
Abstract
Emerging and re-emerging viral diseases occur with regularity within the human population. The conventional ‘one drug, one virus’ paradigm for antivirals does not adequately allow for proper preparedness in the face of unknown future epidemics. In addition, drug developers lack the financial incentives to work on antiviral drug discovery, with most pharmaceutical companies choosing to focus on more profitable disease areas. Safe-in-man broad spectrum antiviral agents (BSAAs) can help meet the need for antiviral development by already having passed phase I clinical trials, requiring less time and money to develop, and having the capacity to work against many viruses, allowing for a speedy response when unforeseen epidemics arise. In this chapter, we discuss the benefits of repurposing existing drugs as BSAAs, describe the major steps in safe-in-man BSAA drug development from discovery through clinical trials, and list several database resources that are useful tools for antiviral drug repositioning.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
12.1 Introduction
Viruses are found ubiquitously in nature, infecting species from all five kingdoms of life. Despite being accepted and studied as a vector of human disease relatively recently, viruses make up for 75% of newly discovered pathogens [1]. This is primarily due to their intrinsically high rate of mutation, allowing them to expand genetically across many environments and hosts. Because of this ability to expand their reach, viruses with established, equilibrated relationships with hosts of one species can frequently gain the ability to infect another, previously unsusceptible species, causing emerging viral diseases [2]. To humans, this constant beat of viral emergence poses a threat to the global economy, public health, and lives.
Unlike bacteria that have highly conserved cell wall structures, enzymes for protein and nucleic acid synthesis, and intermediary metabolism, viruses have fewer unifying features to target. Because of this, antiviral development has classically followed a ‘one drug, one virus’ paradigm. Unlike bacterial infections that could be treated with a broad range of antibiotics, viral infections need to be targeted with specific antivirals, of which there exist only a handful, not nearly enough to cover the 259 known viral pathogens that infect humans [3, 4]. This leads to difficulties in combating viral infection, especially in emerging human diseases where inability to predict the next emerging virus can lead to epidemics and pandemics.
The rise of broad-spectrum antiviral agent (BSAA) development represents a shift in the ‘one drug, one virus’ paradigm. BSAAs act against a range of viruses by targeting conserved processes or structures, such as nucleic acid synthesis or viral protease. Alternatively, BSAAs can target commonly used host factors that assist with viral entry and replication [3]. BSAAs can lessen the impact of emerging diseases, because they can be leveraged as a potential treatment option for emerging viruses as soon as they surface.
Drug repurposing (also known as drug repositioning, drug reprofiling, drug redirecting, or drug re-tasking) is a strategy of drug development that focuses on finding new indications for existing drugs or drug candidates. This holds several advantages over de novo drug discovery, one of the most compelling being that many of these drugs and drug candidates have already been demonstrated to be safe in humans by passing phase I clinical trials. While the first instances of drug repurposing have been based on serendipitous discoveries (such was the case for Viagra and Thalidomide), more systematic approaches have been developed as technological resources become more advanced. However, drug repurposing against viral diseases still represents a small percentage of total drug repurposing efforts [3].
Safe-in-man BSAAs are partially or fully developed compounds that have passed a minimum of phase I clinical trials and have shown antiviral activity across at least two different viral families [5]. By focusing on discovering and expanding the activity of drugs that have already been proven to be safe in humans and have broad antiviral activity, researchers can increase the activity spectrum of our available antiviral agents with less time, money, and resources than would be required of traditional drug discovery.
This chapter will highlight the case for the discovery and development of safe-in-man BSAAs and discuss major steps in the safe-in-man BSAA repositioning process. At the end of this chapter, several knowledge tools that are especially applicable to antiviral drug repositioning will be listed and described.
12.2 The Case for Safe-in-Man Drug Repurposing and Broad-Spectrum Agents
A hugely profitable industry, global pharmaceutical spending is predicted to surpass $1.5 trillion by 2023. For a chance to win a slice of this gargantuan pie, pharmaceutical companies spare no effort in the discovery and development of new drugs, only an average of 54 of which are predicted to be approved and launched per year from 2019 to 2023 [6]. With so much money at stake and a stunningly high failure rate, it should come as no surprise that any measures that reduce cost, time, and other resource use could be highly beneficial.
Conventionally, de novo drug development begins in the discovery phase, where targets are identified and lead compounds are selected and optimized. Following the discovery phase is the preclinical phase, where toxicology, DMPK (drug metabolism and pharmacokinetics), and ADME (absorption, distribution, metabolism, and excretion) studies are performed in animal models in preparation for the first human trials. Finally, phase I, phase II, and phase III clinical trials are conducted, establishing safety and efficacy in humans before it can finally be approved and launched as a new active substance. When a drug is finally approved and launched, it will have cost an average of $1.5 billion to develop and will have taken a bare minimum of 10 years to get from drug discovery to market [7].
Unfortunately, most drugs that undergo clinical trials never continue through to approval and launch. Although pitfalls exist along the entire drug development path, a sizeable portion of this failure lies with clinical trials. Most investigational drugs pass phase I clinical trials, with approximately 70% of drugs entering phase II. However, following phase I, only 8–10% of drugs will pass both phase II and III clinical trials and continue through to approval [8]. The most common reasons for failing these latter phases are safety concerns, disproportionate adverse effects, or lack of efficacy.
Many of the investigational drugs that have fallen off during clinical trials do so because of an inability to demonstrate sufficient efficacy. However, it is possible that some compounds in this ‘Valley of Death’ have several biological targets and exhibit more bioactivity than what was originally investigated. Indeed, many investigational anti-cancer drugs have been shown to exhibit potent antiviral activity in vitro and in vivo [9,10,11,12,13,14,15]. By re-directing drug discovery efforts into repurposing investigational drugs that have passed at least phase I of clinical trials, many efforts can be spared in the drug development process. Because these compounds have already been well-characterized in previous pre-clinical and clinical safety studies, these steps can be largely bypassed, allowing the price tag for the development of these drugs to be decreased to as low as $8.4 million [16].
Viral diseases often disproportionately affect countries with lower income, and thus are overlooked by many pharmaceutical companies due to their low profit potential. However, antiviral drug development is nonetheless crucial to maintaining a functional society at today’s population densities, as viruses continue to emerge from natural reservoirs and cause outbreaks among the human population. One barely needs to consider the overwhelming impact on public health, society, and the global economy brought on by the COVID-19 pandemic to understand why viral outbreak preparedness should have always been a foremost priority for the global community. The dramatically lowered price tag of drug repurposing allows researchers to overcome the often-insurmountable barriers to developing a drug from scratch, thus stimulating more participation and innovation in the field of antiviral research. Ultimately, this will translate to a greater number of therapeutic options against viral disease available to the general public.
In addition to the financial barriers discussed above, the nature of virus-borne disease often requires a level of preparedness against unknown or unspecified emergent viruses that may arise from natural reservoirs at random times. In the event that an emergent disease arises for which there is no effective drug to counter, drug developers must then be able to respond with a level of haste that is usually not required for other diseases. Currently, the conventional model for antiviral drug development fails to support either the requirement of preparedness or haste. However, development of broad-spectrum antivirals can increase pandemic preparedness because they target common biological functions within a wide range of viral families. In this way, they are much better positioned to be frontline drugs in the face of emerging viral diseases. Moreover, by repositioning phase I (or farther) drugs for these purposes, development time can be shortened by at least 5 years, allowing for a faster response to potentially life-threatening epidemics [7]. Importantly, if a potential antiviral agent is repositioned from an already-approved drug, doctors can legally prescribe them for off-label use immediately, thus allowing for the drug to be used even faster [17].
Finally, it is important to acknowledge the toll on other resources that can be spared when focusing on drug repurposing for broad-spectrum antiviral development. In addition to money and time, the drug development process is a major contributor to waste production, energy use, pollution, and CO2 emissions. A study in 2019 estimates that the pharmaceutical industry pumps out 48.55 tons of CO2 equivalents per million dollars, a value that is 55% greater than the automotive industry [18]. Part of this heavy environmental impact can be easily reduced by eliminating redundancies. Pivoting some drug discovery and development efforts towards drug repurposing and focusing on broad-spectrum agents is, therefore, one important way to reduce the strain on finite resources.
12.3 Methods for Safe-in-Man Broad Spectrum Antiviral Development
12.3.1 In Silico Methods for Drug Discovery
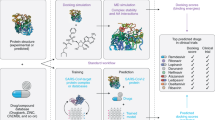
When searching for repurposed BSAA candidates, in silico methods are often used first due to their powerful, high-throughput, and low-cost nature. These techniques often centre on making novel connections between broad concepts like viruses, targets, and drugs, based on shared traits from the pool of existing data. Further experiments in vitro and in vivo are needed to further validate these results, as many of these discovery methods have high false-positive rates by nature. Moreover, these techniques rely on the aggregation and processing of existing data, and therefore may not be suitable for drug discovery endeavours for new drugs or new viruses for which no information is yet known. Common in silico methods include molecular docking studies, network-based modelling, and text mining (Fig. 12.1a).

The preclinical stages of BSAA repositioning. (A) In silico discovery of safe-in-man BSAAs. In molecular docking studies, drug and target structures are analyzed and interactions are simulated. In network-based analyses, networks are constructed of drugs, targets, or both. Drugs and viruses that are connected by few nodes are more likely to be connected themselves. For example, if Virus A is inhibited by Drug 1 and Drug 1 is structurally similar to Drug 2, then Drug 2 may have previously unknown activity against Virus A. Similarly, if Drug 1 is known to work against Virus C and Virus D and E are in the same family, then Drug 1 may have previously unknown activity against Virus D and E. In text mining–based approaches, networks are constructed from concepts taken from text documents such as research articles, abstracts, and press releases. Concepts are then connected in the network. For example, if Virus B is mentioned in connection with Drug 1, and both Drug 1 and Drug 3 are often mentioned in connection with a specific side effect, then Drug 3 may have previously unknown activity against Virus B. (B) In vitro screening of drug candidates. Cell lines, primary cells, or organoids are grown in cell culture and treated with virus. EC50 and CC50 values are determined and SI values are determined. (C) In vivo testing of final drug candidates. Infected animals are either naturally susceptible to the virus under evaluation, immunosuppressed, or transplanted with human tissue to become more susceptible to virus
12.3.1.1 Molecular Docking
Molecular docking is a technique that attempts to simulate interactions between a drug candidate and a drug target [19]. To accomplish this, 3D structural data for a potential drug target and a structurally characterized compound are extracted from a protein or drug database. Then, the interaction is simulated through a variety of docking algorithms. The top scoring drug-target pairs can then be selected for further testing in in vitro studies. This technique is commonly used in drug repurposing studies for all diseases, but is particularly applicable to antiviral drug discovery, where the small number of potential virus and host targets involved allows for a more exhaustive analysis without requiring additional time, computational power, or both.
Because of its applicability to antiviral discovery, molecular docking techniques have already been used extensively for antiviral-directed drug repositioning. For example, in an effort to combat re-emerging chikungunya epidemics, Tripathi et al. [20] screened an FDA-approved drug library for possible interaction with the nsP2, an essential protease in the CHIKV life cycle. From this screening, they identified telmisartan, an approved antihypertensive drug, and novobiocin, an approved antibiotic, for further studies for anti-CHIKV activity. Similarly, this screening strategy has played an important role in identifying drug candidates against SARS-CoV-2. Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir are all pre-existing drugs that have been identified as potential anti-SARS-CoV-2 agents through molecular docking against SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) [21]. Similarly, a molecular docking screen of over 10,000 compounds from the MolPort database against SARS-CoV-2 Main protease (Mpro) of returned nine naturally derived compounds for potential further antiviral development [22].
Antiviral screening can also be done by singling out a single compound and screening against a library of molecular targets. This method has been demonstrated successfully by Rizwana et al. [23], who used molecular docking to test interactions between the antiviral valacyclovir, an anti-herpes medication, against a library of viral proteins. Through this, they were able to identify a viral protein from DENV as a potential target of valacyclovir, suggesting that the previously specific antiviral may indeed have broad-spectrum antiviral qualities.
Several molecular docking tools exist in the literature. The two most widely used program packages are the Amber suite of biomolecular simulation programs [24] and the AutoDOCK suite [25]. Other tools include the AADS (Automated Active Site Detection, Docking, and Scoring) protocol, which combines an automated active site detection algorithm with an attached molecular docking protocol [26]. Additionally, several web-based molecular docking tools such as ParDOCK and Sanjeevini are freely accessible and can be used online [27, 28].
While a useful tool, molecular docking techniques have some limitations. First, molecular interaction simulations are computationally expensive, and large amounts of both computational power and time are required for performing screenings. This may serve has a barrier to entry for researchers without the computational resources to handle the large amount of data processing. Another limitation is the requirement for accurate and detailed structural information for drug targets, which can be difficult to obtain, even from protein structure databases. This lack of information will ultimately lead to high false positive rates, confounding the results of the study [29].
12.3.1.2 Network-Based Modelling
With the advent of network and systems biology, many more computer-based drug discovery methods have focused on the construction of networks. Network-based modelling consists of leveraging existing omics-level data to build and visualize networks that could be used to yield new drug/target connections. A network consists of nodes (representing an individual element such as a gene, protein, virus, or disease) and edges (representing a relationship between two or more nodes), which represent a subset of a biological system. These systems could be gene regulatory networks generated from transcriptomics data, protein–protein interaction (PPi) networks generated from proteomics data, metabolic networks generated from metabolomics data, or other drug- or condition-specific datasets [30].
In general, network-based approaches follow a common framework: (1) gathering information from databases, (2) constructing the network, (3) derive new information from a starting data point, and (4) uncover new biological connections. To derive new information from the network, one can use clustering approaches, which takes advantage of the assumption that proteins or drugs in the same module of a network will likely have the same biological properties; or propagation approaches, in which the starting data point is assumed to be passed along to the connected nodes within the network. Although any kind of network can be used for drug discovery and repositioning, the field of BSAA repositioning derives most use from three main types of networks: PPi networks, drug–target interactions, and inter-drug connections.
Protein–protein interaction network modelling links proteins that are associated with each other and infers that associated proteins can be targeted similarly. For example, if one protein in the network is already a known drug target, other proteins connected to that node would become candidates for new drug target discovery. An innovative use of PPi network was done by Bösl et al. [31], who constructed a PPi network of human and virus proteins from existing databases. They then identified the host subnetworks most commonly targeted by viral proteins, thus returning a list of high-impact proteins that are likely to be antiviral drug targets that can inhibit a broad range of viruses.
Unlike PPi networks which have one type of node, drug–target interaction networks are constructed with two types of nodes: drugs and targets. A connection exists between a drug node and a target node if a drug is known to interact with a target, while a connection exists between two drugs or two targets if there is structural, chemical, or mechanistic similarities between them. The underlying assumption behind drug-target network modelling is that proteins targeted by the same drug may themselves be similar, and that drugs acting on the same protein may also be similar. Taking advantage of this assumption, Yi et al. [32] identified the structurally similar RdRps of DENV and NV as a potential target for BSAA development. Molecular docking was then used to identify entrecitinib, an approved anticancer medication, as a potential BSAA against DENV and NV.
Lastly, inter-drug networks are built with drugs that have shared similarities. For example, if two drugs have similar side effects or similar gene expression profiles, they would be connected. Two drugs could also be connected if their physical properties aligned with one another, such as if two drugs were both small and hydrophobic. These models use the assumption that compounds with similar properties would induce similar biological responses [33]. To cite an example in antiviral development, many amphiphilic drugs that were not originally developed as antivirals have been shown to exhibit antiviral activity towards a wide variety of human viruses, such as flaviviruses, filoviruses, herpesviruses, and coronaviruses [34].
The limitations of network-based modelling are similar to those of molecular docking: these discovery techniques rely on availability of accurate information. A starting target/drug pair is needed to extract information from the network, which means that this tool cannot uncover drug-target connections that is not linked by existing data already. In addition, network modelling only serves as guidance, as our current technology does not allow us to translate a network response into any kind of meaningful, qualitative prediction in vivo.
12.3.1.3 Text Mining–Based Approaches
If construction of networks from public databases leads to no results, text mining methods could also be used to identify potential drug-virus associations. Instead of using protein or drug databases, these techniques gather data from scientific literature databases, news articles, press releases, and other publicly accessible text documents. Like network-based approaches, most text mining approaches build ‘networks’ of terms by connecting related concepts. Most of these methods are based on the Swanson ‘ABC’ model, which states that if two unconnected concepts (‘A’ and ‘C’) have a shared connection to a third concept (‘B’), the original two may also be connected [35].
Especially due to new developments in natural language processing techniques, many drug repurposing efforts have been carried out with text mining methods [36, 37]. To accomplish these, several freely accessible, online-based text mining tools are available. Perhaps one the oldest resources for this is PolySearch, a biomedical text mining tool that returns associations between diseases, genes, drugs, toxins, metabolites, pathways, organs, tissues, organelles, clinical manifestations, drug mechanisms, Gene Ontology and MeSH terms, ICD-10 medical codes, and biological and chemical taxonomies mined from PubMed, Wikipedia, US patents, and text-rich knowledgebases [38]. Other publicly available biological text mining tools include DrugQuest, BEST, and KinderMiner [39,40,41].
12.3.2 In Vitro Studies
Once an existing drug has been identified for potential BSAA activity, in vitro experiments are performed to validate antiviral activity in a real biological setting (Fig. 12.1b). These studies are also often used to determine the potency of antiviral activity of a BSAA, as well as its cellular toxicity. This is often expressed as the concentration of a drug that is needed to achieve 50% of its maximal inhibitory activity (EC50) and the concentration of a drug that is needed to kill 50% of cells in culture (CC50). Strong drug candidates should have high potency (indicated by a low EC50 value) while having low toxicity (indicated by a high CC50 value). This selective ability of an antiviral to inhibit viral activity without harming cells is expressed as the selectivity index (SI = CC50/EC50). Thus, a compound with a higher selectivity value generally indicates a more effective compound.
In vitro studies can assess antiviral efficacy and potency in two main ways: measuring cell viability due to a virus’ cytopathic effect (CPE) and measuring viral infection. To measure viability within a cell population, an indicator that often directly correlates with live or dead cells is used and quantified using colorimetry, chemiluminescence, or fluorescence. Mitochondrial reductase activity could be measured by the addition of resazurin or tetrazolium-based dyes such as MTT, MTS, and XTT, and cell viability could be quantified by a colour change of the dye once it is reduced. Alternatively, metabolic activity could be quantified by commercially available assays for esterase activity, ATP production, glycolytic flux, or oxygen consumption. Membrane integrity is also a commonly used correlate of cell viability, with membrane-impermeable dyes and extracellular LDH detection both being used as an indicator of cell death. Finally, apoptosis-related assays such as TUNEL (measuring DNA fragmentation), Annexin V (measuring phosphatidylserine externalization), and Caspase (detecting the levels of apoptotic caspase activity in cell lysates) can also be used to quantify cell viability.
Although cell viability can be an indicator of viral activity, the two are not perfect correlates. Cells infected by virus may not die immediately, and it is difficult to attribute cell death to viral infection instead of external factors. Because of this, quantifying viral infection could be an alternative approach to studying antiviral activity in vitro. This can be done by detecting parts of viruses using immunoassays, microarrays, or high-throughput sequencing techniques like RT-qPCR and RNA-Seq [42,43,44,45,46]. The standard method of determining virus titers is a plaque assay, which is often used to quantify the amount of infectious viral particles in a sample. To accomplish this, dilutions of a virus sample are incubated on a susceptible cell monolayer and covered with a gel. Then, the resultant patches of dead cells (plaques) are counted to determine the number of virus particles in a sample.
More versatile methods of viral quantification have also been developed, often using engineered viruses that express fluorescent proteins or luciferase, which allow for real-time surveillance and quantification of virus infection [47,48,49,50,51,52]. A similar reporter system can be set up for retroviral infections, in which the host cell is made to express a reporter gene under the control of a promotor that is located on the virus, allowing for the continual detection of infected cells in which a virus is not actively replicating [53, 54]. Perhaps one of the most innovative additions to the antiviral research toolbox is the use of CRISPR-Cas technology to target viral RNA particles and subsequently cleave signal molecules from their quenchers, resulting in signal amplification and detection of very small amounts of virus [55, 56].
There are several different models to study antiviral activity in vitro, including cell lines, primary cells, stem cell-derived cells, and organoids. Typically, a variety of these models are needed to generate a convincing dataset for a drug candidate, and a complete published study of antiviral activity could include data from experiments on a combination of multiple cell lines, primary cells or iPSC-derived cells, and organoids [57].
12.3.2.1 Cell Lines
The first in vitro studies are often done in cell lines, because they are relatively easy to obtain and grow. However, different cell lines can vary in their susceptibility to viral infection, and care needs to be taken in selecting a cell line that will accurately reflect the infection cycle of the virus under investigation. One of the most broadly used cell lines in antiviral research is the Vero cell line and its derivatives, although many cell lines are used specifically for the virus model they are meant to study (to give an example, MDCK cells are a commonly used cell line in experiments involving influenza). It is important to note that large genomic differences exist between cell lines and human primary cells, which can lead to deviations in results between cell lines and primary cells. Moreover, many of the cell lines used for antiviral research originate from non-human species, possibly further confounding results. Therefore, experiments done on cell lines will almost always need further validation.
12.3.2.2 Primary Cells and Stem Cell-Derived Cells
Unlike immortalized cell lines, primary cells are harvested directly from tissues, are usually slower growing, and have a finite lifespan. Additionally, they are often highly differentiated, and can better represent an infected organ system to more accurately reflect a cellular response to infection. For these reasons, they are often used to validate data obtained from cells lines [58,59,60,61]. Importantly, factors such as age, sex, and genetic differences between donors must be considered when choosing primary cells to work with, as these small changes can play a huge role in a cell’s response to viral infection [62, 63]. Primary cells also have their limitations. Like cell lines, not all primary cells are susceptible to all viruses. Moreover, primary cells often have more stringent growing conditions, may require specialized media, have a slower division time, and need to be regularly harvested, all of which increases the cost and effort needed to maintain them.
If primary cells are difficult to obtain, differentiated cell types may also be generated from induced pluripotent stem cells (iPSCs). iPSCs are often derived from an easily obtainable origin, such as blood or skin cells, and are reprogrammed into a pluripotent state. From there, the iPSCs can be supplied with differentiation factors to redirect its differentiation to a cell type of choice. However, generation of iPSC-derived cells can be time-consuming and labor-intensive, and they may respond to viral or drug treatment in unpredictable ways. For example, thymidine kinase of Herpes simplex virus type 1 (HSV-1) is commonly inserted into cells to create an inducible cell death system triggered by ganciclovir. However, Iwasawa et al. [64] reported that expression of thymidine kinase itself was toxic to iPSCs, irrespective of the addition of ganciclovir, suggesting that iPSCs have a markedly different response to small changes in dNTP levels from other cells. Nonetheless, iPSC-derived cells are often used in antiviral studies when attention needs to be focused on a specific tissue type, such as in the case of studying Hepatitis B virus in hepatocytes, or Zika virus in neuronal cells [65,66,67].
12.3.2.3 3D Cultures and Organoids
One further method to mimic true physiological conditions in vitro is by experimenting on 3D cell cultures. It is often criticized that cells grown in 2D monolayers do not sufficiently mimic the complex intercellular interactions that occurs in the body. To alleviate these differences, ECMs and other hydrogels are often used as scaffolds to encourage 3D attachments and geometries in cell cultures [68]. In 2011, Straub et al. demonstrated the first successful in vitro culture of NV was achieved by infecting them in a 3D-cultured colorectal cell line [69]. In addition, keratinocyte raft cultures have been used to study both human papillomavirus (HPV) and human adeno-associated virus-2 (hAAV-2), while more complex 3D cultures of keratinocytes have been used to study Poxviruses [70,71,72].
Organoids are a form of 3D cell culture that is derived from the progenitor cells of a certain organs. They are self-organizing, and therefore can have more complicated structures than man-made 3D scaffolds. Organoids are important models for viral infections that affect whole organ systems, such as Zika virus on brain organoids [67, 73, 74] or rotavirus on intestinal organoids [75,76,77,78,79].
12.3.3 In Vivo Studies
After proof of concept is established in cell culture, in vivo studies are performed to gauge the effect of a drug in animal models (Fig. 12.1c). Animals are an attractive model due to the presence of multiple, complete biological systems that are analogous to human biology, allowing researchers to investigate the effect of a drug in a more complete biological environment. Unfortunately, many human-infecting viruses do not readily infect non-human animals, although some cross-species infections exist [80,81,82,83]. To help with infectivity, many animals are immunocompromised through genetic or chemical manipulation [84]. Additionally, some animals need to be genetically altered to more resemble human physiology or need to be ‘humanized’ though transplantation with human tissue before successful infection can take place [85]. Other animal studies employ species-adapted versions of virus that can infect the species of interest, thus circumventing a need for developing special animals [86, 87].
Although animal models provide a good perspective of drug–virus interactions in the context of a living organism, there are still large differences between animal and human biology that make animals an imperfect model. Thus, caution should be exercised when interpreting results, and potency demonstrated in animals may not translate to potency in humans [88, 89].
12.3.3.1 Mice
Mice are an oft-used animal model in all aspects of human medicine research, and the same is true for antiviral research. Mice are widely used in research on a wide variety of viruses, although different strains must be used due to unpredictable and varied susceptibility to different viruses. For example, it has been established that mice are susceptible to Zika virus infections [90]. However, it has been observed that while the SJL strain of mouse exhibits striking similarity to human response to infections with Zika-infected mothers bearing microcephalic pups, the same microcephaly does not present at all in C57BL/6 pups [91]. Other flaviviruses have also been shown to infect mice to varying degrees of success, with inbred mice being easily infected and wild mice being resistant [92]. A similar phenomenon is seen with influenza, with laboratory strains easily infected and wild mice exhibiting resistance [93]. Additionally, murine cytomegalovirus (a herpesvirus endemic to mice) is known to affect BALB/c mice severely and C57BL/6 mice mildly, whereas Sindbis virus (a togavirus causing fever in humans) is known to affect C57BL/6 mice severely while BALB/c mice are largely resistant [94].
The main shortcoming of the mouse model is the degree of its divergence from human biology, particularly with respect to immunology [95]. This variation often leads to differences in observed symptoms and may lead to discovery of mechanisms that are fundamentally inapplicable to human medicine. To illustrate, influenza infection often causes fever and a rise in body temperature in humans, but the same infection in mice can lead to hypothermia [96]. The fundamental difference between human and mouse virology can be illustrated with CEACAM1, a glycoprotein that is expressed in both species. It has been shown that mice expressing CEACAM1a are susceptible to murine coronavirus, while those that do not express CEACAM1a are not susceptible [97]. This might have been taken as an indication that human CEACAM1 could be a possible antiviral target for antivirals against coronaviruses; unfortunately, human coronaviruses do not interact at all with human CEACAM1. For this reason, the use of mouse models in antiviral research should be carried out with caution, as well as a clear understanding of the differences between mouse and human biology.
12.3.3.2 Non-human Primates
Non-human primates (NHPs) are similar to humans in their adaptive immune system, have high sequence homology for immune genes, and follow the same ABO blood groups that humans have. They represent a very reliable model for antiviral research due to this similarity, although it is associated with serious ethical concerns as well as high maintenance costs.
Much of research on Zika virus has taken place in NHPs. This is in large part due to the fact that the virus was first isolated in rhesus macaques, a common NHP model [98]. Moreover, other NHPs have been known to carry Zika virus in the wild [99]. Because of the similar gestational process, placental barrier, and fetal development between NHPs and humans, these models are often used to study the effect of Zika on gestational development. Indeed, rhesus macaques, cynomolgus macaques, and pigtail macaques have all been used as animal models in the study of Zika virus [100,101,102,103,104,105,106,107,108].
Another area of virus research that has taken advantage of NHP models due to its origin is respiratory syncytial virus (RSV). RSV was first isolated in chimpanzees, and subsequent studies of RSV infection were carried out in chimpanzee models [109]. A wide variety of primate species have been used to study RSV, including owl monkeys, baboons, capuchin monkeys, African green monkeys, rhesus macaques, bonnet monkeys, and cynomolgus macaques [110].
12.3.4 Clinical Trials
In order for a drug to become approved for a new use, it must first pass three phases of clinical trials (Fig. 12.2). Phase I trials are usually limited to under 100 human patients and have the purpose of establishing safety and dosage. To be considered safe in humans, a therapeutic agent must have already passed phase I clinical trials. Because of this, these can often be skipped in the development of existing safe-in-man BSAAs.

The clinical stages of BSAA repositioning versus conventional drug development. Green-shaded areas represent the average proportion of viable drug candidates that remain after each round of trials, while yellow-shaded regions indicate the pool of drug candidates available for repurposing. Safe-in-man BSAAs can often skip phase I clinical trials. Moreover, failed drug candidates from previous trials can often be repurposed for new indications
Phase II clinical trials follow phase I. These can involve several hundreds of patients and last up to 2 years, with the intention of evaluating efficacy and possible adverse side effects. Because study population sizes still may be too small to effectively determine efficacy, a large portion of phase II trial failure is due to safety concerns. Many BSAAs are drug candidates that either have not yet passed phase II trials or have failed a previous phase II trial. Unsurprisingly, failure to pass an earlier phase II trial raises safety concerns, especially if this failure is attributed to adverse effects. However, these drug candidates may be assessed again if the new patient population is disparate from the original population and there is evidence to suggest that the same safety concerns could be dismissed in the new population.
After passing phase II trials, phase III clinical trials can begin. Phase III trials incorporate more patients and take longer than phase II trials, thus allowing drug developers to monitor longer-term adverse effects and evaluate efficacy with greater statistical power against the current standard of care. Compounds may fail phase III trials due to inability to perform comparably or better than contemporary standard treatment, despite an established safety profile. Compounds that either have not yet passed phase III trials or have failed phase III trials due to inability to demonstrate the proper efficacy provide a promising pool of potential safe-in-man BSAAs due to the more stringent safety requirements defined by passing phase II. All repositioned BSAAs, including those agents that have already been approved, must still undergo a phase III trial to demonstrate sufficient efficacy before being approved for use for a new condition.
Once phase III trials have been passed, the drug can finally be approved for use. Upon being approved, many drugs must continue to be subject to continual monitoring to address any further long-term safety concerns in a long-term, phase IV trial. However, BSAAs that have been redirected from already-established drugs which have spent a considerable time on the market will have already undergone some levels of this post-market surveillance, providing a level of security and knowledge before phase IV studies begin.
12.4 Structure–Activity Relationships
As is the case with many drugs, structure–activity relationships (SARs) exist among BSAAs. This is not surprising, as structurally similar compounds are more likely to have similar targets. Because of this, antiviral discovery has often relied on the use of SARs, both to synthesize new antiviral agents and to improve upon existing ones [111, 112].
Interestingly, the breadth of antiviral activity seems to be related to chemical structure. Figure 12.3 shows the structure–activity relationships of known BSAAs. Compounds close to one another on the tree are structurally similar, while the number of viruses targeted by each compound is indicated by a bubble next to the name of the compound. The size of the bubble corresponds to the number of viruses for which antiviral activity has been established for the compound. This relationship between structure and breadth of activity is illustrated clearly by the structurally similar nucleotide and nucleoside analogs favipiravir, brincodofovir, cidofovir, and gemcitabine, all of which have a noticeably broader spectrum of antiviral activity compared to other BSAAs with structurally divergent neighbours.

Structure–activity relationship of known BSAAs. Compounds were clustered based on structural similarity calculated by ECPF4 fingerprints and visualized using the D3 JavaScript library. The broad-spectrum antiviral activities of the compounds are shown as bubbles, with larger bubbles corresponding to a larger number of targeted viruses
This relationship can be used to broaden the antiviral activity of known BSAAs. For example, nitazoxanide, an antiparasitic agent and BSAA that possesses proven antiviral activity against 17 pathogenic human viruses, is structurally related to acetylsalicylic acid, which has only shown antiviral activity in 4 viruses [31, 79, 113,114,115,116,117,118,119,120,121,122,123,124,125]. Because of their structural similarity, it would then be more likely that acetylsalicylic acid could possess previously uncovered, broad antiviral activity as well. Likewise, ementine, remdesivir, and ABT-263 are structurally related BSAAs that have been demonstrated to possess relatively broad antiviral activity, while other structurally related drugs like cepharathine, manidipine, and sofosbuvir have relatively narrow demonstrated antiviral activity. These structural similarities could then pave the way for drugs like cepharanthine, manidipine, and sofosbuvir to be investigated for potential expansion into other viral diseases.
12.5 BSAA Combinations
One Achilles’ heel of antiviral agents lies in the speed and ease by which viruses replicate, leading to a high evolution rate. Since viruses are constantly mutating and evolving, monotherapy treatment using a single antiviral agent can often lead to development of resistance [126]. To combat this, combination therapy can be used to treat viral infections. Combination therapies may be more effective because viruses that are able to evade the mechanism of action of one antiviral agent will likely be inhibited by a second agent with a different mechanism of action, thus requiring two specific mutations in order for a virus to develop any lasting resistance.
Indeed, current treatments for rapidly evolving viruses such as HIV and HCV often involve a combination of antivirals. These include Triumeq (abacavir/dolutegravir/lamivudine – a combination of two viral reverse transcriptase inhibitors and one viral integrase inhibitor) for the treatment of HIV and Harvoni (ledipasvir/sofosbuvir, a combination of an HCV NS5A inhibitor and an HCV RdRp inhibitor) for the treatment of HCV [127, 128]. Other common treatments include Symtuza (darunavir/cobicistat/emtricitabine/tenofovir) and Kaletra (lopinavir/ritonavir) for HIV, as well as Epclusa (sofosbuvir/velpatasvir) for HCV.
Administration of antiviral agents in combination may lead to additive effects, synergism, or antagonism. Additive effects occur when the antiviral activity of a combination is equal to the expected effect of the two separate drugs added together. Synergism occurs when the combined antiviral activity is greater than that of the two individual drugs added together. Synergism between antivirals is advantageous because it can lead to the use of smaller quantities of drug, thereby reducing toxicity decreasing chances for adverse side effects. Finally, antagonism is likely to occur if two antiviral agents have redundant mechanisms of action or interact with each other in a non-productive way. This results in combined antiviral activity that is weaker than that of the two individual drugs combined. Generally, antiviral drugs that have shown antagonistic activity should not be used with each other.
The efficacy of several drug combinations against viral infection has been investigated in clinical trials. Recent clinical trials against COVID-19 have tested combinations of danoprevir/ritonavir (clinicaltrials.gov ID: NCT04291729) and favipirair/tocilizumab (Chinese Clinical Trials Register ID: ChiCTR2000030894). Other clinical trial investigations have included the use of elbasvir/gazoprevir (clinicaltrials.gov ID: NCT03111108), sofosbuvir/ledipasvir (clinicaltrials.gov ID: NCT02480166), and peginterferon-alfa-2b/ribavirin (clinicaltrials.gov ID: NCT00383064 and NCT01045278) against HCV, as well as use of entecavir/adefovir (clinicaltrials.gov ID: NCT01023217) and peginterferon-alfa-2a/adefovir or peginterferon-alfa-2a/entecavir (clinicaltrials.gov ID: NCT00922207) against HBV.
In addition to clinical trials, several pre-clinical research endeavours have also revealed synergistic antiviral activity against a broad range of viruses. For example, in vitro studies have shown that a combination of pimodivir and gemcitabine was shown to exhibit synergism against Influenza A [129]. Similar in vitro studies have also shown that obatoclax and saliphenylhalamide have synergistic activity against Zika virus infection in RPE cells [130]. Most recently, it was demonstrated by Ianevski et al. that salinomycin, amodiaquine, obatoclax, emetine, and homoharringtonine all show synergistic activity when administered with nelfinavir against SARS-CoV-2 infections in cell culture [131]. Interestingly, it was noted that all synergistic combinations that were identified involved one host-directed drug and one virus-directed drug, while most combinations involving two host-directed drugs proved to be antagonistic. This finding suggests that synergism among antivirals may be more likely to exist if a host-directed agent is combined with a virus-directed agent.
When administered correctly, combination antiviral therapies can improve efficacy by simultaneously exhibiting synergistic effects and preventing development of antiviral resistance. This can be especially useful for repositioned BSAAs, because they may inhibit different viruses with different degrees of efficacy. In these cases, the boost in efficacy given by drug synergism can be a valuable tool for antiviral repurposing. Moreover, several BSAAs can be purposefully combined in one formulation to cover the broadest set of viruses possible, which could then be used as a frontline solution to treat emergent viral diseases [132]. For these reasons, the use of BSAA combinations can be a useful tool to maximize the antiviral activity of existing drugs.
12.6 Useful Tools for Antiviral Drug Repurposing Studies
BSAA repositioning strategies are often considered low risk and high reward because initial drug discovery costs are low and chances for success are high. A large part of the reason for this low-risk/high-reward profile is because this strategy benefits from large amounts of pre-existing data that have already been collected from years of previous research. Traditionally, extensive literature reviews were required to collect this data; however, recent acknowledgement by the research community for the need of integrated, comprehensive knowledgebases has led to the establishment of several openly accessible tools that shorten the drug discovery process further. Several useful databases for antiviral repositioning are discussed below and summarized in Table 12.1.
12.6.1 Pharmacological Databases
12.6.1.1 DrugBank
DrugBank is an openly accessible database that contains over 13,000 approved, investigational, experimental, illicit, withdrawn, and nutraceutical drug entries [133, 134]. It contains both chemical information about drugs such as molecular weight and structure, as well as biological information on drug targets such as biological pathways of the target, mechanism of action, PK/PD, and toxicology data. Additionally, information is included on drug–drug interactions, as well as links to source material such as research articles and clinical trials. The database can also be queried using targets, pathways, or indications. Importantly for BSAA research, DrugBank aggregates a list of 151 drugs categorized as antiviral agents along with 690 associated drug targets, which can be browsed or searched individually. This resource can be accessed at: https://www.drugbank.ca/.
12.6.1.2 DrugCentral Database
DrugCentral database is a manually curated online compendium of approved therapeutic agents [135]. It monitors information about drug approvals, as well as collects information on dosage, reported adverse events, indications, and drug formulations. The database can both be browsed from a web browser or downloaded directly for high-throughput analysis. This resource can be accessed at: http://drugcentral.org/.
12.6.1.3 Pharmacogenomic Knowledgebase (PharmGKB)
PharmGKB is an interactive online tool that allows users to query by genes, variants, drugs, diseases, and pathways, focusing on associations between drug phenotype and genetic variants [136]. It is also annotated with clinical and prescription information. A built-in text mining system provides automated annotations of predicted linkages between drugs and genetic variants from PubMed and PubMed Central, and retrieves the sentence from which the information is mined. Each entry also contains links to other molecules, genes, or diseases in the database, with which the entry has some association. This resource can be accessed at: https://www.pharmgkb.org/.
12.6.1.4 DrugVirus Database
The DrugVirus database is a resource specifically focusing on safe-in-man BSAAs [5]. It summarizes and tracks the antiviral activity and developmental status of drugs and drug candidates that have passed at least phase I clinical trials and have shown antiviral activity in at least two viral families. The database includes 816 unique drug-virus combinations, covering 118 safe-in-man drugs that target 83 human viruses. Of these drug-virus combinations, 592 have demonstrated efficacy in cell lines, 14 have demonstrated efficacy in primary cells or organoids, 69 have demonstrated efficacy in an animal model, 18 are currently in phase I clinical trials, 27 are in phase II clinical trials, 28 are in phase III clinical trials, 49 have been approved for use for the virus in question, and 19 are being monitored in phase 4 clinical trials (Fig. 12.4). The graphic interface of the database also highlights BSAA-virus combinations that have not yet been explored. Drug–virus interactions are visualized on a heatmap, which displays viruses of the same family and classification close to one another. Thus, if a drug has been shown to work against one virus in a larger family of viruses, it is more likely that it will have some antiviral activity against the surrounding viruses on the heatmap as well. The DrugVirus database can be accessed at: https://drugvirus.info/.

Summary of drug-virus combination statuses on DrugVirus.info
12.6.2 Proteomics Databases
12.6.2.1 The RCSB Protein Data Bank
Another useful protein database is the RCSB Protein Data Bank (RCSB-PDB) [137]. This database collects 3D structural information for proteins, nucleic acids, and assemblies of both, along with annotations about structural features, function, and links to published articles. The structures can be searched and narrowed down by species, taxonomy, method for structural determination, structural resolution of the entry, release date, protein classification, and symmetry. The RCSB-PDB is a useful resource for molecular docking studies, as it contains comprehensive 3D structural information for candidate drug targets. It can be accessed at: https://www.rcsb.org/.
12.6.2.2 Proteopedia
Similar to other crowd-sourced knowledgebases like Wikipedia, Proteopedia is an editable protein wiki that contains information about structure and function [138]. Like the RCSB-PDB, the main goal of Proteopedia is to provide insight into 3D structure and function. Each protein entry also contains a 3D structural representation, as well as links to literature and associated protein entries within the database. This resource can be accessed at: http://proteopedia.org/.
12.6.2.3 UniProt Knowledgebase
UniProt was developed to be a universal protein resource, consisting of information on protein sequences obtained from nucleic acid sequencing data, functions, and structure [139]. To eliminate redundancy, double entries are merged based on sufficiently redundant protein sequences. The database is divided into two sections: TrEMBL, which contains automatically annotated information, and Swiss-Prot, which contains only manually curated information for each entry. In addition to the knowledgebase itself, UniProt contains the UniProt archive (UniParc), a non-redundant archive of all publicly available sequences, as well as UniProt reference clusters (UniRef), which clusters entries based on sequence similarity. This resource can be accessed at: https://www.uniprot.org/.
12.6.3 Chemical Structure Databases
12.6.3.1 PubChem
PubChem is an editable online repository of bioactive chemicals and substances from the National Institutes of Health [140, 141]. Although it is mainly a chemical database, it includes some large biological macromolecules such as antibodies. It contains physical and chemical information about substances such as 2D, 3D and crystal structure, conformation, boiling point, melting point, density, and solubility. It also includes a wealth of relevant biological information such as proteins, pathways, and bioassay data, patent information, and links to literature. PubChem can be accessed at: https://pubchem.ncbi.nlm.nih.gov/.
12.6.3.2 ChEMBL
A bioinformatic resource provided by the European Bioinformatics Institute (EBI), ChEMBL is a database of bioactive molecules and drugs [142]. It is manually curated to reduce the error rate of information extraction and contains over 1.9 million molecular compounds targeting over 13,000 proteins. The entries include binding and functional information, as well as pharmacological and clinical data. This resource can be accessed at: https://www.ebi.ac.uk/chembl/.
12.6.3.3 ChemDB
ChemDB is a purely chemical database, containing over 5 million commercially available small molecules [143]. This database collects a broad range of chemical compounds, not just focusing on bioactive molecules. However, it provides an extensive resource for experimentally determined physiochemical properties not often covered by other drug discovery-adapted databases. The database also curates annotations from the vendors of the compounds, and allows for searching by these annotations. This resource can be found at: http://cdb.ics.uci.edu/.
12.6.4 Viral Databases
12.6.4.1 Virus Pathogen Resource (ViPR)
ViPR is a virus database funded by the National Institutes of Health [144, 145]. It collects data on known viral pathogens, with focus on genetic sequences, immune epitopes, host factor data, 3D structures, and associated antiviral drugs. In addition to its database, ViPR also provides bioinformatics tools such as phylogenetic tree building, sequence alignment, primer design, and gene annotation for analysis of novel data. Finally, ViPR allows for community contributions and data sharing, which assists in the constant growth of the database. This resource can be accessed at: http://www.viprbrc.org/.
12.6.4.2 ViralZone
ViralZone is a web-based resource from the Swiss Institute of Bioinformatics [146]. It includes information on virions, virus molecular biology, reference sequences, and a downloadable virus thesaurus (virosaurus), which includes complete and partial sequence datasets for eukaryotic and plant viruses. As of June 2020, ViralZone contains 918 pages, covering 128 virus families, 567 virus genera, 7 individual species, and 216 resources on viral molecular biology. The ViralZone resource can be found at: https://viralzone.expasy.org/.
12.7 Conclusion and Future Perspectives
The repositioning of safe-in-man BSAAs represents a paradigm shift from a conventional model of drug development to one that is cheaper, faster, and more efficient. Antiviral research is uniquely positioned to benefit from this strategy, because development of safe-in-man agents can alleviate the lack of financial incentives and funding required to find new antiviral drugs. In the same manner, BSAA repositioning can also provide an avenue for antiviral developers with less funding to take advantage of the vast collection of resources generated by more well-funded areas of drug development, such as cancer research.
There are still hurdles to safe-in-man BSAA development. For instance, while it is both faster and cheaper to develop pre-existing drugs, many research institutions and pharmaceutical companies are less likely to do so when they cannot patent the molecule. This is somewhat mitigated by the both the US and European patent systems, which now both consider sufficiently inventive applications of existing compounds for intellectual property protection [147]. However, achieving ‘sufficiently inventive applications’ may be a hurdle in itself, especially when considering repurposing an existing antiviral for use against another, if different, virus. Patents can also be issued for existing drugs if the new indication requires a different formulation from its original indication. Again, this stipulation does not apply well to repositioning of safe-in-man BSAAs because the goal is to have an antiviral agent with one formulation that can act broadly, much like the case with antibiotics against a broad range of bacteria. Moreover, because it is legal for doctors to prescribe existing drugs for off-label uses, it would be difficult to deter doctors from prescribing the existing or generic drug in that capacity, even if a patent were obtained for a new indication.
Because of these hurdles, interest in drug repositioning studies is falling more and more to pharmaceutical companies with existing drug patents that want to broaden the scope of their own portfolio as much as possible. Even this can prove to be an endeavour unworthy of pursuit because by the time a company successfully identifies a new indication for their drug, the remaining patent time for the drug will likely be too short to make a significant profit [148, 149]. This, in addition to the relative lack of profitability of antivirals, leads to little incentive for many to pursue the study of antiviral repositioning.
However, it is becoming more evident that antiviral development cannot be ignored, and that safe-in-man BSAA can save valuable time and money when an emergency arises. Indeed, three of the most promising antiviral agents against SARS-CoV-2 (remdesivir, favipiravir, and ivermectin) are previously existing, safe-in-man agents that have shown broad-spectrum antiviral activities before the beginning of the COVID-19 global pandemic [150,151,152,153,154].
Focus on discovery and development of safe-in-man BSAAs allows adequate preparedness in the face of constant viral threats and has the capacity to save millions of lives in the process. However, it is imperative that several shifts need to occur in the drug development sphere, including the reworking of pharmaceutical patent systems that have previously focused on the ‘one drug, one disease’ paradigm. In addition, increased funding for antiviral research with special focus on drug repurposing and broadly acting agents, as well as collaborations between academic research and the private sector, are necessary to increase incentives and entice researchers to participate.
Abbreviations
- 2D:
-
2-dimensional
- 3D:
-
3-dimensional
- ADME:
-
Absorption, distribution, metabolism, and excretion
- BSAA:
-
Broad-spectrum antiviral agents
- CC50:
-
Half maximal cytotoxic concentration
- CEACAM1:
-
Carcinoembryonic antigen-related cell adhesion molecule 1
- CHIKV:
-
Chikungunya virus
- COVID-19:
-
Coronavirus disease 2019
- CPE:
-
Cytopathic effect
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- DENV:
-
Dengue virus
- DMPK:
-
Drug metabolism and pharmacokinetics
- dNTP:
-
Deoxyribose nucleotide triphosphate
- EBI:
-
European Bioinformatics Institute
- EC50:
-
Half maximal effective concentration
- ECM:
-
Extracellular matrix
- hAAV-2:
-
Human adeno-associated virus 2
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HIV:
-
Human immunodeficiency virus
- HPV:
-
Human papillomavirus
- HSV-1:
-
Herpes simplex virus 1
- iPSC:
-
Induced pluripotent stem cells
- LDH:
-
Lactate dehydrogenase
- MDCK:
-
Madin-Darby Canine Kidney
- MTS:
-
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- MTT:
-
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- NHP:
-
Non-human primate
- NS5A:
-
Non-structural protein 5A
- NV:
-
Norovirus
- PD:
-
Pharmacodynamics
- PDB:
-
Protein data bank
- PK:
-
Pharmacokinetics
- PPi:
-
Protein–protein interaction
- RCSB:
-
Research Collaboratory for structural bioinformatics
- RdRp:
-
RNA-dependent RNA polymerase
- RPE:
-
Retinal epithelial cells
- RSV:
-
Respiratory syncytial virus
- SAR:
-
Structure–activity relationship
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- SI:
-
Selectivity index
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labelling
- XTT:
-
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
References
Woolhouse M, Gaunt E (2007) Ecological origins of novel human pathogens. Crit Rev Microbiol 33:231–242
Domingo E (2010) Mechanisms of viral emergence. Vet Res 41:38–38
Debing Y, Neyts J, Delang L (2015) The future of antivirals: broad-spectrum inhibitors. Curr Opin Infect Dis 28:596–602
Kapoor G, Saigal S, Elongavan A (2017) Action and resistance mechanisms of antibiotics: a guide for clinicians. J Anaesthesiol Clin Pharmacol 33:300–305
Andersen PI, Ianevski A, Lysvand H, Vitkauskiene A, Oksenych V, Bjørås M, Telling K, Lutsar I, Dumpis U, Irie Y et al (2020) Discovery and development of safe-in-man broad-spectrum antiviral agents. Int J Infect Dis 93:268–276
Aitken M, Kleinrock M, Simorellis A, Nass D (2019) The global use of medicine in 2019 and outlook to 2023: forecasts and areas to watch. Parsippany, NJ:The IQVIA Institute for Human Data Science
Mestre-Ferrandiz J, Sussex J, Towse A (2012) The R&D cost of a new medicine. In: Office of health economics. The Office of Health Economics, London, UK, p 100
US Food Drug Administration (2018) The drug development process: step 3. In: Clinical research. https://www.fda.gov/forpatients/approvals/drugs/ucm405622.htm
Bodiwala HS, Sabde S, Mitra D, Bhutani KK, Singh IP (2011) Synthesis of 9-substituted derivatives of berberine as anti-HIV agents. Eur J Med Chem 46:1045–1049
Dang SS, Jia XL, Song P, Cheng YA, Zhang X, Sun MZ, Liu EQ (2009) Inhibitory effect of emodin and Astragalus polysaccharide on the replication of HBV. World J Gastroenterol 15:5669–5673
Hsiang CY, Ho TY (2008) Emodin is a novel alkaline nuclease inhibitor that suppresses herpes simplex virus type 1 yields in cell cultures. Br J Pharmacol 155:227–235
Hu CJ, Chen YT, Fang ZS, Chang WS, Chen HW (2018) Antiviral efficacy of nanoparticulate vacuolar ATPase inhibitors against influenza virus infection. Int J Nanomedicine 13:8579–8593
Qing M, Zou G, Wang QY, Xu HY, Dong H, Yuan Z, Shi PY (2010) Characterization of dengue virus resistance to brequinar in cell culture. Antimicrob Agents Chemother 54:3686–3695
Todt D, Moeller N, Praditya D, Kinast V, Friesland M, Engelmann M, Verhoye L, Sayed IM, Behrendt P, Dao Thi VL et al (2018) The natural compound silvestrol inhibits hepatitis E virus (HEV) replication in vitro and in vivo. Antiviral Res 157:151–158
Wang P, Rennekamp AJ, Yuan Y, Lieberman PM (2009) Topoisomerase I and RecQL1 function in Epstein-Barr virus lytic reactivation. J Virol 83:8090–8098
Persidis A (2011) The benefits of drug repositioning. In: Drug discovery world. Edify Digital Media Ltd, London, UK
Stafford RS (2008) Regulating off-label drug use — rethinking the role of the FDA. N Engl J Med 358:1427–1429
Belkhir L, Elmeligi A (2019) Carbon footprint of the global pharmaceutical industry and relative impact of its major players. J Clean Prod 214:185–194
Morris GM, Lim-Wilby M (2008) Molecular docking. Methods Mol Biol 443:365–382
Tripathi PK, Soni A, Singh Yadav SP, Kumar A, Gaurav N, Raghavendhar S, Sharma P, Sunil S, Ashish JB et al (2020) Evaluation of novobiocin and telmisartan for anti-CHIKV activity. Virology 548:250–260
Elfiky AA (2020) Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): a molecular docking study. Life Sci 253:117592
Kapusta K, Kar S, Collins JT, Franklin LM, Kolodziejczyk W, Leszczynski J, Hill GA (2020) Protein reliability analysis and virtual screening of natural inhibitors for SARS-CoV-2 main protease (M(pro)) through docking, molecular mechanic & dynamic, and ADMET profiling. J Biomol Struct Dyn 36:1–18
Rizwana BF, Prasana JC, Muthu S, Abraham CS (2019) Molecular docking studies, charge transfer excitation and wave function analyses (ESP, ELF, LOL) on valacyclovir: a potential antiviral drug. Comput Biol Chem 78:9–17
Case DA, Cheatham Iii TE, Darden T, Gohlke H, Luo R, Merz KM Jr, Onufriev A, Simmerling C, Wang B, Woods RJ (2005) The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688
Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ (2016) Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 11:905–919
Singh T, Biswas D, Jayaram B (2011) AADS-an automated active site identification, docking, and scoring protocol for protein targets based on physicochemical descriptors. J Chem Inf Model 51:2515–2527
Gupta A, Gandhimathi A, Sharma P, Jayaram B (2007) ParDOCK: an all atom energy based Monte Carlo docking protocol for protein-ligand complexes. Protein Pept Lett 14:632–646
Jayaram B, Singh T, Mukherjee G, Mathur A, Shekhar S, Shekhar V (2012) Sanjeevini: a freely accessible web-server for target directed lead molecule discovery. BMC Bioinform 13:S7
Lotfi Shahreza M, Ghadiri N, Mousavi SR, Varshosaz J, Green JR (2018) A review of network-based approaches to drug repositioning. Brief Bioinform 19:878–892
Barabási A-L, Gulbahce N, Loscalzo J (2011) Network medicine: a network-based approach to human disease. Nat Rev Genet 12:56–68
Bösl K, Ianevski A, Than TT, Andersen PI, Kuivanen S, Teppor M, Zusinaite E, Dumpis U, Vitkauskiene A, Cox RJ (2019) Common nodes of virus–host interaction revealed through an integrated network analysis. Front Immunol 10:2186
Yi D, Li Q, Pang L, Wang Y, Zhang Y, Duan Z, Liang C, Cen S (2020) Identification of a broad-Spectrum viral inhibitor targeting a novel allosteric site in the RNA-dependent RNA polymerases of dengue virus and Norovirus. Front Microbiol 11:1440–1440
Van Laarhoven T, Marchiori E (2013) Predicting drug-target interactions for new drug compounds using a weighted nearest neighbor profile. PLoS One 8:e66952
Salata C, Calistri A, Parolin C, Baritussio A, Palù G (2017) Antiviral activity of cationic amphiphilic drugs. Expert Rev Anti-Infect Ther 15:483–492
Weeber M, Klein H, De Jong-Van Den Berg LTW, Vos R (2001) Using concepts in literature-based discovery: Simulating Swanson's Raynaud–fish oil and migraine–magnesium discoveries. J Am Soc Inf Sci Technol 52:548–557
Fleuren WWM, Alkema W (2015) Application of text mining in the biomedical domain. Methods 74:97–106
Krallinger M, Erhardt R-A, Valencia A (2005) Text-mining approaches in molecular biology and biomedicine. Drug Discov Today 10:439–445
Cheng D, Knox C, Young N, Stothard P, Damaraju S, Wishart DS (2008) PolySearch: a web-based text mining system for extracting relationships between human diseases, genes, mutations, drugs and metabolites. Nucleic Acids Res 36:W399–W405
Kuusisto F, Steill J, Kuang Z, Thomson J, Page D, Stewart R (2017) A simple text mining approach for ranking pairwise associations in biomedical applications. AMIA Jt Summits Transl Sci Proc 2017:166–174
Lee S, Kim D, Lee K, Choi J, Kim S, Jeon M, Lim S, Choi D, Kim S, Tan AC et al (2016) BEST: next-generation biomedical entity search tool for knowledge discovery from biomedical literature. PLoS One 11:e0164680
Papanikolaou N, Pavlopoulos GA, Theodosiou T, Vizirianakis IS, Iliopoulos I (2016) DrugQuest - a text mining workflow for drug association discovery. BMC Bioinform 17:182
Clark MF, Adams A (1977) Characteristics of the microplate method of enzyme-linked immunosorbent assay for the detection of plant viruses. J Gen Virol 34:475–483
Fischer C, Torres MC, Patel P, Moreira-Soto A, Gould EA, Charrel RN, De Lamballerie X, Nogueira RMR, Sequeira PC, Rodrigues CDS et al (2017) Lineage-specific real-time RT-PCR for yellow fever virus outbreak surveillance, Brazil. Emerg Infect Dis 23:1867–1871
König A, Yang J, Jo E, Park KHP, Kim H, Than TT, Song X, Qi X, Dai X, Park S et al (2019) Efficient long-term amplification of hepatitis B virus isolates after infection of slow proliferating HepG2-NTCP cells. J Hepatol 71:289–300
Laamiri N, Aouini R, Marnissi B, Ghram A, Hmila I (2018) A multiplex real-time RT-PCR for simultaneous detection of four most common avian respiratory viruses. Virology 515:29–37
Landry ML (1990) Nucleic acid hybridization in viral diagnosis. Clin Biochem 23:267–277
Belarbi E, Legros V, Basset J, Desprès P, Roques P, Choumet V (2019) Bioluminescent Ross River virus allows live monitoring of acute and long-term Alphaviral infection by in vivo imaging. Viruses 11:584
De Graaf M, Herfst S, Schrauwen EJ, Van Den Hoogen BG, Osterhaus AD, Fouchier RA (2007) An improved plaque reduction virus neutralization assay for human metapneumovirus. J Virol Methods 143:169–174
Habjan M, Penski N, Spiegel M, Weber F (2008) T7 RNA polymerase-dependent and -independent systems for cDNA-based rescue of Rift Valley fever virus. J Gen Virol 89:2157–2166
Kittel C, Sereinig S, Ferko B, Stasakova J, Romanova J, Wolkerstorfer A, Katinger H, Egorov A (2004) Rescue of influenza virus expressing GFP from the NS1 reading frame. Virology 324:67–73
Lee N, Wong C-K, Chan MCW, Yeung ESL, Tam WWS, Tsang OTY, Choi K-W, Chan PKS, Kwok A, Lui GCY et al (2017) Anti-inflammatory effects of adjunctive macrolide treatment in adults hospitalized with influenza: a randomized controlled trial. Antiviral Res 144:48–56
Perez JT, García-Sastre A, Manicassamy B (2013) Insertion of a GFP reporter gene in influenza virus. Curr Protoc Microbiol 29:15G4.1–15G1.16
Sarzotti-Kelsoe M, Bailer RT, Turk E, Lin CL, Bilska M, Greene KM, Gao H, Todd CA, Ozaki DA, Seaman MS et al (2014) Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J Immunol Methods 409:131–146
Xing L, Wang S, Hu Q, Li J, Zeng Y (2016) Comparison of three quantification methods for the TZM-bl pseudovirus assay for screening of anti-HIV-1 agents. J Virol Methods 233:56–61
Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, Minakhin L et al (2016) C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353:aaf5573
East-Seletsky A, O'connell MR, Knight SC, Burstein D, Cate JH, Tjian R, Doudna JA (2016) Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538:270–273
Sacramento CQ, De Melo GR, De Freitas CS, Rocha N, Hoelz LV, Miranda M, Fintelman-Rodrigues N, Marttorelli A, Ferreira AC, Barbosa-Lima G et al (2017) The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci Rep 7:40920
Denisova OV, Kakkola L, Feng L, Stenman J, Nagaraj A, Lampe J, Yadav B, Aittokallio T, Kaukinen P, Ahola T (2012) Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza a virus infection. J Biol Chem 287:35324–35332
Fink SL, Vojtech L, Wagoner J, Slivinski NSJ, Jackson KJ, Wang R, Khadka S, Luthra P, Basler CF, Polyak SJ (2018) The antiviral drug Arbidol inhibits Zika virus. Sci Rep 8:8989–8989
Rausch K, Hackett BA, Weinbren NL, Reeder SM, Sadovsky Y, Hunter CA, Schultz DC, Coyne CB, Cherry S (2017) Screening bioactives reveals Nanchangmycin as a broad Spectrum antiviral active against Zika virus. Cell Rep 18:804–815
Robinson CL, Chong ACN, Ashbrook AW, Jeng G, Jin J, Chen H, Tang EI, Martin LA, Kim RS, Kenyon RM et al (2018) Male germ cells support long-term propagation of Zika virus. Nat Commun 9:2090
Lee MN, Ye C, Villani AC, Raj T, Li W, Eisenhaure TM, Imboywa SH, Chipendo PI, Ran FA, Slowikowski K et al (2014) Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343:1246980
Zhang Y-H, Zhao Y, Li N, Peng Y-C, Giannoulatou E, Jin R-H, Yan H-P, Wu H, Liu J-H, Liu N (2013) Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nat Commun 4:1–6
Iwasawa C, Tamura R, Sugiura Y, Suzuki S, Kuzumaki N, Narita M, Suematsu M, Nakamura M, Yoshida K, Toda M et al (2019) Increased cytotoxicity of herpes simplex virus thymidine kinase expression in human induced pluripotent stem cells. Int J Mol Sci 20(4):810
Lanko K, Eggermont K, Patel A, Kaptein S, Delang L, Verfaillie CM, Neyts J (2017) Replication of the Zika virus in different iPSC-derived neuronal cells and implications to assess efficacy of antivirals. Antiviral Res 145:82–86
Xia Y, Carpentier A, Cheng X, Block PD, Zhao Y, Zhang Z, Protzer U, Liang TJ (2017) Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J Hepatol 66:494–503
Zhou T, Tan L, Cederquist GY, Fan Y, Hartley BJ, Mukherjee S, Tomishima M, Brennand KJ, Zhang Q, Schwartz RE (2017) High-content screening in hPSC-neural progenitors identifies drug candidates that inhibit Zika virus infection in fetal-like organoids and adult brain. Cell Stem Cell 21 e275:274–283
Padmalayam I, Suto MJ (2012) Chapter twenty-four - 3D cell cultures: mimicking in vivo tissues for improved predictability in drug discovery. In: Desai MC (ed) Annual reports in medicinal chemistry. Academic Press, Cambridge, MA, pp 367–378
Straub TM, Bartholomew RA, Valdez CO, Valentine NB, Dohnalkova A, Ozanich RM, Bruckner-Lea CJ, Call DR (2011) Human norovirus infection of Caco-2 cells grown as a three-dimensional tissue structure. J Water Health 9:225–240
Dollard SC, Wilson JL, Demeter LM, Bonnez W, Reichman RC, Broker T, Chow L (1992) Production of human papillomavirus and modulation of the infectious program in epithelial raft cultures. Genes Dev 6:1131–1142
Koban R, Neumann M, Daugs A, Bloch O, Nitsche A, Langhammer S, Ellerbrok H (2018) A novel three-dimensional cell culture method enhances antiviral drug screening in primary human cells. Antiviral Res 150:20–29
Meyers C, Mane M, Kokorina N, Alam S, Hermonat PL (2000) Ubiquitous human adeno-associated virus type 2 autonomously replicates in differentiating keratinocytes of a normal skin model. Virology 272:338–346
Li C, Deng YQ, Wang S, Ma F, Aliyari R, Huang XY, Zhang NN, Watanabe M, Dong HL, Liu P et al (2017) 25-Hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model. Immunity 46:446–456
Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C (2016) Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165:1238–1254
Watanabe M, Buth JE, Vishlaghi N, De La Torre-Ubieta L, Taxidis J, Khakh BS, Coppola G, Pearson CA, Yamauchi K, Gong D (2017) Self-organized cerebral organoids with human-specific features predict effective drugs to combat Zika virus infection. Cell Rep 21:517–532
Xu Y-P, Qiu Y, Zhang B, Chen G, Chen Q, Wang M, Mo F, Xu J, Wu J, Zhang R-R (2019) Zika virus infection induces RNAi-mediated antiviral immunity in human neural progenitors and brain organoids. Cell Res 29:265–273
Yin Y, Bijvelds M, Dang W, Xu L, Van Der Eijk AA, Knipping K, Tuysuz N, Dekkers JF, Wang Y, De Jonge J (2015) Modeling rotavirus infection and antiviral therapy using primary intestinal organoids. Antiviral Res 123:120–131
Yin Y, Chen S, Hakim MS, Wang W, Xu L, Dang W, Qu C, Verhaar AP, Su J, Fuhler GM (2018) 6-Thioguanine inhibits rotavirus replication through suppression of Rac1 GDP/GTP cycling. Antiviral Res 156:92–101
Yin Y, Wang Y, Dang W, Xu L, Su J, Zhou X, Wang W, Felczak K, Van Der Laan LJ, Pankiewicz KW (2016) Mycophenolic acid potently inhibits rotavirus infection with a high barrier to resistance development. Antiviral Res 133:41–49
Dick GWA (1952) Zika virus (II). Pathogenicity and physical properties. Trans R Soc Trop Med Hyg 46:521–534
Kumar M, Krause KK, Azouz F, Nakano E, Nerurkar VR (2017) A Guinea pig model of Zika virus infection. Virol J 14:75
Miller LJ, Nasar F, Schellhase CW, Norris SL, Kimmel AE, Valdez SM, Wollen-Roberts SE, Shamblin JD, Sprague TR, Lugo-Roman LA et al (2018) Zika virus infection in Syrian Golden hamsters and strain 13 Guinea pigs. Am J Trop Med Hyg 98:864–867
Sit THC, Brackman CJ, Ip SM, Tam KWS, Law PYT, To EMW, Yu VYT, Sims LD, Tsang DNC, Chu DKW et al (2020) Infection of dogs with SARS-CoV-2. Nature 586(7831):776–778
Alves MP, Vielle NJ, Thiel V, Pfaender S (2018) Research models and tools for the identification of antivirals and therapeutics against Zika virus infection. Viruses 10:593
Lai F, Chen Q (2018) Humanized mouse models for the study of infection and pathogenesis of human viruses. Viruses 10:643
Hirst GK (1947) Studies on the mechanism of adaptation of influenza virus to mice. J Exp Med 86:357–366
Raut S, Hurd J, Blandford G, Heath RB, Cureton RJ (1975) The pathogenesis of infections of the mouse caused by virulent and avirulent variants of an influenza virus. J Med Microbiol 8:127–136
Barré-Sinoussi F, Montagutelli X (2015) Animal models are essential to biological research: issues and perspectives. Future Sci OA 1:63
Shanks N, Greek R, Greek J (2009) Are animal models predictive for humans? Philos Ethics Humanit Med 4:2
Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS (2016) A mouse model of Zika virus pathogenesis. Cell Host Microbe 19:720–730
Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JLM, Guimarães KP, Benazzato C, Almeida N, Pignatari GC, Romero S et al (2016) The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534:267–271
Shellam GR, Sangster MY, Urosevic N (1998) Genetic control of host resistance to flavivirus infection in animals. Rev Sci Tech 17:231–248
Lindenmann J (1962) Resistance of mice to mouse-adapted influenza a virus. Virology 16:203–204
Gueńet J-L (2005) Assessing the genetic component of the susceptibility of mice to viral infections. Brief Funct Genomics 4:225–240
Mestas J, Hughes CC (2004) Of mice and not men: differences between mouse and human immunology. J Immunol 172:2731–2738
Bouvier NM, Lowen AC (2010) Animal models for influenza virus pathogenesis and transmission. Viruses 2:1530–1563
Hemmila E, Turbide C, Olson M, Jothy S, Holmes KV, Beauchemin N (2004) Ceacam1a mice are completely resistant to infection by murine coronavirus mouse hepatitis virus A59. J Virol 78:10156–10165
Dick GW, Kitchen SF, Haddow AJ (1952) Zika virus. I. Isolations and serological specificity. Trans R Soc Trop Med Hyg 46:509–520
Bueno MG, Martinez N, Abdalla L, Duarte Dos Santos CN, Chame M (2016) Animals in the Zika virus life cycle: what to expect from Megadiverse Latin American countries. PLoS Negl Trop Dis 10:e0005073
Aliota MT, Dudley DM, Newman CM, Mohr EL, Gellerup DD, Breitbach ME, Buechler CR, Rasheed MN, Mohns MS, Weiler AM et al (2016) Heterologous protection against Asian Zika virus challenge in rhesus macaques. PLoS Negl Trop Dis 10:e0005168
Dudley DM, Aliota MT, Mohr EL, Weiler AM, Lehrer-Brey G, Weisgrau KL, Mohns MS, Breitbach ME, Rasheed MN, Newman CM et al (2016) A rhesus macaque model of Asian-lineage Zika virus infection. Nat Commun 7:12204
Haddow AD, Nalca A, Rossi FD, Miller LJ, Wiley MR, Perez-Sautu U, Washington SC, Norris SL, Wollen-Roberts SE, Shamblin JD et al (2017) High infection rates for adult macaques after Intravaginal or Intrarectal inoculation with Zika virus. Emerg Infect Dis 23:1274–1281
Koide F, Goebel S, Snyder B, Walters KB, Gast A, Hagelin K, Kalkeri R, Rayner J (2016) Development of a Zika virus infection model in Cynomolgus macaques. Front Microbiol 7:2028
Li X-F, Dong H-L, Huang X-Y, Qiu Y-F, Wang H-J, Deng Y-Q, Zhang N-N, Ye Q, Zhao H, Liu Z-Y et al (2016) Characterization of a 2016 clinical isolate of Zika virus in non-human Primates. EBioMedicine 12:170–177
Nguyen SM, Antony KM, Dudley DM, Kohn S, Simmons HA, Wolfe B, Salamat MS, Teixeira LBC, Wiepz GJ, Thoong TH et al (2017) Highly efficient maternal-fetal Zika virus transmission in pregnant rhesus macaques. PLoS Pathog e1006378:13
Osuna CE, Lim S-Y, Deleage C, Griffin BD, Stein D, Schroeder LT, Omange RW, Best K, Luo M, Hraber PT et al (2016) Zika viral dynamics and shedding in rhesus and cynomolgus macaques. Nat Med 22:1448–1455
Rayner JO, Kalkeri R, Goebel S, Cai Z, Green B, Lin S, Snyder B, Hagelin K, Walters KB, Koide F (2018) Comparative pathogenesis of Asian and African-lineage Zika virus in Indian rhesus Macaque’s and development of a non-human primate model suitable for the evaluation of new drugs and vaccines. Viruses 10:229
Waldorf KMA, Stencel-Baerenwald JE, Kapur RP, Studholme C, Boldenow E, Vornhagen J, Baldessari A, Dighe MK, Thiel J, Merillat S (2016) Fetal brain lesions after subcutaneous inoculation of Zika virus in a pregnant nonhuman primate. Nat Med 22:1256–1259
Morris J, Blount R Jr, Savage R (1956) Recovery of cytopathogenic agent from chimpanzees with goryza. Proc Soc Exp Biol Med 92:544–549
Taylor G (2017) Animal models of respiratory syncytial virus infection. Vaccine 35:469–480
Kuz'min VE, Artemenko AG, Muratov EN, Volineckaya IL, Makarov VA, Riabova OB, Wutzler P, Schmidtke M (2007) Quantitative structure−activity relationship studies of [(Biphenyloxy)propyl]isoxazole derivatives. Inhibitors of human rhinovirus 2 replication. J Med Chem 50:4205–4213
Zeng QX, Wang HQ, Wei W, Guo TT, Yu L, Wang YX, Li YH, Song DQ (2020) Synthesis and biological evaluation of berberine derivatives as a new class of broad-spectrum antiviral agents against Coxsackievirus B. Bioorg Chem 95:103490
Allen SJ, Mott KR, Ghiasi H (2014) Inhibitors of signal peptide peptidase (SPP) affect HSV-1 infectivity in vitro and in vivo. Exp Eye Res 123:8–15
Gekonge B, Bardin MC, Montaner LJ (2015) Short communication: Nitazoxanide inhibits HIV viral replication in monocyte-derived macrophages. AIDS Res Hum Retrovir 31:237–241
Hickson SE, Margineantu D, Hockenbery DM, Simon JA, Geballe AP (2018) Inhibition of vaccinia virus replication by nitazoxanide. Virology 518:398–405
La Frazia S, Ciucci A, Arnoldi F, Coira M, Gianferretti P, Angelini M, Belardo G, Burrone OR, Rossignol JF, Santoro MG (2013) Thiazolides, a new class of antiviral agents effective against rotavirus infection, target viral morphogenesis, inhibiting viroplasm formation. J Virol 87:11096–11106
Li Z, Brecher M, Deng YQ, Zhang J, Sakamuru S, Liu B, Huang R, Koetzner CA, Allen CA, Jones SA et al (2017) Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res 27:1046–1064
Mazur I, Wurzer WJ, Ehrhardt C, Pleschka S, Puthavathana P, Silberzahn T, Wolff T, Planz O, Ludwig S (2007) Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF-kappaB-inhibiting activity. Cell Microbiol 9:1683–1694
Pan T, Peng Z, Tan L, Zou F, Zhou N, Liu B, Liang L, Chen C, Liu J, Wu L et al (2018) Nonsteroidal anti-inflammatory drugs potently inhibit the replication of Zika viruses by inducing the degradation of AXL. J Virol 92:e01018-18
Perelygina L, Hautala T, Seppänen M, Adebayo A, Sullivan KE, Icenogle J (2017) Inhibition of rubella virus replication by the broad-spectrum drug nitazoxanide in cell culture and in a patient with a primary immune deficiency. Antiviral Res 147:58–66
Piacentini S, La Frazia S, Riccio A, Pedersen JZ, Topai A, Nicolotti O, Rossignol JF, Santoro MG (2018) Nitazoxanide inhibits paramyxovirus replication by targeting the fusion protein folding: role of glycoprotein-specific thiol oxidoreductase ERp57. Sci Rep 8:10425
Rossignol JF (2014) Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antiviral Res 110:94–103
Shi Z, Wei J, Deng X, Li S, Qiu Y, Shao D, Li B, Zhang K, Xue F, Wang X et al (2014) Nitazoxanide inhibits the replication of Japanese encephalitis virus in cultured cells and in a mouse model. Virol J 11:10
Stachulski AV, Pidathala C, Row EC, Sharma R, Berry NG, Iqbal M, Bentley J, Allman SA, Edwards G, Helm A et al (2011) Thiazolides as novel antiviral agents. 1. Inhibition of hepatitis B virus replication. J Med Chem 54:4119–4132
Wang Y-M, Lu J-W, Lin C-C, Chin Y-F, Wu T-Y, Lin L-I, Lai Z-Z, Kuo S-C, Ho Y-J (2016) Antiviral activities of niclosamide and nitazoxanide against chikungunya virus entry and transmission. Antiviral Res 135:81–90
Fiore AE, Fry A, Shay D, Gubareva L, Bresee JS, Uyeki TM (2011) Antiviral agents for the treatment and chemoprophylaxis of influenza --- recommendations of the advisory committee on immunization practices (ACIP). MMWR Recomm Rep 60:1–24
The Medical Letter (2014) A combination of ledipasvir and sofosbuvir (Harvoni) for hepatitis C. Med Lett Drugs Ther 56:111–112
The Medical Letter (2015) Triumeq--a 3-drug combination for HIV. Med Lett Drugs Ther 57:7–8
Fu Y, Gaelings L, Söderholm S, Belanov S, Nandania J, Nyman TA, Matikainen S, Anders S, Velagapudi V, Kainov DE (2016) JNJ872 inhibits influenza a virus replication without altering cellular antiviral responses. Antiviral Res 133:23–31
Kuivanen S, Bespalov MM, Nandania J, Ianevski A, Velagapudi V, De Brabander JK, Kainov DE, Vapalahti O (2017) Obatoclax, saliphenylhalamide and gemcitabine inhibit Zika virus infection in vitro and differentially affect cellular signaling, transcription and metabolism. Antiviral Res 139:117–128
Ianevski A, Yao R, Fenstad MH, Biza S, Zusinaite E, Reisberg T, Lysvand H, Løseth K, Landsem VM, Malmring JF et al (2020) Potential antiviral options against SARS-CoV-2 infection. Viruses 12:642
Ianevski A, Andersen PI, Merits A, Bjørås M, Kainov D (2019) Expanding the activity spectrum of antiviral agents. Drug Discov Today 24:1224–1228
Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46:D1074–D1082
Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34:D668–D672
Ursu O, Holmes J, Bologa CG, Yang JJ, Mathias SL, Stathias V, Nguyen DT, Schürer S, Oprea T (2019) DrugCentral 2018: an update. Nucleic Acids Res 47:D963–D970
Thorn CF, Klein TE, Altman RB (2013) PharmGKB: the pharmacogenomics knowledge base. Methods Mol Biol 1015:311–320
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242
Hodis E, Prilusky J, Martz E, Silman I, Moult J, Sussman JL (2008) Proteopedia - a scientific 'wiki' bridging the rift between three-dimensional structure and function of biomacromolecules. Genome Biol 9:R121–R121
The Uniprot Consortium (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res 45:D158–D169
Bolton EE, Wang Y, Thiessen PA, Bryant SH (2008) PubChem: integrated platform of small molecules and biological activities. Annu Rep Comput Chem 4:217–241
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B et al (2019) PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 47:D1102–D1109
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, Mcglinchey S, Michalovich D, Al-Lazikani B et al (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40:D1100–D1107
Chen J, Swamidass SJ, Dou Y, Bruand J, Baldi P (2005) ChemDB: a public database of small molecules and related chemoinformatics resources. Bioinformatics 21:4133–4139
Pickett BE, Greer DS, Zhang Y, Stewart L, Zhou L, Sun G, Gu Z, Kumar S, Zaremba S, Larsen CN et al (2012) Virus pathogen database and analysis resource (ViPR): a comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses 4:3209–3226
Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z et al (2012) ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res 40:D593–D598
Hulo C, De Castro E, Masson P, Bougueleret L, Bairoch A, Xenarios I, Le Mercier P (2011) ViralZone: a knowledge resource to understand virus diversity. Nucleic Acids Res 39:D576–D582
Dilly SJ, Morris GS (2017) Pimping up drugs recovered, superannuated and under exploited drugs - an introduction to the basics of drug Reprofiling. Curr Drug Discov Technol 14:121–126
Ashburn TT, Thor KB (2004) Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3:673–683
Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, Doig A, Guilliams T, Latimer J, Mcnamee C et al (2019) Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 18:41–58
Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM (2020) The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res 178:104787
Furuta Y, Komeno T, Nakamura T (2017) Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci 93:449–463
Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K (2005) Mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother 49:981–986
González Canga A, Sahagún Prieto AM, Diez Liébana MJ, Fernández Martínez N, Sierra Vega M, García Vieitez JJ (2008) The pharmacokinetics and interactions of ivermectin in humans--a mini-review. AAPS J 10:42–46
Warren TK, Jordan R, Lo MK, Ray AS, Mackman RL, Soloveva V, Siegel D, Perron M, Bannister R, Hui HC et al (2016) Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 531:381–385
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Yao, R., Ianevski, A., Kainov, D. (2021). Safe-in-Man Broad Spectrum Antiviral Agents. In: Liu, X., Zhan, P., Menéndez-Arias, L., Poongavanam, V. (eds) Antiviral Drug Discovery and Development. Advances in Experimental Medicine and Biology, vol 1322. Springer, Singapore. https://doi.org/10.1007/978-981-16-0267-2_12
Download citation
DOI: https://doi.org/10.1007/978-981-16-0267-2_12
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0266-5
Online ISBN: 978-981-16-0267-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)