
Abstract
Testis-derived spermatogonial stem cells (SSCs) and its in vitro counterpart, male germline stem (GS) cells, can repopulate the empty seminiferous tubules of infertile males and, therefore, are rapidly emerging as newer biotechnological tools for the treatment of male infertility, posthumous reproduction, preservation of elite germplasm and animal transgenesis. It also has potential application in the preservation and restoration of fertility in males with diseases/treatments affecting spermatogenesis. Consequently, the cryopreservation of SSC and GS cells is becoming pivotal in assisted reproductive technology (ART) for male reproduction. Cryopreservation of testis-derived stem cells is particularly indispensable for fertility preservation in pre-pubertal males, who have not yet begun their sperm production. This chapter provides an overview of the indications, applications, and strategies of fertility preservation through the cryopreservation of testicular stem cells. Recent advances on freezing and vitrification techniques, assessment of cryopreservation-induced injuries, and methods of their amelioration are also discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Mammalian testis is known to contain a pool of stem cells, known as spermatogonial stem cells (SSCs), which maintains spermatogenesis throughout the adulthood. These cells can be isolated and propagated in vitro as male germline stem (GS) cells and are capable of initiating donor-derived spermatogenesis in germ cell-depleted testis of infertile males for restoration of fertility. The GS cells can also undergo in vitro spermatogenesis in an organ culture system to produce haploid germ cells that can be used for the production of offspring by in vitro fertilization (IVF), intracytoplasmic sperm injection (ICSI) or round spermatid injection (ROSI). Under an appropriate culture system, the unipotent GSCs can also undergo reprogramming events to generate multipotent adult germline stem (maGS) cells or germline pluripotent stem (GPS) cells. Thus, testicular stem cells have potential application in both infertility management and regenerative medicine. Further, the testicular stem cells, combined with gene manipulation tools, can also be exploited for production of transgenic animal.
The testicular stem cells can be cryopreserved as a purified single-cell suspension or testicular tissue fragments that house SSCs in their niche (Jung et al. 2020a; Onofre et al. 2020). The cryopreservation of purified testicular stem cells can be conducted by conventional freezing or vitrification protocols used for somatic cells and are technically easier. However, SSCs are present in a very low number in adult tissues [~0.03%; (Nakagawa et al. 2007)] and may be difficult to isolate from biopsied specimens of testis. Consequently, newer and effective methods of cryopreservation for these novel cells are necessary. On the other hand, cryopreservation of testicular tissue fragments, enriched in SSC, is more challenging and depends on various factors such as the size of the tissue, permeation efficiency of cryoprotective agents (CPA) and variations in cryobiological properties of their cellular and extracellular matrix (ECM). The cryopreservation of testicular tissue fragments does not require tissue dissociation steps for isolation of cells and can maintain complex interaction between SSCs and testicular somatic cells (Leydig cells, Sertoli cells, and Myoid cells) and cell-to-ECM, which are essential for SSC-derived spermatogenesis. The testicular tissue cryopreservation can also be combined with in vitro spermatogenesis system for in vitro production of spermatozoa and eliminating the need for spermatogonial stem cell transplantation (SSCT). By preserving supporting somatic cells and ECM, the testicular tissue cryopreservation may provide the physical and molecular microenvironment required for survival, self-renewal, proliferation, and differentiation of germ cells to haploid germ cells. However, the superiority of cryopreserving testicular tissue fragments over testicular stem cells remains debatable. Moreover, although in vitro spermatogenesis system has shown promising results in rodents and primates for generating live offspring from cryopreserved testicular tissue fragments, it is still under developing phase for livestock animals and human.
This chapter provides an overview of various strategies for cryopreservation of testicular stem cells, including GS, maGS and GPS cells, and discuss their indications and applications in veterinary science. Suitable references are also drawn from human applications in which the technology is rapidly finding its clinical applications. The details on cryopreservation of testicular tissue fragments is discussed elsewhere (Patra et al. 2021).
2 Methods of Cryopreservation of Testicular Stem Cells
Semen freezing is a well-established procedure in animals for breed improvement programs through artificial insemination. However, cryopreservation of testicular stem cells is relatively recent technology and is still under experimental stage in livestock animals and human. There are very limited systematic studies on the development and optimization of cryopreservation protocols of testicular stem cells. Most of the initial studies have used conventional somatic cell freezing protocols with empirical modifications to cryopreserve the SSC, GS and GPS cells. The post-thaw viability varied from lab-to-lab although the functionality of frozen-thawed SSCs was confirmed by testicular transplantation assays as early as 1996 (Avarbock et al. 1996). Initial studies have also shown that the success of freeze-thawing also varies with animal species, type, and concentration of cryoprotective agents (CPAs), method of freezing and type of cryocontainers (Kanatsu-Shinohara et al. 2003; Wu et al. 2012). However, attempts to directly relate the post-thaw viability of testicular stem cells with variable factors have been very limited.
More recently, ice-free vitrification has been explored as simpler, faster, and cost-effective method for cryopreservation of testicular stem cells. Several strategies ranging from conventional vitrification in plastic straws (PS) to ultrarapid vitrification in microdroplets (MD), electron microscopy (EM) grid, nylon membranes, open pulled straws (OPS), solid surface vitrification (SSV), etc. have been attempted and were reported to support the cryo-survival of both somatic and stem cells in testis (Patra and Gupta 2019, 2020). However, like stem cell freezing, systematic studies on the vitrification of testicular stem cells have been limited. Some studies have suggested vitrification to be superior to freezing due to its simplicity, cost-effectiveness, and better post-warming viability. However, the superiority of one over the other remains debatable (Pukazhenthi et al. 2015). Systematic and controlled studies are needed to optimize the freezing and vitrification protocols by analysing the influence of variable factors such as rate of cooling, type of CPA, size or volume of the sample, cryopreservation device, storage conditions, etc.
3 Freezing of Testicular Stem Cells
Freezing of testicular stem cells involves controlled crystallization of water by their slow cooling in the presence of CPA such as glycerol, dimethyl sulphoxide (DMSO), ethylene glycol (EG) or 1,2-propanediol (PROH). The rate of cooling is so chosen to crystallize the extracellular water with minimal or no intracellular ice crystallization. The optimal rate of cooling varies with the cell type and species but generally range between 0.3 °C and 10 °C/min in most mammalian cell types. In testicular cells and stem cells, a cooling rate of 1 °C/min is considered critical during their cooling from 4 °C to (−)40 °C or lower temperature (optimal window of cooling rate). During this window of cooling, the extracellular water super cool and starts crystallizing between (−)5 °C to (−)10 °C, which results in a gradual increase in extracellular solute concentration and thereby, dehydrates the cells by osmotic-driven efflux of intracellular water. Between (−)10 °C to (−)15 °C, the extracellular ice expands and the sample become increasingly supercooled below (−)40 °C. When the sample temperature reaches (−)40 °C or lower, it can be plunged into a cryogenic agent such as liquid nitrogen or helium for intracellular vitrification without ice crystallization. The frozen samples can then be stored in liquid nitrogen for long-term storage. A working freezing-thawing protocol for cryopreservation of mice SSCs is provided in Box 9.1. In the hands of the authors, the protocol also works for the freezing of putative SSCs in goat.
Box 9.1 A Suggested Protocol for Slow Freezing of Testicular Stem Cells in Mechanical Freezer
Uncontrolled slow freezing of SSCs:
-
1.
Trypsinize the SSC clumps by using trypsin-EDTA to prepare single cell suspension. Note: SSCs survive better in cell clumps of ~15 to 20 cells.
-
2.
Centrifuge the cells at 500 g for 8–10 min and wash the pellets in a serum-free SSC culture medium twice.
-
3.
Count the cell concentration and dilute to a concentration of 2 × 106 cells per ml.
-
4.
Aliquot 0.5 ml of SSC cell suspension in 1.5 ml cryovials and slowly add 0.5 ml of SSC culture medium having 20% (v:v) DMSO.
-
5.
Close the cryovials, rack on an insulated container (e.g. Stryrofoam rack) and keep at −80 °C freezer for overnight (at least 4 h).
-
6.
Quickly plunge the cryovials into liquid nitrogen for cryostorage.
Thawing of SSCs:
-
1.
Identify and retrieve the cryovial from the liquid nitrogen container and place it in a water bath set at 37 °C.
-
2.
Transfer the thawed cell suspension into 9 ml of pre-warmed (37 °C) SSC culture medium in a 15 ml centrifuge tube and centrifuge at 500 g for 8–10 min.
-
3.
Wash the cell pellet with 10 ml of SSC culture medium twice.
-
4.
Count the cell concentration and re-suspend the cells in complete SSC culture medium (supplemented with GDNF) to the desired concentration and plate in culture plates.
The desired rate of cooling for freezing of testicular cells and stem cells can be achieved by uncontrolled slow freezing in a vapour or mechanical freezer or by controlled slow freezing in a programmable controlled rate freezer. Uncontrolled slow freezing can be performed using simple devices that are usually available in most laboratories and does not require expensive programmable freezers. Further, in contrast to controlled rate freezing, it requires a much less volume of liquid nitrogen (cryogenic agent) and the time required for the processing and freezing of the samples. Consequently, uncontrolled slow freezing has been conventionally used in most laboratories for cryopreservation of testicular stem cell suspensions in both human and animals (Pacchiarotti et al. 2013; Sá et al. 2012; Yango et al. 2014). In most conventional protocols, the cells are trypsinized to single-cell suspension, treated with CPAs such as glycerol or DMSO, loaded in cryocontainers such as plastic straws, cryovials or cryobags and cooled at 1 °C/min to (−)70 °C to (−)80 °C, achievable by exposure to liquid nitrogen vapour or (−)80 °C freezer, which is usually available in most laboratories. A number of devices such as Mr. Frosty™, Cryo Cool™, CoolCell™, etc. are also available for maintaining uniform heat transfer during the cooling process, although they are not absolutely necessary. After freezing to (−)70 °C, which typically takes 5–20 min in liquid nitrogen vapour and 4–8 h in the deep freezer, the samples are directly plunged into the liquid nitrogen for storage until further use (Frederickx et al. 2004; Izadyar et al. 2002; Redden et al. 2009). Some researchers have also used multistep slow freezing by equilibration with CPA at 4 °C for 2 h, cooling at (−)20 °C for 2 h and freezing at (−)80 °C before storage in liquid nitrogen (Zhang et al. 2015) but is not considered essential. At the time of use, the cells can be thawed in water bath at 37 °C and cultured in complete culture medium after removal of the CPA. In most freeze-thaw protocols, the post-thaw viability of testicular spermatogonial cells was reported to be as low as 50–60% (Izadyar et al. 2002). Nevertheless, frozen-thawed stem cells retained their self-renewing capability and could restore spermatogenic function to produce normal donor-derived offspring upon transplantation recipient testis without any apparent genetic or epigenetic errors (Wu et al. 2012; Yuan et al. 2009).
In controlled rate slow freezing, the CPA-treated cells are cooled at a controlled rate using a programmable controlled rate freezer. The rates of cooling vary with the species, the volume of cell suspension, and the type and concentration of CPA and may require optimization. Typically, controlled rate freezing of testicular stem cells is performed in 0.25 ml plastic French straws and cooling is done at the rate of (−)1 °C/min to (−)5 °C/min to cool the sample to (−)7 °C to (−)9 °C, following which they are seeded for 7–15 min and then cooled at the rate of (−)0.3 °C/min to (−)0.5 °C/min to (−)40 °C to (−)80 °C before plunging to liquid nitrogen for storage (Frederickx et al. 2004). Seeding induces the ice nucleation by mechanical vibration or rapid temperature reduction in the programmable controlled rate freezer. Some experimenters have found that seeding step is not necessary for controlled rate freezing of testicular stem cells when they were cooled at the rate of (−)1 °C/min to (−)5 °C/min to (−)80 °C, and subsequently at the rate of (−)50 °C/min to (−)120 °C followed by plunging into liquid nitrogen for storage (Izadyar et al. 2002; Pacchiarotti et al. 2013).
Rapid freezing has also been attempted for cryopreservation of testicular cells by treating them with high concentration of CPA in step-wise manner and plunging into liquid nitrogen (Gouk et al. 2011). However, it has met with limited success in terms of post-thaw viability. Besides, rapid freezing lead to random intracellular ice crystal formation that caused extensive cryoinjuries and high cell death. Thus, rapid freezing is not normally used for cryopreservation of testicular cells and stem cells.
4 Vitrification of Testicular Stem Cells
Vitrification is a rapid and simple method of cryopreserving testicular stem cells. It involves the solidification of both extra- and intra-cellular water into amorphous glass-like vitreous state without ice crystal formation. Vitrification of living cells can be achieved by ultrafast cooling (>106 °C/min) below their glass-transition temperature (Tg) to cause an extreme elevation of viscosity to greater than 1015 poise and result in molecular stasis and solidification without ice crystallization of water. Typically, the cells are treated with a very high concentration of CPA for cryoprotective effect, loaded into a cryocontainer such as PS, cryovial, EM grid, OPS, etc. and directly plunged into a cryogenic agent such as liquid nitrogen, liquid helium or nitrogen slush for vitrification and storage. Since high concentration of CPA required for vitrification is toxic to cells, an equilibration step of treating the cells with low concentration of CPA (typically, half of the final concentration) is usually necessary before their exposure to the full concentration of CPA (Gupta and Lee 2010; Jung et al. 2020b). The equilibration period is usually 1–3 min whereas the incubation period in vitrification solution is usually restricted to 25–45 s and are to be optimized for the type and concentration of CPAs, temperature of the solution and volume of the sample (Gupta et al. 2007). A longer (5–15 min) equilibration time with considerably low concentration of permeable CPA in the equilibration solution may be used for high volume samples. At the time of use, the vitrified cells can be warmed and rehydrated in a step-wise manner in the presence of a non-permeating disaccharide (e.g. sucrose, trehalose, etc.) to remove the CPA. The non-permeating disaccharides act as osmotic buffers and prevent the swelling and rupture of cells during cell re-hydration. A working vitrification-warming protocol for cryopreservation of mice SSCs is provided in Box 9.2. In the hands of the authors, the protocol also works for the vitrification of putative SSCs in goat.
Box 9.2 A Suggested Protocol for Vitrification of Testicular Stem Cells by SSV
Vitrification of SSCs:
-
1.
Trypsinize the SSC clumps by using trypsin-EDTA to prepare single cell suspension. Note: SSCs survive better in cell clumps of ~15 to 20 cells.
-
2.
Centrifuge the cells at 500 g for 8–10 min and wash the pellets in a serum-free SSC culture medium twice.
-
3.
Count the cell concentration and dilute to a concentration of 2 × 106 cells per ml in equilibration solution [20% (v:v) EG in calcium- and magnesium-free phosphate buffered saline (PBS)] for 90 s at 37 °C.
-
4.
Expose the equilibrated cells to vitrification solution [40% (v:v) DMSO, 18% (w:v) Ficoll 70 (Sigma) and, 0.3 M Sucrose (Sigma) in calcium- and magnesium-free PBS] at 37 °C.
-
5.
Using a micropipette swiftly drop the cell laden-vitrification solution as microdrop (~10 μl) on the pre-chilled dry surface of a metal block partially (3/4th) submerged into liquid nitrogen. Ensure that the surface of metal block is dry.
-
6.
Upon visual observation of the vitrification, move the vitrified microdrops (complete transparent microdrops) into 1.8 ml cryovials (Nunc cat. No. 343958) using liquid nitrogen-cooled forceps and stored into liquid nitrogen tanks until further analysis. The total duration between treatment with vitrification solution and plunging in liquid nitrogen should be limited to 30-45 s.
Warming and rehydration of vitrified SSCs:
-
1.
Identify and retrieve the cryovial from the liquid nitrogen container and allow the liquid nitrogen, if any, to evaporate.
-
2.
Drop the vitrified microdrops from the cryovial directly into a petri dish containing 1 ml of WS1 solution (0.5 M Sucrose in PBS) at 37 °C for 5 min.
-
3.
Wash the cells through 10 ml of WS2 solution (0.25 M Sucrose in PBS) for 5 min and centrifuged at 300× g for 5 min.
-
4.
Wash the cells through 10 ml of WS3 solution (PBS) for 5 min and centrifuged at 300× g for 5 min.
-
5.
Count the cell concentration and re-suspend the cells in complete SSC culture medium (supplemented with GDNF) to the desired concentration and plate in culture plates.
Vitrification of cells requires a shorter duration of CPA exposure, fewer instruments and therefore is a rapid method of cryopreservation. Further, it does not require any expensive equipment such as programmable controlled rate freezer and therefore, is economical. Moreover, although debatable, the efficiency of vitrification is generally considered superior, or at least similar, to those of slow freezing. Several studies have reported that lack of ice crystal formation reduced cellular damage and improved the post-warming viability of testicular stem cells (Poels et al. 2013). The vitrified-warmed testicular stem cells were shown to be capable of undergoing complete spermatogenesis upon transplantation. Consequently, it has been successfully used for cryopreservation of testicular cells and stem cells in both human and animals (Patra and Gupta 2019, 2020; Yokonishi et al. 2014). However, although the results are encouraging, the vitrification procedure for testicular cells and stem cells remain sub-optimal. Further, the success of vitrification may vary with the experimenter. Several efforts are therefore being made to improve the vitrification of testicular stem cells through optimization of sample volume, type of cryocontainers, type and concentration of CPA, duration of CPA-exposure, use of hydrostatic pressure, optimization of post-warming culture with additives such as antioxidant, etc. (Patra and Gupta 2019, 2020; Saragusty et al. 2010).
4.1 Cryocontainers for Vitrification
Plastic cryovials and straws are the most used cryocontainers for holding the cells during freezing. The same were later used to hold the cell suspension during vitrification. However, it was soon discovered that minimizing the volume of cell suspension, or increasing the surface area, can dramatically improve the post-warming viability of cells. Consequently, several minimum volumes cryocontainers such as EM grid, cryoloop, cryotop, OPS, cryotip, nylop loop, hemi-straw, vitrification spatula, plastic blade, metal mesh, nylon mesh, paper container, etc. became the containers of choice for vitrification for various cell types (Gupta and Lee 2010; Kim et al. 2012; Patra and Gupta 2019). Some of these cryocontainers such as EM grid and OPS allowed direct contact of the sample with cryogenic agent for rapid heat exchange whereas other cryocontainers such as cryotop and closed pulled straw protected the samples from direct contact with cryogenic agent for safety. Accordingly, depending on contact or non-contact of the samples with cryogenic agent during vitrification, the methods of vitrification can be classified as “open vitrification” and “closed vitrification”, respectively (Kumari et al. 2016). The open vitrification methods favour faster heat transfer than closed vitrification methods but poses a risk of contamination with pathogens such as hepatitis B, Brucella spp., which can survive in liquid nitrogen (Bielanski et al. 2000; Fountain et al. 1997; Kumari et al. 2016; Tedder et al. 1995).
While vitrification of testicular stem cells can theoretically be performed by both open and closed vitrification methods using a variety of cryocontainers, most studies have used OPS for their vitrification. In this method, the SSCs, suspended in vitrification solution, are loaded into thin OPS by capillary action and plunged into liquid nitrogen for vitrification and storage. This method of vitrification was found to be superior to both uncontrolled and controlled freezing of testicular cell suspensions (Sá et al. 2012). It was also suggested that the risk of microbial contamination due to direct contact with liquid nitrogen can be minimized using UV-C irradiated liquid nitrogen (Poels et al. 2013) and storing them in a pre-cooled cryotubes (Curaba et al. 2011).
Container-less vitrification methods such as solid surface vitrification (SSV), nylon mesh and MD methods have also been used for open vitrification of testicular cells and tissues (Abrishami et al. 2010; Higaki et al. 2017). The SSV method is a relatively simple method of vitrification wherein the CPA-treated samples are placed on the surface of sterile metal block, kept half-submerged in the liquid nitrogen, for vitrification. Following vitrification, the vitrified samples can be transferred into pre-cooled cryovials for storage in liquid nitrogen. A modified SSV (mSSV) method has also been developed wherein the solid metal block is replaced with a readily available aluminium foil that can be used like a floating boat on the surface of liquid nitrogen (Gupta et al. 2007, 2010). The mSSV method was demonstrated to be successful for vitrification of both testicular cells and tissues (Patra and Gupta 2019; Abrishami et al. 2010; da Silva et al. 2019; Kaneko et al. 2013). The testicular cells, cryopreserved by SSV or mSSV, were viable and could successfully re-initiate the spermatogenesis process upon transplantation (Onofre et al. 2016). In a comparative study, SSV was reported to be superior to OPS and MD vitrification as well as controlled rate freezing for vitrification of testicular tissue (Dumont et al. 2015). Thus, SSV and mSSV may offer excellent alternatives to both slow freezing and OPS vitrification. However, studies on SSV of testicular stem cells are less explored.
More recently, encapsulation-vitrification of testicular cells in alginate hydrogels have also been reported. We have shown that the testicular cells can be encapsulated into alginate microdroplets by mixing them with sodium alginate solution and allowing them to cross-link by dropping into calcium solution. The encapsulated cells can then be vitrified by closed or open vitrification such as SSV or MD (Patra and Gupta 2020). Following vitrification, the encapsulated cells can be retrieved by vortexing them in citric acid solution. We have further shown that the post-warming viability of encapsulation-vitrification varies with the size of microdroplets and can be regulated by adjusting the concentration and flow rate of alginate solution and calcium ion (Noguchi et al. 2006; Patra and Gupta 2020). The encapsulation-vitrification offers several advantages over other methods: (1) encapsulation provides a 3D structure which allows easy handling of cells during vitrification, dilution and rehydration process without requiring a centrifugation step (Patra and Gupta 2020); (2) encapsulation into alginate protects the SSCs against mechanical damages and oxidative stress during vitrification (Pirnia et al. 2017; Poels et al. 2016); (3) encapsulation may support the stemness of SSCs (Pirnia et al. 2017); (4) vitrified-warmed testicular stem cells, encapsulated in alginate microbeads, can be directly cultured in bioreactors for their large-scale expansion for clinical use and; (5) encapsulated cells can be used directly for transplantation (Gül et al. 2020). However, studies on encapsulation-vitrification of testicular cells are very limited. Furthermore, all studies on encapsulation-vitrification of testicular cells so far have used alginate biomaterial for hydrogel formation. A number of other natural protein (fibrin, collagen, gelatine, etc.) and polysaccharides (e.g. chitosan, dextran, etc.) or synthetic organic (e.g. polycaprolactone, polytetrafluoroethylene, etc.) and inorganic (e.g. Titania, silicon, etc.) biocompatible materials may be suitable for encapsulation of testicular cells and are yet to be explored (Vermeulen et al. 2017).
5 Cryoprotective agents (CPA) for Freezing and Vitrification
CPAs are the integral part of freezing and vitrification protocols for prevention of cryoinjury. Two classes viz. permeating (e.g. glycerol, DMSO, EG, PROH) and non-permeating (e.g. sucrose, trehalose, polyethylene glycol, hydroxyethyl starch) CPAs are commonly used for protecting the cells from cryoinjury and improving the cryosurvival during freezing and vitrification. The permeating CPAs can permeate into the cells and modify their cryobiological properties to protect against chilling injury by regulating ice crystallization. On the other hand, non-permeating CPAs are generally used to reduce the requirement of high concentration of permeating CPA and for increasing the membrane stability during dehydration (Lee et al. 2014a). Among various CPAs, glycerol and DMSO have been the most used as CPA for slow freezing whereas DMSO, EG and PROH have been the commonest CPAs for vitrification of testicular stem cells.
In early studies, slow freezing of testicular cells and tissues was achieved using glycerol at a concentration of 1 M, with or without non-permeating CPA such as 0.03–0.1 M sucrose and 10–80% (v:v) serum (Abrishami et al. 2010; Izadyar et al. 2002). However, it required a long duration of glycerization at low temperature and hence, its use became less frequent with the development of more effective permeating CPAs such as DMSO, EG and PROH (Keros et al. 2005; Travers et al. 2011). Among these permeating CPAs, 0.5–1.5 M DMSO (usually, 5–10%, v:v) has been the most commonly used permeating CPA and is considered least cytotoxic for better cell survival during slow freezing. Several studies have shown that SSCs frozen in the presence of DMSO could retain their self-renewing, proliferating and engraftment potential upon transplantation (Hermann et al. 2007). Combined use of DMSO with 200 mM trehalose could further improve the success of SSC cryopreservation (Jung et al. 2020b) and was superior to those of 2.5% polyethylene glycol (PEG) and 200 mM trehalose although number of spermatogonial colonies were higher from SSCs that were frozen thawed in 2.5% PEG with 200 mM trehalose (Lee et al. 2013a, b). Additive effect of trehalose with DMSO was also shown in bovine species (Kim et al. 2015).
The DMSO has also been used very commonly for vitrification of testicular cells at a concentration of 3.0–5.5 M (usually, 30–40%, v:v) (Curaba et al. 2011; Frederickx et al. 2004; Keros et al. 2007; Lee et al. 2014a; Pietzak 3rd et al. 2015; Unni et al. 2012; Wu et al. 2012; Wyns et al. 2007; Yango et al. 2014). DMSO forms transient pores in the plasma membrane, which favours cell dehydration and reduces ice crystal formation during the cooling (Gurtovenko and Anwar 2007). It can also interact with lipid bilayer of plasma membrane to increase its fluidity and protect from mechanical injury caused by ice crystals. Moreover, DMSO can improve the cryosurvival of cells by allowing supercooling of cells below (−)40 °C and preventing intracellular ice crystallization (Mazur 1984). However, high concentration or longer exposure duration of DMSO can be cytotoxic and induce cell differentiation, aberrant DNA methylation and histone modification and apoptosis (Huang et al. 2008; Iwatani et al. 2006; Kawai et al. 2010). Consequently, several alternative CPAs such as EG, PROH, formamide, HES, dextran, etc. have been tried, either alone or in combination to DMSO for reducing its cytotoxicity in various cell types, including testicular cells. The EG has higher cell permeability that DMSO and can weaken the hydrogen bond formation between solutes and water molecules (Weng et al. 2011). It also reduces the osmotic shock during ice crystallization. Thus, EG at a concentration of 1.5–3.0 M or 5.5–6.5 M has been viewed as a good alternative to DMSO for freezing and vitrification, respectively (Frederickx et al. 2004). Nevertheless, till date, DMSO remains to be the most used CPA for both freezing and vitrification of testicular cells. The DMSO and EG can also be used at 1:1 ratio to reduce the toxicity of individual CPAs by reducing their concentration to half.
Natural antifreeze proteins and glycoproteins found in Antarctic fish and plants that have adapted to extreme cold conditions have also been explored for use as CPA (Cheung et al. 2017; Kim et al. 2017). These proteins can cause freezing point depression and prevent the intracellular ice crystallization to reduce cryoinjury. They can also maintain the stability of phospholipids and unsaturated fatty acids in plasma membranes. However, studies on use of antifreeze proteins and glycoproteins have been limited and, their usefulness for cryopreservation of testicular stem cells remains to be explored.
6 CPA Toxicity and Strategies for Its Amelioration
All permeating CPAs are known to have cytotoxicity depending on their concentration and the duration of exposure. Commonly used CPAs such as DMSO, EG and PROH have been reported to cause abnormal spindle morphology and aneuploidy in oocytes and embryos (Huang et al. 2008), although such damage was not observed in testicular cells and tissues (Li et al. 2009; Oblette et al. 2017; Song et al. 2016). DMSO is also known to cause epigenetic aberrations in DNA methylation in testicular cells and tissues (Iwatani et al. 2006; Kawai et al. 2010). Thus, titration of concentration and duration of exposure is required to be optimized for effective cryopreservation with minimal cytotoxicity. Toxicity of CPAs can also be reduced by combining two or more permeating or non-permeating CPAs and reducing the concentration of individual CPA. Experiments have shown that excellent vitrification solutions can be formed by combining a strong glass former such as DMSO with a weak glass former such as EG or formamide (Fahy et al. 2004). Thus, the mixture of DMSO and EG in 1:1 ratio is occasionally used for reducing the concentration of DMSO and its toxicity during vitrification of testicular stem cells and tissues (Abrishami et al. 2010; Baert et al. 2012; Curaba et al. 2011; da Silva et al. 2019; Dumont et al. 2015; Gholami et al. 2013).
Replacing the part of permeating CPA with non-permeating CPAs such as mono- or di-saccharides, polyvinylpyrrolidone (PVP), PEG, Ficoll, dextran and polyvinyl alcohol (PVA) offers another possibility for minimizing the CPA toxicity. In several studies, DMSO was combined with sucrose (Curaba et al. 2011; Goossens et al. 2008; Gouk et al. 2011; Izadyar et al. 2002; Poels et al. 2014; Wyns et al. 2008), trehalose (Lee et al. 2014b, b; Zhang et al. 2015) and dextran (Pacchiarotti et al. 2013) to reduce its cytotoxicity and improve the cryosurvival of testicular cells and tissues during freezing and vitrification. The EG has also been combined with sucrose, trehalose and PVP to reduce its cytotoxicity and improve cryosurvival of testicular cells and tissue (Kaneko et al. 2013; Poels et al. 2012). High concentrations of blood serum, serum albumin, hyaluronan, anti-oxidants, etc. are the other additives that may be used in freezing and vitrification media to improve the success rates of cryopreservation. Since CPAs are known to induce apoptotic cascade, inhibitors such as Z-VAD-FMK, Z-DEVD-FMK, sphingosine-1-phosphate, etc. may also be useful in reducing the CPA-induced apoptosis (Men et al. 2006; Onions et al. 2008).
7 Cryoinjury and Strategies for Its Amelioration
In addition to CPA, the chilling injury during freezing and vitrification procedures can also cause thermal and biochemical stress to the cells and reduce their viability. The chilling injury can occur due to improper cooling rate, osmotic stress and mechanical damage caused by sharp ice crystals. The immediate outcome of these chilling injuries is apoptosis and cell death, which may be observed in the form of cellular degeneration, rupture of cell membrane and leakage of cytoplasmic contents. Mechanism by which freezing and vitrification induce cell death is not fully understood but both canonical and mitochondrial-mediated apoptosis pathways and oxidative stress pathways have been shown to be involved. It may also cause transcriptional stress to cells resulting in increased expression of responsive genes such as Bax, Cirbp, Hsp90ab1 and Sod1 (Bebbere et al. 2019). Further, cryopreservation may also cause dysregulated gene expression and stress resulting in epigenetic aberrations of DNA methylation (Iwatani et al. 2006; Kawai et al. 2010). However, despite these cryoinjuries, the birth of normal and fertile offspring has been reported from the cryopreserved SSCs and testicular cell suspensions (Goossens et al. 2011; Tanaka et al. 2015, 2018; Wu et al. 2012; Yuan et al. 2009). Studies have also shown that cryopreservation had little or no effect on meiotic recombination and fidelity of synapsis formation in testicular cells (Li et al. 2009; Song et al. 2016).
Reactive oxygen species (ROS) such as superoxide anion radical (O2•−), hydroxyl radical (•OH) and hydrogen peroxide (H2O2) are normally generated during mitochondrial respiration and are important for cellular homeostasis. At physiological levels, the ROS also stimulates the self-renewal of testicular stem cells via MAPK and JNK signalling pathways (Morimoto et al. 2013). However, excessive ROS generation or its intracellular accumulation can cause cell death by modifying the functional groups of proteins, lipids and nucleic acids (Finkel and Holbrook 2000). Several studies have shown that the cryopreservation can induce excessive ROS generation to cause oxidative stress and leads to DNA fragmentation, altered cellular metabolism, dysregulation of cell signalling pathways and cell death (Gupta et al. 2010; Thomson et al. 2009; Zhang et al. 2015). Consequently, use of antioxidants in cryopreservation and in vitro culture media has been explored by various researchers to ameliorate the adverse effects of ROS. Several studies have shown the beneficial effects of several antioxidants such as vitamin E, α-tocopherol (an active form of vitamin E), catalase (CAT), reduced glutathione (GSH), β-mercaptoethanol (βME), superoxide dismutase (SOD), melatonin, caffeic acid, selenium, hypotaurine and cysteine on the viability of sperm, oocytes, somatic cells and stem cells, due to their ROS scavenging activity (Aliakbari et al. 2016; Boroujeni et al. 2019; Gupta et al. 2007, 2010; Ha et al. 2016; Navid et al. 2017; Sayed Mahdi et al. 2018). Studies have also shown that supplementation of antioxidants such as vitamin E into the cryopreservation and in vitro culture media reduced the ROS accumulation in frozen-thawed testicular tissue of pre-pubertal mice and lead to better in vitro spermatogenesis (Arkoun et al. 2019). Thus, use of antioxidants in cryopreservation protocols of testicular cells and tissue is generally recommended (Patra and Gupta 2019; Zhang et al. 2015).
Many studies have also shown that apoptosis and necrosis is a common observation during cryopreservation of testicular stem cells by freezing or vitrification. Although the cause of apoptosis could not be precisely ascertained, both canonical and mitochondrial-mediated pathways were found to be involved in cryopreserved testicular stem cells. Studies have also shown that CPA such as DMSO and DM can themselves cause apoptosis. Thus, several studies have also analysed the use of apoptotic inhibitors such as Z-VAD-FMK, Z-DEVD-FMK, sphingosine-1-phosphate, RIPA-56, etc. and reported them to be useful in reducing the CPA- or cryopreservation-induced apoptosis (Men et al. 2006; Onions et al. 2008; Xie et al. 2020).
8 Assessing the Viability of Cryopreserved Testicular Stem Cells
The success of a cryopreservation protocol can be assessed by multiple morphological, biochemical, and molecular methods for assessment of cell viability, apoptosis, ROS activity, ability to initiate spermatogenesis by short-term in vitro culture, and production of fertile and healthy offspring upon SSCT or through in vitro spermatogenesis followed by IVF/ICSI/ROSI. Several biochemical and molecular analytical methods can also be utilized to assess the extent of cellular damage such as chromosomal anomalies, reduced mitochondrial membrane potential, aberrant DNA methylation or histone modifications, free radical formation, etc. Most studies analyse and report the percentage of living cells in frozen-thawed or vitrified-warmed cells as a measure of success in cryopreservation. However, the presence of living cells not necessarily indicates the functionality of the cells. The functionality of cryopreserved testicular cells needs to be verified by transplantation assay or in vitro culture. Successful in vitro spermatogenesis of motile sperm, which could lead to the birth of live offspring by artificial insemination, IVF or ICSI is also a sufficient proof for the success of testicular cell cryopreservation. Alternatively, the success of testicular cryopreservation can be assessed by transplantation of the cryopreserved cells into recipient testis or grafting into a heterologous location such as under skin or orthotropic grafting for monitoring the serum testosterone level or spermatogenesis process (Baert et al. 2012; Kirpatovskii et al. 2018).
9 Biochemical Assays
Biochemical methods are routinely used for evaluating the viability of cryopreserved cells by dye-exclusion Trypan Blue assay, fluorescein diacetate (FDA) assay and lactate dehydrogenase (LDH) release assay. On the other hand, apoptosis in cryopreserved cells can be evaluated by analysing the caspase activity, Annexin V staining or TUNEL assay. Staining for nucleolar organizing region (NOR) and immunocytochemistry for PCNA staining or Ki67 staining can also be utilized to get an idea about the cell proliferation ability of cells. Further, cryoinjuries can also be assessed by evaluating mitochondrial damage by MitoTracker™ or MTT assay, oxidative stress by ROS activity, lipid peroxidation by malondialdehyde (MDA) assay, etc. More recently, Fourier transformed infrared (FTIR) spectroscopy has also been found to be suitable for analysing the general biochemical profile and membrane lipid transition in cryopreserved cells (Meneghel et al. 2019; Wang et al. 2019), although its potential has not yet been fully utilized for testicular stem cells.
10 Molecular Methods
Conventional molecular tools such as polymerase chain reaction (PCR), reverse transcriptase PCR (RT-PCR) and Western blotting can be used for analysis of genes and proteins related to cell proliferation, cell viability, apoptosis, etc. in cryopreserved cells. The stemness of viable SSCs can also be evaluated by analysing the expression of marker genes and proteins such as Oct4, Pgp9.5, VASA, boule, UTF1, UCHL1, GFRα-1, PLZF, etc. (Li et al. 2018). Immunohistochemistry and RNA sequencing (RNAseq) can also be used to evaluate the transcriptome, proteome and epigenome status of the cryopreserved cells. On the other hand, karyotyping and fluorescence in situ hybridization (FISH) can be done to assess the chromosomal configuration of cells and haploidy of spermatozoa obtained from cryopreserved testicular stem cells.
11 Transplantation Assays
Testicular transplantation and donor-derived spermatogenesis is the ‘gold standard’ for true assessment of viability in cryopreserved testicular stem cells. The cells (0.5–2.0 × 105 cells/ml) can be mixed with a dye such as Trypan Blue to visualize the microinjection (Kaul et al. 2010; Onofre et al. 2020) and microinjected into recipient testes prepared by busulfan treatment or gamma irradiation to deplete of endogenous SSCs. The microinjection can be done via rete testes or vas deference or directly into the seminiferous tubules using borosilicate pipettes under microscope. The transplanted SSCs colonize at the basal membrane and re-populate the seminiferous tubules to initiate spermatogenesis and produce fertile spermatozoa. The proof of principle for SSC transplantation (SSCT) came from studies on transplantation of SSCs from infertile Steel (SI/SI) mice to infertile dominant white spotting (W/Wv). The SI mutants are infertile due to Sertoli cell dysfunction whereas W mutant male mice are infertile due to germ cells dysfunction but with normal Sertoli cell function. The testicular transplantation of SSCs from SI mice into W mice restored fertility with donor-derived offspring (Ogawa et al. 2000). Several studies have subsequently shown that fertile donor-derived spermatozoa can be obtained by SSCT in several animal species (Herrid et al. 2019; Shetty et al. 2020).
The transplantation assay for evaluation of cellular viability of testicular stem cells can also be done at an orthotropic location either directly or after encapsulation into alginate matrix (Del Vento et al. 2019; Gül et al. 2020). The functionality of transplanted cells can be evaluated by histological analysis of the transplanted cells for spermatogenesis progression and generation of donor-derived sperm. Studies have also shown that mouse testicular cells, encapsulated into Matrigel™, could self-organize into seminiferous tubules upon subcutaneous injection into the dorsum of the nude mice (Gao et al. 2020). The functioning of testicular cells and SSCs can be tracked by pre-labelling the cells with PKH67 or DiI stain, which retains in the loaded cells for nearly two months (Dong et al. 2019; Mohaqiq et al. 2019). Under experimental settings, genetically modified cells with marker proteins such as enhanced green fluorescent protein (EGFP) under the control of sperm-specific promoters such as Acrosin (Acr) can also be utilized. Transgenic mice expressing EGFP under the control of Acr is already available commercially. Xeno-grafting is also an attractive strategy for fertility preservation in endangered animals and resurrection of extinct animals (Pukazhenthi et al. 2006).
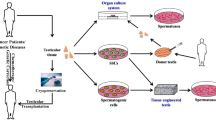
In vitro generation of three-dimensional (3D) testicular organoids (TO) from cryopreserved testicular cells has also emerged as a novel strategy for assessment of cell viability (de Michele et al. 2017; Pendergraft et al. 2017; Vermeulen et al. 2019). The TOs consists of SSCs, germ cells and somatic cells (Sertoli cells, Leydig cells, peritubular Myoid cells) to resemble the testicular structure with interstitial compartments separated by basement membrane and can respond to various environmental cues (de Michele et al. 2017; Sakib et al. 2019; Vermeulen et al. 2018). The functionality of 3D TOs in terms of testosterone production and spermatogenesis events in testicular cells could be a good indicator for the success of cryopreservation (de Michele et al. 2017; Pendergraft et al. 2017).
12 In Vitro Spermatogenesis
In vitro spermatogenesis from SSC could be an excellent alternative to the in vivo SSCT. It was initially applied for mice testicular tissue (Sato et al. 2011) and was found to be useful for verifying the post-thaw viability of frozen-thawed mice testis (Yokonishi et al. 2014). The method involved organ culture of testicular tissue fragments at air-liquid interphase following which the in vitro produced sperm could be used for production of offspring by IVF, ICSI or ROSI. The organotypic air-liquid interphase culture was also proven to be useful for in vitro spermatogenesis from testicular stem cells. The protocol required the introduction of SSCs into an allogenic or cadaver-derived testicular tissue before their organ culture at air-liquid interphase (Gül et al. 2020; Sato et al. 2011). Studies have shown that xenoxenic recipient testis can also be used for in vitro spermatogenesis from testicular stem cells (Mohaqiq et al. 2019) but complete spermatogenesis depends on the compatibility of spermatogenic cycle and phylogenetic distance between the species (Ntemou et al. 2019). Moreover, xenoxenic transplantation of SSCs for in vitro spermatogenesis may pose possible risks of zoonotic disease transmission and immunoreactions (Kaneko et al. 2013; Liu et al. 2016). An alternative to the requirement of cadaver-derived or xenogenic testis could be the use of tissue-engineered testicular constructs (Mohaqiq et al. 2019; Perrard et al. 2016; Rezaei Topraggaleh et al. 2019; Vermeulen et al. 2018). It is possible that the SSCs can be seeded into biomaterial-derived scaffolds and cultured ex vivo in an organotypic air-liquid interphase to achieve in vitro spermatogenesis. Macroporous 3D scaffolds have been developed using decellularized testicular matrix (DTM), which supported the differentiation of spermatogonial cells up to post-meiotic stage (Rezaei Topraggaleh et al. 2019). However, the testicular engineering approach for in vitro spermatogenesis is still at its infancy and, till date, no study has reported the production of fertile spermatozoa in both human and animals. A detailed review on in vitro spermatogenesis can be found elsewhere (Ibtisham and Honaramooz 2020; Pelzman et al. 2020).
13 Application of Testicular Stem Cell Cryopreservation
Semen cryopreservation is the most convenient and the first choice for fertility preservation in both human and animals. However, in instances wherein semen cryopreservation is not possible due to non-availability of sperm (e.g. pre-pubertal males, non-obstructive azoospermia etc.) or non-feasibility (e.g. posthumous reproduction, pre-meiotic barriers to spermatogenesis etc.), cryopreservation of testicular stem cells, or testicular tissue enriched in SSCs, offers alternate and attractive options. The latter is still under the experimental stage with limited demonstrated clinical applications in large animals and human (Valli-Pulaski et al. 2019). Nevertheless, fertility preservation through cryopreservation of testicular stem cells is indicated in both medical and non-medical conditions and, occasionally the only choice (Table 9.1). Testicular stem cell cryopreservation can also be used to preserve genetic resources of wild animals if sperm is not available (e.g. testis from morbid wild animal) or are difficult to cryopreserve. Furthermore, cryopreservation is of utmost importance for preservation of genetically modified testicular stem cells that are meant for transgenic animal production by SSCT or ARTs. The complexity and diversity of important applications of the testicular stem cell cryopreservation are discussed below.
14 Fertility Restoration in Males with Pre- and Post-meiotic Barriers to Spermatogenesis
Among various causes of male infertility such as obstruction to vas deference, orchitis, hormonal disorders, etc., spermatogenic defects contribute ~70 to 90% to male infertility. In livestock animals such as cattle and buffalo, this is generally handled by use of donor-derived frozen-thawed sperm for artificial insemination. However, preservation of male infertility become pertinent in elite bulls, race horses of high genetic merit, pets, and wild animals. In such cases, cryopreservation of testicular stem cells can be envisaged as a powerful tool for preservation and restoration of male fertility. The testicular stem cells may be isolated from the testicular biopsy and cryopreserved by freezing or vitrification to restore the fertility by SSCT or other ARTs. More importantly, cryobanking of testicular stem cells may offer fertility solutions for azoospermic males with defective or immature spermatogenesis due to congenital or acquired conditions. In such cases, the testicular stem cells can be isolated from biopsied testicular tissue and differentiated in vitro to produced spermatozoa for ICSI or ROSI.
Cryptorchidism could be yet another reason that may necessitate cryopreservation of testicular stem cells in males of high genetic merit or transgenic animals. Testis that fails to descent into extra-abdominal scrotum undergoes a progressive reduction in germ cells, defective germ cell maturation and testicular dysfunction due to thermal stress (Docampo and Hadziselimovic 2015; Lee and Coughlin 2001). In such cases, testicular stem cells may be isolated from testis at an early stage, before the herald of the germ cell degeneration, and cryopreserved for restoration of fertility by in vitro spermatogenesis or ectopic grafting (Makala et al. 2015; Wyns et al. 2007). In cryptorchidism, the possibility of fertility restoration by transplantation of cryopreserved testicular stem cells is very bleak because of simultaneous impairment of somatic cell (Leydig cells, Sertoli cells) functionality. Thus, in vitro spermatogenesis is necessary to restore fertility from cryopreserved testicular stem cells.
Castration and vasectomy are effective methods of neutering in animals for making them docile. Vasectomy is also practiced in adult human for sterilization. There may be instances (e.g. death of the offspring) wherein reversal of male fertility is desired. However, the success of vasovasostomy or epididymovasostomy reversal procedure varies with the duration of vasectomy and may even fail (Fuchs and Burt 2002). Semen or testicular tissue freezing before vasectomy could be an option in such instances. The testicular stem cells, isolated from cryopreserved testicular tissue, can be used for restoration of fertility.
15 Male Fertility in Ageing
Cryopreservation of testicular stem cells may also be warranted in non-medical reasons such as ageing. It can offer the opportunity for restoration of fertility in males who have become aged or in cases of postponed parenthood. The later can also be applied for obtaining offspring from animals that have stopped ejaculation or are unsuitable for breeding due to ageing. Recent studies have shown that reduction in sperm count can occur with increasing age (Brandt et al. 2019; Levine et al. 2017) and therefore, can reduce the fertility and fecundity. Sperm from aged individuals also increases the risk of pregnancy complications, accumulation of spontaneous mutations, birth defects and neurological disorders in offspring (Su et al. 2015; Taylor et al. 2019). Thus, preservation of testicular stem cells may become necessary in such cases.
16 Posthumous Reproduction
Cryopreservation of testicular cells offers an opportunity to obtain live offspring from deceased individuals in both human and animals. The sperm or testicular tissue can be collected from peri- or post-mortem males and cryopreserved for future procreation of offspring (Check et al. 2002). It human, it has been applied in rare circumstances of unexpected death of the male partner, and birth of live offspring have been reported (Batzer et al. 2003; Check et al. 2002) although it poses several ethical and legal challenges to both the family and the society at large (Hurwitz and Batzer 2004).
In animals, cryopreservation of testicular specimens offers opportunity for posthumous reproduction in elite bulls or from testicular tissue samples that may have been found accidently from wild animals. The testicular stem cells, combined with in vitro spermatogenesis or testicular transplantation in recipient testis, can restore fertility. The success of such procedure, however, depends on the availability of live testicular stem cells and/or precursor cells and reduces with the increase in the duration of sample collection (Batzer et al. 2003). Depending on the availability of the cell type and their viability, the collected sample may be used for IVF, ICSI, ROSI or in vitro spermatogenesis.
17 Fertility Preservation and Restoration in Pre-pubertals with Oncological Conditions
Testicular dysfunction can occur due to testicular cancer as well as non-testicular neoplasm such as leukaemia. The oncological conditions may either have a direct effect on gonadal function or may indirectly cause testicular dysfunction due to prolonged hyperthermia, cytokine storm, cachexia and nutritional deficiencies of vitamins, minerals and trace elements, etc. (Dohle 2010). While most cancer cases (e.g. leukaemia) remain to have high mortality rate, many neoplastic conditions (e.g. Hodgkin’s lymphoma, testicular tumours) may eventually result in the long-term survival of patients upon treatment (Crha et al. 2009). Unfortunately, radiation and chemotherapy for cancer treatment are generally cytotoxic to rapidly dividing cells, including testicular stem cells, and predispose the surviving males to testicular failure and infertility (Duca et al. 2019). In human, radiation and chemotherapy have been reported to result in azoospermia in ~25% of cancer cases (Green et al. 2010) although such systematic studies on infertility analysis in animals are lacking. Thus, cryopreservation of germ cells is crucial for fertility preservation and restoration in cancer patients expecting gonadotoxic treatment. The semen, cryopreserved prior to initiation of gonadotoxic treatment could be used for treatment of infertility. However, this option is not available in pre-pubertal and paediatric males, which have not yet begun their sperm production. In such cases, cryopreservation of testicular stem cells, or testicular tissue enriched in SSCs, are the only options for fertility preservation and restoration (Lakhoo et al. 2019). The transplantation of cryopreserved testicular stem cells, isolated from the patient’s own testis, allows autologous transplantation, and thereby, not only avoids potential immune rejection but also is ethically acceptable. Thus, isolation, cryopreservation, and testicular transplantation of SSCs are increasingly proposed as a method of choice for restoration of fertility in cancerous individuals in which SSC populations is expected to deplete due to radiation or chemotherapy. Several studies have shown very encouraging results of autologous SSCT in rodents and primates (Ginsberg et al. 2010). However, it is yet to be applied in large animals and human due to several unresolved technical and safety concerns.
One of the biggest safety concerns with SSC cryopreservation and autologous transplantation in cancer cases has been the potential risk of reintroducing cancerous cells upon SSCT. Experiments have shown that presence of 20 cancerous cells in cryopreserved testicular cells from leukemic rats was enough to re-establish malignancy in 3 out of 5 non-leukemic recipient rats (Jahnukainen et al. 2001). Thus, purification of SSCs from contaminating cancerous cells is necessary prior to their SSCT in treated and cured cancer cases (Hermann et al. 2011). Purification of SSCs may be achieved by fluorescence-activated cell sorting (FACS) or magnetic-assisted cell sorting (MACS) using specific antibodies for cancerous cells (e.g. CD4 for T-cell leukaemia) (Fujita et al. 2006; Geens et al. 2007; Hou et al. 2007, 2009; Tian et al. 2019). Long-term culture of testicular cells in SSC-selective media that does not support cancer cell growth has also been tried for elimination of cancerous cells (Sadri-Ardekani and Atala 2014). However, sorting of cells by FACS or MACS or their selective culture in SSC-selective media were met with limited success for clinical use (Geens et al. 2007; Hou et al. 2007, 2009).
A practical and feasible alternative to FACS or MACS-based elimination of cancerous cells for SSCT could be in vitro spermatogenesis of spermatozoa and production of offspring by IVF, ICSI or ROSI (Yokonishi et al. 2014). The cryopreserved SSCs can be seeded into allogenic, xenogenic or a cadaver-derived testicular tissue and organ cultured at air-liquid interphase for in vitro spermatogenesis (Gül et al. 2020; Ntemou et al. 2019; Sato et al. 2011). Unfortunately, this later strategy has not been optimized in most animal species and human. An alternative to the requirement of donor-derived testicular tissue for in vitro spermatogenesis could be the use of biomaterial-derived tissue-engineered testes (Mohaqiq et al. 2019; Perrard et al. 2016; Rezaei Topraggaleh et al. 2019). However, complete in vitro spermatogenesis in tissue engineered testis remains to be demonstrated (Mohaqiq et al. 2019; Portela et al. 2019).
18 Fertility Preservation and Restoration in Non-oncological Diseases
Non-oncological diseases such as autoimmune diseases, haematological disorders, spinal cord injuries, severe trauma to testis, etc. may result in testicular dysfunction and ejaculatory failure. Autoimmune diseases such as systemic lupus erythematosus, multiple sclerosis, and Crohn’s disease are also often associated with abnormal semen parameters, erectile dysfunction and ejaculation failures (Fode et al. 2012). Further, preconditioning chemotherapy or whole-body irradiation before bone marrow transplantation in these patients entails a high risk of gonadal dysfunction (Snowden et al. 2017). In human, ~85% of patients experienced azoospermia after preconditioning chemotherapy or radiation for bone marrow transplantation (Anserini et al. 2002). Similarly, 3 out of 5 pre-pubertal individuals, who underwent hematopoietic stem cell transplantation for sickle cell anaemia, experienced azoospermia (Lukusa et al. 2009). Thus, in such cases, in the absence of semen for cryopreservation, banking of testicular stem cells, or testicular tissue enriched in SSCs, is recommended prior to corresponding therapies.
Cryopreservation of testicular stem cells is also an alternative to semen preservation in individuals affected with serious infectious diseases with potential of transmission via semen or during mating (e.g. immunodeficiency viruses). The repeat sperm washing procedure may reduce the pathogen load but remains a risk to the offspring as well as sero-negative partner, even from males with undetectable serum load of pathogen (Nicopoullos et al. 2011). Further, the duration of infection, patient’s age, immune status, treatment regimen, etc. also reduces the semen volume, sperm concentration, sperm viability and their fertilizability (Bujan et al. 2007; Wang et al. 2014). On the other hand, unlike semen, the testicular stem cells can be screened extensively before in vitro spermatogenesis or SSCT to reduce the risk of disease transmission. The repeat sub-culture steps, differentiation protocols and advanced molecular tools for detection and sorting of purified population of testicular stem cells can significantly reduce or remove the risk of disease transmission altogether.
Cryopreservation of testicular stem cells may also be warranted for fertility preservation prior to pre-conditioning radiation or chemotherapy for bone marrow transplantation in non-infectious genetic diseases such as sickle cell anaemia, thalassemia, Drepanocytosis, idiopathic medulla aplasia, chronic granulomatous disease, etc. It may also be required for early preservation of male fertility in genetic diseases such as Klinefelter syndrome in which degeneration of germ cells is seen at a later stage of life. In Klinefelter syndrome, the loss of SSCs may start at pre-pubertal stage and meiotic arrest is seen at the later stage of life due to aneuploid spermatogonia (Vialard et al. 2012; Wikström and Dunkel 2008). Thus, in such genetic diseases, it becomes imperative to cryopreserve testicular stem cells at an early stage of life. Since individuals with Klinefelter syndrome have fibrosed and hyalinized seminiferous tubules, which may not support spermatogenesis upon SSCT, in vitro spermatogenesis of cryopreserved testicular stem cells will be of immense utility (Braye et al. 2019; Wikström and Dunkel 2008). Importantly, transplantation of autologous cryopreserved testicular stem cells in genetic diseases such as Klinefelter syndrome of AZF mutations will not prevent the vertical transmission of mutations or genetic diseases to the progeny per se. However, genetic correction of GS cells using gene editing technologies such as CRISPR-Cas9 is a possibility in future for generation of mutation-free spermatozoa and healthy fertile offspring.
19 Animal Transgenesis and Genome Editing
Manipulation of testicular stem cells has immense potential for transgenic animal production. This is of particular importance in livestock animal species such as cattle and pigs in which existing methods of transgenesis are not efficient. Moreover, it can be combined with newer genome editing tools such as Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs) and Clustered Regularly Interspaced Short Palindromic Repeats-Cas9 (CRISPR-Cas9) system for knock-in and knock-out animal production for understanding the gene function through gain-of-function and loss-of-loss function studies (Koppes et al. 2020; Park et al. 2017; Zhang et al. 2020). These genetically engineered stem cells are created through a lengthy and cumbersome process and are novel to be cryopreserved for later use. An improper cryopreservation protocol for these genetically modified testicular stem cells can ruin the work of several months and years.
The application of genome editing tools is also expected to broaden the application of testicular stem cell cryopreservation to radically change our approach for infertility treatment. The CRISPR-Cas9 system has been successfully applied to testicular stem cells for genetic correction and production of disease-free individuals or for treatment of infertility through correction of underlying genetic mutation (Wu et al. 2015). Cryopreservation of these gene edited cells is essential for preservation of mutation-free testicular stem cells until the individuals are ready for transplantation. Nevertheless, the genome editing tools require refinements and remain a farfetched goal for both animal and human. The current application of these genome editing is restricted to their application in the generation of model animals for unravelling the gene function.
Gene targeting and genome editing tools can also be used for differentiation and in vitro spermatogenesis from cryopreserved testicular stem cells. Several molecules such as SCF/c-kit system, Dazl RNA binding protein, cyclin D2 and D3 and retinoic acid have been shown to promote differentiation of testicular stem cells. The SCF/c-kit system upregulates cyclin D3 and promotes cell cycle progression in spermatogonia through a rapamycin-sensitive PI3K/p70 S6 kinase pathway (Feng et al. 2000). On the other hand, targeted disruption of Dazl (deleted in azoospermia-like) RNA-binding protein inhibited the differentiation of Aal spermatogonia into A1 (Schrans-Stassen et al. 2001) while transfection of ES cells with Daz gene family (Dazl, Daz and Boule) promoted their in vitro differentiation into germ cell lineage to form haploid gamete in both mouse (Yu et al. 2009) and human (Kee et al. 2009). In another study, in vitro production of haploid sperm cells was shown to occur from male germ cells of fetal cattle when they were treated with retinoic acid (Dong et al. 2010). There are several other factors such as cyclin-dependent kinases (cdks) and cyclins (Sicinski et al. 1996; Tsutsui et al. 1999), Notch-1 signalling molecules (Hofmann et al. 2005), which are involved in the differentiation of spermatogonia but need further investigation for their application in establishing a system for in vitro spermatogenesis from cryopreserved testicular stem cells.
20 Banking for Preservation and International Movement of Animal Genetic Resources
Cryopreservation of testicular stem cells has immense utility in the banking of animal genetic resources, rapid dissemination of elite germplasm and their international transportation at low cost. In last few decades, the genetic diversity of both domestic and wild animals has considerably declined due to rapid urbanization, intensification of agriculture and deforestation (Kristensen et al. 2015). Therefore, several countries have established cryobanks for storage of animal genetic resources wherein cryopreservation of testicular stem cells has been viewed as an important option when semen cryopreservation was not possible. Testicular cell and tissue cryopreservation have also been used for preservation of near-threatened, vulnerable or endangered species (Bashawat et al. 2020; da Silva et al. 2019). Use of sperm from cryopreserved testes allows introduction of genetic variation and minimizes the inbreeding depression in captive breeding. The testicular cells and tissue for cryopreservation can be obtained from living organisms as well as from accidently found dead cadavers for resurrection of the species (Higaki et al. 2017). It has been successfully applied in preserving the testis of few endangered species such as Cheetah (Acinonyx jubatus), Asiatic golden cat (Catopuma temminckii) (Bashawat et al. 2020), Collared peccary (Pecari tajacu) (da Silva et al. 2019), Cyprinid honmoroko (Gnathopogon caerulescens) (Higaki et al. 2017), Hog deer (Hyelaphus porcinus), Barking deer (Muntiacus muntjak), Sambar deer (Rusa unicolor) (Pothana et al. 2017) and Indian spotted mouse deer (Moschiola indica) (Pothana et al. 2015).
21 Challenges and Future Perspectives
Cryopreservation of testicular stem cells suffers from sub-optimal cryopreservation protocols. Most laboratories have borrowed and used the freezing or vitrification protocols from somatic cells or embryonic stem cells to testicular stem cells. Systematic studies on optimization of freezing and vitrification protocols have been very limited and therefore, the viability remains to be low (typically, 50-60%) in most cases. While various factors such as rate of cooling, volume of sample, hydrostatic pressure, type and concentration of CPA, duration of CPA-exposure, use of ROS scavengers or anti-apoptotic agents, epigenetic modifiers, etc. have been reported to affect the cryoinjuries to influence the success of cryopreservation in various cell types, such studies on testicular stem cells have been very limited. Furthermore, fertility preservation and restoration through cryopreservation of testicular stem cells also require costly and lengthy processes of stem cell isolation, characterization and SSCT or in vitro spermatogenesis to be followed by IVF, ICSI or ROSI (Costa et al. 2017; Hermann et al. 2007; Kim et al. 2015; Pacchiarotti et al. 2013; Wu et al. 2012). Several studies have shown successful cryopreservation of testicular stem cells, which could colonize in the empty seminiferous tubules of the recipient testis for donor-derived spermatogenesis and restoration of male fertility (Kanatsu-Shinohara et al. 2003; Wu et al. 2012). However, such studies are yet to be optimized for large animal species such as cattle, buffaloes, pigs, sheep, goats, horses, dogs, cats, and most of the wild animals.
Another challenge with SSC-based fertility preservation is the low number of viable cells in cryopreserved samples, which might be insufficient for restoration of fertility by SSCT. Gonadal ablation of recipient testis by busulfan treatment or irradiation can improve the colonization of transplanted SSCs into recipient testis by depletion of endogenous SSCs (Shetty et al. 2018). However, it not only compromises the viability and functionality of endogenous Sertoli and Leydig cells in recipient testis but also may be lethal to animals due to possible aplacia in bone marrow. Gonadal ablation of endogenous germ cells is also possible by intratesticular injection of less toxic Dolichos biflorus agglutinin or aplantlectin, which specifically binds to spermatogonia and eliminates endogenous germ cells (Herrid et al. 2019) but remains less explored. Currently, gonadal ablation is practiced only in cases where the goal is to verify the stemness of SSCs or to produce transgenic animals via donor-derived spermatogenesis. In other studies, attempts have also been made to improve the success of SSCT by specifically eliminating the endogenous germ cells using gene editing tools, without affecting the stem cell niche. Knock out of Nanos2 by CRISPR/Cas9 system in boars resulted in loss of germ cells without any adverse effect on interstitial cells (Park et al. 2017). However, such knock out-mediated germ cell depletion requires extensive skills and cost, and are not feasible in large animals and human.
In fertility restoration, a possible solution to low number of viable SSCs could be their in vitro expansion prior to SSCT. However, in vitro cultured SSC may pose a risk of genetic or epigenetic instability. Co-transplantation of SSCs with niche components (e.g. Sertoli cells, Leydig cells) or mesenchymal stem cells (MSCs) could also be an alternative to improve the efficiency of SSCT (Kadam et al. 2018, 2019). It has been shown that co-transplantation of SSCs with TGFβ1-treated MSCs could improve the success of SSCT. In fact, co-transplantation of SSCs and MSCs reduced the requirement of SSCs to half of its number (Kadam et al. 2019).
De novo formation of seminiferous tubules and in vitro spermatogenesis are also emerging as a potential alternative to SSCT from cryopreserved testicular stem cells (Patra et al. 2021; Shetty et al. 2018). In vitro spermatogenesis of sperm from cryopreserved testicular stem cells has been demonstrated in rodents and primates (Sato et al. 2011; Yokonishi et al. 2014). However, its usefulness and long-term safety is yet to be confirmed in most of the other large animals and in human.
22 Conclusions
Cryopreservation of testicular stem cells is rapidly emerging as a potential approach for preservation of fertility in both human and animals. It has also shown promising results in preservation of germplasm in endangered wild animals for posthumous reproduction and has become indispensable for preservation of fertility in pre-pubertal or paediatric males. Recent development of air-liquid interphase culture of testicular stem cells has resulted in in vitro spermatogenesis and may eliminate the need of SSCT and potential risk of re-introducing malignant cells into pre-pubertal individuals expecting gonadotoxic treatments. The combination of cryopreservation protocols with in vitro spermatogenesis of testicular stem cells is thus, expected to provide their bench top application in clinics. Although very few systematic studies have been conducted for optimization of freezing or vitrification of testicular stem cells, newer methods and strategies of cryopreservation are rapidly evolving. Results from rodent studies are very promising and future research and clinical trials are expected to provide better results in livestock animals and human. Further, rapid advancements in genome editing tools such as CRISPR-Cas9 will broaden the application of testicular stem cell cryopreservation to radically change our approach for infertility treatment. However, cryopreservation protocols for testicular cells and tissues are still sub-optimal and their success depends on several variable factors, including species variation and lab-to-lab variations. The protocol for in vitro spermatogenesis has also not been validated in human and large animals. Thus, while promising results have been obtained in experimental animals, birth of live offspring from pre-pubertal testes is not yet reported in human and large animals. Further studies are required for optimization of freezing and vitrification protocols including the optimization of cooling rates, finding new substitutes for non-toxic CPAs, standardization of solutions for freezing, equilibration and vitrification, and to assess the long-term safety of the procedures in offspring derived from cryopreserved testicular stem cells.
References
Abrishami M, Anzar M, Yang Y, Honaramooz A (2010) Cryopreservation of immature porcine testis tissue to maintain its developmental potential after xenografting into recipient mice. Theriogenology 73:86–96
Aliakbari F, Gilani MA, Amidi F, Baazm M, Korouji M, Izadyar F, Yazdekhasti H, Abbasi M (2016) Improving the efficacy of cryopreservation of spermatogonia stem cells by antioxidant supplements. Cell Reprogram 18:87–95
Anserini P, Chiodi S, Spinelli S, Costa M, Conte N, Copello F, Bacigalupo A (2002) Semen analysis following allogeneic bone marrow transplantation. Additional data for evidence-based counselling. Bone Marrow Transplant 30:447–451
Arkoun B, Galas L, Dumont L, Rives A, Saulnier J, Delessard M, Rondanino C, Rives N (2019) Vitamin E but not GSH decreases reactive oxygen species accumulation and enhances sperm production during in vitro maturation of frozen-thawed prepubertal mouse testicular tissue. Int J Mol Sci 20:5380
Avarbock MR, Brinster CJ, Brinster RL (1996) Reconstitution of spermatogenesis from frozen spermatogonial stem cells. Nat Med 2:693–696
Baert Y, Goossens E, van Saen D, Ning L, Tournaye H (2012) Orthotopic grafting of cryopreserved prepubertal testicular tissue: in search of a simple yet effective cryopreservation protocol. Fertil Steril 97:1151–1152
Bashawat M, Braun BC, Müller K (2020) Cell survival after cryopreservation of dissociated testicular cells from feline species. Cryobiology 97:191–197
Batzer FR, Hurwitz JM, Caplan A (2003) Postmortem parenthood and the need for a protocol with posthumous sperm procurement. Fertil Steril 79:1263–1269
Bebbere D, Pinna S, Nieddu S, Natan D, Arav A, Ledda S (2019) Gene expression analysis of ovine prepubertal testicular tissue vitrified with a novel cryodevice (E.Vit). J Assist Reprod Genet 36:2145–2154
Bielanski A, Nadin-Davis S, Sapp T, Lutze-Wallace C (2000) Viral contamination of embryos cryopreserved in liquid nitrogen. Cryobiology 40:110–116
Boroujeni MB, Peidayesh F, Pirnia A, Boroujeni NB, Ahmadi SAY, Gholami M (2019) Effect of selenium on freezing-thawing damage of mice spermatogonial stem cell: a model to preserve fertility in childhood cancers. Stem Cell Investig 6:36
Brandt JS, Cruz Ithier MA, Rosen T, Ashkinadze E (2019) Advanced paternal age, infertility, and reproductive risks: a review of the literature. Prenat Diagn 39:81–87
Braye A, Tournaye H, Goossens E (2019) Setting up a cryopreservation programme for immature testicular tissue: lessons learned after more than 15 years of experience. Clin Med Insights Reprod Health 13:1179558119886342
Bujan L, Sergerie M, Moinard N, Martinet S, Porte L, Massip P, Pasquier C, Daudin M (2007) Decreased semen volume and spermatozoa motility in HIV-1-infected patients under antiretroviral treatment. J Androl 28:444–452
Check ML, Check JH, Summers-Chase D, Choe JK, Check DJ, Nazari A (2002) Live birth after posthumous testicular sperm aspiration and intracytoplasmic sperm injection with cryopreserved sperm: case report. Clin Exp Obstet Gynecol 29:95–96
Cheung RCF, Ng TB, Wong JH (2017) Antifreeze proteins from diverse organisms and their applications: an overview. Curr Protein Pept Sci 18:262–283
Costa GMJ, Avelar GF, Lacerda S, Figueiredo AFA, Tavares AO, Rezende-Neto JV, Martins FGP, França LR (2017) Horse spermatogonial stem cell cryopreservation: feasible protocols and potential biotechnological applications. Cell Tissue Res 370:489–500
Crha I, Ventruba P, Zakova J, Huser M, Kubesova B, Hudecek R, Jarkovsky J (2009) Survival and infertility treatment in male cancer patients after sperm banking. Fertil Steril 91:2344–2348
Curaba M, Verleysen M, Amorim CA, Dolmans MM, Van Langendonckt A, Hovatta O, Wyns C, Donnez J (2011) Cryopreservation of prepubertal mouse testicular tissue by vitrification. Fertil Steril 95:1229–1234
da Silva AM, Bezerra LGP, Praxedes ECG, Moreira SSJ, de Souza CMP, de Oliveira MF, Pereira AF, Comizzoli P, Silva AR (2019) Combination of intracellular cryoprotectants preserves the structure and the cells proliferative capacity potential of adult collared peccary testicular tissue subjected to solid surface vitrification. Cryobiology 91:53–60
de Michele F, Poels J, Weerens L, Petit C, Evrard Z, Ambroise J, Gruson D, Wyns C (2017) Preserved seminiferous tubule integrity with spermatogonial survival and induction of Sertoli and Leydig cell maturation after long-term organotypic culture of prepubertal human testicular tissue. Hum Reprod 32:32–45
Del Vento F, Vermeulen M, Ucakar B, Poels J, Rieux A, Wyns C (2019) Significant benefits of nanoparticles containing a necrosis inhibitor on mice testicular tissue autografts outcomes. Int J Mol Sci 20:5833
Docampo MJ, Hadziselimovic F (2015) Molecular pathology of cryptorchidism-induced infertility. Sex Dev 9:269–278
Dohle GR (2010) Male infertility in cancer patients: review of the literature. Int J Urol 17:327–331
Dong WZ, Hua JL, Shen WZ, Dou ZY (2010) In vitro production of haploid sperm cells from male germ cells of foetal cattle. Anim Reprod Sci 118:103–109
Dong L, Gul M, Hildorf S, Pors SE, Kristensen SG, Hoffmann ER, Cortes D, Thorup J, Andersen CY (2019) Xeno-free propagation of spermatogonial stem cells from infant boys. Int J Mol Sci 20:5390
Duca Y, Di Cataldo A, Russo G, Cannata E, Burgio G, Compagnone M, Alamo A, Condorelli RA, La Vignera S, Calogero AE (2019) Testicular function of childhood cancer survivors: who is worse? J Clin Med 8:2204
Dumont L, Arkoun B, Jumeau F, Milazzo JP, Bironneau A, Liot D, Wils J, Rondanino C, Rives N (2015) Assessment of the optimal vitrification protocol for pre-pubertal mice testes leading to successful in vitro production of flagellated spermatozoa. Andrology 3:611–625
Fahy GM, Wowk B, Wu J, Paynter S (2004) Improved vitrification solutions based on the predictability of vitrification solution toxicity. Cryobiology 48:22–35
Feng LX, Ravindranath N, Dym M (2000) Stem cell factor/c-kit up-regulates cyclin D3 and promotes cell cycle progression via the phosphoinositide 3-kinase/p70 S6 kinase pathway in spermatogonia. J Biol Chem 275:25572–25576
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247
Fode M, Krogh-Jespersen S, Brackett NL, Ohl DA, Lynne CM, Sønksen J (2012) Male sexual dysfunction and infertility associated with neurological disorders. Asian J Androl 14:61–68
Fountain D, Ralston M, Higgins N, Gorlin JB, Uhl L, Wheeler C, Antin JH, Churchill WH, Benjamin RJ (1997) Liquid nitrogen freezers: a potential source of microbial contamination of hematopoietic stem cell components. Transfusion 37:585–591
Frederickx V, Michiels A, Goossens E, De Block G, Van Steirteghem AC, Tournaye H (2004) Recovery, survival and functional evaluation by transplantation of frozen-thawed mouse germ cells. Hum Reprod 19:948–953
Fuchs EF, Burt RA (2002) Vasectomy reversal performed 15 years or more after vasectomy: correlation of pregnancy outcome with partner age and with pregnancy results of in vitro fertilization with intracytoplasmic sperm injection. Fertil Steril 77:516–519
Fujita K, Tsujimura A, Miyagawa Y, Kiuchi H, Matsuoka Y, Takao T, Takada S, Nonomura N, Okuyama A (2006) Isolation of germ cells from leukemia and lymphoma cells in a human in vitro model: potential clinical application for restoring human fertility after anticancer therapy. Cancer Res 66:11166–11171
Gao H, Liu C, Wu B, Cui H, Zhao Y, Duan Y, Gao F, Gu Q, Wang H, Li W (2020) Effects of different biomaterials and cellular status on testicular cell self-organization. Adv Biosyst 2020:1900292
Geens M, Van de Velde H, De Block G, Goossens E, Van Steirteghem A, Tournaye H (2007) The efficiency of magnetic-activated cell sorting and fluorescence-activated cell sorting in the decontamination of testicular cell suspensions in cancer patients. Hum Reprod 22:733–742
Gholami M, Saki G, Hemadi M, Khodadadi A, Mohamma-di-Asl J (2013) Effect of melatonin on the expression of apoptotic genes in vitrified-thawed spermatogonia stem cells type A of 6-day-old mice. Iran J Basic Med Sci 16:906–909
Ginsberg JP, Carlson CA, Lin K, Hobbie WL, Wigo E, Wu X, Brinster RL, Kolon TF (2010) An experimental protocol for fertility preservation in prepubertal boys recently diagnosed with cancer: a report of acceptability and safety. Hum Reprod 25:37–41
Goossens E, Frederickx V, Geens M, De Block G, Tournaye H (2008) Cryosurvival and spermatogenesis after allografting prepubertal mouse tissue: comparison of two cryopreservation protocols. Fertil Steril 89:725–727
Goossens E, Bilgec T, Van Saen D, Tournaye H (2011) Mouse germ cells go through typical epigenetic modifications after intratesticular tissue grafting. Hum Reprod 26:3388–3400
Gouk SS, Loh YF, Kumar SD, Watson PF, Kuleshova LL (2011) Cryopreservation of mouse testicular tissue: prospect for harvesting spermatogonial stem cells for fertility preservation. Fertil Steril 95:2399–2403
Green DM, Kawashima T, Stovall M, Leisenring W, Sklar CA, Mertens AC, Donaldson SS, Byrne J, Robison LL (2010) Fertility of male survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Clin Oncol 28:332–339
Gül M, Dong L, Wang D, Diri MA, Andersen CY (2020) Surrogate testes: allogeneic spermatogonial stem cell transplantation within an encapsulation device may restore male fertility. Med Hypotheses 139:109634
Gupta MK, Lee HT (2010) Cryopreservation of oocytes and embryos by vitrification. Clin Exp Reprod Med 37:267–291
Gupta MK, Uhm SJ, Lee HT (2007) Cryopreservation of immature and in vitro matured porcine oocytes by solid surface vitrification. Theriogenology 67:238–248
Gupta MK, Uhm SJ, Lee HT (2010) Effect of vitrification and beta-mercaptoethanol on reactive oxygen species activity and in vitro development of oocytes vitrified before or after in vitro fertilization. Fertil Steril 93:2602–2607
Gurtovenko AA, Anwar J (2007) Modulating the structure and properties of cell membranes: the molecular mechanism of action of dimethyl sulfoxide. J Phys Chem B 111:10453–10460
Ha SJ, Kim BG, Lee YA, Kim YH, Kim BJ, Jung SE, Pang MG, Ryu BY (2016) Effect of antioxidants and apoptosis inhibitors on cryopreservation of murine germ cells enriched for spermatogonial stem cells. PLoS One 11:e0161372
Hermann BP, Sukhwani M, Lin CC, Sheng Y, Tomko J, Rodriguez M, Shuttleworth JJ, McFarland D, Hobbs RM, Pandolfi PP, Schatten GP, Orwig KE (2007) Characterization, cryopreservation, and ablation of spermatogonial stem cells in adult rhesus macaques. Stem Cells 25:2330–2338
Hermann BP, Sukhwani M, Salati J, Sheng Y, Chu T, Orwig KE (2011) Separating spermatogonia from cancer cells in contaminated prepubertal primate testis cell suspensions. Hum Reprod 26:3222–3231
Herrid M, Nagy P, Juhasz J, Morrell JM, Billah M, Khazanehdari K, Skidmore JA (2019) Donor sperm production in heterologous recipients by testis germ cell transplantation in the dromedary camel. Reprod Fertil Dev 31:538–546
Higaki S, Kuwata N, Tanaka K, Tooyama I, Fujioka Y, Sakai N, Takada T (2017) Successful vitrification of whole juvenile testis in the critically endangered cyprinid honmoroko (Gnathopogon caerulescens). Zygote 25:652–661
Hofmann MC, Braydich-Stolle L, Dym M (2005) Isolation of male germ-line stem cells; influence of GDNF. Dev Biol 279:114–124
Hou M, Andersson M, Zheng C, Sundblad A, Söder O, Jahnukainen K (2007) Decontamination of leukemic cells and enrichment of germ cells from testicular samples from rats with Roser’s T-cell leukemia by flow cytometric sorting. Reproduction 134:767–779
Hou M, Andersson M, Zheng C, Sundblad A, Söder O, Jahnukainen K (2009) Immunomagnetic separation of normal rat testicular cells from Roser’s T-cell leukaemia cells is ineffective. Int J Androl 32:66–73
Huang JY, Chen HY, Park JY, Tan SL, Chian RC (2008) Comparison of spindle and chromosome configuration in in vitro- and in vivo-matured mouse oocytes after vitrification. Fertil Steril 90:1424–1432
Hurwitz JM, Batzer FR (2004) Posthumous sperm procurement: demand and concerns. Obstet Gynecol Surv 59:806–808
Ibtisham F, Honaramooz A (2020) Spermatogonial stem cells for in vitro spermatogenesis and in vivo restoration of fertility. Cell 9:745
Iwatani M, Ikegami K, Kremenska Y, Hattori N, Tanaka S, Yagi S, Shiota K (2006) Dimethyl sulfoxide has an impact on epigenetic profile in mouse embryoid body. Stem Cells 24:2549–2556
Izadyar F, Matthijs-Rijsenbilt JJ, den Ouden K, Creemers LB, Woelders H, de Rooij DG (2002) Development of a cryopreservation protocol for type A spermatogonia. J Androl 23:537–545
Jahnukainen K, Hou M, Petersen C, Setchell B, Söder O (2001) Intratesticular transplantation of testicular cells from leukemic rats causes transmission of leukemia. Cancer Res 61:706–710
Jung SE, Ahn JS, Kim YH, Kim BJ, Won JH, Ryu BY (2020a) Effective cryopreservation protocol for preservation of male primate (Macaca fascicularis) prepubertal fertility. Reprod Biomed Online 41(6):1070–1083
Jung SE, Kim M, Ahn JS, Kim YH, Kim BJ, Yun MH, Auh JH, Ryu BY (2020b) Effect of equilibration time and temperature on murine spermatogonial stem cell cryopreservation. Biopreserv Biobank 18:213–221
Kadam P, Ntemou E, Baert Y, Van Laere S, Van Saen D, Goossens E (2018) Co-transplantation of mesenchymal stem cells improves spermatogonial stem cell transplantation efficiency in mice. Stem Cell Res Ther 9:317
Kadam P, Ntemou E, Onofre J, Van Saen D, Goossens E (2019) Does co-transplantation of mesenchymal and spermatogonial stem cells improve reproductive efficiency and safety in mice? Stem Cell Res Ther 10:310
Kanatsu-Shinohara M, Ogonuki N, Inoue K, Ogura A, Toyokuni S, Shinohara T (2003) Restoration of fertility in infertile mice by transplantation of cryopreserved male germline stem cells. Hum Reprod 18:2660–2667
Kaneko H, Kikuchi K, Nakai M, Somfai T, Noguchi J, Tanihara F, Ito J, Kashiwazaki N (2013) Generation of live piglets for the first time using sperm retrieved from immature testicular tissue cryopreserved and grafted into nude mice. PLoS One 8:e70989
Kaul G, Kaur J, Rafeeqi TA (2010) Ultrasound guided transplantation of enriched and cryopreserved spermatogonial cell suspension in goats. Reprod Domest Anim 45:e249–e254
Kawai K, Li YS, Song MF, Kasai H (2010) DNA methylation by dimethyl sulfoxide and methionine sulfoxide triggered by hydroxyl radical and implications for epigenetic modifications. Bioorg Med Chem Lett 20:260–265
Kee K, Angeles VT, Flores M, Nguyen HN, Reijo Pera RA (2009) Human DAZL, DAZ and BOULE genes modulate primordial germ-cell and haploid gamete formation. Nature 462:222–225
Keros V, Rosenlund B, Hultenby K, Aghajanova L, Levkov L, Hovatta O (2005) Optimizing cryopreservation of human testicular tissue: comparison of protocols with glycerol, propanediol and dimethylsulphoxide as cryoprotectants. Hum Reprod 20:1676–1687
Keros V, Hultenby K, Borgström B, Fridström M, Jahnukainen K, Hovatta O (2007) Methods of cryopreservation of testicular tissue with viable spermatogonia in pre-pubertal boys undergoing gonadotoxic cancer treatment. Hum Reprod 22:1384–1395
Kim YM, Uhm SJ, Gupta MK, Yang JS, Lim JG, Das ZC, Heo YT, Chung HJ, Kong IK, Kim NH, Lee HT, Ko DH (2012) Successful vitrification of bovine blastocysts on paper container. Theriogenology 78:1085–1093
Kim KJ, Lee YA, Kim BJ, Kim YH, Kim BG, Kang HG, Jung SE, Choi SH, Schmidt JA, Ryu BY (2015) Cryopreservation of putative pre-pubertal bovine spermatogonial stem cells by slow freezing. Cryobiology 70:175–183
Kim HJ, Lee JH, Hur YB, Lee CW, Park SH, Koo BW (2017) Marine antifreeze proteins: structure, function, and application to cryopreservation as a potential cryoprotectant. Mar Drugs 15:27
Kirpatovskii VI, Efremov GD, Frolova EV (2018) Ectopic organogenesis after allotransplantation of freshly removed or cryopreserved neonatal testicle under the renal capsule in rats. Bull Exp Biol Med 166:268–273
Koppes EA, Redel BK, Johnson MA, Skvorak KJ, Ghaloul-Gonzalez L, Yates ME, Lewis DW, Gollin SM, Wu YL, Christ SE, Yerle M, Leshinski A, Spate LD, Benne JA, Murphy SL, Samuel MS, Walters EM, Hansen SA, Wells KD, Lichter-Konecki U, Wagner RA, Newsome JT, Dobrowolski SF, Vockley J, Prather RS, Nicholls RD (2020) A porcine model of phenylketonuria generated by CRISPR/Cas9 genome editing. JCI Insight 5:141523
Kristensen TN, Hoffmann AA, Pertoldi C, Stronen AV (2015) What can livestock breeders learn from conservation genetics and vice versa? Front Genet 6:38
Kumari N, Gupta MK, Singh RK (2016) Open encapsulation-vitrification for cryopreservation of algae. Cryobiology 73:232–239
Lakhoo K, Davies J, Chakraborty S, Berg S, Tennyson R, Fowler D, Manek S, Verrill C, Lane S (2019) Development of a new reproductive tissue cryopreservation clinical service for children: the Oxford programme. Pediatr Surg Int 35:1271–1278
Lee PA, Coughlin MT (2001) Fertility after bilateral cryptorchidism. Evaluation by paternity, hormone, and semen data. Horm Res 55:28–32
Lee YA, Kim YH, Kim BJ, Jung MS, Auh JH, Seo JT, Park YS, Lee SH, Ryu BY (2013a) Cryopreservation of mouse spermatogonial stem cells in dimethylsulfoxide and polyethylene glycol. Biol Reprod 89:109
Lee YA, Kim YH, Kim BJ, Kim BG, Kim KJ, Auh JH, Schmidt JA, Ryu BY (2013b) Cryopreservation in trehalose preserves functional capacity of murine spermatogonial stem cells. PLoS One 8:e54889
Lee YA, Kim YH, Ha SJ, Kim BJ, Kim KJ, Jung MS, Kim BG, Ryu BY (2014a) Effect of sugar molecules on the cryopreservation of mouse spermatogonial stem cells. Fertil Steril 101:1165–1175
Lee YA, Kim YH, Ha SJ, Kim KJ, Kim BJ, Kim BG, Choi SH, Kim IC, Schmidt JA, Ryu BY (2014b) Cryopreservation of porcine spermatogonial stem cells by slow-freezing testis tissue in trehalose. J Anim Sci 92:984–995
Levine H, Jørgensen N, Martino-Andrade A, Mendiola J, Weksler-Derri D, Mindlis I, Pinotti R, Swan SH (2017) Temporal trends in sperm count: a systematic review and meta-regression analysis. Hum Reprod Update 23:646–659
Li J, Leng M, Ma T, Yu D, Shi H, Shi Q (2009) Cryopreservation has no effect on meiotic recombination and synapsis in testicular tissues. Fertil Steril 91:1404–1407
Li H, Bian YL, Schreurs N, Zhang XG, Raza SHA, Fang Q, Wang LQ, Hu JH (2018) Effects of five cryoprotectants on proliferation and differentiation-related gene expression of frozen-thawed bovine calf testicular tissue. Reprod Domest Anim 53:1211–1218
Liu Z, Nie YH, Zhang CC, Cai YJ, Wang Y, Lu HP, Li YZ, Cheng C, Qiu ZL, Sun Q (2016) Generation of macaques with sperm derived from juvenile monkey testicular xenografts. Cell Res 26:139–142
Lukusa AK, Vermylen C, Vanabelle B, Curaba M, Brichard B, Chantrain C, Dupont S, Ferrant A, Wyns C (2009) Bone marrow transplantation or hydroxyurea for sickle cell anemia: long-term effects on semen variables and hormone profiles. Pediatr Hematol Oncol 26:186–194
Makala H, Pothana L, Sonam S, Malla A, Goel S (2015) Regeneration of Leydig cells in ectopically autografted adult mouse testes. Reproduction 149:259–268
Mazur P (1984) Freezing of living cells: mechanisms and implications. Am J Phys 247:C125–C142
Men H, Agca Y, Riley LK, Critser JK (2006) Improved survival of vitrified porcine embryos after partial delipation through chemically stimulated lipolysis and inhibition of apoptosis. Theriogenology 66:2008–2016
Meneghel J, Kilbride P, Morris JG, Fonseca F (2019) Physical events occurring during the cryopreservation of immortalized human T cells. PLoS One 14:e0217304
Mohaqiq M, Movahedin M, Mazaheri Z, Amirjannati N (2019) In vitro transplantation of spermatogonial stem cells isolated from human frozen-thawed testis tissue can induce spermatogenesis under 3-dimensional tissue culture conditions. Biol Res 52:16
Morimoto H, Iwata K, Ogonuki N, Inoue K, Atsuo O, Kanatsu-Shinohara M, Morimoto T, Yabe-Nishimura C, Shinohara T (2013) ROS are required for mouse spermatogonial stem cell self-renewal. Cell Stem Cell 12:774–786
Nakagawa T, Nabeshima Y, Yoshida S (2007) Functional identification of the actual and potential stem cell compartments in mouse spermatogenesis. Dev Cell 12:195–206
Navid S, Rastegar T, Baazm M, Alizadeh R, Talebi A, Gholami K, Khosravi-Farsani S, Koruji M, Abbasi M (2017) In vitro effects of melatonin on colonization of neonate mouse spermatogonial stem cells. Syst Biol Reprod Med 63:370–381
Nicopoullos JD, Almeida P, Vourliotis M, Gilling-Smith C (2011) A decade of the sperm-washing programme: correlation between markers of HIV and seminal parameters. HIV Med 12:195–201
Noguchi J, Ohnuma K, Ozawa M, Karja NW, Fahrudin M, Somfai T, Kikuchi K, Kaneko H (2006) Successful long term culture of immature porcine sertoli cells in the reconstructed testicular cell cord. J Reprod Dev 52:383–389
Ntemou E, Kadam P, Van Saen D, Wistuba J, Mitchell RT, Schlatt S, Goossens E (2019) Complete spermatogenesis in intratesticular testis tissue xenotransplants from immature non-human primate. Hum Reprod 34:403–413
Oblette A, Rives N, Dumont L, Rives A, Verhaeghe F, Jumeau F, Rondanino C (2017) Assessment of sperm nuclear quality after in vitro maturation of fresh or frozen/thawed mouse pre-pubertal testes. Mol Hum Reprod 23:674–684
Ogawa T, Dobrinski I, Avarbock MR, Brinster RL (2000) Transplantation of male germ line stem cells restores fertility in infertile mice. Nat Med 6:29–34
Onions VJ, Mitchell MR, Campbell BK, Webb R (2008) Ovarian tissue viability following whole ovine ovary cryopreservation: assessing the effects of sphingosine-1-phosphate inclusion. Hum Reprod 23:606–618
Onofre J, Baert Y, Faes K, Goossens E (2016) Cryopreservation of testicular tissue or testicular cell suspensions: a pivotal step in fertility preservation. Hum Reprod Update 22:744–761
Onofre J, Kadam P, Baert Y, Goossens E (2020) Testicular tissue cryopreservation is the preferred method to preserve spermatogonial stem cells prior to transplantation. Reprod Biomed Online 40:261–269
Pacchiarotti J, Ramos T, Howerton K, Greilach S, Zaragoza K, Olmstead M, Izadyar F (2013) Developing a clinical-grade cryopreservation protocol for human testicular tissue and cells. Biomed Res Int 2013:930962
Park KE, Kaucher AV, Powell A, Waqas MS, Sandmaier SE, Oatley MJ, Park CH, Tibary A, Donovan DM, Blomberg LA, Lillico SG, Whitelaw CB, Mileham A, Telugu BP, Oatley JM (2017) Generation of germline ablated male pigs by CRISPR/Cas9 editing of the NANOS2 gene. Sci Rep 7:40176
Patra T, Gupta MK (2019) Cryopreservation of murine testicular Leydig cells by modified solid surface vitrification with supplementation of antioxidants. Cryobiology 88:38–46
Patra T, Gupta MK (2020) Evaluation of sodium alginate for encapsulation-vitrification of testicular Leydig cells. Int J Biol Macromol 153:128–137
Patra T, Pathak D, Gupta MK (2021) Strategies for cryopreservation of testicular cells and tissues in cancer and genetic diseases. Cell Tissue Res 385:1–19. https://doi.org/10.1007/s00441-021-03437-4
Pelzman DL, Orwig KE, Hwang K (2020) Progress in translational reproductive science: testicular tissue transplantation and in vitro spermatogenesis. Fertil Steril 113:500–509
Pendergraft SS, Sadri-Ardekani H, Atala A, Bishop CE (2017) Three-dimensional testicular organoid: a novel tool for the study of human spermatogenesis and gonadotoxicity in vitro. Biol Reprod 96:720–732
Perrard MH, Sereni N, Schluth-Bolard C, Blondet A, Plotton I, Morel-Journel N, Lejeune H, David L, Durand P (2016) Complete human and rat ex vivo spermatogenesis from fresh or frozen testicular tissue. Biol Reprod 95:89
Pietzak EJ 3rd, Tasian GE, Tasian SK, Brinster RL, Carlson C, Ginsberg JP, Kolon TF (2015) Histology of testicular biopsies obtained for experimental fertility preservation protocol in boys with cancer. J Urol 194:1420–1424
Pirnia A, Parivar K, Hemadi M, Yaghmaei P, Gholami M (2017) Stemness of spermatogonial stem cells encapsulated in alginate hydrogel during cryopreservation. Andrologia 49:12650
Poels J, Van Langendonckt A, Dehoux JP, Donnez J, Wyns C (2012) Vitrification of non-human primate immature testicular tissue allows maintenance of proliferating spermatogonial cells after xenografting to recipient mice. Theriogenology 77:1008–1013
Poels J, Van Langendonckt A, Many MC, Wese FX, Wyns C (2013) Vitrification preserves proliferation capacity in human spermatogonia. Hum Reprod 28:578–589
Poels J, Abou-Ghannam G, Herman S, Van Langendonckt A, Wese FX, Wyns C (2014) In search of better spermatogonial preservation by supplementation of cryopreserved human immature testicular tissue xenografts with N-acetylcysteine and testosterone. Front Surg 1:47
Poels J, Abou-Ghannam G, Decamps A, Leyman M, Rieux A, Wyns C (2016) Transplantation of testicular tissue in alginate hydrogel loaded with VEGF nanoparticles improves spermatogonial recovery. J Control Release 234:79–89
Portela JMD, de Winter-Korver CM, van Daalen SKM, Meißner A, de Melker AA, Repping S, van Pelt AMM (2019) Assessment of fresh and cryopreserved testicular tissues from (pre)pubertal boys during organ culture as a strategy for in vitro spermatogenesis. Hum Reprod 34:2443–2455
Pothana L, Makala H, Devi L, Varma VP, Goel S (2015) Germ cell differentiation in cryopreserved, immature, Indian spotted mouse deer (Moschiola indica) testes xenografted onto mice. Theriogenology 83:625–633
Pothana L, Devi L, Goel S (2017) Cryopreservation of adult cervid testes. Cryobiology 74:103–109
Pukazhenthi BS, Neubauer K, Jewgenow K, Howard J, Wildt DE (2006) The impact and potential etiology of teratospermia in the domestic cat and its wild relatives. Theriogenology 66:112–121
Pukazhenthi BS, Nagashima J, Travis AJ, Costa GM, Escobar EN, França LR, Wildt DE (2015) Slow freezing, but not vitrification supports complete spermatogenesis in cryopreserved, neonatal sheep testicular xenografts. PLoS One 10:e0123957
Redden E, Davey R, Borjigin U, Hutton K, Hinch G, Hope S, Hill J, Herrid M (2009) Large quantity cryopreservation of bovine testicular cells and its effect on enrichment of type A spermatogonia. Cryobiology 58:190–195
Rezaei Topraggaleh T, Rezazadeh Valojerdi M, Montazeri L, Baharvand H (2019) A testis-derived macroporous 3D scaffold as a platform for the generation of mouse testicular organoids. Biomater Sci 7:1422–1436
Sá R, Cremades N, Malheiro I, Sousa M (2012) Cryopreservation of human testicular diploid germ cell suspensions. Andrologia 44:366–372
Sadri-Ardekani H, Atala A (2014) Testicular tissue cryopreservation and spermatogonial stem cell transplantation to restore fertility: from bench to bedside. Stem Cell Res Ther 5:68
Sakib S, Uchida A, Valenzuela-Leon P, Yu Y, Valli-Pulaski H, Orwig K, Ungrin M, Dobrinski I (2019) Formation of organotypic testicular organoids in microwell culture. Biol Reprod 100:1648–1660
Saragusty J, Walzer C, Petit T, Stalder G, Horowitz I, Hermes R (2010) Cooling and freezing of epididymal sperm in the common hippopotamus (Hippopotamus amphibius). Theriogenology 74:1256–1263
Sato T, Katagiri K, Gohbara A, Inoue K, Ogonuki N, Ogura A, Kubota Y, Ogawa T (2011) In vitro production of functional sperm in cultured neonatal mouse testes. Nature 471:504–507
Sayed Mahdi N, Azarbani F, Pirnia A, Abbaszadeh A, Gholami M (2018) The effect of caffeic acid on spermatogonial stem cell-type a cryopreservation. Rep Biochem Mol Biol 7:85–93
Schrans-Stassen BH, Saunders PT, Cooke HJ, de Rooij DG (2001) Nature of the spermatogenic arrest in Dazl −/− mice. Biol Reprod 65:771–776
Shetty G, Mitchell JM, Lam TNA, Wu Z, Zhang J, Hill L, Tailor RC, Peters KA, Penedo MC, Orwig KE, Meistrich ML (2018) Donor spermatogenesis in de novo formed seminiferous tubules from transplanted testicular cells in rhesus monkey testis. Hum Reprod 33:2249–2255
Shetty G, Mitchell JM, Meyer JM, Wu Z, Lam TNA, Phan TT, Zhang J, Hill L, Tailor RC, Peters KA, Penedo MC, Hanna C, Orwig KE, Meistrich ML (2020) Restoration of functional sperm production in irradiated pubertal rhesus monkeys by spermatogonial stem cell transplantation. Andrology 8:1428–1441
Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD, Eppig JJ, Bronson RT, Elledge SJ, Weinberg RA (1996) Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature 384:470–474
Snowden JA, Badoglio M, Labopin M, Giebel S, McGrath E, Marjanovic Z, Burman J, Moore J, Rovira M, Wulffraat NM, Kazmi M, Greco R, Snarski E, Kozak T, Kirgizov K, Alexander T, Bader P, Saccardi R, Farge D (2017) Evolution, trends, outcomes, and economics of hematopoietic stem cell transplantation in severe autoimmune diseases. Blood Adv 1:2742–2755
Song W, Zhao W, Yang Q, Wang X, Jin H, Yao G, Peng Z, Shi S, Yang H, Sun Y (2016) Effect of rapid cryopreservation on meiotic recombination in human spermatocytes. Microsc Res Tech 79:923–928
Su XJ, Yuan W, Huang GY, Olsen J, Li J (2015) Paternal age and offspring congenital heart defects: a national cohort study. PLoS One 10:e0121030
Tanaka A, Nagayoshi M, Takemoto Y, Tanaka I, Kusunoki H, Watanabe S, Kuroda K, Takeda S, Ito M, Yanagimachi R (2015) Fourteen babies born after round spermatid injection into human oocytes. Proc Natl Acad Sci U S A 112:14629–14634
Tanaka A, Suzuki K, Nagayoshi M, Takemoto Y, Watanabe S, Takeda S, Irahara M, Kuji N, Yamagata Z, Yanagimachi R (2018) Ninety babies born after round spermatid injection into oocytes: survey of their development from fertilization to 2 years of age. Fertil Steril 110:443–451
Taylor JL, Debost JPG, Morton SU, Wigdor EM, Heyne HO, Lal D, Howrigan DP, Bloemendal A, Larsen JT, Kosmicki JA, Weiner DJ, Homsy J, Seidman JG, Seidman CE, Agerbo E, McGrath JJ, Mortensen PB, Petersen L, Daly MJ, Robinson EB (2019) Paternal-age-related de novo mutations and risk for five disorders. Nat Commun 10:3043
Tedder RS, Zuckerman MA, Goldstone AH, Hawkins AE, Fielding A, Briggs EM, Irwin D, Blair S, Gorman AM, Patterson KG et al (1995) Hepatitis B transmission from contaminated cryopreservation tank. Lancet 346:137–140
Thomson LK, Fleming SD, Aitken RJ, De Iuliis GN, Zieschang JA, Clark AM (2009) Cryopreservation-induced human sperm DNA damage is predominantly mediated by oxidative stress rather than apoptosis. Hum Reprod 24:2061–2070
Tian J, Ma K, Pei CB, Zhang SH, Li X, Zhou Y, Yan B, Wang HY, Ma LH (2019) Relative safety of various spermatogenic stem cell purification methods for application in spermatogenic stem cell transplantation. Stem Cell Res Ther 10:382
Travers A, Milazzo JP, Perdrix A, Metton C, Bironneau A, Macé B, Rives N (2011) Assessment of freezing procedures for rat immature testicular tissue. Theriogenology 76:981–990
Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, Kiyokawa H (1999) Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol 19:7011–7019
Unni S, Kasiviswanathan S, D'Souza S, Khavale S, Mukherjee S, Patwardhan S, Bhartiya D (2012) Efficient cryopreservation of testicular tissue: effect of age, sample state, and concentration of cryoprotectant. Fertil Steril 97:200–208
Valli-Pulaski H, Peters KA, Gassei K, Steimer SR, Sukhwani M, Hermann BP, Dwomor L, David S, Fayomi AP, Munyoki SK, Chu T, Chaudhry R, Cannon GM, Fox PJ, Jaffe TM, Sanfilippo JS, Menke MN, Lunenfeld E, Abofoul-Azab M, Sender LS, Messina J, Klimpel LM, Gosiengfiao Y, Rowell EE, Hsieh MH, Granberg CF, Reddy PP, Sandlow JI, Huleihel M, Orwig KE (2019) Testicular tissue cryopreservation: 8 years of experience from a coordinated network of academic centers. Hum Reprod 34:966–977
Vermeulen M, Poels J, de Michele F, Rieux A, Wyns C (2017) Restoring fertility with cryopreserved prepubertal testicular tissue: perspectives with hydrogel encapsulation, nanotechnology, and bioengineered scaffolds. Ann Biomed Eng 45:1770–1781
Vermeulen M, Del Vento F, de Michele F, Poels J, Wyns C (2018) Development of a cytocompatible scaffold from pig immature testicular tissue allowing human sertoli cell attachment, proliferation and functionality. Int J Mol Sci 19:227
Vermeulen M, Del Vento F, Kanbar M, Pyr Dit Ruys S, Vertommen D, Poels J, Wyns C (2019) Generation of organized porcine testicular organoids in solubilized hydrogels from decellularized extracellular matrix. Int J Mol Sci 20:5476
Vialard F, Bailly M, Bouazzi H, Albert M, Pont JC, Mendes V, Bergere M, Gomes DM, de Mazancourt P, Selva J (2012) The high frequency of sperm aneuploidy in klinefelter patients and in nonobstructive azoospermia is due to meiotic errors in euploid spermatocytes. J Androl 33:1352–1359
Wang D, Li L, Xie Q, Hou Z, Yu X, Ma M, Huang T (2014) Factors affecting sperm fertilizing capacity in men infected with HIV. J Med Virol 86:1467–1472
Wang M, Karlsson JOM, Aksan A (2019) FTIR analysis of molecular changes associated with warming injury in cryopreserved leukocytes. Langmuir 35:7552–7559
Weng L, Chen C, Zuo J, Li W (2011) Molecular dynamics study of effects of temperature and concentration on hydrogen-bond abilities of ethylene glycol and glycerol: implications for cryopreservation. J Phys Chem A 115:4729–4737
Wikström AM, Dunkel L (2008) Testicular function in Klinefelter syndrome. Horm Res 69:317–326
Wu X, Goodyear SM, Abramowitz LK, Bartolomei MS, Tobias JW, Avarbock MR, Brinster RL (2012) Fertile offspring derived from mouse spermatogonial stem cells cryopreserved for more than 14 years. Hum Reprod 27:1249–1259
Wu Y, Zhou H, Fan X, Zhang Y, Zhang M, Wang Y, Xie Z, Bai M, Yin Q, Liang D, Tang W, Liao J, Zhou C, Liu W, Zhu P, Guo H, Pan H, Wu C, Shi H, Wu L, Tang F, Li J (2015) Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Res 25:67–79
Wyns C, Curaba M, Martinez-Madrid B, Van Langendonckt A, François-Xavier W, Donnez J (2007) Spermatogonial survival after cryopreservation and short-term orthotopic immature human cryptorchid testicular tissue grafting to immunodeficient mice. Hum Reprod 22:1603–1611
Wyns C, Van Langendonckt A, Wese FX, Donnez J, Curaba M (2008) Long-term spermatogonial survival in cryopreserved and xenografted immature human testicular tissue. Hum Reprod 23:2402–2414
Xie Y, Chen H, Luo D, Yang X, Yao J, Zhang C, Lv L, Guo Z, Deng C, Li Y, Liang X, Sun X, Liu G (2020) Inhibiting necroptosis of spermatogonial stem cell as a novel strategy for male fertility preservation. Stem Cells Dev 29:475–487
Yango P, Altman E, Smith JF, Klatsky PC, Tran ND (2014) Optimizing cryopreservation of human spermatogonial stem cells: comparing the effectiveness of testicular tissue and single cell suspension cryopreservation. Fertil Steril 102:1491–1498
Yokonishi T, Sato T, Komeya M, Katagiri K, Kubota Y, Nakabayashi K, Hata K, Inoue K, Ogonuki N, Ogura A, Ogawa T (2014) Offspring production with sperm grown in vitro from cryopreserved testis tissues. Nat Commun 5:4320
Yu Z, Ji P, Cao J, Zhu S, Li Y, Zheng L, Chen X, Feng L (2009) Dazl promotes germ cell differentiation from embryonic stem cells. J Mol Cell Biol 1:93–103
Yuan Z, Hou R, Wu J (2009) Generation of mice by transplantation of an adult spermatogonial cell line after cryopreservation. Cell Prolif 42:123–131
Zhang XG, Wang YH, Han C, Hu S, Wang LQ, Hu JH (2015) Effects of trehalose supplementation on cell viability and oxidative stress variables in frozen-thawed bovine calf testicular tissue. Cryobiology 70:246–252
Zhang B, Wang C, Zhang Y, Jiang Y, Qin Y, Pang D, Zhang G, Liu H, Xie Z, Yuan H, Ouyang H, Wang J, Tang X (2020) A CRISPR-engineered swine model of COL2A1 deficiency recapitulates altered early skeletal developmental defects in humans. Bone 137:115450
Acknowledgments
This work was supported by grant from Indian Council of Medical Research (ICMR), Government of India.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Patra, T., Bhaskar, R., Gupta, M.K. (2021). Cryopreservation of Testicular Stem Cells and Its Application in Veterinary Science. In: Choudhary, R.K., Choudhary, S. (eds) Stem Cells in Veterinary Science. Springer, Singapore. https://doi.org/10.1007/978-981-16-3464-2_9
Download citation
DOI: https://doi.org/10.1007/978-981-16-3464-2_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-3463-5
Online ISBN: 978-981-16-3464-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)