526. https://doi.org/10.1371/journal.ppat.1009526
Badani H, Garry RF, Wimley WC (2014) Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity. Biochim Biophys Acta Biomembr 1838(9):2180–2197. https://doi.org/10.1016/j.bbamem.2014.04.015
Baggiolini M (2001) Chemokines in pathology and medicine. J Intern Med 250(2):91–104. https://doi.org/10.1046/j.1365-2796.2001.00867.x
Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med 13(5):587–596. https://doi.org/10.1038/nm1567
Bharucha JP, Sun L, Lu W, Gartner S, Garzino-Demo A (2021) Human beta-defensin 2 and 3 inhibit HIV-1 replication in macrophages. Front Cell Infect Microbiol 11(July):1–18. https://doi.org/10.3389/fcimb.2021.535352
Blain KY, Kwiatkowski W, Zhao Q, La Fleur D, Naik C, Chun T-W, Tsareva T, Kanakaraj P, Laird MW, Shah R, George L, Sanyal I, Moore PA, Demeler B, Choe S (2007) Structural and functional characterization of CC Chemokine CCL14. Biochemistry 46(35):10008–10015. https://doi.org/10.1021/bi700936w
Blanpain C, Migeotte I, Lee B, Vakili J, Doranz BJ, Govaerts C, Vassart G, Doms RW, Parmentier M (1999) CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood 94(6):1899–1905. https://doi.org/10.1182/blood.V94.6.1899
Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA (1996) The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382(6594):829–833. https://doi.org/10.1038/382829a0
Boskey ER, Telsch KM, Whaley KJ, Moench TR, Cone RA (1999) Acid production by vaginal flora in vitro is consistent with the rate and extent of vaginal acidification. Infect Immun 67(10):5170–5175. https://doi.org/10.1128/IAI.67.10.5170-5175.1999
Brambilla A, Villa C, Rizzardi G, Veglia F, Ghezzi S, Lazzarin A, Cusini M, Muratori S, Santagostino E, Gringeri A, Louie LG, Sheppard HW, Poli G, Michael NL, Pantaleo G, Vicenzi E (2000) Shorter survival of SDF1-3′A/3′A homozygotes linked to CD4 + T cell decrease in advanced human immunodeficiency virus type 1 infection. J Infect Dis 182(1):311–315. https://doi.org/10.1086/315650
Brantly ML, Paul LD, Miller BH, Falk RT, Wu M, Crystal RG (1988) Clinical features and history of the destructive lung disease associated with alpha-1-antitrypsin deficiency of adults with pulmonary symptoms. Am Rev Respir Dis 138(2):327–336. https://doi.org/10.1164/ajrccm/138.2.327
Brelot A, Chakrabarti LA (2018) CCR5 revisited: how mechanisms of HIV entry govern AIDS pathogenesis. J Mol Biol 430(17):2557–2589. https://doi.org/10.1016/j.jmb.2018.06.027
Brender JR, McHenry AJ, Ramamoorthy A (2012) Does cholesterol play a role in the bacterial selectivity of antimicrobial peptides? Front Immunol 3:1–4. https://doi.org/10.3389/fimmu.2012.00195
Bulet P, Stocklin R, Menin L (2004) Anti-microbial peptides: from invertebrates to vertebrates. Immunol Rev 198(1):169–184. https://doi.org/10.1111/j.0105-2896.2004.0124.x
Burkhardt AM, Tai KP, Flores-Guiterrez JP, Vilches-Cisneros N, Kamdar K, Barbosa-Quintana O, Valle-Rios R, Hevezi PA, Zuñiga J, Selman M, Ouellette AJ, Zlotnik A (2012) CXCL17 is a mucosal chemokine elevated in idiopathic pulmonary fibrosis that exhibits broad antimicrobial activity. J Immunol 188(12):6399–6406. https://doi.org/10.4049/jimmunol.1102903
Buske C, Kirchhoff F, Münch J (2015) EPI-X4, a novel endogenous antagonist of CXCR4. Oncotarget 6(34):35137–35138. https://doi.org/10.18632/oncotarget.6037
Cartwright A, Schmutz C, Askari A, Kuiper JH, Middleton J (2014) Orphan receptor GPR15/BOB is up-regulated in rheumatoid arthritis. Cytokine 67(2):53. https://doi.org/10.1016/j.cyto.2014.02.015
Cerini F, Gaertner H, Madden K, Tolstorukov I, Brown S, Laukens B, Callewaert N, Harner JC, Oommen AM, Harms JT, Sump AR, Sealock RC, Peterson DJ, Johnson SK, Abramson SB, Meagher M, Offord R, Hartley O (2016) A scalable low-cost cGMP process for clinical grade production of the HIV inhibitor 5P12-RANTES in Pichia pastoris. Protein Expr Purif 119:1–10. https://doi.org/10.1016/j.pep.2015.10.011
Cerini F, Offord R, McGowan I, Hartley O (2017) Stability of 5P12-RANTES, a candidate rectal microbicide, in human rectal lavage. AIDS Res Hum Retroviruses 33(8):768–777. https://doi.org/10.1089/aid.2016.0199
Chen B (2019) Molecular mechanism of HIV-1 entry. Trends Microbiol 27(10):878–891. https://doi.org/10.1016/j.tim.2019.06.002
Choi W-T, Nedellec R, Coetzer M, Colin P, Lagane B, Offord RE, Hartley O, Mosier DE (2012) CCR5 mutations distinguish N-terminal modifications of RANTES (CCL5) with agonist versus antagonist activity. J Virol 86(18):10218–10220. https://doi.org/10.1128/JVI.00353-12
Clayton F, Kotler DP, Kuwada SK, Morgan T, Stepan C, Kuang J, Le J, Fantini J (2001) Gp120-induced Bob/GPR15 activation: a possible cause of human immunodeficiency virus enteropathy. Am J Pathol 159(5):1933. https://doi.org/10.1016/S0002-9440(10)63040-4
Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P (1995) Identification of RANTES, MIP-1alpha, and MIP-1beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 270(5243):1811–1815. https://doi.org/10.1126/science.270.5243.1811
Cole AM, Hong T, Boo LM, Nguyen T, Zhao C, Bristol G, Zack JA, Waring AJ, Yang OO, Lehrer RI (2002) Retrocyclin: a primate peptide that protects cells from infection by T- and M-tropic strains of HIV-1. Proc Natl Acad Sci U S A 99(4):1813–1818. https://doi.org/10.1073/pnas.052706399
Cole AL, Yang OO, Warren AD, Waring AJ, Lehrer RI, Cole AM (2006) HIV-1 adapts to a retrocyclin with cationic amino acid substitutions that reduce fusion efficiency of gp41. J Immunol 176(11):6900–6905. https://doi.org/10.4049/jimmunol.176.11.6900
Cole AM, Patton DL, Rohan LC, Cole AL, Cosgrove-Sweeney Y, Rogers NA, Ratner D, Sassi AB, Lackman-Smith C, Tarwater P, Ramratnam B, Ruchala P, Lehrer RI, Waring AJ, Gupta P (2010) The formulated microbicide RC-101 was safe and antivirally active following intravaginal application in pigtailed macaques. PLoS One 5(11):e15111. https://doi.org/10.1371/journal.pone.0015111
Compton AA, Malik HS, Emerman M (2013) Host gene evolution traces the evolutionary history of ancient primate lentiviruses. Philos Trans R Soc B Biol Sci 368(1626):20120496. https://doi.org/10.1098/rstb.2012.0496
Connell BJ, Hermans LE, Wensing AMJ, Schellens I, Schipper PJ, van Ham PM, de Jong DTCM, Otto S, Mathe T, Moraba R, Borghans JAM, Papathanasopoulos MA, Kruize Z, Venter FWD, Kootstra NA, Tempelman H, Tesselaar K, Nijhuis M (2020) Immune activation correlates with and predicts CXCR4 co-receptor tropism switch in HIV-1 infection. Sci Rep 10(1):15866. https://doi.org/10.1038/s41598-020-71699-z
Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR (1997) Change in coreceptor use correlates with disease progression in HIV-1 infected individuals. J Exp Med 185(4):621–628. https://doi.org/10.1084/jem.185.4.621
Crump MP (1997) Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J 16(23):6996–7007. https://doi.org/10.1093/emboj/16.23.6996
Daher KA, Selsted ME, Lehrer RI (1986) Direct inactivation of viruses by human granulocyte defensins. J Virol 60(3):1068–1074. https://doi.org/10.1128/jvi.60.3.1068-1074.1986
De Jong JJ, De Ronde A, Keulen W, Tersmette M, Goudsmit J (1992) Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J Virol 66(11):6777–6780. https://doi.org/10.1128/JVI.66.11.6777-6780.1992
Detheux M, Ständker L, Vakili J, Münch J, Forssmann U, Adermann K, Pöhlmann S, Vassart G, Kirchhoff F, Parmentier M, Forssmann WG (2000) Natural proteolytic processing of hemofiltrate CC chemokine 1 generates a potent CC chemokine receptor (CCR)1 and CCR5 agonist with anti-HIV properties. J Exp Med 192(10):1501. https://doi.org/10.1084/jem.192.10.1501
Diamond G, Beckloff N, Weinberg A, Kisich KO (2009) The roles of antimicrobial peptides in innate host defense. Curr Pharm Des 15(21):2377–2392
Ding J, Tasker C, Valere K, Sihvonen T, Descalzi-Montoya DB, Lu W, Chang TL (2013) Anti-HIV activity of human defensin 5 in primary CD4+ T cells under serum-deprived conditions is a consequence of defensin-mediated cytotoxicity. PLoS One 8(9):1–11. https://doi.org/10.1371/journal.pone.0076038
Epand RM (2003) Fusion peptides and the mechanism of viral fusion. Biochim Biophys Biomembr 1614(1):116–121. https://doi.org/10.1016/S0005-2736(03)00169-X
Faber A, Roderburg C, Wein F, Saffrich R, Seckinger A, Horsch K, Diehlmann A, Wong D, Bridger G, Eckstein V, Ho AD, Wagner W (2007) J Biomed Biotechnol 2007:1–10. https://doi.org/10.1155/2007/26065
Fan H, Wong D, Ashton SH, Borg KT, Halushka PV, Cook JA (2012) Beneficial effect of a CXCR4 agonist in murine models of systemic inflammation. Inflammation 35(1):130–137. https://doi.org/10.1007/s10753-011-9297-5
Feng Z, Dubyak GR, Lederman MM, Weinberg A (2006) Cutting edge: human β defensin 3—a novel antagonist of the HIV-1 coreceptor CXCR4. J Immunol 177(2):782–786. https://doi.org/10.4049/jimmunol.177.2.782
Feng Z, Dubyak GR, Jia X, Lubkowski JT, Weinberg A (2013) Human β-defensin-3 structure motifs that are important in CXCR4 antagonism. FEBS J 280(14):3365–3375. https://doi.org/10.1111/febs.12328
Fischer A, Zundler S, Atreya R, Rath T, Voskens C, Hirschmann S, López-Posadas R, Watson A, Becker C, Schuler G, Neufert C, Atreya I, Neurath MF (2016) Differential effects of α4β7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut 65(10):1642. https://doi.org/10.1136/gutjnl-2015-310022
Forssmann W-G, The Y-H, Stoll M, Adermann K, Albrecht U, Tillmann H-C, Barlos K, Busmann A, Canales-Mayordomo A, Giménez-Gallego G, Hirsch J, Jiménez-Barbero J, Meyer-Olson D, Münch J, Pérez-Castells J, Ständker L, Kirchhoff F, Schmidt RE (2010) Short-term monotherapy in HIV-infected patients with a virus entry inhibitor against the gp41 fusion peptide. Sci Transl Med 2(63):63re3. https://doi.org/10.1126/scitranslmed.3001697
Francis SC, Hou Y, Baisley K, van de Wijgert J, Watson-Jones D, Ao TT, Herrera C, Maganja K, Andreasen A, Kapiga S, Coulton GR, Hayes RJ, Shattock RJ (2016) Immune activation in the female genital tract: expression profiles of soluble proteins in women at high risk for HIV infection. PLoS One 11(1):e0143109. https://doi.org/10.1371/journal.pone.0143109
Fumakia M, Yang S, Gu J, Ho EA (2016) Protein/peptide-based entry/fusion inhibitors as anti-HIV therapies: challenges and future direction. Rev Med Virol 26(1):4–20. https://doi.org/10.1002/rmv.1853
Furci L, Tolazzi M, Sironi F, Vassena L, Lusso P (2012) Inhibition of HIV-1 infection by human α-defensin-5, a natural antimicrobial peptide expressed in the genital and intestinal mucosae. PLoS One 7(9):1–10. https://doi.org/10.1371/journal.pone.0045208
Gaertner H, Cerini F, Escola J-M, Kuenzi G, Melotti A, Offord R, Rossitto-Borlat I, Nedellec R, Salkowitz J, Gorochov G, Mosier D, Hartley O (2008) Highly potent, fully recombinant anti-HIV chemokines: reengineering a low-cost microbicide. Proc Natl Acad Sci 105(46):17706–17711. https://doi.org/10.1073/pnas.0805098105
Gallo SA, Wang W, Rawat SS, Jung G, Waring AJ, Cole AM, Lu H, Yan X, Daly NL, Craik DJ, Jiang S, Lehrer RI, Blumenthal R (2006) θ-Defensins prevent HIV-1 Env-mediated fusion by binding gp41 and blocking 6-helix bundle formation. J Biol Chem 281(27):18787–18792. https://doi.org/10.1074/jbc.M602422200
Gardner MS, Rowland MD, Siu AY, Bundy JL, Wagener DK, Stephenson JL (2009) Comprehensive defensin assay for saliva. Anal Chem 81(2):557–566. https://doi.org/10.1021/ac801609r
Gifford RJ, Katzourakis A, Tristem M, Pybus OG, Winters M, Shafer RW (2008) A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proc Natl Acad Sci U S A 105(51):20362. https://doi.org/10.1073/pnas.0807873105
Gilbert PB, McKeague IW, Eisen G, Mullins C, Guéye-NDiaye A, Mboup S, Kanki PJ (2003) Comparison of HIV-1 and HIV-2 infectivity from a prospective cohort study in Senegal. Stat Med 22(4):573. https://doi.org/10.1002/sim.1342
Gilg A, Harms M, Olari L-R, Urbanowitz A-K, Bonig H, Münch J (2021) Absence of the CXCR4 antagonist EPI-X4 from pharmaceutical human serum albumin preparations. J Transl Med 19(1). https://doi.org/10.1186/s12967-021-02859-6
Gleichmann M, Gillen C, Czardybon M, Bosse F, Greiner-Petter R, Auer J, Müller HW (2000) Cloning and characterization of SDF-1γ, a novel SDF-1 chemokine transcript with developmentally regulated expression in the nervous system. Eur J Neurosci 12(6):1857–1866. https://doi.org/10.1046/j.1460-9568.2000.00048.x
González N, Bermejo M, Calonge E, Jolly C, Arenzana-Seisdedos F, Pablos JL, Sattentau QJ, Alcamí J (2010) SDF-1/CXCL12 production by mature dendritic cells inhibits the propagation of X4-tropic HIV-1 isolates at the dendritic cell-T-cell infectious synapse. J Virol 84(9):4341–4351. https://doi.org/10.1128/JVI.02449-09
Gonzalez E, Ballana E, Clotet B, Esté JA (2011) Development of resistance to VIR-353 with cross-resistance to the natural HIV-1 entry virus inhibitory peptide (VIRIP). AIDS 25(13):1575–1583. https://doi.org/10.1097/QAD.0b013e328348a733
Grivel J-C, Shattock RJ, Margolis LB (2010) Selective transmission of R5 HIV-1 variants: where is the gatekeeper? J Transl Med 9(suppl 1):S6. https://doi.org/10.1186/1479-5876-9-S1-S6
Grubb A, Simonsen O, Sturfelt G, Truedsson L, Thysell H (1985) Serum concentration of cystatin C, factor D and β2-microglobulin as a measure of glomerular filtration rate. Acta Med Scand 218(5):499. https://doi.org/10.1111/j.0954-6820.1985.tb08880.x
Harms M, Gilg A, Ständker L, Beer AJ, Mayer B, Rasche V, Gruber CW, Münch J (2020a) Microtiter plate-based antibody-competition assay to determine binding affinities and plasma/blood stability of CXCR4 ligands. Sci Rep 10(1):16036. https://doi.org/10.1038/s41598-020-73012-4
Harms M, Habib MMW, Nemska S, Nicolò A, Gilg A, Preising N, Sokkar P, Carmignani S, Raasholm M, Weidinger G, Kizilsavas G, Wagner M, Ständker L, Abadi AH, Jumaa H, Kirchhoff F, Frossard N, Sanchez-Garcia E, Münch J (2020b) An optimized derivative of an endogenous CXCR4 antagonist prevents atopic dermatitis and airway inflammation. Acta Pharm Sin B 11:2694. https://doi.org/10.1016/j.apsb.2020.12.005
Hartley O, Dorgham K, Perez-Bercoff D, Cerini F, Heimann A, Gaertner H, Offord RE, Pancino G, Debré P, Gorochov G (2003) Human immunodeficiency virus type 1 entry inhibitors selected on living cells from a library of phage chemokines. J Virol 77(12):6637–6644. https://doi.org/10.1128/JVI.77.12.6637-6644.2003
Hayn M, Blötz A, Rodríguez A, Vidal S, Preising N, Ständker L, Wiese S, Stürzel CM, Harms M, Gross R, Jung C, Kiene M, Jacob T, Pöhlmann S, Forssmann WG, Münch J, Sparrer KMJ, Seuwen K, Hahn BH, Kirchhoff F (2021) Natural cystatin C fragments inhibit GPR15-mediated HIV and SIV infection without interfering with GPR15L signaling. Proc Natl Acad Sci U S A 118(3):e2023776118. https://doi.org/10.1073/pnas.2023776118
Heveker N, Montes M, Germeroth L, Amara A, Trautmann A, Alizon M, Schneider-Mergener J (1998) Dissociation of the signalling and antiviral properties of SDF-1-derived small peptides. Curr Biol 8(7):369–376. https://doi.org/10.1016/S0960-9822(98)70155-1
Hu H, Di B, Tolbert WD, Gohain N, Yuan W, Gao P, Ma B, He Q, Pazgier M, Zhao L, Lu W (2019) Systematic mutational analysis of human neutrophil α-defensin HNP4. Biochim Biophys Acta Biomembr 1861(4):835–844. https://doi.org/10.1016/j.bbamem.2019.01.007
Ikegawa M, Yuan J, Matsumoto K, Herrmann S, Iwamoto A, Nakamura T, Matsushita S, Kimura T, Honjo T, Tashiro K (2001) Elevated plasma stromal cell-derived factor 1 protein level in the progression of HIV type 1 infection/AIDS. AIDS Res Hum Retroviruses 17(7):587–595. https://doi.org/10.1089/088922201300119680
Ioannidis JPA (2001) Effects of CCR5-Δ 32, CCR2-64I, and SDF-1 3′A alleles on HIV-1 disease progression: an international meta-analysis of individual-patient data. Ann Intern Med 135(9):782. https://doi.org/10.7326/0003-4819-135-9-200111060-00008
Janssens R, Struyf S, Proost P (2018) The unique structural and functional features of CXCL12. Cell Mol Immunol 15(4):299–311. https://doi.org/10.1038/cmi.2017.107
Joseph SB, Swanstrom R, Kashuba ADM, Cohen MS (2015) Bottlenecks in HIV-1 transmission: insights from the study of founder viruses. Nat Rev Microbiol 13(7):414–425. https://doi.org/10.1038/nrmicro3471
Kaiser LM, Harms M, Sauter D, Rawat VP, Glitscher M, Hildt E, Döhner K, Döhner H, Münch J, Buske C (2021) Acute myeloid leukemia cells are targeted by the naturally occurring CXCR4 antagonist EPI-X4. BioRxiv 2021.03.11.434944. https://doi.org/10.1101/2021.03.11.434944
Karlsson I, Antonsson L, Shi Y, Öberg M, Karlsson A, Albert J, Olde B, Owman C, Jansson M, FenyÖ EM (2004) Coevolution of RANTES sensitivity and mode of CCR5 receptor use by human immunodeficiency virus type 1 of the R5 phenotype. J Virol 78(21):11807–11815. https://doi.org/10.1128/JVI.78.21.11807-11815.2004
Kawamura T, Bruce SE, Abraha A, Sugaya M, Hartley O, Offord RE, Arts EJ, Zimmerman PA, Blauvelt A (2004) PSC-RANTES blocks R5 human immunodeficiency virus infection of langerhans cells isolated from individuals with a variety of CCR5 diplotypes. J Virol 78(14):7602–7609. https://doi.org/10.1128/JVI.78.14.7602-7609.2004
Kiene M, Marzi A, Urbanczyk A, Bertram S, Fisch T, Nehlmeier I, Gnirß K, Karsten CB, Palesch D, Münch J, Chiodi F, Pöhlmann S, Steffen I (2012) The role of the alternative coreceptor GPR15 in SIV tropism for human cells. Virology 433(1):73. https://doi.org/10.1016/j.virol.2012.07.012
Kinter A, Catanzaro A, Monaco J, Ruiz M, Justement J, Moir S, Arthos J, Oliva A, Ehler L, Mizell S, Jackson R, Ostrowski M, Hoxie J, Offord R, Fauci AS (1998) CC-chemokines enhance the replication of T-tropic strains of HIV-1 in CD4+ T cells: role of signal transduction. Proc Natl Acad Sci 95(20):11880–11885. https://doi.org/10.1073/pnas.95.20.11880
Kramer HB, Lavender KJ, Qin L, Stacey AR, Liu MKP, di Gleria K, Simmons A, Gasper-Smith N, Haynes BF, McMichael AJ, Borrow P, Kessler BM (2010) Elevation of intact and proteolytic fragments of acute phase proteins constitutes the earliest systemic antiviral response in HIV-1 infection. PLoS Pathog 6(5):e1000893. https://doi.org/10.1371/journal.ppat.1000893
Kwa D, Vingerhoed J, Boeser B, Schuitemaker H (2003) Increased in vitro cytopathicity of CC chemokine receptor 5–restricted human immunodeficiency virus type 1 primary isolates correlates with a progressive clinical course of infection. J Infect Dis 187(9):1397–1403. https://doi.org/10.1086/374650
Lakshmaiah Narayana J, Golla R, Mishra B, Wang X, Lushnikova T, Zhang Y, Verma A, Kumar V, Xie J, Wang G (2021) Short and robust anti-infective lipopeptides engineered based on the minimal antimicrobial peptide KR12 of human LL-37. ACS Infect Dis 7:1795. https://doi.org/10.1021/acsinfecdis.1c00101
Lederman MM (2004) Prevention of vaginal SHIV transmission in rhesus macaques through inhibition of CCR5. Science 306(5695):485–487. https://doi.org/10.1126/science.1099288
Lieberman-Blum SS, Fung HB, Bandres JC (2008) Maraviroc: a CCR5-receptor antagonist for the treatment of HIV-1 infection. Clin Ther 30(7):1228–1250. https://doi.org/10.1016/S0149-2918(08)80048-3
Lin G, Baribaud F, Romano J, Doms RW, Hoxie JA (2003) Identification of gp120 binding sites on CXCR4 by using CD4-independent human immunodeficiency virus type 2 Env proteins. J Virol 77(2):931–942. https://doi.org/10.1128/JVI.77.2.931-942.2003
Loetscher P, Gong J-H, Dewald B, Baggiolini M, Clark-Lewis I (1998) N-terminal peptides of stromal cell-derived factor-1 with CXC chemokine receptor 4 agonist and antagonist activities. J Biol Chem 273(35):22279–22283. https://doi.org/10.1074/jbc.273.35.22279
Longenecker CT, Kitch D, Sax PE, Daar ES, Tierney C, Gupta SK, McComsey GA (2015) Reductions in plasma cystatin c after initiation of antiretroviral therapy are associated with reductions in inflammation: ACTG A5224s. J Acquir Immune Defic Syndr 69(2):168. https://doi.org/10.1097/QAI.0000000000000557
Luo J, Luo Z, Zhou N, Hall JW, Huang Z (1999a) Attachment of C-terminus of SDF-1 enhances the biological activity of its N-terminal peptide. Biochem Biophys Res Commun 264(1):42–47. https://doi.org/10.1006/bbrc.1999.1476
Luo Z, Zhou N, Luo J, Hall JW, Huang Z (1999b) The role of positively charged residues in CXCR4 recognition probed with synthetic peptides. Biochem Biophys Res Commun 263(3):691–695. https://doi.org/10.1006/bbrc.1999.1441
Lusso P (2006) HIV and the chemokine system: 10 years later. EMBO J 25(3):447–456. https://doi.org/10.1038/SJ.EMBOJ.7600947
Mack M, Luckow B, Nelson PJ, Cihak J, Simmons G, Clapham PR, Signoret N, Marsh M, Stangassinger M, Borlat F, Wells TNC, Schlöndorff D, Proudfoot AEI (1998) Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J Exp Med 187(8):1215–1224. https://doi.org/10.1084/jem.187.8.1215
Majetschak M (2011) Extracellular ubiquitin: immune modulator and endogenous opponent of damage-associated molecular pattern molecules. J Leukoc Biol 89(2):205–219. https://doi.org/10.1189/jlb.0510316
Mayor U, Peng J (2012) Deciphering tissue-specific ubiquitylation by mass spectrometry. Methods Mol Biol 832:65–80. https://doi.org/10.1007/978-1-61779-474-2_3
McBride JW, Dias N, Cameron D, Offord RE, Hartley O, Boyd P, Kett VL, Malcolm RK (2017) Pharmacokinetics of the protein microbicide 5P12-RANTES in sheep following single-dose vaginal gel administration. Antimicrob Agents Chemother 61(10):e00965. https://doi.org/10.1128/AAC.00965-17
McBride JW, Boyd P, Dias N, Cameron D, Offord RE, Hartley O, Kett VL, Malcolm RK (2019a) Vaginal rings with exposed cores for sustained delivery of the HIV CCR5 inhibitor 5P12-RANTES. J Control Release 298:1–11. https://doi.org/10.1016/j.jconrel.2019.02.003
McBride JW, Malcolm RK, Dias N, Cameron D, Offord RE, Hartley O, Kett VL, Devlin B, Boyd P (2019b) Development and pharmacokinetics of a combination vaginal ring for sustained release of dapivirine and the protein microbicide 5P12-RANTES. Int J Pharm 564:207–213. https://doi.org/10.1016/j.ijpharm.2019.04.040
McBrien JB, Kumar NA, Silvestri G (2018) Mechanisms of CD8 + T cell-mediated suppression of HIV/SIV replication. Eur J Immunol 48(6):898–914. https://doi.org/10.1002/eji.201747172
McMahon HT, Gallop JL (2005) Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 438(7068):590–596. https://doi.org/10.1038/nature04396
Mehlotra RK, Hall NB, Bruse SE, John B, Blood Zikursh MJ, Stein CM, Siba PM, Zimmerman PA (2015) CCR2, CCR5, and CXCL12 variation and HIV/AIDS in Papua New Guinea. Infect Genet Evol 36:165–173. https://doi.org/10.1016/j.meegid.2015.09.014
Modi WS, Scott K, Goedert JJ, Vlahov D, Buchbinder S, Detels R, Donfield S, O’Brien SJ, Winkler C (2005) Haplotype analysis of the SDF-1 (CXCL12) gene in a longitudinal HIV-1/AIDS cohort study. Genes Immun 6(8):691–698. https://doi.org/10.1038/sj.gene.6364258
Mohr KB, Zirafi O, Hennies M, Wiese S, Kirchhoff F, Münch J (2015) Sandwich enzyme-linked immunosorbent assay for the quantification of human serum albumin fragment 408–423 in bodily fluids. Anal Biochem 476:29–35. https://doi.org/10.1016/j.ab.2015.01.023
Moore JP, Kitchen SG, Pugach P, Zack JA (2004) The CCR5 and CXCR4 coreceptors—central to understanding the transmission and pathogenesis of human immunodeficiency virus type 1 infection. AIDS Res Hum Retroviruses 20(1):111–126. https://doi.org/10.1089/088922204322749567
Mori T, Yoshida M, Hazekawa M, Ishibashi D, Hatanaka Y, Nagao T, Kakehashi R, Kojima H, Uno R, Ozeki M, Kawasaki I, Yamashita T, Nishikawa J, Uchida T (2021) Antimicrobial activities of ll-37 fragment mutant-poly (lactic-co-glycolic) acid conjugate against Staphylococcus aureus, Escherichia coli, and Candida albicans. Int J Mol Sci 22(10). https://doi.org/10.3390/ijms22105097
Mörner A, Björndal Å, Albert J, KewalRamani VN, Littman DR, Inoue R, Thorstensson R, Fenyö EM, Björling E (1999) Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. J Virol 73(3):2343. https://doi.org/10.1128/jvi.73.3.2343-2349.1999
Mosier DE (2008) How HIV changes its tropism: evolution and adaptation? Curr Opin HIV AIDS 4(2):1. https://doi.org/10.1097/COH.0b013e3283223d61
Mosier DE, Picchio GR, Gulizia RJ, Sabbe R, Poignard P, Picard L, Offord RE, Thompson DA, Wilken J (1999) Highly potent RANTES analogues either prevent CCR5-using human immunodeficiency virus type 1 infection in vivo or rapidly select for CXCR4-using variants. J Virol 73(5):3544. https://doi.org/10.1128/jvi.73.5.3544-3550.1999
Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, Zlotnik A (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410(6824):50–56. https://doi.org/10.1038/35065016
Müller JA, Zirafi O, Roan NR, Lee SJ, Münch J (2016) Evaluation of EPI-X4 as a urinary peptide biomarker for diagnosis and prognosis of late acute GvHD. Bone Marrow Transplant 51(8):1137–1139. https://doi.org/10.1038/bmt.2016.65
Müller JA, Glöckle A, Gawanbacht A, Geyer M, Münch J, Kirchhoff F (2018) Reduced susceptibility to VIRIP-based HIV-1 entry inhibitors has a high genetic barrier and severe fitness costs. J Virol 92(17):e00733. https://doi.org/10.1128/jvi.00733-18
Münch J, Ständker L, Pöhlmann S, Baribaud F, Papkalla A, Rosorius O, Stauber R, Sass G, Heveker N, Adermann K, Escher S, Klüver E, Doms RW, Forssmann WG, Kirchhoff F (2002) Hemofiltrate CC chemokine 1[9-74] causes effective internalization of CCR5 and is a potent inhibitor of R5-tropic human immunodeficiency virus type 1 strains in primary T cells and macrophages. Antimicrob Agents Chemother 46(4):982. https://doi.org/10.1128/AAC.46.4.982-990.2002
Münch J, Ständker L, Adermann K, Schulz A, Schindler M, Chinnadurai R, Pöhlmann S, Chaipan C, Biet T, Peters T, Meyer B, Wilhelm D, Lu H, Jing W, Jiang S, Forssmann W-G, Kirchhoff F (2007) Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell 129(2):263–275. https://doi.org/10.1016/j.cell.2007.02.042
Münk C, Wei G, Yang OO, Waring AJ, Wang W, Hong T, Lehrer RI, Landau NR, Cole AM (2003) The θ-defensin, retrocyclin, inhibits HIV-1 entry. AIDS Res Hum Retroviruses 19(10):875–881. https://doi.org/10.1089/088922203322493049
Nagasawa T (2007) The chemokine CXCL12 and regulation of Hsc and B lymphocyte development in the bone marrow niche. Adv Exp Med Biol 602:69–75. https://doi.org/10.1007/978-0-387-72009-8_9
Nardese V, Longhi R, Polo S, Sironi F, Arcelloni C, Paroni R, DeSantis C, Sarmientos P, Rizzi M, Bolognesi M, Pavone V, Lusso P (2001) Structural determinants of CCR5 recognition and HIV-1 blockade in RANTES. Nat Struct Biol 8(7):611–615. https://doi.org/10.1038/89653
Nedellec R, Coetzer M, Lederman MM, Offord RE, Hartley O, Mosier DE (2011) Resistance to the CCR5 inhibitor 5P12-RANTES requires a difficult evolution from CCR5 to CXCR4 coreceptor use. PLoS One 6(7):e22020. https://doi.org/10.1371/journal.pone.0022020
Nguyen TX, Cole AM, Lehrer RI (2003) Evolution of primate θ-defensins: a serpentine path to a sweet tooth. Peptides 24(11):1647–1654. https://doi.org/10.1016/j.peptides.2003.07.023
Nguyen LP, Pan J, Dinh TT, Hadeiba H, O’Hara E, Ebtikar A, Hertweck A, Gökmen MR, Lord GM, Jenner RG, Butcher EC, Habtezion A (2015) Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat Immunol 16(2):207. https://doi.org/10.1038/ni.3079
Nishiyama Y, Murakami T, Kurita K, Yamamoto N (1999) Low-molecular-weight anti-HIV-1 peptides from the amino-terminal sequence of RANTES: possible lead compounds for coreceptor-directed anti-HIV-1 agents. Bioorg Med Chem Lett 9(10):1357–1360. https://doi.org/10.1016/S0960-894X(99)00204-8
Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, Loetscher M, Baggiolini M, Moser B (1996) The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 382(6594):833. https://doi.org/10.1038/382833a0
Ocón B, Pan J, Dinh TT, Chen W, Ballet R, Bscheider M, Habtezion A, Tu H, Zabel BA, Butcher EC (2017) A mucosal and cutaneous chemokine ligand for the lymphocyte chemoattractant receptor GPR15. Front Immunol 8:1111. https://doi.org/10.3389/fimmu.2017.01111
Okajima F (2013) Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal 25(11):2263. https://doi.org/10.1016/j.cellsig.2013.07.022
Onopiuk A, Tokarzewicz A, Gorodkiewicz E (2015) Cystatin C. A kidney function biomarker. Adv Clin Chem 68:57. https://doi.org/10.1016/bs.acc.2014.11.007
Oren Z, Lerman JC, Gudmundsson GH, Agerberth B, Shai Y (1999) Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem J 341(3):501–513. https://doi.org/10.1042/0264-6021:3410501
Owen SM, Rudolph DL, Wang W, Cole AM, Waring AJ, Lal RB, Lehrer RI (2004) RC-101, a retrocyclin-1 analogue with enhanced activity against primary HIV type 1 isolates. AIDS Res Hum Retroviruses 20(11):1157–1165. https://doi.org/10.1089/aid.2004.20.1157
Pahar B, Madonna S, Das A, Albanesi C, Girolomoni G (2020) Immunomodulatory role of the antimicrobial LL-37 peptide in autoimmune diseases and viral infections. Vaccines 8(3):517. https://doi.org/10.3390/vaccines8030517
Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP, Bjorkman PJ, Wilen CB, Doms RW, O’Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH (2013) Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci 110(17):6626–6633. https://doi.org/10.1073/pnas.1304288110
Pan B, Wang X, Nishioka C, Honda G, Yokoyama A, Zeng L, Xu K, Ikezoe T (2017) G-protein coupled receptor 15 mediates angiogenesis and cytoprotective function of thrombomodulin. Sci Rep 7(1):692. https://doi.org/10.1038/s41598-017-00781-w
Pan WL, Wang Y, Hao Y, Wong JH, Chan WC, Wan DCC, Ng TB (2018) Overexpression of CXCR4 synergizes with LL-37 in the metastasis of breast cancer cells. Biochim Biophys Acta Mol Basis Dis 1864(11):3837–3846. https://doi.org/10.1016/j.bbadis.2018.09.008
Park M, Kim J, Lee I, Park S, Bae J, Park M (2018) Towards the application of human defensins as antivirals. Biomol Ther 26(3):242–254. https://doi.org/10.4062/BIOMOLTHER.2017.172
Petersen DC, Glashoff RH, Shrestha S, Bergeron J, Laten A, Gold B, van Rensburg EJ, Dean M, Hayes VM (2005) Risk for HIV-1 infection associated with a common CXCL12 (SDF1) polymorphism and CXCR4 variation in an African population. JAIDS J Acqui Immune Defic Syndr 40(5):521–526. https://doi.org/10.1097/01.qai.0000186360.42834.28
Podaza E, Palacios F, Croci DO, Risnik D, Yan XJ, Almejún MB, Colado A, Elías EE, Borge M, Morande PE, Bezares R, Fernández-Grecco H, Rabinovich GA, Gamberale R, Chiorazzi N, Giordano M (2020) Expression and function of cathelicidin hCAP18/LL-37 in chronic lymphocytic leukemia. Haematologica 105(9):e465–e469. https://doi.org/10.3324/haematol.2019.227975
Pollakis G, Paxton WA (2012) Use of (alternative) coreceptors for HIV entry. Curr Opin HIV AIDS 7(5):440–449. https://doi.org/10.1097/COH.0b013e328356e9f3
Popper SJ, Sarr AD, Travers KU, Guèye-Ndiaye A, Mboup S, Essex ME, Kanki PJ (1999) Lower human immunodeficiency virus (HIV) type 2 viral load reflects the difference in pathogenicity of HIV-1 and HIV-2. J Infect Dis 180(4):1116. https://doi.org/10.1086/315010
Potthoff AV, Münch J, Kirchhoff F, Brockmeyer NH (2007) HIV infection in a patient with alpha-1 antitrypsin deficiency: a detrimental combination? AIDS 21(15):2115–2116. https://doi.org/10.1097/QAD.0b013e3282f08b97
Pozzobon T, Goldoni G, Viola A, Molon B (2016) CXCR4 signaling in health and disease. Immunol Lett 177:6–15. https://doi.org/10.1016/j.imlet.2016.06.006
Quiñones-Mateu ME, Lederman MM, Feng Z, Chakraborty B, Weber J, Rangel HR, Marotta ML, Mirza M, Jiang B, Kiser P, Medvik K, Sieg SF, Weinberg A (2003) Human epithelial β-defensins 2 and 3 inhibit HIV-1 replication. AIDS 17(16):F39. https://doi.org/10.1097/00002030-200311070-00001
Reeves JD, Hibbitts S, Simmons G, McKnight Á, Azevedo-Pereira JM, Moniz-Pereira J, Clapham PR (1999) Primary human immunodeficiency virus type 2 (HIV-2) isolates infect CD4-negative cells via CCR5 and CXCR4: comparison with HIV-1 and simian immunodeficiency virus and relevance to cell tropism in vivo. J Virol 73(9):7795. https://doi.org/10.1128/jvi.73.9.7795-7804.1999
Regis EG, Barreto-de-Souza V, Morgado MG, Bozza MT, Leng L, Bucala R, Bou-Habib DC (2010) Elevated levels of macrophage migration inhibitory factor (MIF) in the plasma of HIV-1-infected patients and in HIV-1-infected cell cultures: a relevant role on viral replication. Virology 399(1):31–38. https://doi.org/10.1016/j.virol.2009.12.018
Regoes RR, Bonhoeffer S (2005) The HIV coreceptor switch: a population dynamical perspective. Trends Microbiol 13(6):269–277. https://doi.org/10.1016/j.tim.2005.04.005
van Rij RP, Broersen S, Goudsmit J, Coutinho RA, Schuitemaker H (1998) The role of a stromal cell-derived factor-1 chemokine gene variant in the clinical course of HIV-1 infection. AIDS (London, England) 12(9):F85–F90
Rodríguez A, Webster P, Ortego J, Andrews NW (1997) Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol 137(1):93. https://doi.org/10.1083/jcb.137.1.93
Saini V, Marchese A, Majetschak M (2010a) CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J Biol Chem 285(20):15566–15576. https://doi.org/10.1074/jbc.M110.103408
Saini V, Romero J, Marchese A, Majetschak M (2010b) Ubiquitin receptor binding and signaling in primary human leukocytes. Commun Integr Biol 3(6):608–610. https://doi.org/10.4161/cib.3.6.13375
Saini V, Staren DM, Ziarek JJ, Nashaat ZN, Campbell EM, Volkman BF, Marchese A, Majetschak M (2011) The CXC chemokine receptor 4 ligands ubiquitin and stromal cell-derived factor-1α function through distinct receptor interactions. J Biol Chem 286(38):33466–33477. https://doi.org/10.1074/jbc.M111.233742
Sancho-Vaello E, Gil-Carton D, François P, Bonetti E-J, Kreir M, Pothula KR, Kleinekathöfer U, Zeth K (2020) The structure of the antimicrobial human cathelicidin LL-37 shows oligomerization and channel formation in the presence of membrane mimics. Sci Rep 10(1):17356. https://doi.org/10.1038/s41598-020-74401-5
Schröder JM, Harder J (1999) Human beta-defensin-2. Int J Biochem Cell Biol 31(6):645–651. https://doi.org/10.1016/S1357-2725(99)00013-8
Schuitemaker H, van’t Wout AB, Lusso P (2011) Clinical significance of HIV-1 coreceptor usage. J Transl Med 9(suppl 1):S5. https://doi.org/10.1186/1479-5876-9-S1-S5
Secchi M, Longhi R, Vassena L, Sironi F, Grzesiek S, Lusso P, Vangelista L (2012) Enhancement of anti-HIV-1 activity by hot spot evolution of RANTES-derived peptides. Chem Biol 19(12):1579–1588. https://doi.org/10.1016/j.chembiol.2012.10.007
Shafee TMA, Lay FT, Hulett MD, Anderson MA (2016) The defensins consist of two independent, convergent protein superfamilies. Mol Biol Evol 33(9):2345–2356. https://doi.org/10.1093/MOLBEV/MSW106
Shahmiri M, Enciso M, Adda CG, Smith BJ, Perugini MA, Mechler A (2016) Membrane core-specific antimicrobial action of cathelicidin LL-37 peptide switches between pore and nanofibre formation. Sci Rep 6(1):38184. https://doi.org/10.1038/srep38184
Shao H, Crnogorac MM, Kong T, Chen S-Y, Williams JM, Tack JM, Gueriguian V, Cagle EN, Carnevali M, Tumelty D, Paliard X, Miranda LP, Bradburne JA, Kochendoerfer GG (2005) Site-specific polymer attachment to a CCL-5 (RANTES) analogue by oxime exchange. J Am Chem Soc 127(5):1350–1351. https://doi.org/10.1021/ja043096w
Sokkar P, Harms M, Stürzel C, Gilg A, Kizilsavas G, Raasholm M, Preising N, Wagner M, Kirchhoff F, Ständker L, Weidinger G, Mayer B, Münch J, Sanchez-Garcia E (2021) Computational modeling and experimental validation of the EPI-X4/CXCR4 complex allows rational design of small peptide antagonists. Commun Biol 4(1):113. https://doi.org/10.1038/s42003-021-02638-5
Söllner TH (2004) Intracellular and viral membrane fusion: a uniting mechanism. Curr Opin Cell Biol 16(4):429–435. https://doi.org/10.1016/j.ceb.2004.06.015
Sørensen OE, Borregaard N, Cole AM (2008) Antimicrobial peptides in innate immune responses. Contrib Microbiol 15:61–77. https://doi.org/10.1159/000136315
Steinstraesser L, Tippler B, Mertens J, Lamme E, Homann HH, Lehnhardt M, Wildner O, Steinu HU, Überla K (2005) Inhibition of early steps in the lentiviral replication cycle by cathelicidin host defense peptides. Retrovirology 2:1–12. https://doi.org/10.1186/1742-4690-2-2
Sun L, Finnegan CM, Kish-Catalone T, Blumenthal R, Garzino-Demo P, La Terra Maggiore GM, Berrone S, Kleinman C, Wu Z, Abdelwahab S, Lu W, Garzino-Demo A (2005) Human β-defensins suppress human immunodeficiency virus infection: potential role in mucosal protection. J Virol 79(22):14318–14329. https://doi.org/10.1128/JVI.79.22.14318-14329.2005
Suply T, Hannedouche S, Carte N, Li J, Grosshans B, Schaefer M, Raad L, Beck V, Vidal S, Hiou-Feige A, Beluch N, Barbieri S, Wirsching J, Lageyre N, Hillger F, Debon C, Dawson J, Smith P, Lannoy V et al (2017) A natural ligand for the orphan receptor GPR15 modulates lymphocyte recruitment to epithelia. Sci Signal 10(496):eaal0180. https://doi.org/10.1126/scisignal.aal0180
Trumpfheller C, Tenner-Racz K, Racz P, Fleischer B, Frosch S (1998) Expression of macrophage inflammatory protein (MIP)-1α, MIP-1β, and RANTES genes in lymph nodes from HIV + individuals: correlation with a Th1-type cytokine response. Clin Exp Immunol 112(1):92–99. https://doi.org/10.1046/j.1365-2249.1998.00555.x
Tsou CL, Gladue RP, Carroll LA, Paradis T, Boyd JG, Nelson RT, Neote K, Charo IF (1998) Identification of C-C chemokine receptor 1 (CCR1) as the monocyte hemofiltrate C-C chemokine (HCC)-1 receptor. J Exp Med 188(3):603. https://doi.org/10.1084/jem.188.3.603
Tudan C, Willick GE, Chahal S, Arab L, Law P, Salari H, Merzouk A (2002) C-terminal cyclization of an SDF-1 small peptide analogue dramatically increases receptor affinity and activation of the CXCR4 receptor. J Med Chem 45(10):2024–2031. https://doi.org/10.1021/jm0104015
Unutmaz D, KewalRamani VN, Littman DR (1998) G protein-coupled receptors in HIV and SIV entry: new perspectives on lentivirus-host interactions and on the utility of animal models. Semin Immunol 10(3):225. https://doi.org/10.1006/smim.1998.0134
Vangelista L, Vento S (2018) The expanding therapeutic perspective of CCR5 blockade. Front Immunol 8:1981. https://doi.org/10.3389/FIMMU.2017.01981
Vangelista L, Longhi R, Sironi F, Pavone V, Lusso P (2006) Critical role of the N-loop and β1-strand hydrophobic clusters of RANTES-derived peptides in anti-HIV activity. Biochem Biophys Res Commun 351(3):664–668. https://doi.org/10.1016/j.bbrc.2006.10.090
Veazey RS, Ling B, Green LC, Ribka EP, Lifson JD, Piatak M Jr, Lederman MM, Mosier D, Offord R, Hartley O (2009) Topically applied recombinant chemokine analogues fully protect macaques from vaginal simian-human immunodeficiency virus challenge. J Infect Dis 199(10):1525–1527. https://doi.org/10.1086/598685
Venken T, Krnavek D, Münch J, Kirchhoff F, Henklein P, de Maeyer M, Voet A (2011) An optimized MM/PBSA virtual screening approach applied to an HIV-1 gp41 fusion peptide inhibitor. Proteins 79(11):3221–3235. https://doi.org/10.1002/prot.23158
Vigant F, Santos NC, Lee B (2015) Broad-spectrum antivirals against viral fusion. Nat Rev Microbiol 13(7):426–437. https://doi.org/10.1038/nrmicro3475
Vilas Boas LCP, Campos ML, Berlanda RLA, de Carvalho Neves N, Franco OL (2019) Antiviral peptides as promising therapeutic drugs. Cell Mol Life Sci 76(18):3525–3542. https://doi.org/10.1007/s00018-019-03138-w
Vyroubalova EC, Hartley O, Mermod N, Fisch I (2006) Identification of peptide ligands to the chemokine receptor CCR5 and their maturation by gene shuffling. Mol Immunol 43(10):1573–1578. https://doi.org/10.1016/j.molimm.2005.09.025
Wang W, Cole AM, Hong T, Waring AJ, Lehrer RI (2003) Retrocyclin, an antiretroviral θ-defensin, is a lectin. J Immunol 170(9):4708–4716. https://doi.org/10.4049/jimmunol.170.9.4708
Wang G, Watson KM, Buckheit RW (2008) Anti-human immunodeficiency virus type 1 activities of antimicrobial peptides derived from human and bovine cathelicidins. Antimicrob Agents Chemother 52(9):3438–3440. https://doi.org/10.1128/AAC.00452-08
Wang G, Mishra B, Epand RF, Epand RM (2014) High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim Biophys Acta Biomembr 1838(9):2160–2172. https://doi.org/10.1016/j.bbamem.2014.01.016
Watanabe MAE, de Oliveira Cavassin GG, Orellana MD, Milanezi CM, Voltarelli JC, Kashima S, Covas DT (2003) SDF-1 gene polymorphisms and syncytia induction in Brazilian HIV-1 infected individuals. Microb Pathog 35(1):31–34. https://doi.org/10.1016/S0882-4010(03)00088-3
Wei M, Rong C, Zhao J, Liu X, Yang F, Zeng J (2018) Role of SDF-1 3′A polymorphism in HIV-1 disease progression: a systematic review and meta-analysis. Gene 677:182–188. https://doi.org/10.1016/j.gene.2018.07.058
Wetzel KS, Elliott STC, Collman RG (2018) SIV coreceptor specificity in natural and non-natural host infection: implications for cell targeting and differential outcomes from infection. Curr HIV Res 16(1):41–51. https://doi.org/10.2174/1570162X15666171124121805
White JM, Delos SE, Brecher M, Schornberg K (2009) Structures and mechanisms of viral membrane fusion proteins. Crit Rev Biochem Mol Biol 43(3):189–219. https://doi.org/10.1080/10409230802058320.Structures
White CW, Pfleger K, Hill SJ (2019) Inhibition of CXCR4 signalling and ligand binding by CXCL17. FASEB J 33(S1):503.8. https://doi.org/10.1096/fasebj.2019.33.1_supplement.503.8
White CW, Kilpatrick LE, Dale N, Abhayawardana RS, Dekkers S, Stocks MJ, Pfleger KDG, Hill SJ (2021) CXCL17 is an endogenous inhibitor of CXCR4 via a novel mechanism of action. BioRxiv 2021.07.05.451109. https://doi.org/10.1101/2021.07.05.451109
Wilken J, Hoover D, Thompson DA, Barlow PN, McSparron H, Picard L, Wlodawer A, Lubkowski J, Kent SB (1999) Total chemical synthesis and high-resolution crystal structure of the potent anti-HIV protein AOP-RANTES. Chem Biol 6(1):43–51. https://doi.org/10.1016/S1074-5521(99)80019-2
Winkler C (1998) Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. Science 279(5349):389–393. https://doi.org/10.1126/science.279.5349.389
Wong JH, Legowska A, Rolka K, Ng TB, Hui M, Cho CH, Lam WWL, Au SWN, Gu OW, Wan DCC (2011) Effects of cathelicidin and its fragments on three key enzymes of HIV-1. Peptides 32(6):1117–1122. https://doi.org/10.1016/j.peptides.2011.04.017
Wu Z, Cocchi F, Gentles D, Ericksen B, Lubkowski J, DeVico A, Lehrer RI, Lu W (2005) Human neutrophil α-defensin 4 inhibits HIV-1 infection in vitro. FEBS Lett 579(1):162–166. https://doi.org/10.1016/j.febslet.2004.11.062
Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330(6007):1066–1071. https://doi.org/10.1126/science.1194396
Wu W, Kim CH, Liu R, Kucia M, Marlicz W, Greco N, Ratajczak J, Laughlin MJ, Ratajczak MZ (2012) The bone marrow-expressed antimicrobial cationic peptide LL-37 enhances the responsiveness of hematopoietic stem progenitor cells to an SDF-1 gradient and accelerates their engraftment after transplantation. Leukemia 26(4):736–745. https://doi.org/10.1038/leu.2011.252
Xu L, Li Y, Sun H, Li D, Hou T (2013) Structural basis of the interactions between CXCR4 and CXCL12/SDF-1 revealed by theoretical approaches. Mol Biosyst 9(8):2107. https://doi.org/10.1039/c3mb70120d
Yamamoto K, Kawakubo T, Yasukochi A, Tsukuba T (2012) Emerging roles of cathepsin e in host defense mechanisms. Biochim Biophys Acta 1824(1):105. https://doi.org/10.1016/j.bbapap.2011.05.022
Yu L, Cecil J, Peng S-B, Schrementi J, Kovacevic S, Paul D, Su EW, Wang J (2006) Identification and expression of novel isoforms of human stromal cell-derived factor 1. Gene 374(1–2):174–179. https://doi.org/10.1016/j.gene.2006.02.001
Zhao L, Tolbert WD, Ericksen B, Zhan C, Wu X, Yuan W, Li X, Pazgier M, Lu W (2013) Single, double and quadruple alanine substitutions at oligomeric interfaces identify hydrophobicity as the key determinant of human neutrophil alpha defensin HNP1 function. PLoS One 8(11):e78937. https://doi.org/10.1371/journal.pone.0078937
Zheng Y, Han G, Abagyan R, Wu B, Stevens R, Cherezov V, Kufareva I, Handel T (2017) Structure of CC chemokine receptor 5 with a potent chemokine antagonist reveals mechanisms of chemokine recognition and molecular mimicry by HIV. Immunity 46(6):1005–1017. https://doi.org/10.1016/j.immuni.2017.05.002
Zhong R, Law P, Wong D, Merzouk A, Salari H, Ball ED (2004) Small peptide analogs to stromal derived factor–1 enhance chemotactic migration of human and mouse hematopoietic cells. Exp Hematol 32(5):470–475. https://doi.org/10.1016/j.exphem.2004.01.011
Zi M, Xu Y (2018) Involvement of cystatin C in immunity and apoptosis. Immunol Lett 196:80. https://doi.org/10.1016/j.imlet.2018.01.006
Zirafi O, Kim KA, Ständker L, Mohr KB, Sauter D, Heigele A, Kluge SF, Wiercinska E, Chudziak D, Richter R, Moepps B, Gierschik P, Vas V, Geiger H, Lamla M, Weil T, Burster T, Zgraja A, Daubeuf F et al (2015) Discovery and characterization of an endogenous CXCR4 antagonist. Cell Rep 11(5):737. https://doi.org/10.1016/j.celrep.2015.03.061
Zirafi O, Hermann PC, Münch J (2016) Proteolytic processing of human serum albumin generates EPI-X4 an endogenous antagonist of CXCR4. J Leukoc Biol 99(6):863–868. https://doi.org/10.1189/jlb.2MR1115-521RR
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Harms, M., Hayn, M., Zech, F., Kirchhoff, F., Münch, J. (2022). Endogenous Peptide Inhibitors of HIV Entry. In: Jiang, S., Lu, L. (eds) Virus Entry Inhibitors. Advances in Experimental Medicine and Biology, vol 1366. Springer, Singapore. https://doi.org/10.1007/978-981-16-8702-0_5
Download citation
DOI: https://doi.org/10.1007/978-981-16-8702-0_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-8701-3
Online ISBN: 978-981-16-8702-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)