
Abstract
Histone deacetylase (HDAC) inhibitors represent a novel class of therapeutic agents for cancer treatment. These enzymes play a pivotal regulatory role on chromatin epigenetics by deacetylation of histone proteins and lead to the production of reactive oxygen species (ROS) via activation of apoptotic pathway and autophagy. Surprisingly, the HDAC inhibitor-mediated ROS production in various cancer cells such as leukemia was found to be elevated by a combination of DNA damaging agents and proteasome inhibitors. In contrast, cancer cells possess proteins such as Trx and Trx reductase, as well as GSH peroxidase (Gpx) that help in preventing the oxidative stress of the cells by reducing the H2O2. Various bioinformatics tools can be utilized to understand the HDAC inhibitor-mediated differential gene expression data obtained by using the Affymetrix platform as well as the Illumina platform. Further, Gene Ontology (GO) and pathway analyses tools reveal pro-apoptotic gene expression signature and intrinsic apoptotic pathway during ROS generation. The differentially expressed genes were further subjected to Ingenuity Pathway Analysis (IPA) tool to study the associations of various molecular and cellular functions. ReMap (regulatory map of TF binding sites) analysis tool demonstrated the chromatin occupancy by proteins such as bromodomain-containing protein-4 (BRD4) and MYC proteins at their binding sites during HDAC inhibitor-treated cancer cells. Studies on HDAC inhibitor-mediated ROS generation and the tumor-suppressive effects can be better studied by using a combination of various molecular and bioinformatics methods that would help in better therapy against cancer.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

Keywords
Introduction
Histone deacetylases (HDACs) are the proteins that regulate the expression of several proteins involved in cancer. HDACs remove the acetyl groups and help in conversion of compacted chromatin to open confirmation. HDAC inhibitors were found to inhibit cancer initiation and progression. Several HDAC inhibitors are in phase I, II, and III clinical trials. These are promising anticancer drugs that modulate chromatin epigenetics and there by regulate gene expression. Studies have found that reversing epigenetic changes by targeting HDAC is a potent therapeutic strategy. Recent studies have identified the initiation, as well as the progression, of cancer that involves various types of epigenetic modifications. The HDAC inhibitors (HDACi) modulate several processes such as intrinsic apoptosis, extrinsic apoptosis, autophagy, cell cycle arrest, and tumor immunogenicity (Newbold et al. 2016). Treatment with HDAC inhibitors causes intrinsic apoptosis by upregulating the genes such as Bid and Bim and by decreasing Bcl-2 and Bcl-xL. Similarly, HDAC inhibitor-mediated extrinsic apoptosis involves the increased expression of DR4, FAS, and TRAIL (Bolden et al. 2013). HDACi induces G1/S phase cell cycle arrest by upregulating genes such as p21 and p15 and downregulating cyclin D1, cyclin E1, CDK4, and CDK6. Interestingly, HDAC inhibitors induce G2/M phase cell cycle arrest by the downregulation of cyclin A and cyclin B. These molecules also upregulate the FOXO1 gene, essential for autophagy (Lee et al. 2012) as well as decrease the natural killer cells’ ligand expression (Murakami et al. 2008).
HDAC Inhibitors and ROS Production
The HDACi can induce apoptosis by upregulating the pro-apoptotic gene expression like Bam and Bax or by downregulating the anti-apoptotic genes like Bcl-2 and Bcl-xL selectively in cancer cells at the transcriptional level (Minucci and Pelicci 2006). They also elevate the reactive oxygen species (ROS) levels and activate various death receptors such as TRAIL, DR5, FAS, and TNF-α in cancer cells. Further, the high amount of cellular ROS affects the mitochondrial membrane potential and induces apoptosis via an intrinsic pathway (Rosato et al. 2003).
Petruccelli et al. (2011) reported that the SAHA induces double-strand breaks and enhances the cellular ROS levels in acute myeloid leukemia (AML) cancer cells, and also increases the caspase-3/7 activity as well as cell cycle arrest at the G2/M phase. PCI24781, a novel HDACi, induces apoptosis in a ROS-dependent manner and decreases the nuclear factor kappa-light-chain-enhancer of activated B cell (NF-KB) expression (Sholler et al. 2013). LAQ-824, an HDACi, in combination with fludarabine promotes high intracellular ROS production and induces DNA damage and apoptosis in the leukemia cells. It also triggers pro-apoptotic protein expression and activates caspase-2 release from the mitochondria to the cytosol to initiate the apoptosis pathway (Rosato et al. 2008). Trichostatin A, an HDACi, downregulates the anti-apoptotic protein Bcl-2 and induces apoptosis in an oxidative stress-dependent manner in human cervical cancer cells.
HDACi induces the expression of Bid, a pro-apoptotic protein that disrupts the mitochondrial membrane potential and elevates the cellular ROS levels, thus inducing apoptosis (Ruefli et al. 2001). They also alter the antioxidant levels in cancer cells, by upregulating thioredoxin-binding protein-2 (TBP-2) which inhibits the activity of antioxidant protein thioredoxin (Trx) leading to enhanced ROS production at cytosol, thus inducing apoptosis (Butler et al. 2002). Apart from apoptosis induction, the HDACi can also induce cell cycle arrest by upregulating CDKN1A that codes p21WAF1/CIP (Richon et al. 2000), repressing the cyclin A and cyclin D gene expression. HDAC inhibitors also downregulate the vascular endothelial growth factor (VEGF) and endothelial nitric oxide synthase (eNOS) (Qian et al. 2006).
T315I mutation is the most common BCR/ABL mutation which is the main causative factor for the imatinib resistance. The combination of HDACi with ROS-inducing agents will enhance the efficacy in the treatment of cancer. Adaphostin (NSC680410) is a tyrosine kinase inhibitor that inhibits p210Bcr/abl expression, and activity in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) cancers (Chandra et al. 2006). It also elevates intracellular ROS levels, peroxide, superoxide, and glutathione and induces apoptosis along with the inhibition of electron transport in mitochondria of cancer cells (Nilsa Rivera De Valle et al. 2018). β-Phenylethyl isothiocyanate (PEITC), a redox modulatory agent, induces apoptosis and cell cycle arrest by downregulation of anti-apoptotic proteins like Bcl-2 and Bcl-xL in prostate cancer cells (Xiao et al. 2004). It also inhibits complex III in the electron transport chain inside the mitochondria and cytochrome p450 enzymes. Studies also indicated the ROS-enhancing activity of PEITC in cancer cells. Combination of a class of HDAC inhibitors such as MS-275, apicidin, and romidepsin killed nasopharyngeal cancer (NPC) cells with proteasome inhibitors such as bortezomib-induced apoptosis of nasopharyngeal cancer cells via reactive oxygen species (ROS) and caspase-dependent pathway (Hui and Chiang 2014). Recent studies have identified that the combination of HDACi with ROS-generating agents, proteasome inhibitors, methylation modulators, and DNA-damaging agents can be effective against cancers cells by inducing cell cycle arrest, apoptosis via high ROS production in mitochondria, histone hyperacetylation, and alteration in gene expression, ultimately killing the cancer cells (Miller et al. 2011) (Fig. 1).

HDAC inhibitor-mediated ROS generation in cancer cells. (a) In cancer cells, HDAC inhibitors induce Bid expression and enhance TBP-2 with a concomitant decrease in Trx protein level. (b) Combination therapy using HDAC inhibitors along with proteasome inhibitors (bortezomib), DNA-damaging agents (fludarabine), adaphostin, and methylation modulators induces enhanced ROS production
HDAC Inhibitors and Protein Quality Control Systems
Eukaryotic cells possess efficient protein quality control (PQC) systems that include molecular chaperones, the ubiquitin proteasome system (UPS), and autophagy for the effective sensing of the proper folding and refolding of proteins. Interestingly, the PQC system modifies the epigenetic readout of HDAC inhibitor-treated cells (Michelle kulla et al. 2020). Further, PQCs help in the recognition of acetylation after HDAC inhibitor treatment and provide some more targets for effective treatment.
Molecular Chaperons
The heat shock proteins (HSPs) such as HSP90 family, HSP70 family, HSP60/GroEL family, and small heat shock proteins (sHSPs) act as vital molecular chaperons that regulate protein folding and were found to be affected by the acetylation (Jeng et al. 2015). HSP90 regulates protein folding and plays a major role in the activation of cell proliferation and signal transduction, thus promoting cancer, and interestingly, this protein directly binds to chromatin which makes this protein interplay in both epigenetics and molecular chaperones. The HSP90 activity was found to be impaired when it is treated with panobinostat (LBH589) or hyperacetylated by knockdown of histone deacetylase 6 (HDAC6) (Kovacs et al. 2005). HSP70 is involved in the protein folding and refolding, and high expression of HSP70 was associated with the methylation patterns of histone H3 in oral squamous cell carcinoma.
Ubiquitin Proteasome System
The proteasome recognizes the polyubiquitin chain of the proteins like metabolic enzymes, transcription factors, cyclins, and CDK inhibitors and degrades it by unfolding. These proteins play a vital role in cancer progression by proliferation and nutrient availability. The p53, a tumor suppressor gene, is reported to be degraded by the mouse double minute 2 homolog (MDM2) via poly-ubiquitination followed by proteasome degradation. This degradation can be prevented by inhibiting the proteasome activity of cancer cells where the p53 gene was mutated. Studies on the combination of bortezomib (proteasome inhibitor) and vorinostat or SAHA (HDAC inhibitor) induced synergic effects in inhibiting cancer cell proliferation (Johnson 2015).
Autophagy
Autophagy is acellular process that degrades cytoplasmic constituents misfolded, aggregated, and dysfunctional proteins when these proteins are very high amounts and can’t be handled by the UPS and molecular chaperone PQC systems (Chun and Kim 2018). The inhibition of autophagy genes like Atg7 can induce apoptosis in colon and prostate cancer (Li et al. 2018). In imatinib-resistant CLL and colon cancer, it is observed that the HDACi induce autophagy, and the autophagy pathway induces back the pro-apoptotic and cytostatic effects of HDACi when used for combination treatment. Many HDACi such as Marbostat-100, trichostatin A, sodium butyrate, and YCW1 in a combination of various compounds have been reported to have significant effects on the PQC systems in various cancer cells like AML, breast cancer, colon cancer, and ovarian cancer (Noack et al. 2017).
Endogenous ROS Activation
Suberoylanilide hydroxamic acid (SAHA), an FDA-approved drug, was approved for cutaneous T-cell lymphoma. Recent studies with various prodrug and synthetic analog strategies have increased the selectivity against cancer cells. The abundant amount of endogenous ROS (H2O2) in cancer cells removes the OBP cap from the SAHA-OBP (novel SAHA prodrug), and thus an active SAHA drug is released in the cancer cells (Bhagat et al. 2018). The released SAHA reduces the HDAC6 protein levels and induces apoptosis by tubulin hyperacetylation. SAHA-OBP prodrug provides better efficacy that directs SAHA drug treatment toward cancer cells in terms of selectivity and stability.
Thioredoxin Role in ROS Regulation
In mammalian cells, the thioredoxin (Trx) system acts as an antioxidant mechanism by reducing the oxidized proteins. The Trx has two isoforms, namely, Trx1 (found in the cytosol) and Trx2 (found in mitochondria), and its family consists of various highly conserved thiol group proteins such as protein disulfide isomerases, glutaredoxins (Grxs), and quiescin sulfhydryl oxidase (Lee et al. 2013). It can maintain a reducing environment inside a cancer cell microenvironment by catalyzing the electron flux from NADPH to Trx in a two-step process. The N-terminal cysteine of Trx forms a disulfide bond with the substrate protein and then the Trx is oxidized and the substrate protein is reduced, and then the Trx is further reduced by Trx reductase (TrxR) using NADPH as an electron source to enhance the ROS scavenging activity.
In cancer cells, the components of the thioredoxin system such as Trx and Trx reductase were found to be overexpressed and Trx-interacting protein (TXNIP), which is under-expressed (Jia et al. 2019). The amount of Trx alters the ROS levels and can play a crucial role in cancer progression. It also involves the ROS scavenging activity and gene expression of transcription factors such as redox factor-1 (Ref-1), hypoxia-inducible factor-1α (HIF-1α), nuclear factor kappa B (NF-κB), and tumor suppressor genes such as p53 and PTEN (Bassi and Stambolic 2013).
The high mRNA expression of Trx and TrxR is associated with various cancers such as breast, colorectal (CRC), oral, and prostate cancers and NSCLC, while the low expression of TXNIP mRNA levels is associated with breast, liver, and pancreatic cancers (Jia et al. 2019). The overexpression of Trx-1 promotes metastasis, and invasion in CRC shows resistance to docetaxel and cisplatin (Yamada et al. 1996), increases the transcriptional activity of forkhead box protein O1 (FOXO1) (Wang et al. 2015), and activates protein kinase B (Akt) (Li et al. 2012).
Tax inhibitors such as 4-benzothiazole-substituted quinol (PMX464), 1-methylpropyl-2-imidazolyl disulfide (PX-12), and suberoylanilide hydroxamic acid (SAHA) are reported to induce apoptosis and cell cycle arrest at the G2/M phase (You and Park 2017).
Glutathione Role in Antioxidation
Glutathione (GSH) is a tripeptide of glutamic acid (E), cysteine (C), and glycine (C) which is involved in the xenobiotic metabolism with the conjugation of GSH-S-transferases (GST) (Meister 1988). Nearly 90% of the GSH is present in the cytosol, followed by mitochondria and ER. In eukaryotic cells, GSH is present in both thiol-reduced (GSH) and disulfide-oxidized (GSSG) forms, and GSH is found to be 10–100-fold greater than the GSSG form that reacts with ROS and escapes apoptosis in cancer cells (Lushchak 2012). The elevated levels of GSH were found to be associated with cancer cell proliferation, invasion, and metastasis, and also the depleted levels of GSH in cancer cells seem to be more sensitive to treatment like chemotherapy (Marengo et al. 2010). GSH can act as a chemopreventive agent by detoxifying carcinogens and induction of antioxidant and anti-inflammatory responses as well as can also lead to carcinogenesis by evading apoptosis, drug resistance, and maintenance of redox levels in the cells.
The high expression of GSH-related enzymes, such as 𝛾-glutamylcysteine ligase (GCL) and 𝛾-glutamyl transpeptidase (GGT), is also associated with cancer cell growth. Buthionine sulfoximine (BSO), a GCL inhibitor, reduces the GSH levels and induces apoptosis and arrest cell cycle, and even it is also studied as a combination with various drugs for neuroblastoma (Marengo et al. 2011). The GST is overexpressed in cancer cells and directly binds to MAPK pathway kinases and alters its levels which leads to cancer drug resistance.
GSH peroxidase (Gpx) prevents the oxidative stress of the cells by reducing H2O2. Currently, eight GSH peroxidases (Gpx) are reported to be found in various parts of the body and are involved in cancer cell growth promotion by providing a strong antioxidant mechanism to the cells. Gpx1 is downregulated in breast cancer cells and its expression is regulated by the direct binding of the transcription factor AP-2 gamma (TFAP2C) to the Gpx1 promoter in the AP-2 regulatory region (Zhang et al. 2020). The upregulation of Gpx8 is directly associated with EMT via regulation of IL-6 -STAT3, thus promoting stem cell nature in breast cancer. Gpx8 reduces the IL-6 production, leading to low cytokine production, thus resulting in the cancer phenotype.
Cancer cells overexpressed GSH-S-transferases (GST) that conjugate with a variety of anticancer drugs such as cisplatin, busulfan, and dichloroacetate, thereby showing drug resistance in multiple solid cancers. The direct inhibition of GST by ethacrynic acid, ethacraplatin, 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio) hexanol (NBDHEX), auranofin, and piperlongumine can help to overcome drug resistance (Allocati et al. 2018).
Reactive Oxygen Species: Types, Source, and Detoxification
Reactive oxygen species (ROS) are highly reactive, unstable, and partially reduced oxygen derivatives (ions, radicals, or molecules) produced as a by-product of cellular respiration and metabolic process. ROS is classified into free oxygen radicals and non-radicals and its detailed types were given in Table 1. Among all, superoxide (O2•−), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH) are highly expressed and well-studied in various cancers (Liou and Storz 2010).
During the cellular respiration, the superoxide (O2•−) radicals are produced during the electron transport chain in the inner mitochondrial membrane and then released to the cytosol or mitochondrial matrix where they are scavenged by Cu/ZnSOD (zinc/copper superoxide dismutase) and MnSOD (manganese superoxide dismutase) into hydrogen peroxide (H2O2), respectively. Then H2O2 is reduced into H2O by antioxidant enzymes such as catalase, glutathione peroxidase (Gpx), and peroxiredoxins (Prx).
ROS in Normal, Cancer, and Cancer Stem Cells
The intracellular ROS plays a major role in cell proliferation, differentiation, and vesicle trafficking, and elevated ROS levels lead to senescence and tumor formation. The high levels of ROS are scavenged by antioxidants such as superoxide dismutases (SODs), glutathione peroxidase (Gpx), glutathione reductase (GR), peroxiredoxin, and catalase present inside the cell (Forman et al. 2014). High ROS levels alter the lipid bilayer structure by inducing oxidative stress by peroxidation of fatty acids, affects protein function by oxidation of redox-reacting cysteine and/or tyrosine residues of signaling proteins, and induces mitochondrial DNA mutations of the gene that encodes for electron transport chain complexes. A result of high metabolic activity and mitochondrial dysfunction in cancer cells leads to high ROS production which can’t be counteracted by the antioxidants.
In normal cells, glucose and glutamine were uptaken to produce ATP by anaerobic glycolysis followed by the Krebs cycle and oxidative phosphorylation in the mitochondria. Pyruvate produced at the end of glycolysis is catalyzed by pyruvate dehydrogenase (PDH) to produce acetyl-CoA to be used in the TCA cycle. Further, the ROS levels are maintained by antioxidants, and the mitochondrial membrane potential (ΔΨm) and oxygen consumption rate (OCR) seem to be higher and with high production of ATP (Lleonart et al. 2018).
In cancer cells, ATP is produced from glycolysis rather than oxidative phosphorylation, thus resulting in a higher metabolic rate at mitochondria and endoplasmic reticulum (ER) (Dickinson and Chang 2011). Here pyruvate dehydrogenase (PDH) is inhibited by hypoxia-driven enzyme PDH kinase 1 (PDK1) and lactate dehydrogenase A (LDHA), thus converting pyruvate into lactate rather than acetyl-CoA (i.e., Warburg effect) which leads to the higher production rate of nucleic acids, amino acids, and fatty acids (Warburg 1956). Due to the mitochondrial membrane potential (ΔΨm), the ROS levels were increased with a higher rate than the ROS scavengers, thereby leading to a malignant state.
In cancer stem cells (CSCs), the ATP production is similar to normal cells with high mitochondrial membrane potential (ΔΨm) and oxygen consumption rate (OCR) with a high production of ATP (Song et al. 2015). The mediated ROS levels in the CSC show drug resistance with the cancer cell survival potency. It also upregulates FOXO1 (forkhead transcription factor), glutathione synthetases, and other antioxidant enzymes to maintain the intracellular ROS levels as a feedback loop process.
ROS Production
In normal somatic cells, ROS production helps in immune defense. In cancer cells, the production of ROS is increased due to environmental factors, such as smoking and UV, and intrinsic factors, such as increased metabolism; expression of various oncogenes such as c-Myc, Ras, and BRCA1; hypoxia condition, integrin activation; etc. (Yang et al. 2018).
ROS in Cancer and Role of Signaling Pathways
Superoxide (O2•−), hydrogen peroxide (H2O2) outcompetes the MnSOD levels and enables high cellular proliferation in cancer as a result of decreased expression of antioxidants. ROS mediates various signaling pathways such as MAPK/ERK1/2 pathway, PI3K/Akt pathway, and IKK/NF-κB pathways. In the MAPK pathway, H2O2 acts in the Cys118 residue of RAS and inhibits the GDP/GTP exchange or directly acts on ERK1/2 (downstream kinase of RAS), thus increasing the cell survival and growth (Steelman et al. 2008). ROS upregulates the mRNA of cyclin B2, cyclin D3, cyclin E1, and cyclin E2 that enables G1/S phase transition, thus leading to increased cell proliferation (Felty et al. 2015). ROS at high intracellular levels induce apoptosis by caspase activation via cytochrome c release from the mitochondria and also activate c-Jun N-terminal kinases (JNKs) that downregulate Bcl-2 and Bcl-XL (anti-apoptotic proteins) and upregulates Bax (apoptotic protein). Though low ROS levels promote cell survival and proliferation by regulating the cell cycle proteins (Dhanasekaran and Reddy 2017), elevated ROS levels activate NF-κB and nuclear factor (erythroid-derived 2)-like-2 factor (Nrf2) transcription factors that lead to apoptotic evasion, proliferation, and metastasis.
High ROS modulates the levels of β-catenin/Wnt, activator protein 1, HIF-1α (hypoxia-inducible factor-1 alpha), inflammatory cytokines, and growth factors leading to inflammation via activation of inducible nitric oxide synthase (iNOS) and downregulation of cyclooxygenase 2 (COX2) enzymes. ROS induces cytokine secretion via caspase1 activation and activator protein 1 protein levels (Forrester et al. 2018). ROS levels also play a major role in epithelial-mesenchymal transition (EMT) and migration by regulating uPA (urokinase-type plasminogen activator) via TGF-β1 upregulation and induction of hypoxia-mediated matrix metalloproteinases (MMPs), respectively. Moreover, high ROS levels upregulate C-X-C motif chemokine 14 (CXCL14) and enable cell motility during EMT (Liao et al. 2019). The ROS effects on gene expression in various cancers are given in Table 2.
Foxo Protein Signaling in ROS Generation in Lung Cancer
Foxo1, Foxo3, and Foxo4 are critically involved in cellular oxidative stress. FoxoM1 gene was found to be amplified in lung cancer (Leone et al. 2017). FoXo competes with TCF and binds at the TCF binding site of β-catenin and suppresses proliferation by inhibiting Wnt pathways. In addition, the FoXo M gene was found to be amplified in lung cancer.
ROS-Dependent Gene Expression in Cancer Cells
ROS is the reactive oxygen molecule involved in a variety of diseases such as cancer, and diabetes, neurodegeneration, etc. ROS is classified in free radical ROS with unpaired electrons or non-radical ROS such as H2O2 that can be converted to radical ROS. ROS is produced by the activation of enzymes cytochrome P450, NADPH oxidase, and cyclooxygenase and also by the activation of nonenzymatic enzymes involving mitochondrial-mediated respiratory chain. Excessive ROS can produce damage to DNA, proteins, and lipids and lead to an increase in ROS that results in cell death (Gorrini et al. 2013). In cells, the most important and widely studied antioxidant ROS scavengers include thioredoxin (Trx), nuclear factor erythroid 2-related factor2 (NRF2), catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase.
ROS was found to be involved in initiation of cancer by activating various pathways including the ERK pathway as well as tumor angiogenesis (Fiaschi and Chiarugi 2012). Studies have shown ROS might induce certain oncogenes such as c-Myc and Ras leading to the stability of nuclear as well as mitochondrial stability. To counteract the changed ROS pattern, cancer cells alter the metabolic pathway that ultimately led to the tumor metastasis. RNA sequencing and DNA mutation status have identified the role of glutathione peroxidase (Gpx), peroxiredoxins (TPx), as well as several genes that are involved in dual-specificity phosphatase-1 (DUSP1), FoxM1, HMoX1, and superoxide dismutase (SOD). The correlation between oxidative gene signature and overall survival has identified gene expression changes involved in ROS metabolisms such as FoxM1, TXNRD1, DUSP1, EPHX2, NUDT1, RNF7, and SEPP1. DUSP1 is a dual-specificity phosphatase involved in the inactivation of MAPK and acts as redox sensitizer (Kim et al. 2012). NUDT1 is a nudix hydrolase that functions in hydrolyzing the oxidized nucleotide leading to DNA damage (Waz et al. 2017). Ring finger protein 7 (RNF7) was found to be highly expressed in cancer cells which acts as a scavenger of ROS in cancer cells (Sun and Li 2012). EPHX2 is a cytosolic epoxide hydrolase involved in cancer metastasis and was found to be upregulated during oxidative stress (Bracalente et al. 2016). SEPP is a selenoprotein that possesses antioxidant properties and is a target of NRF2 family. Bioinformatic analysis by using STRING that predicts protein-protein interaction identified Gpx, SOD, and Trx pathways play an important role in dictating the cancer cell fate.
Anti-oxidative Stress (AOS) Genes
Anti-oxidative stress (AOS) scavenges ROS that arises during cellular metabolism. Gene set enrichment (GO), network, and pathway analysis have identified thioredoxin and glutathione pathways that are tightly associated with cancer. The key downstream targets of AOS are NRF2, NF-kappa B, FoxM1, etc. (Rotblat et al. 2014). Studies using the bio-profiling de GENE SRV tool reveals about particular gene signatures that enriched in various cancers and prediction of patient outcome. The key genes such as BTG3, CASP3, CDC2, G6PD, peroxiredoxin (PRDX4), NUDT1, PRDX4, HMOX1, GAPDH, PSMMB5, SEL, ECT2, EGLN1, LONP1 are involved in poor prognosis. The genes such as COL1A1, GAPDH, GCLC, GSS, NAD(P)H dehydrogenase quinone 1 (NQ01), RNF7, STK24, thioredoxin (TXN), and thioredoxin reductase 1 (TXNRD1) involved in prognosis in lung cancer. Genes that are important for good prognosis are PON2 and SIRT1 in breast cancer and NF-kB1 in lung cancer
HDAC Inhibition and ROS
HDAC inhibition results in tumor cell death by inducing reactive oxygen species (ROS). Interestingly, pretreatment with antioxidants results in the prevention of ROS as well as apoptosis. Cancer cells treated with HDAC inhibitors such as vorinostat and MS-275 induce ROS and caspase activation specifically in cancer cells but not in normal cells (Ungerstedt et al. 2005). Furthermore, treatment with non-hydroxamate NCH-51 resulted in enhancement in ROS level in leukemia cells and cytotoxicity by modulating the genes involved in antioxidation when compared with FDA-approved drug SAHA (Sanda et al. 2007). Surprisingly, SAHA-induced cytotoxicity was enhanced by using small interfering RNA against thioredoxin (Trx). A study by El-Naggar et al. (2019) found that class I HDAC inhibitors enhance Y-box binding protein1 (YB-1) acetylation and induce oxidative stress. Cancers usually inhibit ROS induction partly by the activation of nuclear factor erythroid 2-related factor 2 (NRF2). During oxidative stress, it was found that NRF2 protein gets stabilized and gets dissociated from kelch-like ECH-associated protein 1 (Keap1). The possible ways of NRF2 activation include NRF2 activating mutations, Keap1 inactivating mutations, and oncogene activation.
HDAC Inhibitor and YB1 Relation in ROS Production
MS-275 HDAC inhibitors induce translational activation of NFE2L2 by YB1 (El-Naggar et al. 2019). HDAC inhibitors induce ROS in melanoma cells and acute myeloid leukemia (AML) (Petruccelli et al. 2011). MS-275 specifically targets HDAC-1 and HDAC-3 and induces ROS in leukemia (Rosato et al. 2003). MS-275 function increases YB1 acetylation (K81acetylation). Interestingly, mutation that converts K to E was observed in several cancers. YB1 was found to associate with nuclear protein filaments and bind to mRNA in the cytoplasm, thereby enhancing protein translational efficiency. It was found that YB1 protein is upregulated in breast cancer, colorectal cancer, sarcoma, etc. In breast cancers, it enhances the expression of genes that drives the metastasis such as SNAIL, ZEB2, and Twist1 (Evdokimova et al. 2009). In colorectal cancers (CRCs), YB1 regulates IGF1R. Y-box binding protein 1 is an RNA binding protein that binds to the 5′-untranslated region (5′-UTR) or 3′-untranslated region (3′-UTR) of various genes via its cold shock domain (CSD) (Eliseeva et al. 2011). Interestingly, in sarcomas, YB1 binds to 5′-UTR of the HIF1A gene and activates the HIF-1α mRNA synthesis (El-Naggar et al. 2015). Thus, YB1 regulation by HDAC inhibitor has a huge potential in controlling cancer cell metastasis.
HDAC Inhibitors and ROS-Mediated Effects on Cancer Cells
Studies have identified HDAC inhibitor-induced apoptosis is mediated by ROS-dependent mechanism. Hydroxamate pan-HDAC inhibitor LAQ-824 (40 nM) treatment at lower concentrations induces enhanced ROS generation and sustained DNA damage and upregulation of γ-H2AX foci. Interestingly, the apoptosis was not significant in those cells before treatment with LAQ-824 followed by fludarabine (0.4μM) results in enhanced apoptosis in leukemia cells via a drastic increase in γH2AX, phosphor ATM, ROS, and Bak expression (Rosato et al. 2008). The LAQ-824-induced ROS was found to be diminished by the treatment of ROS scavengers such as N-acetyl cysteine (NAC) or manganese (III) tetra kis-4-benzoic acid porphyrin (mn-TBAP). Interestingly, the treatment of HDAC inhibitor also involves an inhibitory effect on DNA repair genes via decreased expression on Ku86 and RAD50, BRCA1, RAD51, etc. Occurrence of this event also decreased the DNA binding activity of DNA repair proteins, activation of caspase-2, and release of histone H1.2 into the cytoplasm. Studies by Reczek and Chandel (2017) have indicated that oncogene, loss of tumor suppressor, enhanced metabolism, hypoxia, and low glucose play an important role in ROS in various cancers.
GBM vs. ROS
HDAC inhibitors have been shown to improve the outcomes by regulation of acetylation process in the preclinical setting. SAHA was found to be efficient in apoptosis induction in GBM cells (Premkumar et al. 2013). Furthermore, the combination of bortezomib and SAHA usage leads to effective therapy by enhancing the activation of Bax, Bak, cytochrome release (pro-apoptotic mitochondria injury), and γ-H2Ax foci with concomitant downregulation of Rad51 (Jane et al. 2011). HDAC inhibitors such as LBH589, LAQ 824, and trichostatin A combined with AEE 78 (inhibitor of MAPK, Akt) cause enhanced apoptosis in non-small cell lung cancer, ovarian cancer, and leukemia cells via ROS generation. This indicates HDACi have potential ROS inducers. Glioma cancer (GBM) isocitrate dehydrogenase IDH1 (R132H) causes resistance to HDAC inhibitors (Kim et al. 2019). Also, glioma with IDH mutations inhibits IDH catalytic activity and enhances hypoxia-inducible factor-1 α (HIF-α). IDH-R132H mutation led to HIF-1-α expression. Also, FAT1 (a typical cadherin 1), the upstream regulator of ROS, transcriptionally enhances HIF-α, and in turn, IDH R13H regulates FAT1 (Kumar et al. 2020). Studies have shown ROS production is regulated mainly through the hedgehog pathway.
ROS and reactive nitrogen are linked with the redox system in the tumor microenvironment. Chidamide (HDACi) treatment in GBM cancer cells leads to the upregulation of microRNA miR-338-5p located in intron 8 of apoptosis-associated tyrosine kinase (Lei et al. 2017). Studies have indicated that miR-338-5p mimics decrease U87, HS683, and MiR-338-5p. Researchers have found in gastrointestinal cancer cells, namely GI and GU, that the treatment of HDAC inhibitor induces the expression of Fas ligand (CD95L) and its receptor CD95 R (FAS receptor) leading to cancer cell death. Similarly, a combination of sorafenib and HDAC inhibitors kills GBM and medulloblastoma cells (Tang et al. 2012).
HDAC Inhibitors in Bladder Cancer
HDAC inhibitors (HDACi) romidepsin, trichostatin A, and vorinostat were found to be the most important chemotherapeutic agents for bladder cancer. Treatment with HDAC inhibitors resulted in the upregulation of protein expression of 2472 genes as well as downregulation of protein expression of 2049 proteins when compared to the untreated control cells. The bioinformatics analysis study has identified the involvement of these differentially expressed proteins in the regulation of cell cycle, cell death, free radical generation, and immune regulatory pathways. Proteomic analysis has also confirmed the role of HDAC inhibitors such as TSA and romidepsin on cell cycle, oxidative stress, apoptosis, etc.
Resistance Against HDAC Inhibitors in Gastric Cancer
Studies by Zhu et al. (2014) using a panel of gastric cancer cells have identified the important role of ribonuclease inhibitor known as RNH1 in driving the chemoresistance during HDAC inhibitor treatment (trichostatin A, SAHA). In addition, overexpression of the RNH1 gene in gastric cancer cells prevents ROS production and inhibits cancer cell apoptosis. This study has revealed the importance of ROS generation for effective apoptosis in cancer cells treated with HDAC inhibitors. The differential gene expression due to HDAC inhibitor was studied insensitively as well as resistant cell line using Affymetrix platform as well as Illumina platform. Studies have observed that seven gees such as ribonuclease inhibitor (RNH1), signal transducer and activator of transcription 1 (STAT1), C-X-C motif chemokine ligand (CXCL5), RAB40B (a member of Ras oncogene family), bladder cancer-associated protein (BLCAP), sphingosine-1-phosphate phosphatase-2 (SGPP2), and ELF
HDAC Inhibitors in Rhabdomyosarcoma
HDAC inhibitors such as TSA and SAHA induce myogenic differentiation and also inhibit tumor growth in embryonal rhabdomyosarcoma (ERMS), the most common soft tissue cancer in children, which is characterized by poor prognosis. Loss- or gain-of-function studies have identified the role of NOTCH1 and Ephrin B1 pathways that were regulated by HDACs to drive the tumor cell migration and inhibit the differentiation.
HDAC Inhibitors and Autophagy
Suberoylanilide hydroxamic acid (SAHA) stimulates autophagy in T-cell leukemia as evident by the accumulation of autophagic vacuoles and conversion of LC-III -I. SAHA upregulated Beclin-1 and Atg7 and promote Atg12-Atg5 (Li et al. 2010). Also, several HDAC inhibitors modulate apoptosis and autophagy in various cancer cells (Table 3).
Systemic Approaches
Rosenwasser et al. (2013) developed a bioinformatics tool named ROSMETER for the identification of transcriptomic imprints related to ROS (reactive oxygen species) in Arabidopsis thaliana. In ROSMETER, the transcriptome was given as query and the ROS signature profiles of biotic and abiotic stress-induced plants. This platform helps to identify the molecular-level mechanism of ROS in A. thaliana.
Cancer cells produce more antioxidants to scavenge ROS by the exploitation of anti-oxidative stress (AOS) response genes. Rotblat et al. (2013) examined the expression of 285 oxidative stress genes from 994 tumors and 353 normal tissues and found that 116 oxidative stress genes overexpressed in multiple types of cancers and under-expressed in normal cells. They also used Gene-set enrichment, Gene Ontology, network, and pathway analysis and found that the thioredoxin and glutathione pathway genes were correlated with cancer.
Yang et al. (2019) developed OxidizeME, a multiscale description of stress response induced by ROS in E. coli. It addresses major oxidative stress responses which include ROS-induced auxotrophy, nutrient-dependent sensitivity of growth, ROS-specific differential gene expression, and coordinated expression of an iron-sulfur cluster (ISC) and sulfur assimilation (SUF) systems.
Posen et al. (2005) developed a precise tool photo-switch to analyze the ROS type and regulation at different doses in cell culture models. This tool enables us to exactly predict the ROS generation and regulation in cellular mechanisms in detail even in viable cells. They used hydroethidine as a detector and determined light-dependent generation of ROS in situ photogeneration of a nontoxic bacteriochlorophyll-based sensitizer in cell culture models. Even, it is helpful in the study of the ROS effects on various protein kinases involved in the cell cycle regulation and function.
Topf et al. (2015) used a quantitative proteomics approach to identify redox switches modulated by ROS. They performed a global proteomic analysis of nearly 2200 proteins in yeast redoxome, to map redox-active thiols influenced by ROS by various approaches that include isotope-coded affinity tag (ICAT) and state-of-the-art liquid chromatography tandem MS (LC-MS/MS). From this approach, they found that the high intracellular ROS levels cause mitochondrial dysfunction, which further leads to regulation of protein synthesis inside the cells.
System Biology Approach on Regulation of Molecular Pathways by HDAC Inhibitors in Cancer
Wittenburg et al. (2012) have employed Affymetrix canine v2.0 genechip in canine osteosarcoma cells treated with valproic acid (VPA) and have identified differential expression of various genes and the pathways by Meta Core software version 6.4 and have identified the involvement in cell cycle, cytoskeleton remodeling, the ubiquitin proteasome system, and oxidative phosphorylation. The significance of various genes involved in a particular pathway was confirmed by Fisher’s exact test. Furthermore, the validation of microarray results was carried out by a real-time PCR study in which average threshold values (Ct) were used to confirm the gene expression changes using Relative Expression Software Tool (REST) v2.0.13 (Qiagen).
Computational Approach in Drug Discovery
It is well-known that Food and Drug Administration (FDA) drugs as well as the drug repurposing strategy reduce the cost and time to discover new drugs against a particular disease condition. Many of the recent day drugs can reverse the gene expression present in cancer cells, but researchers usually failed to consider the various functions and their dependencies’ system level. In this regard, computational approaches that analyze the transcriptional data would help in the drug repurposing strategy and for effective drug discovery (Peyvandipour et al. 2018).
Potential Application of HDAC Inhibitors in Inducing Apoptosis
The normal fibroblast is transformed to a cancerous state by using various genes such as SV40-T antigens, hTERT, and mutant Ras (H-Ras G12V). The transformed fibroblast cells with oncogenes such as Ras (BJ LTSTERas) and normal (BJ) fibroblast cells that were treated with romidepsin (HDAC inhibitor) have indicated that HDAC inhibitors induce tumor-selective apoptosis. Furthermore, the differential expression of various genes was studied by using Affymetrix microarray analysis. In addition, Gene Ontology (GO) and pathway analyses have shown the involvement of apoptotic pathway. Further, IPA tool was employed to identify the impact of HDAC inhibitors on various molecular and cellular functions (Lamb et al. 2006).
Network analysis involving transcriptional profiling, metabolic flux analysis (MFA), and biochemical analysis was also employed to understand the enhanced glycolysis, tricarboxylic acid cycle (TCA), and use of glutamine in cancer cells. A recent clinical study has shown abexinostat (PCI24781) was known to inhibit class I and class II HDAC and induce apoptosis through the induction of reactive oxygen species (ROS) in B-cell lymphoma (Teodori et al. 2020). RNA sequencing studies in synovial sarcoma cells treated with quinostat that inhibit class I and class II HDACs have indicated an altered gene expression related to cell cycle arrest, neuronal differentiation, and reactive oxygen species generation (Laporte et al. 2017). In addition, microarray studies in thyroid cancer cells treated with PDX101 and LBH589 have shown common changes in cell cycle regulatory genes, DNA damage, as well as apoptosis-related genes. Also, pituitary tumor cell line AtT20 treated with SAHA has demonstrated downregulation of LXRα and upregulation of apoptosis-related genes (Lu et al. 2017). Interestingly, pancreatic ductal carcinoma cell lines treated with the HDAC inhibitor domatinostat (4SC-202) were subjected to RNA sequencing (RNA-seq) and ChIP sequencing, and the results have indicated an enhanced histone acetylation status, particularly H3K27ac in the promoter regions of the upregulated genes due to HDAC inhibitor treatment. Further studies by using ReMap (regulatory map of TF binding sites) analysis tool demonstrated the chromatin occupancy by bromodomain-containing protein-4 (BRD4) and MYC proteins at their binding sites (Mishra et al. 2017).
Gene expression profiling (GEP) and Gene Ontology enrichment analyses have indicated that 35 gene signatures are associated with the actin cytoskeleton and protein processing in the endoplasmic reticulum that are responsible for decreasing sensitivity of multiple myeloma (MM) cancer cells to HDACi (Mithraprabhu et al. 2013). Zhu et al. (2016) have studied global proteome and lysine acetylome, 3-plex SILAC-based quantitative proteomics technique, high-resolution LC-MS/MS, and bioinformatics analysis have identified a total of 1124 lysine acetylation sites in valproic acid and SAHA-treated AML cells. Surprisingly, the acetylome changes mediated by both the HDAC inhibitors were different from each other.
Conclusions and Future Directions
In summary, this chapter focuses on the role of HDAC inhibitors in the generation of ROS and apoptotic induction in cancer cells via intrinsic as well as extrinsic pathways. The ROS modulatory activity of HDAC inhibitors such as MS-275, vorinostat, and valproic acid in various cancers helps in effectively killing cytotoxicity. Furthermore, various proteins such as NF-kB and Foxo play a vital role in ROS generation. HDAC inhibitor-mediated ROS generation involves activation of Notch and Wnt pathways. In addition to epigenomics, mRNA expression profiling, microRNA expression study, and proteomics, metabolomics, and chemo-proteomics approaches have revealed the importance of the complete understanding of therapeutic response in cancer and various diseases. Studies using system biology and molecular biology approaches during HDAC inhibitor-mediated ROS will help in effective drug discovery, therapeutic response, and drug repurposing, as well as diagnostic aspects against cancer (Fig. 2).

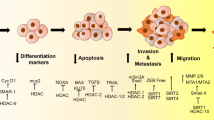
Various approaches that are involved in personalized therapy and disease diagnosis
Abbreviations
- 4HNE:
-
4-Hydroxynonenal
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- ApoE−/− mice:
-
Apolipoprotein E knockout mice
- ATG7:
-
Autophagy related 7
- BAM:
-
β-Barrel assembly machinery
- BAX:
-
BCL2-associated X, apoptosis regulator
- Bcl-2:
-
B-cell lymphoma 2
- Bcl-xL:
-
B-cell lymphoma-extra large
- Bid:
-
BH3-interacting domain death agonist
- Bim:
-
Bcl-2-like protein 11
- CD45:
-
Lymphocyte common antigen
- CD68:
-
Cluster of differentiation 68
- CDKN1A:
-
Cyclin-dependent kinase inhibitor 1A
- CDKs:
-
Cyclin-dependent kinases
- CLL:
-
Chronic lymphocytic leukemia
- CTCL:
-
Cutaneous T-cell lymphoma
- CVD:
-
Cardiovascular diseases
- DR4:
-
Death receptor 4
- DR5:
-
Death receptor 5
- eNOS:
-
Endothelial nitric oxide synthase
- FAS:
-
Fas cell surface death receptor
- FOXO1:
-
Forkhead box protein O1
- GSH:
-
Glutathione
- H2O2:
-
Hydrogen peroxide
- HD:
-
Huntington’s disease
- HDAC:
-
Histone deacetylase
- HDACi:
-
HDAC inhibitor
- HSP70:
-
Heat shock protein 90
- HSP90:
-
Heat shock protein 70
- HSPs:
-
Heat shock proteins
- LAQ-824:
-
Dacinostat
- LBH589:
-
Panobinostat
- MDM2:
-
Mouse double minute 2 homolog
- mHTT:
-
Mutant huntingtin
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NOX1:
-
NADPH oxidase 1
- NOX4:
-
NADPH oxidase 4
- PCI24781:
-
Abexinostat
- PDH:
-
Pyruvate dehydrogenase
- PDKs:
-
Pyruvate dehydrogenase kinases
- PEITC:
-
β-Phenylethyl isothiocyanate
- PQC:
-
Protein quality control
- ROS:
-
Reactive oxygen species
- SAHA:
-
Suberoylanilide hydroxamic acid
- sHSPs:
-
Small heat shock proteins
- TNF-α:
-
Tumor necrosis factor alpha
- TRIAL:
-
NF-related apoptosis-inducing ligand
- UPS:
-
Ubiquitin proteasome system
- VEGF:
-
Vascular endothelial growth factor
- AKT:
-
Protein kinase B
- BSO:
-
Buthionine sulfoximine
- CRC:
-
Colorectal cancer
- GCL:
-
𝛾-Glutamylcysteine ligase
- Gpx:
-
Glutathione peroxidase
- Grxs:
-
Glutaredoxins
- GST:
-
GSH-S-transferases
- HIF-1α:
-
Hypoxia-inducible factor-1α
- IL-6:
-
Interleukin-6
- MAPK:
-
Mitogen-activated protein kinase
- mRNA:
-
Messenger RNA
- NBDHEX:
-
Ethacraplatin, 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio) hexanol
- NSCLC:
-
Non-small cell lung carcinoma
- PMX464:
-
4-Benzothiazole-substituted quinol
- PTEN:
-
Phosphatase and tensin homolog deleted on chromosome 10
- PX-12:
-
1-Methylpropyl-2-imidazolyl disulfide
- Ref-1:
-
Redox factor-1
- STAT3:
-
Signal transducer and activator of transcription 3
- TFAP2C:
-
Transcription factor AP-2 gamma
- Trx:
-
Thioredoxin
- TrxE:
-
Trx reductase
- TXNIP:
-
Trx-interacting protein
- ΔΨm:
-
Mitochondrial membrane potential
- Cav-1:
-
Caveolin-1
- COX2:
-
Cyclooxygenase 2
- CSCs:
-
Cancer stem cells
- Cu/ZnSOD:
-
Zinc/copper superoxide dismutase
- CXCL14:
-
C-X-C motif chemokine 14
- CYGB:
-
Cytoglobin
- EMT:
-
Epithelial-mesenchymal transition
- ER:
-
Endoplasmic reticulum
- FoxA1:
-
Forkhead box protein A1
- FOXM1:
-
Forkhead box M1
- GR:
-
Glutathione reductase
- iNOS:
-
Inducible nitric oxide synthase
- JNKs:
-
c-Jun N-terminal kinases
- LDHA:
-
Lactate dehydrogenase A
- MMPs:
-
Matrix metalloproteinases
- MnSOD:
-
Manganese superoxide dismutase
- NOX5:
-
NADPH oxidase
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- NSCLC:
-
Non-small cell lung cancer
- NUDT1:
-
(Nucleoside diphosphate linked moiety X)-type motif 1
- OCR:
-
Oxygen consumption rate
- PDH1:
-
PDH kinase 1
- Prx:
-
Peroxiredoxins
- SEPP1:
-
Selenoprotein P1
- SNAIl:
-
Zinc finger protein SNAI1
- SODs:
-
Superoxide dismutases
- TGF-β1:
-
Transforming growth factor beta 1
- UCP2:
-
Mitochondrial uncoupling protein 2
- uPA-:
-
Urokinase-type plasminogen activator
- VEGF:
-
Vascular endothelial growth factor
- AOS:
-
Anti-oxidative stress
- ICAT:
-
Isotope-coded affinity tag
- ISC:
-
Iron-sulfur cluster
- LC-MS/MS:
-
Liquid chromatography tandem MS
- SUF:
-
Sulfur assimilation
- ATM:
-
Ataxia telangiectasia mutated
- Bax:
-
BCL2-associated X, apoptosis regulator
- Bcl-2:
-
B-cell lymphoma 2
- Bid:
-
BH3-interacting domain death agonist
- Bmi1:
-
Polycomb group ring finger protein 4
- GSH:
-
Glutathione
- NADPH oxidase:
-
Nicotinamide adenine dinucleotide phosphate oxidase
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- SAHA:
-
Suberanilohydroxamic acid (vorinostat)
- SLC7A11:
-
Cystine/glutamate antiporter xCT
- SOD2:
-
Superoxide dismutases 2
- STAT3:
-
Signal transducer and activator of transcription 3
- TSA:
-
Trichostatin A
- VPA:
-
Valproic acid
- 4SC-202:
-
Domatinostat
- BLCAP:
-
Bladder cancer-associated protein
- BRCA1:
-
Breast cancer type 1 susceptibility protein
- BRD4:
-
Bromodomain-containing protein-4
- BTG3:
-
BTG anti-proliferation factor 3
- CASP3:
-
Caspase-3
- CAT:
-
Catalase
- CDC2:
-
Cyclin-dependent kinase 1
- Cmap:
-
Connectivity map
- COL1A1:
-
Collagen, type I, alpha 1
- CSD:
-
Cold shock domain
- Ct:
-
Threshold values
- DUSP1:
-
Dual-specificity phosphatase-1
- Ect2:
-
Epithelial cell transforming 2
- EGLN1:
-
Hypoxia-inducible factor prolyl hydroxylase 2
- EPHX2:
-
Epoxide hydrolase 2
- ERMS:
-
Embryonal rhabdomyosarcoma
- FOXM1:
-
Forkhead box protein M1
- Foxo:
-
Forkhead box transcription factors
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GBM:
-
Glioblastoma
- GCLC:
-
Glutamate-cysteine ligase catalytic subunit
- GEP:
-
Gene expression profiling
- GO:
-
Gene Ontology
- GSS:
-
Glutathione synthetase
- HMOX1:
-
Heme oxygenase 1
- IDH1:
-
Isocitrate dehydrogenase
- Keap1:
-
Kelch-like ECH-associated protein 1
- LONP1:
-
Lon protease homolog, mitochondrial precursor
- MFA:
-
Metabolic flux analysis
- MM:
-
Multiple myeloma
- NAC:
-
N-acetyl cysteine
- NFE2L2:
-
Nuclear factor, erythroid 2-like 2
- NPC:
-
Nasopharyngeal cancer
- NQ01:
-
NAD(P)H dehydrogenase [quinone] 1
- NUDT1:
-
Nudix hydrolase 1
- PCI24781:
-
Abexinostat
- PON2:
-
Paraoxonase 2
- PR DX4:
-
Peroxiredoxin
- PSMMB5:
-
Proteasome subunit beta type-5
- RAD50:
-
RAD50 double-strand break repair protein
- ReMap:
-
Regulatory map of TF binding sites
- REST:
-
Relative expression software tool
- RNA-seq:
-
RNA sequencing
- RNF7:
-
Ring finger protein 7
- RNF7:
-
Ring finger protein 7
- Sepp1:
-
Selenoprotein P
- SGPP2:
-
Sphingosine-1-phosphate phosphatase-2
- SIRT1:
-
Sirtuin 1
- STAT1:
-
Signal transducer and activator of transcription 1
- STK24:
-
Serine/threonine-protein kinase 24
- TPx:
-
Peroxiredoxins
- TSA:
-
Trichostatin A
- TXNRD1:
-
Thioredoxin reductase 1
- TXNRD1:
-
Thioredoxin reductase 1
- YB1:
-
Y-box binding protein1
References
Allocati N, Masulli M, Di Ilio C et al (2018) Glutathione transferases: substrates, inhibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 7(1):8. https://doi.org/10.1038/s41389-017-0025-3
Bassi C, Stambolic V (2013) PTEN, here, there, everywhere. Cell Death Differ 20(12):1595–1596. https://doi.org/10.1038/cdd.2013.127
Bhagat SD, Singh U, Mishra RK et al (2018) An endogenous reactive oxygen species (ROS)-activated histone deacetylase inhibitor prodrug for cancer chemotherapy. ChemMedChem 13(19):2073–2079. https://doi.org/10.1002/cmdc.201800367
Bolden JE, Shi W, Jankowski K et al (2013) HDAC inhibitors induce tumor-cell selective pro-apoptotic transcriptional responses. Cell Death Dis 4:e519. https://doi.org/10.1038/cddis.2013.9
Bracalente C, Salguero N, Notcovich C et al (2016) Reprogramming human A375 amelanotic melanoma cells by catalase overexpression: reversion or promotion of malignancy by inducing melanogenesis or metastasis. Oncotarget 7(27):41142–41153. https://doi.org/10.18632/oncotarget.9220
Butler LM, Zhou X, Xu WS et al (2002) The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A 99(18):11700–11705. https://doi.org/10.1073/pnas.182372299
Chandra J, Tracy J, Loegering et al (2006) Adaphostin-induced oxidative stress overcomes BCR/ABL mutation-dependent and -independent imatinib resistance. Blood 107(6):2501–2506. https://doi.org/10.1182/blood-2005-06-2302
Chun Y, Kim J (2018) Autophagy: an essential degradation program for cellular homeostasis and life. Cells 7(12):278. https://doi.org/10.3390/cells7120278
Dhanasekaran DN, Reddy EP (2017) JNK-signaling: a multiplexing hub in programmed cell death. Genes Cancer 8(9-10):682–694. https://doi.org/10.18632/genesandcancer.155
Dickinson BC, Chang CJ (2011) Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 7(8):504–511. https://doi.org/10.1038/nchembio.607
Eliseeva IA, Kim ER, Guryanov SG et al (2011) Y-box-binding protein 1 (YB-1) and its functions. Biochemistry (Mosc) 76(13):1402–1433. https://doi.org/10.1134/S0006297911130049
El-Naggar AM, Veinotte CJ, Cheng H et al (2015) Translational activation of HIF1α by YB-1 promotes sarcoma metastasis. Cancer Cell 27(5):682–697. https://doi.org/10.1016/j.ccell.2015.04.003
El-Naggar AM, Somasekharan SP, Wang Y et al (2019) Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep 20(12):e48375. https://doi.org/10.15252/embr.201948375
Evdokimova V, Tognon C, Ng T et al (2009) Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell 15(5):402–415. https://doi.org/10.1016/j.ccr.2009.03.017
Felty Q, Singh KP, Roy D (2015) Estrogen-induced G1/S transition of G0-arrested estrogen-dependent breast cancer cells is regulated by mitochondrial oxidant signaling. Oncogene 24(31):4883–4893. https://doi.org/10.1038/sj.onc.1208667
Fiaschi T, Chiarugi P (2012) Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int J Cell Biol 2012:762825. https://doi.org/10.1155/2012/762825
Forman HJ, Ursini F, Maiorino M (2014) An overview of mechanisms of redox signaling. J Mol Cell Cardiol 73:2–9. https://doi.org/10.1016/j.yjmcc.2014.01.018
Forrester SJ, Kikuchi DS, Hernandes MS et al (2018) Reactive oxygen species in metabolic and inflammatory signaling. Circ Res 122(6):877–902. https://doi.org/10.1161/CIRCRESAHA.117.311401
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12(12):931–947. https://doi.org/10.1038/nrd4002
Hui KF, Chiang AK (2014) Combination of proteasome and class I HDAC inhibitors induces apoptosis of NPC cells through an HDAC6-independent ER stress-induced mechanism. Int J Cancer 135(12):2950–2961. https://doi.org/10.1002/ijc.28924
Jane EP, Premkumar DR, Pollack IF (2011) Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-{kappa}B signaling pathway. Mol Cancer Ther 10(1):198–208. https://doi.org/10.1158/1535-7163.MCT-10-0725
Jeng W, Lee S, Sung N et al (2015) Molecular chaperones: guardians of the proteome in normal and disease states. F1000Res 4:214. https://doi.org/10.12688/f1000research.7214.1
Jia JJ, Geng WS, Wang ZQ et al (2019) The role of thioredoxin system in cancer: strategy for cancer therapy. Cancer Chemother Pharmacol 84(3):453–470. https://doi.org/10.1007/s00280-019-03869-4
Johnson DE (2015) The ubiquitin-proteasome system: opportunities for therapeutic intervention in solid tumors. Endocr Relat Cancer 22:T1–T17. https://doi.org/10.1530/ERC-14-0005
Kim HS, Ullevig SL, Zamora D et al (2012) Redox regulation of MAPK phosphatase 1 controls monocyte migration and macrophage recruitment, Proc. Natl. Acad Sci 109(41):E2803–E2812. https://doi.org/10.1073/pnas.1212596109
Kim JH, Jang WY, Jung TY et al (2019) Recurrent glioma with lineage conversion from oligodendroglioma to astrocytoma in two cases. Front Oncol 9:828. https://doi.org/10.3389/fonc.2019.00828
Kovacs JJ, Murphy PJ, Gaillard S et al (2005) HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 18(5):601–607. https://doi.org/10.1016/j.molcel.2005.04.021
Kumar S, Banerjee J, Tripathi M et al (2020) IDH1-R132H-FAT1-ROS-HIF-1α signaling pathway inhibits glioblastoma tumor progression. Neurol India 68(5):1059–1060. https://doi.org/10.4103/0028-3886.294541
Lamb J, Crawford ED, Peck D et al (2006) The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313(5795):1929–1935. https://doi.org/10.1126/science.1132939
Laporte AN, Poulin NM, Barrott JJ et al (2017) Death by HDAC inhibition in synovial sarcoma cells. Mol Cancer Ther 16(12):2656–2667. https://doi.org/10.1158/1535-7163.MCT-17-0397
Lee YJ, Won AJ, Lee J et al (2012) Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifenresistant MCF-7 breast cancer cells. Int J Med Sci 9:881–893. https://doi.org/10.7150/ijms.5011
Lee S, Kim SM, Lee RT (2013) Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid Redox Signal 18(10):1165–1207. https://doi.org/10.1089/ars.2011.4322
Lei D, Zhang F, Yao D et al (2017) MiR-338-5p suppresses proliferation, migration, invasion, and promote apoptosis of glioblastoma cells by directly targeting EFEMP1. Biomed Pharmacother 89:957–965. https://doi.org/10.1016/j.biopha.2017.01.137
Leone A, Roca MS, Ciardiello C et al (2017) Oxidative stress gene expression profile correlates with cancer patient poor prognosis: identification of crucial pathways might select novel therapeutic approaches. Oxid Med Cell Longev 2017:2597581. https://doi.org/10.1155/2017/2597581
Li J, Liu R, Lei Y et al (2010) Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 6(6):711–724. https://doi.org/10.4161/auto.6.6.12397
Li C, Thompson MA, Tamayo AT et al (2012) Over-expression of thioredoxin-1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B-cell lymphoma. Oncotarget 3(3):314–326. https://doi.org/10.18632/oncotarget.463
Li T, Zhang C, Hassan S et al (2018) Histone deacetylase 6 in cancer. J Hematol Oncol 11(1):111. https://doi.org/10.1186/s13045-018-0654-9
Liao Z, Chua D, Tan NS (2019) Reactive oxygen species: a volatile driver of field cancerization and metastasis. Mol Cancer 18(1):65. https://doi.org/10.1186/s12943-019-0961-y
Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44(5):479–496. https://doi.org/10.3109/10715761003667554
Lleonart ME, Abad E, Graifer D et al (2018) Reactive oxygen species-mediated autophagy defines the fate of cancer stem cells. Antioxid Redox Signal 28(11):1066–1079. https://doi.org/10.1089/ars.2017.7223
Lu J, Chatain GP, Bugarini A et al (2017) Histone deacetylase inhibitor SAHA is a promising treatment of Cushing disease. J Clin Endocrinol Metab 102(8):2825–2835. https://doi.org/10.1210/jc.2017-00464
Lushchak V (2012) Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids 736837. https://doi.org/10.1155/2012/736837
Marengo B, De Ciusis C, Ricciarelli R et al (2010) DNA oxidative damage of neoplastic rat liver lesions. Oncol Rep 23(5):1241–1246. https://doi.org/10.3892/or_00000756
Marengo B, De Ciucis C, Ricciarelli R et al (2011) PKCδ sensitizes neuroblastoma cells to L-buthionine-sulfoximine and etoposide inducing reactive oxygen species overproduction and DNA damage. PLoS One 6(2):e14661. https://doi.org/10.1371/journal.pone.0014661
Meister A (1988) On the discovery of glutathione. Trends Biochem Sci 13(5):185–188. https://doi.org/10.1016/0968-0004(88)90148-x
Miller CP, Singh MM, Rivera-Del Valle N et al (2011) Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J Biomed Biotechnol 2011:514261. https://doi.org/10.1155/2011/514261
Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6:38–51. https://doi.org/10.1038/nrc1779
Mishra VK, Wegwitz F, Kosinsky RL et al (2017) Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res 45(11):6334–6349. https://doi.org/10.1093/nar/gkx212
Mithraprabhu S, Khong T, Jones SS et al (2013) Histone deacetylase (HDAC) inhibitors as single agents induce multiple myeloma cell death principally through the inhibition of class I HDAC. Br J Haematol 162(4):559–562. https://doi.org/10.1111/bjh.12388
Murakami T, Sato A, Chun NAL et al (2008) Transcriptional modulation using HDACi depsipeptide promotes immune cell-mediated tumor destruction of murine B16 melanoma. J Invest Dermatol 128:1506–1516. https://doi.org/10.1038/sj.jid.5701216
Newbold A, Falkenberg KJ, Prince HM et al (2016) How do tumor cells respond to HDAC inhibition? FEBS J 283(22):4032–4046. https://doi.org/10.1111/febs.13746
Noack K, Mahendrarajah N, Hennig D et al (2017) Analysis of the interplay between all-trans retinoic acid and histone deacetylase inhibitors in leukemic cells. Arch Toxicol 91(5):2191–2208. https://doi.org/10.1007/s00204-016-1878-5
Petruccelli LA, Dupere-Richer D, Pettersson F et al (2011) Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One 6:e20987. https://doi.org/10.1371/journal.pone.0020987
Peyvandipour A, Saberian N, Shafi A et al (2018) A novel computational approach for drug repurposing using systems biology. Bioinformatics 34(16):2817–2825. https://doi.org/10.1093/bioinformatics/bty133
Posen Y, Kalchenko V, Seger R et al (2005) Manipulation of redox signaling in mammalian cells enabled by controlled photogeneration of reactive oxygen species. J Cell Sci 118(Pt 9):1957–1969. https://doi.org/10.1242/jcs.02323
Premkumar DR, Jane EP, Agostino NR et al (2013) Bortezomib-induced sensitization of malignant human glioma cells to vorinostat-induced apoptosis depends on reactive oxygen species production, mitochondrial dysfunction, Noxa upregulation, Mcl-1 cleavage, and DNA damage. Mol Carcinog 52(2):118–133. https://doi.org/10.1002/mc.21835
Qian DZ, Kato Y, Shabbeer S et al (2006) Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res 12(2):634–642. https://doi.org/10.1158/1078-0432.CCR-05-1132
Reczek CR, Chandel NS (2017) The two faces of reactive oxygen species in cancer. Annu Rev Cancer Biol 1:79–98. https://doi.org/10.1146/annurev-cancerbio-041916-065808
Richon VM, Sandhoff TW, Rifkind RA et al (2000) Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A 97(18):10014–10019. https://doi.org/10.1073/pnas.180316197
Rivera-Del Valle N, Cheng T, Irwin ME et al (2018) Combinatorial effects of histone deacetylase inhibitors (HDACi), vorinostat and entinostat, and adaphostin are characterized by distinct redox alterations. Cancer Chemother Pharmacol 81(3):483–495. https://doi.org/10.1007/s00280-017-3509-0
Rosato RR, Almenara JA, Grant S (2003) The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21. Cancer Res 63(13):3637–3645
Rosato RR, Almenara JA, Maggio SC et al (2008) Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol Cancer Ther 7(10):3285–97. https://doi.org/10.1158/1535-7163.MCT-08-0385
Rosenwasser S, Fluhr R, Joshi JR et al (2013) ROSMETER: a bioinformatic tool for the identification of transcriptomic imprints related to reactive oxygen species type and origin provides new insights into stress responses. Plant Physiol 163(2):1071–1083. https://doi.org/10.1104/pp.113.218206
Rotblat B, Grunewald TG, Leprivier G et al (2013) Anti-oxidative stress response genes: bioinformatic analysis of their expression and relevance in multiple cancers. Oncotarget 4(12):2577–2590. https://doi.org/10.18632/oncotarget.1658
Rotblat B, Southwell AL, Ehrnhoefer DE et al (2014) HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc Natl Acad Sci U S A 111(8):3032–3037. https://doi.org/10.1073/pnas.1314421111
Ruefli AA, Ausserlechner MJ, Bernhard D et al (2001) The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A 98(19):10833–10838. https://doi.org/10.1073/pnas.191208598
Sanda T, Okamoto T, Uchida Y et al (2007) Proteome analyses of the growth inhibitory effects of NCH-51, a novel histone deacetylase inhibitor, on lymphoid malignant cells. Leukemia 21(11):2344–2353. https://doi.org/10.1038/sj.leu.2404902
Sholler GS, Currier EA, Dutta A et al (2013) PCI-24781 (abexinostat), a novel histone deacetylase inhibitor, induces reactive oxygen species-dependent apoptosis and is synergistic with bortezomib in neuroblastoma. J Cancer Ther Res 2:21. https://doi.org/10.7243/2049-7962-2-21
Song IS, Jeong YJ, Jeong SH et al (2015) FOXM1-induced PRX3 regulates stemness and survival of colon cancer cells via maintenance of mitochondrial function. Gastroenterology 149(4):1006–1016. https://doi.org/10.1053/j.gastro.2015.06.007
Steelman LS, Abrams SL, Whelan J et al (2008) Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 22(4):686–707. https://doi.org/10.1038/leu.2008.26
Sun Y, Li H (2012) Functional characterization of SAG/RBX2/ROC2/RNF7, an antioxidant protein and an E3 ubiquitin ligase. Protein Cell 4(2):103–116. https://doi.org/10.1007/s13238-012-2105-7
Tang Y, Yacoub A, Hamed HA et al (2012) Sorafenib and HDAC inhibitors synergize to kill CNS tumor cells. Cancer Biol Ther 13(7):567–574. https://doi.org/10.4161/cbt.19771
Teodori L, Sestili P, Madiai V et al (2020) MicroRNAs bioinformatics analyses identifying HDAC pathway as a putative target for existing anti-COVID-19 therapeutics. Front Pharmacol 11:582003. https://doi.org/10.3389/fphar.2020.582003
Topf U, Suppanz I, Samluk L et al (2015) Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat Commun 9:324. https://doi.org/10.1038/s41467-017-02694-8
Ungerstedt JS, Sowa Y, Xu WS et al (2005) Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A 102(3):673–678. https://doi.org/10.1073/pnas.0408732102
Wang J, Yang H, Li W et al (2015) Thioredoxin 1 upregulates FOXO1 transcriptional activity in drug resistance in ovarian cancer cells. Biochim Biophys Acta 1852(3):395–405. https://doi.org/10.1016/j.bbadis.2014.12.002
Warburg O (1956) On the origin of cancer cells. Science 123(3191):309–314. https://doi.org/10.1126/science.123.3191.309
Waz S, Nakamura T, Hirata K et al (2017) Structural and kinetic studies of the human Nudix hydrolase MTH1 reveal the mechanism for its broad substrate specificity. J Biol Chem 292(7):2785–2794. https://doi.org/10.1074/jbc.M116.749713
Wittenburg LA, Ptitsyn AA, Thamm DH (2012) A systems biology approach to identify molecular pathways altered by HDAC inhibition in osteosarcoma. J Cell Biochem 113(3):773–783. https://doi.org/10.1002/jcb.23403
Xiao D, Johnson CS, Trump DL et al (2004) Proteasome-mediated degradation of cell division cycle 25C and cyclin-dependent kinase 1 in phenethyl isothiocyanate-induced G2-M-phase cell cycle arrest in PC-3 human prostate cancer cells. Mol Cancer Ther 3(5):567–575
Yamada M, Tomida A, Yoshikawa H et al (1996) Increased expression of thioredoxin/adult T-cell leukemia-derived factor in cisplatin-resistant human cancer cell lines. Clin Cancer Res 2(2):427–432
Yang H, Villani RM, Wang H et al (2018) The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res 37(1):266. https://doi.org/10.1186/s13046-018-0909-x
Yang L, Mih N, Anand A et al (2019) Cellular responses to reactive oxygen species are predicted from molecular mechanisms. Proc Natl Acad Sci U S A 116(28):14368–14373. https://doi.org/10.1073/pnas.1905039116
You BR, Park WH (2017) Suberoylanilide hydroxamic acid induces thioredoxin1-mediated apoptosis in lung cancer cells via up-regulation of miR-129-5p. Mol Carcinog 56(12):2566–2577. https://doi.org/10.1002/mc.22701
Zhang ML, Wu HT, Chen WJ et al (2020) Involvement of glutathione peroxidases in the occurrence and development of breast cancers. J Transl Med 18(1):247. https://doi.org/10.1186/s12967-020-02420-x
Zhu Y, Das K, Wu J et al (2014) RNH1 regulation of reactive oxygen species contributes to histone deacetylase inhibitor resistance in gastric cancer cells. Oncogene 33(12):1527–1537. https://doi.org/10.1038/onc.2013.104
Zhu X, Liu X, Cheng Z et al (2016) Quantitative analysis of global proteome and lysine acetylome reveal the differential impacts of VPA and SAHA on HL60 cells. Sci Rep 6:19926. https://doi.org/10.1038/srep19926yang
Acknowledgments
Corresponding author Dr. M. Janaki Ramaiah would like to thank KLEF for the excellent research facilities, constant encouragement, and support, and the author would also like to thank the funding agency DBT/BR/PR20836/MED/30/1727/2016 for the financial support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Section Editor information
Rights and permissions
Copyright information
© 2022 Springer Nature Singapore Pte Ltd.
About this entry

Cite this entry
Mekala, J.R., Ramalingam, P., Moparthi, N.R., Kutala, V.K. (2022). ROS Modulatory Role of HDAC Inhibitors in Cancer Cells. In: Chakraborti, S. (eds) Handbook of Oxidative Stress in Cancer: Therapeutic Aspects. Springer, Singapore. https://doi.org/10.1007/978-981-16-5422-0_250
Download citation
DOI: https://doi.org/10.1007/978-981-16-5422-0_250
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-5421-3
Online ISBN: 978-981-16-5422-0
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences