
Abstract
Synthetic lethality happens between two genes that the mutation of either gene is feasible but the mutation of both concurrently leads to viability loss. The key to the use of synthetic lethality during cancer therapy is the identification of stable synthetic lethal genetic interactions. The transformation of synthetic lethality to clinical practice will be aided by the integration of genetic interaction data in vitro and in vivo. Lung cancer, especially non-small cell lung cancer (NSCLC), has recently been treated with molecular targeted therapeutic drugs. Despite the development of third-generation inhibitors, dealing with resistance to epidermal growth factor receptor inhibitors (EGFRi) still has an outstanding challenge. Moreover, advances of KRAS-driven lung cancers have been hindered by the diversity in the targetable mechanisms, with unanticipated resistance mechanisms to other effective targeted therapies. Synthetic lethality ensures that targets are identified in this situation to find novel therapeutic targets and address acquired and intrinsic resistance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
5.1 Introduction
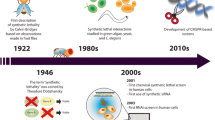
Synthetic lethality was first discovered in 1922, when Calvin Bridges found that simultaneous mutations in two genes in a single fruit fly were fatal, but single mutations in each gene did not cause individual death (Bridges 1922). This laboratory finding has also been observed in single cells, where simultaneous loss of function of two genes can lead to cell lethality, while cells can survive with loss of function of either gene alone (Novick et al. 1989; Boone et al. 2007). This creates a novel and intriguing concept of tumors, because loss of mutated function of tumor suppressor genes is common, but they are often “undruggable” (Epstein 2013). Targeted targets that are available for drug therapy may lead to drug resistance due to delayed compensation pathways, so it is necessary to search for synthetic lethal genetic interactions for drug resistance.
Recent advances in genome sequencing have made it possible to rapidly identify genetic mutations that distinguish tumor from non-tumor cells (Hyman et al. 2017). Tumor-specific genetic changes reveal the biological changes which drive tumor progression, and reveal targets that can be used selectively to treat tumors. Individualized or precision gene-targeted therapy for tumors is characterized by providing highly specific treatment to reduce adverse reactions and overtreatment. Currently, the frontier of oncology therapy is using this individualized oncogenomic approach to successfully treat patients with tumors that cannot respond to standard therapies (Stockley et al. 2016; Swanton et al. 2016).
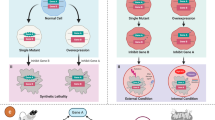
Generally, most gene-targeted cancer therapies take advantage of a phenomenon known as “oncogene addiction,” in which a tumor relies on an oncogene or its pathway to survive. Unfortunately, although antibody-based or small molecule oncogene inhibitors have been shown to be effective against certain tumor genotypes (Pagliarini et al. 2015), not all tumors have functional acquired targeting of oncogenes, and resistance to treatment is also a common result. In these tumors, oncogenic and non-oncogenic mutations can be exploited to further treat tumors by identifying secondary targets that lead to synthetic lethality (Fig. 5.1).

The loss or the inhibition of either of the protein products of gene A or B alone is viable. Mutation or pharmacological inhibition of the protein product of gene B in cells with a mutation of gene A results in synthetic lethality
More than a decade ago, it was discovered that non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations can respond to EGFR inhibitors (EGFRi) selectively. This great discovery has led to individualized treatment for these difficult-to-treat tumors (Lynch et al. 2004). Although EGFRi have achieved some degree of achievement, most patients who responded to EGFRi treatment initially relapsed within 16 months because of acquired drug resistance (Mok et al. 2009). A deeper research of microcosmic mechanisms of acquired EGFRi resistance could lead to the progress of next-generation medicine to overcome resistance and thus delay tumor recurrence.
KRAS mutations have been discovered in NSCLC tumors for over 20 years (Bos 1989), but we are just starting to recognize the value of identifying KRAS tumor character. Mutations in EGFR and KRAS seem to be mutually exclusive. Recent studies have shown that patients with mutated KRAS genes have difficulty benefiting from traditional or adjuvant chemotherapy and even fail to respond to EGFR inhibitors. Obviously, there is a requirement to develop new therapeutic targets specifically for patients with KRAS-mutated NSCLC.
In this review, we emphasize some of the recent researches and subsequent challenges in using synthetic lethality to find novel potential therapy targets, including its clinical transformation. Then, in view of the latest progress of synthetic lethality in lung cancer, we focus on the mechanism of oncogene signaling network in EGFRi-resistant NSCLC and the research progress in synthetic lethality related to KRAS-driven lung cancer.
5.2 Expanding Definition of Synthetic Lethality
Theoretically, a synthetic lethal-based approach could be extended beyond the loss of function at the tumor target. Cancer cells often have the characteristics of gene overexpression, which may be due to changes in somatic copy number (Zack et al. 2013) or due to epigenetic changes which can increase gene transcription. Genes that are overexpressed in tumors can be identified by identifying the interaction gene pairs that cause “synthetic dosage lethality” (SDL). SDL is a kind of genetic interaction in which one gene is overexpressed related to the reduction in function of another gene, leading to lethality. The concept of SDL was first reported in yeast (Yan et al. 1991). The overexpression of MAD2 in several types of tumors has been studied extensively in recent years, demonstrating an SDL effect with PP2A inhibition (Bian et al. 2014).
In addition, tumor cells are found to be heterogeneous usually and in different microenvironments, both of which can influence genetic interactions, and most of them are condition dependent. These conditions may be inherent, such as the metabolic state and genetic background, or they are extrinsic, such as the extracellular microenvironment and therapeutic medication. And some genetic interactions need the mutation of three or more genes to get synthetic lethality (Tong et al. 2004). On the contrary, background mutations can inhibit synthetic lethality and thus generate synthetic viability. Thus, the genetic background of a tumor, such as the activation of an oncogene or the loss of p53, can reveal or inhibit synthetic lethality. It is possible to take advantage of condition-dependent synthetic lethality to exploit tumor-related conditions such as continuous stress, increased mutational load, changed metabolism, and exposure to standard antitumor therapy, to promote the extent of synthetic lethal interactions (O’Neil et al. 2017). For instance, hypoxia reduces the effect of homologous recombination (Bindra et al. 2004), leading to a damaged state of DNA repair, making cells sensitive to suppression of the repair protein PARP1 (Chan et al. 2010). So screening for phenotypes that synthesize lethal interactions under hypoxia or metabolic conditions can reveal the tumor microenvironment, which can discover novel condition-dependent synthetic lethal interactions with higher tumor cell killing specificity.
5.3 Translating Synthetic Lethality into Therapeutics
Clinical treatment based on synthetic lethality is an intriguing concept, but few mature therapeutic strategies have been used clinically to date. One of the main obstacles is the difficulty of finding a stable, clinically useful synthetic lethal interaction.
There are three main challenges to screen for synthetic lethal interactions. First of all, these genetic interactions lead to lethality, so this makes it difficult to recover and identify these mutants. Second, most synthetic lethal interactions are conditional-dependent interactions and may be unstable across all genetic backgrounds or in different conditions. And third, synthetic lethal interaction pairs are relatively rare, and a huge number of mutated gene pairs need to be researched to identify synthetic lethality, which is a huge workload. Considering all above, most synthetic lethal genetic interaction screening is performed only in yeast, as it is easier to obtain techniques that accelerate high-throughput production and analysis of mutated gene pairs under laboratory conditions (O’Neil et al. 2017).
Theoretically, using a treatment strategy based on synthetic lethality also presents three benefits. First, synthetic lethal interactions are selective for tumor-specific genetic mutations, which can easily identify responders in patients. Second, considering there is a large treatment window, treatment side effects based on synthetic lethality are smaller, and lower drug doses are effective. Last, this strategy can be used to any type of tumor mutation, including tumor suppressors and those therapeutic targets that are not thought to be undruggable (O’Neil et al. 2017). Practically, PARP and BRCA1/BRCA2, the only one synthetic lethal interaction, has been successfully transferred from laboratory discovery (Bryant et al. 2005) to the clinical application (Fong et al. 2009). However, synthetic lethality databases, such as SynlethDB (Guo et al. 2016), contain a huge number of interactions according to the published literature.
In addition, there is a need to evaluate the potential clinical value of synthetic lethal interactions, to include what can be used for tumor prevention (Walcott et al. 2016). Evidence for such chemoprophylaxis strategies based on synthetic lethality is still inchoate, gathered mainly using animal models. Theoretically, a strategy based on synthetic lethality may be more effective if it is implemented before the precancerous lesion becomes highly heterogeneous. It is also necessary to determine whether intermittent administration or lower doses will be effective by this strategy in order to avoid drug toxicity related to continuous high-dose chemotherapy (Wu and Lippman 2011).
5.4 Synthetic Lethality and EGFRi-Resistant NSCLC
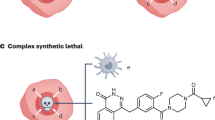
In recent years, a lot of studies have been conducted to explore the “oncogene addiction” of EGFRi-resistant NSCLC, based on the notion of synthetic lethality. For EGFR-mutated lung tumor, synthetic lethality is usually used to describe two types of gene interactions: first, genes that are particularly needed in EGFR-mutated cells compared to EGFR wild-type cells, and second, genes that, when damaged, can be combined with EGFRi therapy to cause synergistic death. For the latter, potential targets may focus on the genes that regulate sensitivity to EGFRi. Construction of candidate gene libraries to detect synthetic lethal interactions related to EGFRi resistance can be actuated by hypothesis. For instance, some researches have focused on the analysis of the EGFR “interaction group,” which is a network of molecules that interact with EGFR directly or indirectly as a group of possible targets (Fig. 5.2) (Astsaturov et al. 2010; Li et al. 2013; Yoshida et al. 2016; Saafan et al. 2016). Alternatively, phosphorylproteomic analysis of EGFR-mutated signal transduction networks in lung tumor models could afford pathway-specific candidates to be included in targeted screening (Yoshida et al. 2014; Huang and White 2008). In recent years, key pathways associated with EGFRi resistance have been reported, including nuclear factor-κB (NF-κB), signal transducer and activator of transcription 3 (STAT3), and Wnt signaling.

The loss of gene X in normal cell alone is viable, as the mutation of gene X in EGFR mutant tumor cell can result in synthetic lethality. Lung cancer cells that carry mutated EGFR but are insensitive to EGFRi may rely on gene X to survive in the presence of EGFRi
Among them, activation of NF-κB pathway is associated with chemotherapy resistance, exhibiting its potential as a clinical candidate (Godwin et al. 2013). Bivona et al. performed a pooled shRNA screening using a library of over 2000 genes in the H1650 cell line. By shRNA screening in the presence of erlotinib, the authors identified genes that promote EGFR dependence and recover EGFR sensitivity when silenced (Bivona et al. 2011). From this screening, 18 genes associated with NF-κB and Fas were revealed as death receptor signaling pathways that sensitized cells to erlotinib. When stimulated with Fas ligand, tumor cells initiated caspase-mediated apoptosis and activates NF-κB pathway (Godwin et al. 2013). Recently, Sudo et al. have shown that the NF-κB pathway is involved in resistance to EGFRi (Sudo et al. 2015). Synthetic lethal screening of genomic shRNA with gefitinib combination was performed in NSCLC H1975 cell lines, which contained both L858R and T790M EGFR mutations. In fact, one of the barriers to investigating NF-κB signaling in EGFRi resistance is the shortage of inhibitors that target NF-κB directly (Gilmore and Herscovitch 2006).
The STAT3 is another main driver of cancer signaling and drug resistance in various types of cancer. As portion of the research into the mechanisms why NF-κB promotes EGFRi resistance, Bivona’s previous study also observed activation of NF-κB in erlotinib-treated cells leading to the production of IL-6 and follow-up activation of STAT3 pathway (Blakely et al. 2015). Therefore, inhibition of the IL-6-STAT3-NF-κB signaling axis appears to be the key to restoring EGFRi sensitivity in other drug-resistant NSCLC. In the study by Astsaturov et al., STAT3 was considered as a potential synthetic lethal target itself for EGFRi therapy (Astsaturov et al. 2010). The authors constructed a computer network of EGFR-centric proteins by integrating lots of databases to identify molecules that functionally interact with EGFR. Unfortunately, the lack of STAT3 inhibitors in current clinical trials makes this strategy difficult to carry out in clinical settings.
Ligands in the Wnt signaling family affect a variety of cellular processes, and disorder of the Wnt/β-catenin pathway is also known to occur in different types of cancer, such as colorectal cancer (Zhan et al. 2017). Casas-Selves et al. reported several classic Wnt signaling pathways leading to the regulating factor in EGFR inhibitor can promote cell survival by whole genome synthetic lethal shRNA screening (Casas-Selves et al. 2012). After bioinformatics analysis and second-generation sequencing, they identified multiple shRNA hits associated with the Wnt/Tankyrase/β-catenin signaling pathway, such as genes encoding poly-ADP-Tankyrase1 (TNKS1) and Tankyrase 2 (TNKS2). Mice carrying tumor xenografts showed more growth inhibition significantly in response to gefitinib treatment with shRNA targeting TNKS1 compared with controls.
Although there is an understanding base of synthetic lethality related to first-generation inhibitor resistance, the only agent approved for patients with advanced EGFR mutation is osimertinib. There is still a lack of available remedies for patients who are resistant to first-generation EGFRi but do not have the T790M mutation. There are still significant challenges that limit the transformation the large volume of synthetic lethal interactions described above into an effective treatment for NSCLC patient.
5.5 Synthetic Lethality and KRAS-Driven NSCLC
Mutations of KRAS gene are also usually found in different tumors, such as lung cancer, colorectal cancer, and pancreatic cancer (Bos 1989). Approximately 15–25% of patients with NSCLC are with KRAS mutations meanwhile (Brose et al. 2002). Some researches have illustrated that KRAS and EGFR mutations are mutually exclusive to a certain extent, suggesting that they have functionally identical roles in lung tumor progress (Shigematsu et al. 2005; Kosaka et al. 2004; Taron et al. 2005). However, patients with EGFR mutations responded better to inhibitors (Fukuoka et al. 2011). Although several strategies have been explored to inhibit KRAS, the pursuit of inhibitors for KRAS therapy has fallen to meet expectations. A barrier to the research of specific KRAS inhibitors is that the mutant KRAS protein has lost its normal enzymatic function, and this loss of the mutant enzymatic function is more difficult to inhibit. The most effective method for KRAS-mutated NSCLC is a combination of targeted oncogenic KRAS and other therapeutics that are specific to the tumor molecular spectrum. However, realizing this prospect requires an understanding of these abnormalities and the compensatory mechanisms of tumor cells. In recent years, the synthetic lethal effects associated with KRAS-mutated NSCLC have been reported mainly including phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MEK, also known as MAPK) pathways, exportin 1 (XPO1, also known as CRM1) pathways, Polo-like kinase 1 (PLK1), and RhoA/Rho kinase (ROCK) pathways.
Targeting the pathway downstream of KRAS is a potentially promising approach. Inhibitors of the PI3K/mTOR pathway are active in cancer with PI3K mutations and may be effective in treating KRAS-mutated lung cancers when combined with MEK inhibitors (Engelman et al. 2008). This elucidates the reason for the remarkable efficacy of receptor tyrosine kinase (RTK) targeted cancer therapy when RTK inhibition results in loss of PI3K and ERK signaling (She et al. 2005; Mellinghoff et al. 2005; Engelman 2007; Sharma et al. 2006). Engelman et al. (Engelman et al. 2008) treated mice with KRAS-mutated lung tumors with PI3K and MEK inhibitors simultaneously and found that this strategy resulted in significant combined tumor regression. In addition, serine threonine kinase 11 (STK11), which is also called liver kinase B1 (LKB1), is a multitask tumor suppressor kinase (Mahoney et al. 2009). LKB1 is inactivated by somatic cells in about 30% of NSCLC, and the homozygous loss of the gene combined with KRAS leads to a more aggressive tumor phenotype than that of KRAS alone (Ding et al. 2008; Makowski and Hayes 2008). NSCLC with both LKB1 inactivation and KRAS activation have been reported to be functionally different from other lung cancers, showing sensitivity to single dose therapy with the MEK inhibitor rapamycin or CI-1040 (Mahoney et al. 2009). In recent years, there have been reports that the combination of ataxia-telangiectasia mutation (ATM) dysfunction with MEK1/2 inhibitors has a synthetic lethal effect on KRAS-driven lung cancer, because ATM mediates the pro-survival interaction between MEK/ERK and AKT/mTOR pathways (Smida et al. 2016).
Recently, Kim J et al. reported that exportin 1 (XPO1, also known as CRM1) is a drug target for KRAS-mutated lung cancer. The authors demonstrate that KRAS-mutated NSCLC is dependent on nuclear output for KRAS-specific cellular autonomic addiction, and that chemical interference with XPO1 inhibitors produces a strong synthetic lethal interaction with KRAS. The primary mechanism by which XPO1 inhibitors are sensitive to KRAS-mutated NSCLC may be intolerance to the accumulation of nuclear factor IκBα (also known as NF-κBα), thereby inhibiting transcription factor activity of NF-κB. These findings suggest that clinically available XPO1 inhibitors combined with genome to guide patient selection are a promising treatment strategy for a significant number of patients with lung tumor (Kim et al. 2016).
In another research, Wang J et al. reported that combined inhibition of Polo-like kinase 1 (PLK1) and RhoA/Rho kinase (ROCK) leads to synthetic lethality in KRAS-mutated NSCLC. Research showed that the combined inhibition significantly increased the transcription and activity of p21, a cyclin-dependent kinase inhibitor, resulting in specific G2/M phase arrest in KRAS mutant cells. Overexpressed p21 can preferentially impair the growth of KRAS mutant cells through cDNA transfection or clinical drugs, suggesting that there is a synthetic lethality interaction between KRAS and p21 that can be used medicinally. Application of BI-2536, a selective inhibitor of PLK1, and fasudil, a ROCK inhibitor, inhibited tumor growth and significantly prolonged mouse survival in a KRAS mouse model, suggesting a strong synergistic effect and potential for the therapy of KRAS-mutated cancers in vivo (Wang et al. 2016).
In addition, neurofibromatosis type 1 (NF1) pathway (Johannessen et al. 2008), Wilms tumor gene 1 (WT1) (Vicent et al. 2010), and cyclin CDK4 (Puyol et al. 2010) have also been reported to be involved in the synthetic lethality of KRAS-mutated NSCLC. However, much remains to be learned about the molecular details of KRAS proteins’ function. We should summarize the tumor protein abnormalities that lead to the oncogene-specific synthetic lethal interactions, thus providing additional targeted therapy chances for KRAS-mutated NSCLC, and developing appropriate alternative markers to monitor medicine action, which is critical for the treatment of these patients.
5.6 Conclusion
Synthetic lethality as a concept does not seem to meet its promise of defining new clinical strategies to deal with tumors and their drug resistance due to the lack of some reproducibility between different researches and the difficulty in transforming any identified targets into clinical practice (Downward 2015). It is essential that practical steps be taken to deal with these difficulties and challenges. It is our view that for most of the identified synthetic lethal interactions, the underlying mechanisms how these genes contribute to lethality are mostly unknown. In order to exploit synthetic lethality fully, there is an urgent requirement to ascertain the effect of those genes on signaling networks. Furthermore, many identified synthetic lethal interactions are background dependent, which is referred to as a “soft” interaction (Ashworth et al. 2011; Lord and Ashworth 2013). The establishment of these background-dependent molecular bases enables the recognition and translation of these “soft” interactions into more powerful “hard” synthetic lethal effects. At the same time, understanding this background dependence can also provide messages for biomarker discovery to recognize the patients who will benefit from therapeutic strategy based on synthetic lethality. Our understanding of how synthetic lethality promotes tumor heterogeneity is also inadequate. Given that drug resistance is now confirmed to be driven by various resistance mechanisms in heterogeneous tumors (Hata et al. 2016; Ramirez et al. 2016; Bivona and Doebele 2016), it is imaginable that different subpopulations of tumor cells may need various synthetic lethality strategies to minimize tumor heterogeneity and avoid the growth of major components.
Therefore, synthetic lethality is a genetic notion that has had an important influence on cancer research. Bioinformatics analysis of synthetic lethal interaction data from experiments in vitro and in vivo will facilitate the screening of potential therapeutic targets for tumors. However, these data also highlight the mechanistic complexity of tumor phenotypes, which need to be untangled in order to successfully find specific targets that can be used to selectively kill tumor cells, thus realizing the full potential of individualized anticancer therapies.
References
Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell. 2011;145(1):30–8.
Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal. 2010;3:ra67140.
Bian Y, Kitagawa R, Bansal PK, Fujii Y, Stepanov A, Kitagawa K. Synthetic genetic array screen identifies PP2A as a therapeutic target in Mad2-overexpressing tumors. Proc Natl Acad Sci U S A. 2014;111(4):1628–33.
Bindra RS, Schaffer PJ, Meng A, Woo J, Maseide K, Roth ME, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol. 2004;24(19):8504–18.
Bivona TG, Doebele RC. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat Med. 2016;22(5):472–8.
Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, et al. FAS and NF-kappa B signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471(7339):523–6.
Blakely CM, Pazarentzos E, Olivas V, Asthana S, Yan JJ, Tan I, et al. NF-kappa B-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015;11(1):98–110.
Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nat Rev Genet. 2007;8(6):437–49.
Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–9.
Bridges CB. The origin of variations in sexual and sex-limited characters. Am Nat. 1922;56:51–63.
Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62(23):6997–7000.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7.
Casas-Selves M, Kim J, Zhang Z, Helfrich BA, Gao D, Porter CC, et al. Tankyrase and the canonical wnt pathway protect lung cancer cells from EGFR inhibition. Cancer Res. 2012;72(16):4154–64.
Chan N, Pires IM, Bencokova Z, Coackley C, Luoto KR, Bhogal N, et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010;70(20):8045–54.
Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–75.
Downward J. RAS synthetic lethal screens revisited: still seeking the elusive prize? Clin Cancer Res. 2015;21(8):1802–9.
Engelman JA. The role of phosphoinositide 3-kinase pathway inhibitors in the treatment of lung cancer. Clin Cancer Res. 2007;13S(152):4637S–40S.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6.
Epstein RJ. The unpluggable in pursuit of the undruggable: tackling the dark matter of the cancer therapeutics universe. Front Oncol. 2013;3:304.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-Ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34.
Fukuoka M, Wu Y, Thongprasert S, Sunpaweravong P, Leong S, Sriuranpong V, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol. 2011;29(21):2866–74.
Gilmore TD, Herscovitch M. Inhibitors of NF-kappa B signaling: 785 and counting. Oncogene. 2006;25(51):6887–99.
Godwin P, Baird AM, Heavey S, Barr MP, O'Byrne KJ, Gately K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front Oncol. 2013;3:120.
Guo J, Liu H, Zheng J. SynLethDB: synthetic lethality database toward discovery of selective and sensitive anticancer drug targets. Nucleic Acids Res. 2016;44(D1):D1011–7.
Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262–9.
Huang PH, White FM. Phosphoproteomics: unraveling the signaling web. Mol Cell. 2008;31(6):777–81.
Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell. 2017;168(4):584–99.
Johannessen CM, Johnson BW, Williams SMG, Chan AW, Reczek EE, Lynch RC, et al. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18(1):56–62.
Kim J, McMillan E, Kim HS, Venkateswaran N, Makkar G, Rodriguez-Canales J, et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature. 2016;538(7623):114–7.
Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64(24):8919–23.
Li J, Bennett K, Stukalov A, Fang B, Zhang G, Yoshida T, et al. Perturbation of the mutated EGFR interactome identifies vulnerabilities and resistance mechanisms. Mol Syst Biol. 2013;9(1):705.
Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–8.
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39.
Mahoney CL, Choudhury B, Davies H, Edkins S, Greenman C, van Haaften G, et al. LKB1/KRAS mutant lung cancers constitute a genetic subset of NSCLC with increased sensitivity to MAPK and mTOR signalling inhibition. Br J Cancer. 2009;100(2):370–5.
Makowski L, Hayes DN. Role of LKB1 in lung cancer development. Br J Cancer. 2008;99(5):683–8.
Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu SJ, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353(19):2012–24.
Mok TS, Wu Y, Thongprasert S, Yang C, Chu D, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–57.
Novick P, Osmond BC, Botstein D. Suppressors of yeast actin mutations. Genetics. 1989;121(4):659–74.
O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18(10):613–23.
Pagliarini R, Shao W, Sellers WR. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015;16(3):280–96.
Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18(1):63–73.
Ramirez M, Rajaram S, Steininger RJ, Osipchuk D, Roth MA, Morinishi LS, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun. 2016;7(1):1–8.
Saafan H, Foerster S, Parra-Guillen ZP, Hammer E, Michaelis M Jr, Cinatl J, et al. Utilising the EGFR interactome to identify mechanisms of drug resistance in non-small cell lung cancer—proof of concept towards a systems pharmacology approach. Eur J Pharm Sci. 2016;94(SI):20–32.
Sharma SV, Fischbach MA, Haber DA, Settleman J. “Oncogenic shock”: explaining oncogene addiction through differential signal attenuation. Clin Cancer Res. 2006;12S(142):4392S–5S.
She QB, Solit DB, Ye Q, O'Reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005;8(4):287–97.
Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97(5):339–46.
Smida M, Fece DLCF, Kerzendorfer C, Uras IZ, Mair B, Mazouzi A, et al. MEK inhibitors block growth of lung tumours with mutations in ataxia-telangiectasia mutated. Nat Commun. 2016;7:13701.
Stockley TL, Oza AM, Berman HK, Leighl NB, Knox JJ, Shepherd FA, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. 2016;8(1):1–12.
Sudo M, Mori S, Madan V, Yang H, Leong G, Koeffler HP. Short-hairpin RNA library: identification of therapeutic partners for gefitinib-resistant non-small cell lung cancer. Oncotarget. 2015;6(2):814–24.
Swanton C, Soria JC, Bardelli A, Biankin A, Caldas C, Chandarlapaty S, et al. Consensus on precision medicine for metastatic cancers: a report from the MAP conference. Ann Oncol. 2016;27(8):1443–8.
Taron M, Ichinose Y, Rosell R, Mok T, Massuti B, Zamora L, et al. Activating mutations in the tyrosine kinase domain of the epidermal growth factor receptor are associated with improved survival in gefitinib-treated chemorefractory lung adenocarcinomas. Clin Cancer Res. 2005;11(16):5878–85.
Tong A, Lesage G, Bader GD, Ding HM, Xu H, Xin XF, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303(5659):808–13.
Vicent S, Chen R, Sayles LC, Lin C, Walker RG, Gillespie AK, et al. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J Clin Investig. 2010;120(11):3940–52.
Walcott FL, Patel J, Lubet R, Rodriguez L, Calzone KA. Hereditary cancer syndromes as model systems for chemopreventive agent development. Semin Oncol. 2016;43(1):134–45.
Wang J, Hu K, Guo J, Cheng F, Lv J, Jiang W, et al. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat Commun. 2016;7:11363.
Wu X, Lippman SM. An intermittent approach for cancer chemoprevention. Nat Rev Cancer. 2011;11(12):879–85.
Yan H, Gibson S, Bk T. MCM2 and MCM3, 2 proteins important for ars activity, are related in structure and function. Genes Dev. 1991;5(6):944–57.
Yoshida T, Zhang G, Smith MA, Lopez AS, Bai Y, Li J, et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clin Cancer Res. 2014;20(15):4059–74.
Yoshida T, Song L, Bai Y, Kinose F, Li J, Ohaegbulam KC, et al. ZEB1 mediates acquired resistance to the epidermal growth factor receptor-tyrosine kinase inhibitors in non-small cell lung cancer. PLoS One. 2016;11:e01473441.
Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45(10SI):1134–40.
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–73.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Huang, J., Qiao, T., Wang, X. (2022). Synthetic Lethality and Lung Cancer. In: Shen, H., Zeng, Y., Li, L., Wang, X. (eds) Regionalized Management of Medicine. Translational Bioinformatics, vol 17. Springer, Singapore. https://doi.org/10.1007/978-981-16-7893-6_5
Download citation
DOI: https://doi.org/10.1007/978-981-16-7893-6_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-7892-9
Online ISBN: 978-981-16-7893-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)