
Abstract
Raman spectroscopy is based on the Raman effect, which refers to inelastic scattering of monochromatic light (in the visible, near-infrared, or ultraviolet range) incident on molecules, which results in a small fraction of the scattered light undergoing changes in frequency relative to the frequency of the incident photons. The frequency change corresponds to the frequency of transition between internal energy levels (usually molecular vibrational levels) of the scattering molecule.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


11.1 Introduction
11.1.1 Raman Effect and Its Discovery
Raman spectroscopy is based on the Raman effect, which refers to inelastic scattering of monochromatic light (in the visible, near-infrared, or ultraviolet range) incident on molecules, which results in a small fraction of the scattered light undergoing changes in frequency relative to the frequency of the incident photons [1, 2]. The frequency change corresponds to the frequency of transition between internal energy levels (usually molecular vibrational levels) of the scattering molecule.
The seminal discovery of Raman effect was made on February 28, 1928, by Dr. C.V. Raman (then Palit Professor of Physics at the University of Calcutta) with his research student-collaborator K.S. Krishnan. The experiments were carried out in Raman’s laboratory (which was equipped with simple, rudimentary facilities) at the Indian Association for the Cultivation of Science (IACS), which was then located at 210 Bowbazar Street, Kolkata (then Calcutta), India. For this great discovery, Raman won the Nobel Prize in Physics in 1930, becoming the first Asian recipient of the Nobel Prize in a field of science. Figure 11.1 presents a simple pictorial depiction of the Raman scattering process.

Raman and Rayleigh scattering processes. (A photograph of Prof. C.V. Raman is included)
The emergence of Raman spectroscopy as an analytical tool signified an important new addition to the repertoire of vibrational spectroscopy, providing an important and potentially more versatile alternative to infrared (IR) spectroscopy (based on absorption of IR radiation, inducing transitions between vibrational energy levels). Raman envisioned the immense future potential of the new spectroscopic technique he had just discovered. Indeed, in his Nobel lecture delivered at Stockholm on December 11, 1930, he made the prophetic remark ‘… the new field of spectroscopy has practically unrestricted scope in the study of problems relating to the structure of matter’. In a little over nine decades that have elapsed since Raman’s original discovery, with many important technological breakthroughs having taken place, Professor Raman’s dreams have been amply fulfilled.
11.1.2 Historical Perspective
Professor Raman’s fascination for light scattering started in 1921 during a voyage through the Mediterranean Sea on his return trip to India from Europe, where he went to participate in the Congress of Universities of the British Empire held at Oxford, England. Raman, who possessed a lifelong interest in natural phenomena, especially natural colors, was curious about the origin of the beautiful blue opalescence of the Mediterranean. The prevailing idea then was based on Lord Rayleigh’s belief that the blue color of the sea was due to reflection of the blue sky, which had been previously explained by Lord Rayleigh as being due to elastic (Rayleigh) scattering by atmospheric gas molecules. Raman was skeptic about Lord Rayleigh’s explanation regarding the role of the sea water ‘simply as a reflecting mirror for the blue sky’, and since it was Raman’s practice to carry some small optical equipment with him, he could conduct some simple experiments on board the ship. Using a Nicol prism polarizer, he was able to quench the surface reflection of the sky, while the blue color of the sea became more vivid. Hence, he concluded that contrary to Lord Rayleigh’s belief, the color of the sea was fundamentally due to the same reason as that of the sky, namely, in case of the sea, due to elastic scattering of light by water molecules. Upon his return to Kolkata, Raman and his students immediately switched the emphasis of their research activities to the scattering of light by different liquids.
In 1923, the Austrian physicist Adolf Smekal theoretically predicted the change of frequency of light due to scattering by molecules. However, experimental observation of such a phenomenon presented a formidable challenge because of the extreme feebleness of inelastically scattered light. In fact, only 1 in about 106 photons participates in inelastic scattering, while the bulk of the photons (10−4–10−3 parts of the incident light) undergo elastic scattering (Rayleigh scattering). While Raman’s work on the scattering of light by liquids was in progress, A.H. Compton (USA) discovered that X-rays lose energy and thus shift to longer wavelengths [3], due to inelastic collisions with electrons (Compton Effect), and for this discovery, Compton received the Nobel Prize in Physics in 1927. Compton’s success acted as a strong incentive for Raman to intensify his quest for the ‘optical analogue of the Compton effect’, due to scattering of visible light by molecules.
The set-up that Raman used for his initial findings [1, 2, 4] was striking in simplicity and elegance, and stands out as a remarkable testimony to his ingenuity and experimental prowess. Raman chose bright sunny days and used sunlight (brought to a strong focus by a telescope objective combined with an additional lens) as the source of light, complementary optical filters (which comprised a combination of blue-violet and yellow-green filters) as the analyzer to detect the wavelength change of the scattered light, some 60 liquid samples (freed from dust and fluorescent impurity by careful filtration and repeated distillation), which he took one by one in a glass container, while his own dark-adapted eyes served as the detector. As expected, the complementary filter combination completely extinguished the track of light through the sample, but to Raman’s great delight, for all of the samples, the light track reappeared when the yellow-green filter was transferred to a place between the sample and the observer’s eyes. This unequivocally proved modification of the frequency of light due to scattering by the liquids. Thus, after nearly 7 years of dedicated work on light scattering, Raman succeeded in making the first experimental observation of the inelastic scattering of photons in the optical region, and described his novel observations in a one-page paper (entitled ‘A New Type of Secondary Radiation’) which he immediately cabled to Nature [1]. In order to obtain quantitative measurements of the spectral shifts, for his subsequent studies, Raman used an improved set-up which now consisted of filtered light from a mercury arc lamp, as the source of monochromatic radiation and a baby Hilger spectrograph involving photographic recording of the spectra, permitting measurements of the Raman spectral shifts [2]. Raman and his colleagues showed the applicability of the Raman scattering method to materials in various states, solids, liquids, and gases, proving the universality of the technique, with the ability to provide unique fingerprints of a broad range of molecules of interest.
11.1.3 Invention of Lasers and Other Technological Breakthroughs: Impact on the Growth and Development of Raman Spectroscopy
Owing to the initial lack of adequate technology, especially the nonavailability of intense light sources, for well over the first three decades since the original discovery of the Raman Effect (in 1928), the application of Raman spectroscopy was limited to relatively simple molecules. Strong Rayleigh background scattering from complex molecules prevented detection of the feeble Raman signals for most of their vibrational modes. The invention of laser sources, beginning with the ruby laser in 1960, followed by more efficient lasers, namely gas lasers and compact miniaturized diode lasers (which are excellent sources of highly intense, nearly monochromatic light covering ranges from the UV, spanning the visible range, and extending to near infrared), changed the scenario and revolutionized Raman spectroscopy. This aspect, along with other factors, namely the development of high-quality monochromators and sensitive light detectors comprising photomultiplier tubes (PMTs) and more recently, highly sensitive charged coupled detectors (CCDs), and inclusion of microscopy and imaging techniques have enormously advanced the scope and range of applications of Raman spectroscopy [5]. At least 25 variants of Raman spectroscopy have been developed with various goals [6], e.g., (a) to enhance the signal sensitivity (Resonance Raman and Surface-Enhanced Raman (SERS) techniques), (b) to selectively probe specific sites (e.g. functional groups of macromolecules (Resonance Raman)), and (c) to improve spatial resolution (Raman microscopy). Comprehensive, recent reviews have been published highlighting the theory and methodology of advances in various Raman spectroscopy techniques, with relevant applications [6,7,8,9,10]. Because of its noninvasive nature, the use of Raman spectroscopy has proliferated to diverse fields encompassing broad areas of chemistry, physics, and life sciences, nanotechnology, materials science, biomedical applications including diagnosis of malignancies in cancer cells and tissues and other diseases, pharmaceutical science, agricultural and food technology, and forensic science.
11.1.4 Scope of This Chapter
Since Raman spectroscopy is based on scattering of light, it is noninvasive in nature. Therefore, the field of biology, including the more recently developing biomedical field, has probably most significantly benefited from developments of Raman spectroscopy. The ease of studying solutions of biomolecules in water (for which Raman scattering is relatively weak, thus posing negligible interference in signal gathering), and opportunities for a wide range of applications, encompassing in vitro, ex vivo, and in vivo studies, have opened up new frontiers. The object of this chapter is to provide an overview on Raman spectroscopy, including theory and basic instrumentation, along with representative applications in biology, emphasizing biophysics, structural biology, and related aspects.
11.2 Principles: Basic Mechanisms of Photon–Molecule Interactions in Rayleigh Scattering Different Types of Raman Scattering and in IR Absorption
Raman and infrared spectroscopy are two alternative approaches of vibrational spectroscopy, which are useful for gaining information on molecular vibrations. A nonlinear molecule containing N atoms possesses 3N-6 fundamental vibrations. This number is 3N-5 for a linear molecule. The energies of these vibrations correspond to the infrared region of the electromagnetic spectrum.
Raman spectroscopy involves inelastic scattering of light, usually in the visible, ultraviolet, or near-infrared region. On the other hand, infrared (IR) spectroscopy involves direct absorption of electromagnetic radiation in the infrared region. In order for a molecular vibration to be Raman active, the vibration must be accompanied by a change in polarizability, α (which can be looked upon as the deformability of the electron cloud of the molecule by the electric field) so that the first derivative of α with respect to the normal coordinate Q (representing stretching or deformation vibration) must be nonzero. Thus, what happens in a Raman process is a change in the induced dipole moment, P, which is related to the electric field vector E as follows:
In order to absorb infrared radiation, a molecular vibration must cause a change in the dipole moment (μ), and accordingly, the first derivative of μ with respect to the normal coordinate, Q, must be nonzero. Since the presence of a dipole moment and its change are essential for a molecular vibration to be IR active, the vibrational spectra of homonuclear diatomic (like O2, N2, H2) could not be studied via IR absorption, owing to the absence of dipole moments. The discovery of the Raman technique in 1928 opened the door to exploration of the vibrational spectra of such molecules.
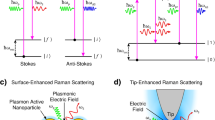
The essential differences between IR and Raman processes, Rayleigh scattering, and different types of the basic Raman scattering process (normal, pre-resonance, and Resonance Raman) are shown in the energy level diagram (Fig. 11.2) depicting the photon–molecule interactions involved.

At ambient temperature, the bulk of the molecules are in the ground vibrational level of the ground electronic state. For Rayleigh scattering, an incident photon with the monochromatic frequency, νo, momentarily raises the molecule to a virtual level represented by a dashed line (the level is ‘virtual’ in the sense that it is not a molecular eigenstate and has an energy corresponding to the energy of the exciting photon). A second scattered photon with the same frequency as that of the incident photon appears, and therefore, this is an elastic scattering process (Rayleigh scattering). The scattered photons with modified frequencies, νo − νvib and νo + νvib are called Stokes and anti-Stokes Raman lines, respectively. For IR absorption and a particular vibration with frequency νvib, a photon with energy hνvib is absorbed, raising the molecule to a vibrationally excited level in the electronic ground state. Both elastic and inelastic scattering processes are two photon processes. Owing to the vastly greater population of molecules in the ground vibrational level (compared to those in vibrationally excited state), Stokes lines are significantly stronger than the anti-Stokes line, and therefore generally used in conventional Raman spectra. In Resonance Raman spectroscopy, the exciting frequency falls in the region of the electronic absorption spectrum (matching the absorption frequency νa of the molecule). In this situation, the polarizability, α, becomes frequency dependent, resulting in tremendous (>103 fold) increase in intensity of the Raman scattered light, resulting in its enhanced analytical sensitivity as compared to the weak normal Raman spectra. Besides higher sensitivity, another advantage of Resonance Raman is selectivity, permitting selective excitation of functional groups in macromolecules (e.g. the retinal part of the visual pigment rhodopsin, or specific components of molecular components like drug–protein or drug–DNA complexes), enabling selectively probing the structural changes in the component of particular interest [11,12,13].
11.3 Instrumentation
11.3.1 Basic Set-Up Used in a Raman Spectrometer
Two different approaches are used to collect basic Raman spectra, and consequently, two major types of Raman spectrometers are commercially available, and the choice is largely dependent on user requirement and preferences. These two types of instruments are: (a) Dispersive Raman spectrometer, and (b) Fourier Transform Raman spectrometer.
11.3.1.1 Dispersive Raman Spectrometer
A block diagram showing the basic components of a conventional, dispersive Raman spectrometer is shown in Fig. 11.3.

The components are: source of monochromatic light (Laser), sample compartment, monochromator (to analyze the Raman scattered light), and detector (Photomultiplier (PMT) or charge coupled detector (CCD)), computer, and recorder. The collections as well as processing of the spectra are controlled by a computer (PC). A variety of laser sources can be used as the excitation source. These include: argon ion (514.5 and 488.0 nm), Krypton ion (530.9 and 547.1 nm), He: NE (632.8 nm), Nd: YAG (1064 nm fundamental, with second, third, and fourth harmonics being available), and diode laser (630 and 780 nm).While in previous decades gas lasers were predominantly used for visible range excitation, they are being gradually replaced by solid state diode-based lasers, in view of the superior spectral quality and stability of these new generation lasers. Generally, the following three different types of CW (continuous wave) solid-state, diode laser light sources are commercially available: (1) Diode-pumped single longitudinal mode (SLM) lasers, which are readily available in compact form with a choice of a wide range of wavelengths from UV to near-IR range, (2) single-mode diode lasers, and (3) Volume Bragg grating (VBG) frequency stabilized diode lasers. Photomultiplier tubes, used in earlier decades, have been largely replaced by CCD detectors in modern Raman spectrometers.
11.3.1.2 Fourier Transform Raman Spectrometer
As an alternative to conventional Raman spectrometers, which have an older history, Fourier transform Raman Spectrometers have been an important part of the repertoire for workers in the field of Biological Raman spectroscopy. Motivation for the development of Fourier Transform Raman (FT-Raman) systems, which were first developed in the 1980s, largely came from the need to avoid ambient fluorescence from the sample, which can be avoided by near-IR excitation, using the 1064 nm line of an Nd: YAG laser. Since not too many species have electronic absorption band in the near-IR, and therefore, excitation in this region precludes the possibility of an interfering fluorescence background, which can swamp the intrinsically weak Raman signals. However, owing to the inverse fourth power dependence of the scattering intensity on the incident wavelength (which is true for both Raman and Rayleigh scattering), the Raman signals obtained with near-IR excitation are extremely weak. To overcome this limitation, a Michelson interferometer is used, and the interferograms generated are Fourier transformed to generate the spectra. FT-Raman spectrometers have proved to be especially useful for exploring biological samples where background fluorescence poses a major obstacle in collecting meaningful Raman data.
11.3.2 Advanced Raman Techniques
With advances in laser technology and optics, at least 25 different variants of Raman spectroscopy have been developed. The impetus for such developments stemmed from a number of motivations, especially to enhance the sensitivity (e.g. surface-enhanced Raman), to acquire specific information of selective sites of macromolecules or molecular complexes (Resonance Raman), and to obtain spatial resolution (Raman microscopy) [5,6,7]. Here, we describe two important techniques which have proved to be particularly useful in biological applications. These are surface-enhanced Raman Spectroscopy (SERS) and coherent anti-Stokes Raman Spectroscopy (CARS). Brief descriptions of these two advanced techniques are presented here:
11.3.2.1 Surface Enhanced Raman Spectroscopy (SERS)
SERS is a surface-sensitive technique involving enhancement of Raman scattering for molecules adsorbed on rough metal surfaces or by nanostructures like plasmonic-magnetic silica nanotubes. The first observation of SERS was serendipitous and was made in 1974 by Fleischman et al. [15] in course of their attempts to distinguish two types of a heterocyclic molecule on the surface of a silver electrode which was used to reduce absorption effects. The increase in Raman signal by SERS has been attributed to two principal mechanisms [6]: (a) Electromagnetic enhancement where local surface plasmons concentrate the local field proximal to the surface of the metal in ‘hot spots’ situated on the sharp edges of the nanostructures. (b) Chemical enhancement via charge transfer (CT) between the metal surface and the analyte, which enhances the Raman signal by 102–104 times. In this context, it is noteworthy that while the CT mechanism applies to specific molecules, the electromagnetic mechanism applies for all analytes. The most common materials used to fabricate SERS are silver and gold, because of their superior plasmonic response. The basic optical set-up for an SERS experiment is presented in Fig. 11.4.

11.3.2.2 Coherent Anti-Stokes Raman Spectroscopy (CARS)
CARS involves a third-order nonlinear four-wave optical mixing process. A pump beam and probe beam of frequencies, ὡp and ὡprb, respectively, are mixed with a third beam of frequency ὡs (Stokes beam) and are allowed to impinge on the sample. The frequency difference (ὡp – ὡs) must match the frequency associated with the Raman-active vibrational mode, ὡ. The frequency of the Stokes beam is normally tuned to fulfill this condition, that is, ὡ = ὡp − ὡs. Next, a probe photon of frequency ὡprb acts as a perturbation for the anti-Stokes scattering process to occur at the frequency ὡAS = ὡp − ὡs + ὡprb. The basic optical set-up used for a CARS experiment [6, 7] is depicted in Fig. 11.5.

Coherent Anti-Stokes Raman scattering (CARS) process. CARS involves a four-wave mixing of pump, Stokes, probe, and anti-Stokes beams. (Adapted with some modification from Ref. [6])
11.4 Applications to Biomolecules: Overview
Biomolecules are produced by living systems and are associated with various physiologically relevant functions. The four major classes of biomolecules are proteins, nucleic acids, lipids, and carbohydrates. Since Raman spectroscopy is based on light scattering, it is of noninvasive nature and is therefore suitable for exploring biomolecular systems under in vitro, in vivo, and ex vivo situations, leading to its wide applications as an analytical tool in biology. Each molecule has its characteristic Raman spectroscopic signature, which is determined by the three-dimensional structure of the molecule. An analysis of this spectrum can therefore yield a signature of the overall molecular structure as well as on intramolecular and intermolecular interactions. Since lasers were invented in the 1960s, followed by other technological breakthroughs, there has been a continuing outburst on Raman studies on different types of biomolecules. The initial developments were reviewed in comprehensive texts by A.T. Tu and P.R. Carey, published in 1982 [4, 11], and later additional books and monographs have appeared on specific areas of applications including SERS and confocal microscopy, detailing subsequent progress in biological and biomedical applications [14, 16,17,18]. In this chapter, we shall present perspectives from an historical context, highlighting the applications of Raman spectroscopy in the conformational analyses of the two most important classes of biomacromolecules, namely, proteins and DNA.
11.4.1 Proteins: Conformational and Related Studies
Proteins, together with amino acids and peptides, were probably the first group of biorelevant molecules to which Raman spectroscopy was applied, beginning with the pioneering efforts by J.T. Edsall’s group at Harvard University, USA, which started within a decade of the discovery of the Raman effect in 1928. Even in the pre-laser era, Edsall recorded a Raman spectrum of the protein (an enzyme) lysozyme as far back as 1958, using Hg line excitation. However, the feebleness of the spectrum permitted assignment of only six lines [19]. Following the invention of lasers in the 1960s, laser-excited Raman spectra were published during the 1970s onward by various workers, including Lord et al., Yu et al., Frushour and Koenig, Tu et al., Fasman, Peticolas, and others for a wide range of peptides, polypeptides, and proteins. For reviews of these early ventures, the reader is referred to excellent texts by Tu and by Carey [4, 11]. Systematic analyses of Raman (together with IR) spectra of synthetic polypeptides facilitated spectra-secondary structure correlations in critical detail [4, 11, 20].
What is the protocol followed for the use of Raman spectroscopy to analyze the secondary structure of proteins? This is essentially based on the use of the vibrational modes of the peptide bond –CONH–, which is associated with nine normal modes of vibration, called amide bands [4, 11, 20, 21]. These are designated as amide A and B, and amide I–VII modes, in decreasing order of frequency. The typical frequencies and vibrational assignments of these modes are as follows: amides A and B (3300 and 3100 cm−1, respectively), the doublet arising due to Fermi resonance interaction between NH stretching and the first overtone of amide II), amide I (1600–1690 cm−1 mainly due to C=O stretching), amide II (1480–1575 cm−1, C–N stretch, NH in plane bending), amide III (1230–1300 cm−1, C–N stretching, NH in plane bending, involving a different phase relationship compared to amide II), amide IV (625–767 cm−1, OCN bending), amide V (640–800 cm−1, mainly NH out of plane bending), amide VI (540–600 cm−1, mainly C=O out of plane bending), amide VII (200 cm−1, mainly CN torsion). Assignments of the amide bands and spectra-structure correlations were originally established by detailed spectroscopic measurements (via Raman and IR), together with normal mode calculations on the peptide model, N-methylacetamide, and various homo-polypeptide models. Pioneers in this field of research were Miyazawa, Elliott and Ambrose, Krimm, and others [20]. Among the amide bands, amide I and amide III are most prominent in the Raman spectra of proteins, and their frequencies are useful for detecting the presence of different secondary structures (alpha helix, beta sheet, beta turns, disordered structure). The approximate frequency (cm−1) ranges for the most intense amide I and amide III Raman bands are listed in Table 11.1.
In addition to the identification of different secondary structures from frequency data, the intensities of these bands (amide I and amide III) have also proved to be useful in quantitative analyses of secondary structure [23], which is reminiscent of protocols developed earlier for applications of far UV CD spectroscopy for estimating secondary structure compositions of proteins via multicomponent analysis.
Apart from the importance of amide bands, which provide information on protein conformation, the side chain Raman frequencies are a rich source of information for the microenvironments of the side chains of proteins. These include the Fermi resonance doublet of tyrosine (850/830 cm−1 intensity ratio) reflecting buried vs. exposed Tyr residues, the 1360/1340 cm−1 intensity ratio of tryptophan which can be correlated with the environment of the indole group and is useful as a hydrophobicity indicator, that is, for diagnosing buried versus exposed tryptophan residues, and vibrational modes of sulfur containing residues, especially changes in the S–S stretching band around 510 cm−1, which provides details on protein folding, aggregation, and other aspects of relevance to protein structure and function [4, 11, 21].
Use of advanced Raman methodologies has opened up new dimensions in structural studies on proteins. For example, using chiroptical measurements based on Raman optical activity (ROA) motifs and folds in proteins can be explored [24, 25]. High analytical sensitivity can be achieved with surface-enhanced resonance Raman spectroscopy (SERRS). This was demonstrated, for example, for the enzyme glucose oxidase at a silver electrode, where release of free flavin adenine dinucleotide (FAD) was detected from enhanced Raman signals for a solution with extremely low concentrations (10−10 M). Interesting studies have been reported on the use of 2D Raman spectroscopy to explore pH-induced changes in protein conformation and dynamics [39]. Baranska and coworkers published an exhaustive vibrational spectra library, incorporating Raman data on as many as 26 proteins of different structure and function, using different laser lines (488, 532, and 1064 nm). The proteins included different structural classes, encompassing alpha-helical, beta-sheet, and mixed structures [21]. Another important direction which has been pursued by S. Krimm and coworkers, and others, concerns detailed vibrational analysis on polypeptides using Raman (along with infrared) spectroscopic measurements combined with theoretical analysis involving normal mode calculations [20]. Studies on conformational polymorphism in calcium and magnesium salts of polyglutamic acid revealed an interesting correlation of the Raman-active CC skeletal sketching band (900–1000 cm−1 region) with the Ramachandran φ angle. Also, dependence of the conformationally sensitive amide III frequency on side chain composition was demonstrated [26,27,28,29]. Most particularly, based on Raman and Normal mode analysis data, strong support was obtained for an earlier proposal (stemming from far UV CD studies) for the existence of a left-handed three-fold ‘extended helical’ conformation in polypeptides with charged side chains [29]. UV resonance Raman studies by Asher’s group (published in 2001) have been of special interest. Asher’s work revealed an interesting dependence of amide III on the dihedral angle ψ, thus providing a unique and sensitive UV resonance Raman structural probe [30].
11.4.2 Nucleic Acids: Conformational and Drug–DNA Interaction Studies
Raman spectroscopic studies on nucleic acids and its constituents were initiated by Lord and Thomas in the late 1960s, soon after the availability of lasers. A great deal of attention has been devoted to the study of the interactions of DNA and RNA with polypeptides, proteins, metal ions, and drugs, for which Raman spectroscopy turned out to be a highly sensitive technique, providing critical insights regarding the binding sites of the ligands in the nucleic acid matrix, as well as conformational changes involved. Since Raman bands provide insights into local structural details, in contrast to electronic CD, where gross structural changes can only be probed (owing to the delocalized nature of electronic transitions), the superiority of Raman spectroscopy as an analytical tool in Structural Biology and Biophysics became immediately apparent.
Raman bands of nucleic acids arise from the in-plane vibrations of the bases (adenine, guanine, thymine, cytosine, and uracil) and from the phosphodiester backbone. The phosphodiester stretching vibration (at ca. 815 cm−1) is sensitive to conformational changes, while the phosphoionic bond stretching vibration (at ca. 1100 cm−1) is relatively insensitive to structural changes and serves as an intensity standard. Raman spectra of DNA or RNA provide valuable insights regarding base stacking and interbase H-bonding interactions. B–Z transitions of synthetic polynucleotides due to drug binding have been an active area of exploration. Resonance Raman spectroscopy (with resonance enhancement of nucleic acid base vibrational modes by using far UV excitation, where the bases selectively absorb and undergo resonance enhancement) provides a useful means of selectively probing the DNA and drug vibrations, especially if the electronic absorption of the drug occurs in the near-UV or visible range. While duplex DNA structures (right-handed B and A forms, and left-handed Z form) were the exclusive focus of attention in earlier studies, much recent attention centers on 4-stranded, noncanonical G-quadruplex DNA structures, in view of their role in downregulating high telomerase activity in cancer cells, as well as their relevance to neurodegenerative and other human diseases. In this context, a detailed Raman study by Palacky et al. [31] on the polymorphism of human telomeric quadruplexes is noteworthy. Very recently, Vorlickova et al. performed a critical assessment to establish the potential of Raman spectroscopy in recognizing different G-quadruplex topologies [32]. For their exhaustive research, they chose as model structures a 22-mer human telomeric fragment AG3(TTAG3) and its two variants with adenine a basic lesions at positions 19 and 7, respectively, characterized by three contrasting G4 folds: a basket form, a (3 + 1) hybrid form, and a parallel G4. This study revealed that the reliability of Raman spectroscopy in this context is comparable to that of electronic CD, and moreover, Raman studies have the potential to provide additional useful structural information of significant interest.
Table 11.2 lists Raman marker band frequencies for different DNA structures, including duplex DNA (B, A, and Z forms) as well as quadruplex DNA (for parallel as well as antiparallel quadruplex).
11.4.3 Drug–DNA Interaction Probed via Raman Spectroscopy
Interaction of DNA with therapeutically important small molecules has been a topic of continuing attention in relation to the biophysical applications of Raman spectroscopy. Recent interest in quadruplex DNA as drug design targets for the treatment of a variety of human disorders, and the application of Raman spectroscopy in the quest for drugs targeting quadruplex DNA of different topologies represents a promising field of investigation. Recent years have witnessed considerable interest on plant flavonoids as novel therapeutics having high potency and low cytotoxicity, making them viable alternatives to conventional drugs. Various spectroscopic methods are useful for exploring the interactions of such compounds with their biorelevant target molecules. With this scenario in mind, we embarked on the exploration of the prospect of the dietary flavonoid fisetin as a quadruplex DNA ligand, with its potential utility as an anticancer drug [34]. Various spectroscopic and other biophysical techniques were used in this work. We used Raman spectroscopy as an important part of the repertoire of our experimental tools. Salient aspects of this research are presented here since it exemplifies the application of Raman spectroscopy in a novel research direction, namely drug discovery involving natural product-based drugs targeting quadruplex DNA, significantly complementing insights from studies via other common spectroscopic techniques such as electronic absorption, fluorescence, and circular dichroism (CD).
Raman spectroscopy (using 632.8 nm excitation) was used to examine the binding of fisetin (3, 4, 3′, 4′-OH flavone, structure shown in Fig. 11.6) with the quadruplex forming sequence d (T2AG4)4. The data are presented in Fig. 11.7. The intense Raman bands at 1084, 1478, and 1582 cm−1 are consistent with the quadruplex structure, in general, while the antiparallel topology is evidenced by the presence of the shoulders around 684 and 1330 cm−1. Interestingly, the Raman spectrum of fisetin–DNA complex shows distinct changes indicating fisetin–G4 DNA interaction. In particular, the line at 1455 cm−1 for the free ligand (due to C–O and C–C stretching of the B ring of fisetin) is totally absent in the presence of DNA, suggesting the absence of free movement of the B ring, which is indicative of the interaction of the B ring with DNA via H-bonding. Most particularly, since the major spectral changes are found to occur for the thymine and adenine marker lines (at 1660, 725, and 825 cm−1), it appears that fisetin binds the DNA at the loop region which is made up of adenine and thymine bases. Thus, this particular study exemplifies the strength of Raman spectroscopy about the drug-binding site in DNA in specific detail.

Chemical structure of fisetin, a therapeutically important plant flavonoid

Raman spectral profiles showing studies on quadruplex DNA-fisetin interaction. Blue: 1 mM fisetin; red: 1 mM (T2AG4)4 DNA; green: (T2AG4)4 DNA interacting with 1 mM fisetin; pink: 10 mM Tris buffer (pH 7) with 26 mM NaCl, in which the solutions were prepared. (Figure adapted from Ref. [34] in modified form)
11.5 Conclusions and Future Outlook
In a little over nine decades since Raman effect was discovered by Professor C.V. Raman, Raman spectroscopy has developed into a powerful research tool in structural biology and biophysics, especially in relation to the elucidation of the structure, function, and dynamics of proteins and nucleic acids and other important biomolecules such as lipids and carbohydrates. The noninvasive nature of the technique, lack of the need for labels, and the intrinsically high chemical specificity, which are the hallmarks of Raman spectroscopy, permit its widespread applications in a broad range of situations in vitro, ex vivo, and in vivo. Raman spectroscopy coupled with microscopy has enabled enormously powerful prospects of live cell imaging and associated studies. Latest developments in Raman spectroscopy and confocal Raman microscopy are opening up vast possibilities in biomedical applications, especially in the diagnosis of cancers, and other diseases such as atherosclerosis and osteoarthritis. Such diagnostic potential stems from the exquisitely high sensitivity of Raman techniques to detect subtle biochemical changes in cells underlying various diseases. The reliability and thresholds for cancer diagnosis are being greatly augmented by refinements in the SERS technique, permitting detection and discrimination among different types of cancers at the level of single cells. Great advances can be foreseen leading to routine and versatile applications of Raman techniques in diagnostic medicine.
References
Raman CV, Krishnan KS (1928) A new type of secondary radiation. Nature 121:501
Raman CV, Krishnan KS (1928) A new class of spectra due to secondary radiation. Part I. Indian J Phys 2:399–419
Compton AH (1923) A quantum theory of the scattering of X-rays by light elements. Phys Rev 21:483
Tu AT (1982) Raman spectroscopy in biology: principles and applications. Wiley, New York
Adar F, Delhaye M, Da Silva E (2007) Evolution of the instrumentation for detection of the Raman effect as driven by available technologies and by developing applications. J Chem Educ 84:50–60
Jones RR, Hooper DC, Zhang L, Wolverson D, Valev VK (2019) Raman techniques: fundamentals and frontiers. Nanoscale Res Lett 14(1):231
Das RS, Agrawal YK (2011) Raman spectroscopy: recent advancements, techniques and applications. Vib Spectrosc 57:163–176
Paudel A, Raijada D, Rantanen J (2015) Raman spectroscopy in pharmaceutical product design. Adv Drug Deliv Rev 89:3–20
Smith R, Wright KL, Ashton L (2016) Raman spectroscopy: an evolving technique for live cell studies. Analyst 141:3500–3600
Jia M, LiS ZL, Lu X, Zhang H (2018) Analysis of biomolecules based on the surface enhanced Raman spectroscopy. Nano 8(9):730
Carey PR (1982) Biochemical applications of Raman and resonance Raman spectroscopies. Academic Press, Boston
Smekal A (1923) Zur Quantentheorie der Dispersion. Naturwissenschaften 11:873–875
Long DA (2002) The Raman effect. Wiley, New York
Rostron P, Gaber S, Gaber D (2016) Raman spectroscopy, review. Int J Eng Tech Res 6:50–64
Fleischmann M, Hendra PJ, McQuillan AJ (1974) Raman spectra of pyridine adsorbed at a silver electrode. Chem Phys Lett 26:163–166
Spiro TG (1987) Biological applications of Raman spectroscopy. Wiley, New York
Peticolas WL (2004) Raman spectroscopy of DNA and proteins. Methods Enzymol 246:189–436
Thomas GJ Jr (1999) Raman spectroscopy of proteins and nucleic acid assemblies. Annu Rev Biophys Biomol Struct 28:1027
Garfinkel D, Edsall JT (1958) Raman spectra of amino acids and related compounds X. the Raman spectra of certain peptides and of lysozyme. J Am Chem Soc 80(15):3818–3823
Krimm S, Bandekar J (1986) Vibrational spectroscopy and conformation of peptides, polypeptides and proteins. Adv Protein Chem 38:181–364
Rygula A, Majzner, Marzec KM, Kaczor A, Pilarczyk M, Baranska M (2008) Raman spectroscopy of proteins. J Raman Spectrosc 44:1061–1076
Ishizaki H, Balaram P, Nagaraj R, Venkatachalapathi YV, Tu AT (1981) Determination of beta-turn conformation by laser Raman spectroscopy. Biophys J 36:509–517
Williams RW (1986) Protein secondary structure analysis using Raman amide I and amide III spectra. Methods Enzymol 130:311–331
Zhu F, Isaacs NW, Hecht L, Barron LD (2005) Raman optical activity: a tool for protein structure analysis. Structure 13:1409–1419
Blanch EW, Hecht L, Barron LD (2006) Raman optical activity. In: Busch KW, Busch MA (eds) Chiral analysis. Elsevier, Amsterdam, pp 545–594
Sengupta PK, Krimm S (1985) Vibrational analysis of peptides, polypeptides and proteins. XXXII. α-poly (L-glutamic acid). Biopolymers 24:1479–1491
Sengupta PK, Krimm S (1984) Vibrational analysis of peptides, polypeptides and proteins. XXI. β-calcium-poly (L-glutamate). Biopolymers 23:1565–1594
Sengupta PK, Krimm S (1987) Raman and normal-mode studies of the extended-helix conformation in polypeptide chains. Biopolymers 26(S0):S99–S107
Sengupta PK, Krimm S (1985) Raman and normal mode studies of the polypeptide chain conformations in crystalline magnesium and calcium poly(glutamate)s. Spectrochim Acta Part A Mol Spectrosc 41(1–2):205–207
Asher SA, Ianoul A, Mix G, Boyden MN, Karnoup A, Diem M, Schweitzer-Stenner R (2001) Dihedral psi angle dependence of the amide III vibration: a uniquely sensitive UV resonance Raman structural probe. J Am Chem Soc 123:11775–11781
Palacky J, Vorlickova M, Kejnovska I, Mojzes P (2013) Polymerphism of human telomere quadruplex structure controlled by DNA concentration: a Raman study. Nucleic Acid Res 41:1005–1016
Palacky J, Mojzes P, Kejnovska I, Vorlickova M (2020) Does Raman spectroscopy recognize different G-quadruplex arrangements. J Raman Spectrosc 51:301–312
Hildebrandt P, Lecomte S (1999) Encyclopedia of spectroscopy and spectrometry, 2nd edn. Elsevier, Amsterdam
Sengupta B, Pahari B, Blackmon L, Sengupta PK (2013) Prospect of bioflavonoid Fisetin as a quadruplex DNA ligand. PLoS One 8(6):65383
Acknowledgments
The author is deeply grateful to Prof. Sandip K. Sengupta for insightful discussions and many valuable suggestions. He wishes to express his heartfelt thanks to the Department of Biophysics, Molecular Biology and Bioinformatics for infrastructural support. He also wishes to thank Dr. Snehasish Bhattacharjee and Mr. Niladri Bhattacharyya for their kind assistance.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sengupta, P.K. (2022). Raman Spectroscopy in Biology: Perspectives and Emerging Frontiers. In: Sahoo, H. (eds) Optical Spectroscopic and Microscopic Techniques. Springer, Singapore. https://doi.org/10.1007/978-981-16-4550-1_11
Download citation
DOI: https://doi.org/10.1007/978-981-16-4550-1_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-4549-5
Online ISBN: 978-981-16-4550-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)