
Abstract
Cancer still exists as one of the most alarming disease throughout the world. Predictions put forward a count of 13 million fatalities due to cancer by the year 2030 which shows the indispensable need for scientific interventions against cancer. In recent years, dysregulation of the redox balance has been demonstrated as an important cause for cancer development, progression, and subsequent metastasis in the human cells. This disruption in redox homeostasis is mediated through the upregulation of free radicals which are predominantly found to be reactive oxygen species (ROS). ROS plays a crucial role in tissue homeostasis, regulation of cellular signaling, differentiation, and survival. In addition, ROS regulates cellular homeostasis and also acts as a chief modulator in the process of cellular dysfunction resulting in disease pathophysiology. Elevated levels of the dysregulated ROS subsidize the detrimental processes like tumorigenesis, cancer progression, and spreading. On the other hand, excessive levels of ROS will subsequently result in an anti-tumorigenic effect facilitated by the promotion of cell-death, induction of cell-cycle arrest, and senescence. Thus, ROS acts as a double-edged sword with both pro and anti-tumorigenic effects leading to a duo of detrimental and beneficial role in cancer biology. Chronic inflammation is often associated with elevated levels of ROS and RNS that give rise to several epigenetic changes, DNA mutations, and genomic instability which in turn promotes tumor initiation, development, progression, metastatic dissemination, and treatment resistance. This chapter will summarize in detail the production of ROS, the link between oxidative-stress-induced inflammation in cancer, chronic inflammation and cancer, the role of ROS in the tumor microenvironment, and regulation of ROS.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
Introduction
Regardless of continuous efforts in advancements and development of novel treatment strategies, cancer still exists as one of the most alarming disease throughout the world. Being a dreadful disease, cancer remains an unconquerable elusive target for scientists in both developing and developed countries. Predictions put forward a count of 13 million fatalities due to cancer by the year 2030 which shows the indispensable need for scientific interventions against cancer. Also, the multifaceted presentation of cancer further worsens the complexity of the disease. In recent years, dysregulation of the redox balance has been demonstrated as an important cause for cancer development, progression, and subsequent metastasis in the human cells. This disruption in redox homeostasis is mediated through the upregulation of free radicals which are predominantly found to be reactive oxygen species (ROS) (The Lancet 2018; Aggarwal et al. 2019).
ROS is a group of highly reactive molecules that is produced as a result of aerobic metabolism. ROS plays a crucial role in tissue homeostasis, regulation of cellular signaling, differentiation, and survival. In addition, ROS regulates cellular homeostasis and also acts as a chief modulator in the process of cellular dysfunction resulting in disease pathophysiology (Forrester et al. 2018; Harris and Denicola 2020; Jia et al. 2020; Perillo et al. 2020). Disparities in the basal levels of ROS lead to detrimental effects in cells, provoking several disease conditions. Substantial research during the past two decades has unraveled the essentiality of ROS for the process of initiation, progression, angiogenesis, and metastasis of cancer. Elevated levels of the dysregulated ROS subsidize the detrimental processes like tumorigenesis, cancer progression, and spreading. On the other hand, excessive levels of ROS will subsequently result in an anti-tumorigenic effect facilitated by the promotion of cell death, induction of cell-cycle arrest, and senescence. Thus, ROS acts as a double-edged sword with both pro- and anti-tumorigenic effects leading to a duo of detrimental and beneficial role in cancer biology (Liao et al. 2019; Harris and Denicola 2020; Kirtonia et al. 2020). This chapter will summarize in detail about the production of ROS, the link between oxidative-stress-induced inflammation in cancer, chronic inflammation and cancer, the role of ROS in the tumor microenvironment, and regulation of ROS.
Production of ROS
ROS can potentially mediate both pathophysiological and physiological signal transduction. Production of ROS is dependent upon several enzymes and subcellular compartments that are linked to numerous metabolic regulations. Further, any changes in the redox balance can directly influence the diseases that are associated with metabolic dysfunction, proving the importance of controlled levels of ROS. ROS are series of molecules that include superoxide (O2−) – which is moderately reactive and short-lived, hydrogen peroxide (H2O2) – moderately reactive and long-lived, hydroxyl radical (•OH) – extremely short-lived, and the most potent oxidizing species, and peroxyl radical (ROO•) – the most reactive ROS (Mittal et al. 2014; Forrester et al. 2018; Harris and Denicola 2020).
ROS from Mitochondria
Mitochondria are the central source of endogenous ROS. A subtle balance between pro- and antioxidants is a necessitating factor for the proper functioning of the respiratory chain. Mitochondrial respiration, which is relied on a proton gradient and electron transfer, plays a pivotal role in oxidative ATP production. During this process, water is acquired as a resultant product by the reduction of molecular oxygen (O2) in the electron transport chain (ETC). Improper mitochondrial ROS (mitoROS) productions are associated with several detrimental metabolic diseases and inflammatory reactions. MitoROS production generates intracellular ROS like O2˙− and H2O2 at various sites of mitochondria (Forrester et al. 2018; Snezhkina et al. 2019). For example, O2˙− are produced at complex I, complex III, pyruvate dehydrogenase, glycerol 3-phosphate dehydrogenase, 2-oxoglutarate dehydrogenase, and Q oxidoreductase (Brand 2010).
Complex I act as an access point for electrons from NADH to get into the mitochondrial respiratory chain. Interaction of O2 with flavin mononucleotide (FMN) in the presence of a high ratio of matrix NADH/NAD+ results in the production and subsequent release of O2˙− into the mitochondrial matrix (MM). Further, mitoROS are also produced through a two-step process of reverse electron transfer (RET) in complex I. Complex III provides another crucial platform for mitoROS production. Even though complex III produces a low amount of O2˙−, the Qi site of mitochondrial complex III gets inhibited in the presence of antimycin A, which in turn upregulates the production of O2˙− from Qo site. This upregulation results from the interaction of O2 with ubisemiquinone bound to the Qo site. Apart from complex I and III, complex II flavin site also assists in mitoROS production. ROS generated from complex III and glycerol 3-phosphate dehydrogenase is mainly released into intermembrane mitochondrial space (IMS), but further dismutation of O2˙− into H2O2 by Manganese superoxide dismutase (Mn-SOD) leads to the diffusion of H2O2 into MM. Further,mitochondrial aconitase converts H2O2 into •OH through a Fenton reaction in MM (Forrester et al. 2018; Snezhkina et al. 2019). Thus, the indecorous functioning of the powerhouse of the cell powers-up the production of detrimental ROS.
ROS from Oxidase Activity
Apart from mitochondria, numerous oxidases acts as pivotal producers of ROS, among which NADPH oxidases (NOX) play the chief role. One of the most common sources of cytoplasmic ROS (cytoROS) is the NOX family, which includes seven members, namely, NOX1, NOX2, NOX3, NOX4, NOX5, dual oxidase 1 (DUOX1), and DUOX2 (Landry and Cotter 2014). As integral membrane proteins, NOX proteins generate O2− via NADPH electron exchange (transference of electron from NADPH to FAD). Dysfunctional NOX activity can potentially uplift ROS production which in turn enhances cell transformation, tumorigenesis, angiogenesis, tumor growth, and metastasis. Together with NOX activity, enzymes like xanthine oxidase (XOD), nitric oxide synthase (NOS), cyclooxygenase (COX), lipoxygenase (LOX), monoamine oxidases A and B (MAOA, MOAB), diamine oxidase (DAO), acetylpolyamine oxidase (APOA), spermine oxidase (SMO), cytochrome P450 (CYP) oxidase, and lysyl oxidase also produce ROS in their own respective mechanism (Forrester et al. 2018; Liao et al. 2019; Jia et al. 2020; Snezhkina et al. 2019). The aforementioned enzymatic production of ROS plays an indispensable disadvantageous role that leads to several diseases and inflammatory responses .
ROS from Peroxisomes
Peroxisomes play wide-ranging important roles in the living cells which include fatty acid β-oxidation, α-oxidation, amino acid catabolism, ketogenesis, pentose phosphate pathway (PPP), polyamine oxidation, glyoxylate metabolism, and cholesterol and isoprenoid metabolism (Wanders and Waterham 2006). Peroxisomes play an indispensable role in the maintenance of cellular oxidative balance; any disruption or dysfunctionality in its role will facilitate carcinogenesis. Peroxisomes generate a broad range of ROS: O2˙−, H2O2, •OH, and reactive nitrogen species (RNS): nitric oxide (NO•), peroxynitrite (ONOO−). Peroxisomes are one of the chief producers of H2O2 despite the presence of catalase (CAT, which detoxifies H2O2). Disparate to mitochondria, electron transfer in peroxisome will not result in ATP generation, instead, H2O2 is produced from the transfer of the free electrons to H2O. Further, the catalytic activity of various peroxisomal enzymes and spontaneous dismutation of the O2˙− leads to the generation of H2O2 (Forrester et al. 2018; Snezhkina et al. 2019).
Peroxisome generates O2˙− in both membrane and matrix. Xanthine oxidoreductase (XOR) and urate oxidase (UO) are the two enzymes that are responsible for the generation of O2˙− in the matrix and ETC in the peroxisomal membrane acts as an alternative source for O2˙−. XOR catalyzes the reduction of nitrites and nitrates to NO• which reacts with O2˙− to produce the highly reactive ONOO−. Thus, dysregulation of the peroxisome activity and the reduced CAT activity can potentially promote the generation of ROS and oxidative-stress leading to genome instability and DNA damage which in turn facilitates cancer development (Forrester et al. 2018; Snezhkina et al. 2019).
Chronic Inflammation and Cancer
Chronic inflammation is often associated with elevated levels of ROS and RNS that give rise to several epigenetic changes, DNA mutations, and genomic instability which in turn promotes tumor initiation, development, progression, metastatic dissemination, and treatment resistance. Inflammation has various tumor-promoting effects in the tumor microenvironment and is also considered as a recognized hallmark of cancer. This crucial correlation of inflammation and cancer was first documented by Rudolf Virchow which got numerous mechanistic and epidemiological supports in the past decades (Salman and Ashraf 2013; Shalapour et al. 2015).
Inflammation is caused by wide-spread sources including viral and microbial infections, exposure to allergens, toxic chemicals, radiation, consumption of tobacco, and alcohol. In general, inflammation is a sort of protective mechanism, but prolonged persistent inflammation can potentially cause harmful effects and damages to the body cells and tissues. The two phases of inflammation are acute and chronic inflammation. Acute inflammation is often a short-time beneficial inflammatory response mediated by the innate immune system. Whereas, in the second phase, prolonged inflammation referred to chronic inflammation can potentially persuade chronic illness like cancer. In the process of inflammation, recruitment of mast cells and leukocytes to the damage-site leads to a condition of “respiratory burst” which is a resultant event of elevated uptake of oxygen that further upshots an upregulated release and buildup of ROS at the damage site (Reuter et al. 2010; Salman and Ashraf 2013).
ROS is involved in all three stages of cancer – cancer initiation, promotion, and progression. In the initial stage of cancer (initiation), ROS causes gene mutation, structural alterations, and damages to DNA. During the promotion stage, ROS leads to upregulated cell proliferation or a reduction in apoptosis due to its detrimental contribution to the blockade of cell-to-cell communication, abnormal gene expression, alteration of second-messenger systems. Toward the final stage, ROS worsens the initiated cell population by adding DNA alternations. Along with this, ROS potentially triggers some of the signaling pathways including numerous transcription factors like nuclear factor κB (NF-κB), activator protein-1 (AP-1), specificity protein (Sp-1), p53, and mitogen-activated protein kinase phosphatases (MKP). Activation of these transcriptional factors leads to angiogenesis, cell proliferation, and metastasis. Similarly, inflammatory cells also release a diverse set of soluble mediators and metabolites like arachidonic acid, chemokines, and cytokines which further recruit more inflammatory cells to the damage-site and ultimately produce an elevated level of reactive species. These inflammatory mediators trigger changes in transcriptional factors and activate several deleterious signal transduction cascades. Numerous transcriptional factors are involved in this process which includes NF-κB, hypoxia-inducible factor-1α (HIF1-α), signal transducer and activator of transcription 3 (STAT3), nuclear factor of activated T cells (NFAT), AP-1, and NF-E2-related factor-2 (Nrf2), which are responsible for facilitating instantaneous cellular responses. Additionally, abnormal expressions of inflammatory chemokines (CXC chemokine receptor-4 (CXCR4), Interleukin-8 (IL-8)), and cytokines (IL-1, IL-6, tumor necrosis factor (TNF)) have also been reported for its critical role in this process of oxidative-stress induced inflammation. Thus, the scenario of sustained oxidative-inflammatory environment leads to a pernicious cycle of events which not only damages the same cell but also damages the healthy neighboring cells and thereby leads to carcinogenesis (Reuter et al. 2010; Salman and Ashraf 2015).
Oxidative-Stress-Induced Inflammation
Inflammation is a normal host response as a result of infections or other stimuli. It is a primary reaction of a tissue to eliminate pathogen or infected tissue components to restore the normal physiological functions (Wu et al. 2014). Immune cells such as phagocytic macrophages, Polymorph nuclear neutrophils (PMNs), eosinophil are the integral part of innate immune response and play a vital role in fighting against the infection through the generation of various reactive species such as Hydrogen Peroxide (H2O2), Superoxide, Nitrous oxide, Hydroxyl radical, and peroxynitrite (Azad et al. 2008). The species help in invading and eliminating the pathogen. Often, inflammation subsides after the removal of infectious agents or on the completion of tissue repair mechanism. However, continuous tissue injuries and regeneration will increase the production of ROS from the inflammatory cells causing damage to the healthy cells (Walser et al. 2008). ROS interacts with epithelial DNA resulting in mutation and chromosomal alterations. As a response to DNA damage, the cells activate p53 genes associated with cell cycle and DNA repair process. But when the rate of ROS is increased, it leads to chronic inflammation. The chronic inflammation provides a platform for recurring DNA damage, rich inflammatory cells, increased ROS, proliferating growth factors, and other growth-inducing factors that ultimately increase the frequency of mutation. All put together facilitates the progression of transforming the cells into their malignant state therby increasing the risk of tumorigenesis (Li et al. 2013).
Molecular Mechanisms of ROS-Induced Carcinogenesis
ROS was found to be the key component in inducing inflammation. Over the past few years, it was reported that the expression and synthesis of nitric oxide were upregulated by various cytotoxins during cell division and cell repair, further complicating the process of inflammation (Azad et al. 2008). With all the comprehensive understanding and available studies, it can be stated that the effects of ROS over inflammation may be beneficial or harmful depending on the cell type and physiological conditions involved (Walser et al. 2008). The molecular mechanisms involved in inflammation-induced carcinogenesis are analogous to the ROS generating potential of the cells.
The elevated risk of developing cancer is due to the behavior of damaged cells with an imbalance in cell division and cell repair mechanisms (Salman and Ashraf 2013). Generally, all cancer cells develop permanent functional changes to the DNA leading to oxidative damage in the bases, also attacking the proteins and lipids of the cells. Oxidative modification of DNA polymerase or inhibition of DNA repair enzymes indirectly promotes mutagenesis. DNA adduct formation mediated by ROS elevates the risk of carcinogenesis, further stimulating the oncogenes such as jun and fos (Azad et al. 2008).
The inflammatory cells produce certain growth factors and transcription factors including NF-κB, STAT3, AP-1, hypoxia-inducible factor-1 (HIF-1), as well as altered expression of specific microRNAs in cancer cells that promote the expression of genes related to cell growth, apoptosis, and invasion (Li et al. 2013).
DNA Damage
Extensive studies have been conducted to show that DNA damage including alterations in the native structure such as base pair insertion/deletion, base modifications, chromosomal changes, and microsatellite instability and translocation of segments was due to the direct interaction of ROS to the DNA. Other than ROS-induced damage, DNA is subjected to more than 100 different oxidative modifications (Walser et al. 2008). Most importantly, the hydroxyl radical (OH•) is the major source of DNA damage that influences the phosphates, bases, and deoxyribose ultimately resulting in strand damage. Interaction of hydroxyl radicals with the deoxyribose of the DNA can give rise to single and double-stranded breaks. The most frequent base modifications observed in DNA are 8-oxo-7,8-dihydroguanine (8-oxoG) 2,6-diamino-4-hydroxy-5-formamidopyrimidine resulting from the addition of Hydroxyl radical to the eighth position of the guanine ring producing oxidized end products (Srinivas et al. 2018). The single-strand breaks and the oxidized bases of DNA give rise to chromosomal instability and can act as major contributors in tumorigenesis. Further, alterations in the DNA methylation patterns of genes suggest that ROS is also responsible for the epigenetic changes and tumorigenic effects due to the oxidative stress undergone by various sites in the genome (Reuter et al. 2010).
Role of ROS in DNA Damage Induced by Replication Stress/Other Factors
Among the various sources of endogenous oxidative DNA damage, replication stress induced by oncogenes plays a key role. Any unusual replication fork during DNA replication results in replication stress to which the oncogenes are often associated (Salman and Ashraf 2013). Frequent alterations in the pro-oncogenes cause replication stress in the DNA inducing genomic instability that helps in tumor development (Sesti et al. 2012). As the oncogene is activated and modified, the risk of ROS is hiked to influence the DNA replication. ROS rich environment does not allow the cells to recruit antioxidant proteins for the recovery of cellular stress (Srinivas et al. 2018). Recent studies suggest that the source of most ROS accompanying acute or chronic inflammation is the NADPH Oxidase (NOX) factor. In a study performed with human tumor cell lines, it was found that the expression of NOX protein to generate ROS increases the risk of inflammation by recruitment of several inflammatory mediators (Wu et al. 2014). ROS production by NOX has a critical role in modifying the inflammatory responses and DNA damage with respect to cancer progression. It was also implicated that a multifold increase in p53 mutation causing loss of DNA repair mechanism is due to the overexpression of NOX1 complex (Yang et al. 2018).
Cell Signaling Cascades in ROS-Mediated Inflammation and Cancer
ROS functions as an important modulator of signaling mechanisms in cancer initiation and progression. A series of pathological events including cell proliferation, apoptosis, infiltration of inflammatory mediators and growth factors, and transcription of genes that are encoded for inflammatory proteins are activated as a result of oxidative stress mediated by ROS and RNS (Reuter et al. 2010). The signaling cascade mediated by ROS induces the activation and phosphorylation of mitogen-activated protein kinase (MAPK), which results in the activation of the NF-κB, AP-1, and STAT. These factors ultimately make the genes such as jun and fos (immediate early genes) to get activated, which involves inflammatory influx, cell proliferation, transformation, and differentiation. When the mechanisms associated with the oxidative upregulation of these factors are persistent, the molecular changes resulting from these pathways elicit the progression of cells eventually leading to cancer (Gào and Schöttker 2017).
Endogenous free radical arises as secondary messengers form the inflammatory neutrophils or macrophage activation during various cell signaling pathways or as by-products of P50 metabolism, NADPH oxidase activity, and peroxisome activities. Besides the aforementioned transcription factors, ROS also finds a role in regulating the growth factors and certain Kinases or phosphates. The effect of ROS is partly mediated by a subfamily of MAKPs – Extracellular signal-regulated Kinases (ERKs) that mediate the cell proliferation, stress responses, and apoptosis throughout the signaling cascade (Sesti et al. 2012).
Transcription Factors – NF-κB, STAT, AP-1, HIF-1
AP-1 and NF-κB are the most important factors which are sensitive to ROS. Oxidants such as H2O2 and inflammatory cytokines such as TNF-α activate AP-1 and NF- κB and were reported to modulate the expression of pro-inflammatory genes leading to initiation of neoplastic transformation and recruitment of several genes involved in cell proliferation, differentiation, inflammation, and tumor development (Azad et al. 2008). Activation of NF-κB by phosphorylation-dependent proteasome degradation of IκBα facilitates the accumulation of the NF-κB in the nucleus. Upon accumulation, NF-κB binds to the κB elements in the promoter regions of genes encoding pro-inflammatory cytokines, INOX, and COX-2, which are involved in the inflammation-associated carcinogenesis. NF-κB regulated genes were found to play a vital role in the modulation of intracellular levels. Conversely, ROS was shown to activate NF-κB by direct oxidation of the transcription factor (Zhang et al. 2016).
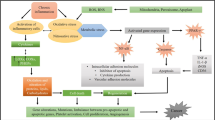
STAT is a redox-sensitive factor that mainly acts as a switch between the inflammation and cancer. Once activated, STAT-3 dimers get translocated to the nucleus and bind to the promoter regions of genes encoding inflammatory and cell cycle regulatory proteins. Oxidation of STAT-3 by H2O2 enhances the proliferation of tumor-initiating cells; protects the normal and premalignant cells from apoptosis thereby promoting tumor progression (Wu et al. 2014). Hypoxia Inducing Factor-1 (HIF-1) mediates the responses to chronic hypoxia characterized by reduced oxygen availability to the cells. NOX-derived ROS have been shown to influence the hypoxic conditions by increasing the HIF-1α synthesis and involving in the regulation o inflammatory cytokines (VEGF and IL-6), ultimately resulting in cell aggregation and tumor development (Aggarwal et al. 2019) (Fig. 1).

Schematic illustration of key molecular events and cell signaling cascades induced by ROS, leading chronic inflammation and cancer
Apoptosis and Survival
Apoptosis is a normal physiological process by which unwanted or damaged cells are eliminated from the body during normal biological processes. It is characterized by cell shrinkage, membrane blebbing, and breakdown of DNA (Sesti et al. 2012). Imbalance in the rate of cell division and cell death in normal tissues favors the neoplastic development and cancer cells use increased survival and decreased death strategies to sustain proliferation and evade apoptosis (Azad et al. 2008). ROS such as H2O2 has been associated with the cell survival responses at lower doses whereas higher doses of ROS activate apoptosis. High ROS doses activate tumor suppressor gene p53 which plays a key role in oxidative-induced stress or other cellular stress responses. This induces cell cycle arrest to promote DNA repair and cell death by apoptosis (Ivanova et al. 2016).
Apoptosis induced by H2O2 is linked with the increased protein expression of p53, PUMA, NOXA, and Bax and phosphorylation of p53 in several cancer types. The mitochondrial pathway of apoptosis is induced as a response to cellular stresses including DNA damage, growth factor deprivation, hypoxia, and oxidative-stress. ROS have been strictly associated with the mitochondrial apoptosis. Mitochondria are the habitat of intracellular ROS and are produced by leakage from the respiratory ETC (Kuwabara et al. 2008). Mitochondrial ROS targets the mtDNA and causes impaired transcription of proteins in the ETC which further increases ROS generation leading to loss of membrane potential and loss of ATP synthesis (Redza-Dutordoir and Averill-Bates 2016). These series of events finally result in apoptosis. Superoxide and H2O2 can cause cytochrome release from the mitochondria and initiates the mitochondrial apoptosis. In a study with HeLa cells, oxidative-stress induced apoptosis was mediated by the upstream regulation of p53 through caspase-dependent and caspase-independent mechanisms involving loss of membrane permeability and release of apoptosis-inducing factor from the mitochondria, respectively (Redza-Dutordoir and Averill-Bates 2016).
ROS function both as supporting and opposing factor in tumor development. Activation of pro-apoptotic signaling molecules such as apoptosis regulating kinase (ASK1), c-jun N-terminal Kinase (JNK), and p38, mediated by ROS can initiate apoptosis. As a denial, NF-κB induced by ROS comes into play to inhibit the process of apoptosis leading to neoplastic development in various cell types (Kuwabara et al. 2008). Different ROS with varying doses involves antiapoptotic roles by inactivating caspases, induction of p53 gene expression, and upregulation of proteins such as Flip, Bcl-2, and Bcl-X. Hence, ROS exhibits pro and antiapoptotic roles depending on the cellular redox state and the types of cells involved. ERK, a member of MAPK, and Protein kinase B (PKB) are the leading factors in cell survival induced by ROS (Redza-Dutordoir and Averill-Bates 2016). It has been reported that when cells are exposed to H2O2, phosphoinositide 3-kinase (PI3K) is upregulated, and PKB is activated bringing about the cell survival pathways. Several studies have shown that the oxidative stress by different sources of ROS stimulates two opposite pathways leading to cell death and cell survival and can simultaneously be suppressed by antioxidizing agents (Kuwabara et al. 2008) (Fig. 2).

Oxidative-stress induced by ROS leading to apoptosis and cell survival by activation of the MAPK pathway
Inflammatory Markers and ROS
Some of the notable inflammatory proteins associated with the NOX activation and ROS overproduction are matrix metalloproteinase-9 (MMP-9), intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule (VAM), COX2, cytosolic phospholipase 2 (cPLA2). It has been known that cells release inflammatory mediators, cytokines, and chemokines such as IL-1β, IL-6, IL-8, and TNF-α, as a response to oxidative stress of various sources. The inflammatory mediators have a crucial role to play in the process of chronic inflammation and are capable of directing the nature of the inflammatory responses by selective recruitment and activation of inflammatory cells. Direct or indirect sources of ROS activate the epithelial cells to induce pro-inflammatory cells. The genes of the inflammatory mediators are regulated by the redox-sensitive transcription factors including AP-1 and NF-κB (Lee and Yang 2012).
The relationship between the role of ROS in cancer and prolonged inflammation has been experimentally validated by several studies. The experimental data showed that the inflammatory responses regulate cancer based on the release of cytokines, chemokines in the tumor microenvironment either by providing an antitumor response or by inducing cell transformation and malignancy (Forrester et al. 2018). ROS can influence the inflammatory mediators creating crosstalk between the chronic inflammation and cancer progression through their endogenous accumulation. The inflammatory cells will eventually lead to a massive generation of ROS by upregulating oxidant-associated enzymes (Policastro and Notcovich 2013). In general, ROS activates NF-κB in response to inflammatory signals through amino acid phosphorylation mechanisms. Specific makers of inflammation use ROS as a part of the signaling pathways. IL-1β involves NOX2-derived ROS that induces the recruitment of endosomal recruitment and TNF-α induced NF-κB activation increases antioxidant expression, thereby lowering the apoptotic signaling through the JNK pathway (Lee and Yang 2012).
Many evidences prove the role of mitochondrial ROS in mediating the inflammatory signaling on pathogen exposure and also during the malignant state of tumors. Mitochondrial H2O2 production contributes to the activation of NF-κB and inhibits Hypoxia-induced NF-κB activation and IL-6 secretion leading to tumor growth. Mitochondrial ROS signaling is considered primary in regulating the inflammasome activation involving IL-1β and IL-18 transcription and apoptosis regulating Caspase1 (Lee and Yang 2012). Fully activated inflammasome with the lysosomal destabilization governs the IL-1β maturation, which in turn reflects in the apoptosis of damaged cells. Lysosomal fusion requires translocation of the microtubule-associated protein to the phagosome, induced by mitochondrial ROS. The TLR signaling induces the recruitment of mitochondrial proteins to the phagosome where ROS kills phagocytosed pathogens and increases the NOX-dependent ROS production. ROS production also plays a role in the activation of NF-κB in T-cells and regulates their metabolic programming. Overall, ROS acts as primary regulators of inflammatory response with respect to NF-κB activation and inflammasome signaling (Forrester et al. 2018).
Tumor Microenvironment
The tumor microenvironment is a complex mixture of multiple cell types, tumor supporting matrix, and several additional factors that can aid and assist in tumor growth. Recruitment of fibroblasts, immune cells, and vasculature associated cells by the malignant cells initiate tumors and drive tumor progression (Gu et al. 2018). As the tumor progresses, the extracellular matrix (ECM) being a part of the tumor mass, provides structural support for tumor development and modulates the tumor microenvironment as well. The components of tumor microenvironment include malignant cells and non-cancer stromal cells, ECM components, tumor lymphatic and vessels, and inflammatory cells as well. The characteristics of the tumor microenvironment may vary according to the redox state of the cells including Hypoxia, angiogenesis, tumor metabolism, and cell signaling (Weinberg et al. 2019).
It is widely recognized that tumor microenvironments are thoroughly influenced by ten important characteristics of cancer. The characteristics include the following:
-
(i)
Unlimited multiplication
-
(ii)
Escaping from growth suppressors
-
(iii)
Maintaining proliferative signaling
-
(iv)
Resisting apoptosis
-
(v)
Genome instability and mutation
-
(vi)
Promoting invasion and metastasis
-
(vii)
Stimulating angiogenesis
-
(viii)
Eliminating cell energy limitations
-
(ix)
Tumor-enhanced inflammation
-
(x)
Evading immune destruction of foreign bodies
Components and Characteristics of TME
ECM and stroma form a basement membrane that can serve as a storage reservoir of growth factors and chemokines to stimulate tumorigenesis. Normal fibroblasts are responsible for tissue homeostasis and are primarily in the process of wound healing. In contrast, cancer-associated fibroblasts (CAFs) are infiltrating into tumor cells establishing crucial roles in cancer initiation, progression, and metastasis. Alpha smooth muscle actin myofibroblasts are found to be the major subtype of CAFs present in tumors. CAFs are more proliferative than the normal fibroblasts and can activate specific signaling pathways significant for the promotion and progression of cancer (Whiteside 2008; Wang et al. 2017; Weinberg et al. 2019).
Desmoplasia is an important marker of tumor growth and can generate mechanical forces that will limit the blood supply to the tumor by compressing the vessels thereby creating a hypoxic environment. Excess production of CAFs identified in the aggressive tumors express smooth muscle actin (α-SMA) and is termed as myofibroblasts whose main function is wound healing and tissue repair. ROS is an important factor in the differentiation of fibroblasts to myofibroblasts. TGF-β1 also plays a major role in the transition of fibroblast to myofibroblasts (Wang et al. 2017). It is known that the mitochondrial ROS activates TGF-β1 in the signaling of inflammation-induced cancer. When fibroblasts become devoid of mitochondrial ROS, the expression of TGF-β1 is reduced, wherein cancer progression is eventually subsided. Several studies have suggested that ROS shows an impact on the subtypes of fibroblasts. One such subtype affected by ROS is the platelet-derived growth factor β (PDGF- β) and ROS could be an integral part of the fibroblast proliferation and migration. ROS produced by the tumor cell can facilitate the reprogramming of CAFs. ROS finds a role in disturbing the routine of tumor-infiltrating T cells depending on the levels of ROS (Policastro and Notcovich 2013).
The main function of immune cells is to maintain the tissue homeostasis, to protect against the invading pathogens and to eliminate damaged cells. However, the immune-inflammatory cells persist in the sites of chronic inflammation leading to diverse tissue pathologies and neoplasia (Whiteside 2008). Few studies on immune-system research have suggested that the infiltration of immune-inflammatory cells may be the early initiation of cancer. The involvement of immune cells in cancer development can be divided into 3 stages: elimination, equilibrium, and escape. In the elimination phase, the immune system defeats nascent tumors, achieved with the help of several signaling molecules and inflammatory factors. Once the cancer cells are eliminated, the active immune cells have an additional role called “immunoediting” wherein the equilibrium stage keeps the tumor growth under control. However, the tumor cells are completely eliminated, and in order to escape the immune surveillance, cancer cells tend to adapt certain phenotypic changes including EMT (Endothelial mesenchyme transition) . With the advantage of surviving, the cancer cells would develop into solid tumors. The immune system helps the cancer cells to accompany the dominant cells so that they grow at the fastest rate in the limited environment. In the escape phase, immune-inflammatory cells can help the cancer cells in altering the tumor immune escape mechanisms and can reduce the antitumor protein or cytokines to promote tumor development (Wang et al. 2017; Gu et al. 2018).
Hypoxia, Angiogenesis, and Metastasis
ROS is viewed as an important signaling molecule rather than merely being a by-product of cellular metabolism (Wu et al. 2014). At low levels, ROS participate in the process of hypoxia adaptation by regulating the stability of HIF-1α. Moderate levels of ROS are involved in the production of inflammatory cytokines and their regulation by directly inactivating MAKP and higher levels of ROS are capable of inducing apoptosis and autophagy (Waris and Ahsan 2006).
Oxygen radicals and insufficiency (hypoxia) cooperatively promote tumor angiogenesis (Xia et al. 2007). Tumor cells generally outgrow their blood supply leading to oxygen deprivation causing hypoxia, which leads to degradation of DNA to its constituent bases. The release of thymidine is catabolized by thymidine phosphorylase which is an important overexpressing enzyme in tumor cells and can cause oxygen radical production (Navaneetha Krishnan et al. 2019). The aforementioned fact was substantiated in Breast Cancer wherein reoxygenation of the tumor after hypoxia will drive additional oxygen radical formation and they are stressed by metabolic alteration and macrophage infiltrations. The accumulation of HIF1 in hypoxia promotes the transcription of Vascular endothelial growth factor (VEGF) resulting in angiogenesis. Oxygen radicals increase the production of VEGF and HIF-1 (Kumari et al. 2018) (Fig. 3).

High levels of ROS leading to cancer metastasis and induction of angiogenesis by ROS via hypoxia-dependent and -independent pathways
ROS in Cancer Metastasis
Metastasis is the state where primary tumor cells spread to distant organs and are considered to be the main cause of cancer morbidity and mortality (Aggarwal et al. 2019). Studies on tumor microenvironment have revealed that the tumor metastasis is not an autonomous process but a complex event, occurring due to the bidirectional interaction between the malignant and nonmalignant cells and the mutational burden of cancer cells. The upregulation of NF-κB, metalloproteases, and transforming growth factor beta (TGF-β) causes tumor metastasis (Brown and Bicknell 2001). Epithelial to mesenchymal transition (EMT) is the key component in causing metastasis wherein the epithelial cells tend to lose their cell ashesion, polarity and gain entry into the circulation thereby reaching different tissues at distant site. It has been proved that ROS is majorly involved in causing EMT. TGF-β facilitates cell migration and invasion through ROS-dependent mechanisms. Another study has revealed that ROS can increase tumor migration by inducing hypoxia-mediated protein expression leading to the rapid mobility of the malignant cells (Liao et al. 2019).
Angiogenesis and ROS
In the initial stages of tumorigenesis, new blood vessels are formed from the preexisting vasculatures, popularly called as angiogenesis (Xia et al. 2007). ROS-mediated angiogenesis is initiated by cancer proliferation thereby increasing the metabolic rate leading to increased ROS levels. Elevation of ROS levels in the tumor microenvironment causes oxidative-stress, initiating secretion of angiogenic modulators (Aggarwal et al. 2019). Endogenous and exogenous ROS stimulates growth factors such as VEGF- and HIF-1α-promoting tumor migration and ROS-dependent cellular signaling. ROS mediates the VEGF secretion and activates the PI3K pathway and is additionally modulating the cancer progression (Xia et al. 2007). It has been reported that the epidermal growth factor (EGF) leads to increased production of H2O2 activating the PI3K pathway resulting in overexpression of VEGF. Further NADPH oxidase 2 (NOX-2)-derived ROS was reported to induce cancer progression and migration that are regulated by the proto-oncogene tyrosine protein kinase (tpk) pathway, inducing angiogenesis (Ushio-Fukai and Nakamura 2008). In human endothelial cells, it was reported that Angiotensin 1 induced the release of ROS by activating the endothelial-specific tyrosine kinase receptor leading to vascular remodeling. The hypoxia-dependent pathway mediated by the phosphoinositide-3-kinase regulatory subunit or serine-threonine kinase mechanisms increase the expression of VEGF via the activation of MAPK involving HIF-1α and causes upregulation of matrix metalloproteinase (MMP2 & MMP9) ultimately leading to angiogenesis. Hypoxia independent pathway results in angiogenesis through the activation of NF-κB via Toll-like receptors (TLRs) where the membrane lipid ligands are oxidized exogenous ROS (Aggarwal et al. 2019).
Regulation of ROS
ROS homeostasis is essential for proper cell signaling and cell survival, where the optimal levels of ROS trigger numerous signaling pathways that are responsible for the regulation of several crucial functions like differentiation, cellular proliferation, and metabolic adaptations in an organized manner. The increased production of spatially localized ROS in the cancerous cells hyperactivates various cell signaling pathways, which are required for detrimental cell transformation and tumorigenesis. In the process of ROS production, levels of ROS are delimited by several factors in the tumor microenvironment. Accumulation of ROS can either be the result of increased ROS production or decreased ROS elimination (Trachootham et al. 2009; Sena and Chandel 2012; Sabharwal and Schumacker 2014; Chen et al. 2016).
ROS production is restricted by several checkpoints of NOXs, following the activation of receptors through ligands like insulin, transforming growth factor, platelet-derived growth factor, fibroblast growth factor, nerve growth factor, epidermal growth factor, and tumor necrosis factor-α (TNF-α). A tumor tissue becomes hypoxic when the tumor diameter reaches 200 μm, wherein the state of hypoxia regulates the transcription of Nrf2 and thereby reduces the ROS accumulation. Matrix metalloproteinases (MMPs) are found to be a crucial regulator of the mitochondrial respiratory chain and intracellular ROS production. Furthermore, cellular metabolism, glucose metabolism, and mitochondrial respiratory chain are also associated with regulation and generation of ROS (Chen et al. 2016).
To maintain the level of ROS and to protect the cells from ROS-induced damages, cells use their extensive antioxidant defense system, either enzymatic (peroxidases, dismutases, and catalases) or nonenzymatic (glutathione (GSH), vitamin A, C, and E). Superoxide dismutases (SODs) are placed in several cellular compartments and play a crucial role in the rapid conversion of O2− to H2O2. In order to avoid cellular toxicity, levels of H2O2 are maintained in an optimal range for proper cell signaling. Numerous antioxidants like peroxiredoxins (PRXs), catalases (CAT), and glutathione peroxidases (GPXs) are responsible for this task, wherein these antioxidants convert the intracellular H2O2 into water (H2O). Thus, for the purpose of the regulation of intracellular ROS levels, cells have a robust antioxidant system where SODs are responsible for the dismutation of O2− into H2O2 which in turn removed/converted by CAT, PRXs, and GPXs to generate H2O (Chen et al. 2016; Reczek and Chandel 2017).
Both, generation and detoxification of ROS can be promoted by NADPH, where NADPH can be generated through multiple generative mechanisms in both cytosol and mitochondria. In that line, one-carbon metabolism, a pathway centered around folate, where carbon units from serine and glycine feed to the folate cycle generates NADPH in both cytosol and mitochondria. Unsurprisingly, cancer cell proliferation is enhanced by the upregulated one-carbon metabolism, where the serine catabolism mediated through mitochondrial one-carbon metabolism sustains the redox balance and thereby allows cancer cell proliferation in tumor hypoxia. Hypoxia-inducible factor (HIF) collaborates with Myc in the Myc-transformed cells during the process of hypoxia to persuade the expression of serine hydroxymethyltransferase 2 (SHMT2, mitochondrial one-carbon metabolism enzyme) which in turn elevates the production of NADPH. Furthermore, this increase of NADPH generation leads to the counterbalance of hypoxia-induced escalation in mitoROS by maintaining the antioxidant capacity (Reczek and Chandel 2017).
Conclusion
ROS can influence the inflammatory mediators creating crosstalk between the chronic inflammation and cancer progression through their endogenous accumulation. The inflammatory cells will eventually lead to a massive generation of ROS by upregulating oxidant-associated enzymes. The relationship between the role of ROS in cancer and prolonged inflammation has been experimentally validated by several studies. The inflammatory mediators selectively recruit inflammatory cells and serves as the key players in mediating the inflammatory responses. During chronic inflammation, the immune-inflammatory cells persist in the sites of inflammation causing intense tissue pathologies and neoplasia. Several studies have been conducted to understand the infiltration of inflammatory cells, when ROS causes obstruction to the normal cellular signaling, which would eventually result in the early initiation of cancer. Low levels of ROS induce the activation of HIF-1α and regulates it stability to participate in the adaptation of hypoxia. On the other hand, moderate levels of ROS are involved in transcription of inflammatory genes resulting in cell survival and cancer development and high levels of ROS are responsible for inducing apoptosis and autophagy. Oxygen radicals and insufficiency (hypoxia) cooperatively promote tumor angiogenesis. Overall, the extensive research during the past two decades have substantially revealed the role of oxidative stress, and its mediators can lead to chronic inflammation, which in turn can activate multiple inflammation pathways suggesting a close link between oxidative stress, chronic inflammation, and cancer.
References
Aggarwal V, Tuli HS, Varol A et al (2019) Role of reactive oxygen species in cancer progression: molecular mechanisms and recent advancements. Biomol Ther 9. https://doi.org/10.3390/biom9110735
Azad N, Rojanasakul Y, Vallyathan V (2008) Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev 11:1–15. https://doi.org/10.1080/10937400701436460
Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45:466–472. https://doi.org/10.1016/j.exger.2010.01.003
Brown NS, Bicknell R (2001) Oxidative stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res 3:323–327. https://doi.org/10.1186/bcr315
Chen X, Song M, Zhang B, Zhang Y (2016) Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxidative Med Cell Longev 2016:11–16. https://doi.org/10.1155/2016/1580967
Forrester SJ, Kikuchi DS, Hernandes MS et al (2018) Reactive oxygen species in metabolic and inflammatory signaling:877–902. https://doi.org/10.1161/CIRCRESAHA.117.311401
Gào X, Schöttker B (2017) Reduction-oxidation pathways involved in cancer development: a systematic review of literature reviews. Oncotarget 8:51888–51906. https://doi.org/10.18632/oncotarget.17128
Gu H, Huang T, Shen Y et al (2018) Review article reactive oxygen species-mediated tumor microenvironment transformation: the mechanism of radioresistant gastric cancer. 2018. https://doi.org/10.1155/2018/5801209
Harris IS, Denicola GM (2020) The complex interplay between antioxidants and ROS in Cancer:1–12. https://doi.org/10.1016/j.tcb.2020.03.002
Ivanova D, Zhelev Z, Aoki I et al (2016) Overproduction of reactive oxygen species – obligatory or not for induction of apoptosis by anticancer drugs. Chin J Cancer Res 28:383–396. https://doi.org/10.21147/j.issn.1000-9604.2016.04.01
Jia P, Dai C, Cao P et al (2020) The role of reactive oxygen species in tumor treatment. RSC Adv 10:7740–7750. https://doi.org/10.1039/c9ra10539e
Kirtonia A, Sethi G, Garg M (2020) The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol Life Sci. https://doi.org/10.1007/s00018-020-03536-5
Kumari S, Badana AK, Murali Mohan G et al (2018) Reactive oxygen species: a key constituent in cancer survival. Biomark Insights 13. https://doi.org/10.1177/1177271918755391
Kuwabara M, Asanuma T, Niwa K, Inanami O (2008) Regulation of cell survival and death signals induced by oxidative stress. J Clin Biochem Nutr 43:51–57. https://doi.org/10.3164/jcbn.2008045
Landry WD, Cotter TG (2014) ROS signalling, NADPH oxidases and cancer. Biochem Soc Trans 42:934–938. https://doi.org/10.1042/BST20140060
Lee I, Yang C (2012) Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol 84:581–590. https://doi.org/10.1016/j.bcp.2012.05.005
Li X, Fang P, Mai J et al (2013) Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol 6:1–19. https://doi.org/10.1186/1756-8722-6-19
Liao Z, Chua D, Tan NS (2019) Reactive oxygen species: a volatile driver of field cancerization and metastasis. Mol Cancer 18:1–10. https://doi.org/10.1186/s12943-019-0961-y
Mittal M, Siddiqui MR, Tran K et al (2014) Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20:1126–1167. https://doi.org/10.1089/ars.2012.5149
Navaneetha Krishnan S, Rosales JL, Lee KY (2019) ROS-mediated cancer cell killing through dietary phytochemicals. Oxidative Med Cell Longev 2019. https://doi.org/10.1155/2019/9051542
Perillo B, Di Donato M, Pezone A et al (2020) ROS in cancer therapy: the bright side of the moon. Exp Mol Med 52:192–203. https://doi.org/10.1038/s12276-020-0384-2
Policastro LL, Notcovich C (2013) The tumor microenvironment: characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxid Redox Signal 19(8):854–895. https://doi.org/10.1089/ars.2011.4367
Reczek CR, Chandel NS (2017) The two faces of reactive oxygen species in cancer. Annu Rev Cancer Biol 1:79–98. https://doi.org/10.1146/annurev-cancerbio-041916-065808
Redza-Dutordoir M, Averill-Bates DA (2016) Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta, Mol Cell Res 1863:2977–2992. https://doi.org/10.1016/j.bbamcr.2016.09.012
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49:1603–1616. https://doi.org/10.1016/j.freeradbiomed.2010.09.006
Sabharwal SS, Schumacker PT (2014) Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14:709–721. https://doi.org/10.1038/nrc3803
Salman KA, Ashraf S (2013) Reactive oxygen species: a link between chronic inflammation and cancer. Asia Pac J Mol Biol Biotechnol 22(2):42–49
Salman KA, Ashraf S (2015) Reactive oxygen species: a link between chronic inflammation and cancer. Asia Pac J Mol Biol Biotechnol 22:42–49
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48:158–167. https://doi.org/10.1016/j.molcel.2012.09.025
Sesti F, Tsitsilonis OE, Kotsinas A, Trougakos IP (2012) Oxidative stress-mediated biomolecular damage and inflammation in tumorigenesis. 402:395–402
Shalapour S, Karin M, Shalapour S, Karin M (2015) Immunity, inflammation, and cancer: an eternal fight between good and evil find the latest version: immunity, inflammation, and cancer : an eternal fight between good and evil. J Clin Invest 125:3347–3355. https://doi.org/10.1172/JCI80007.The
Snezhkina AV, Kudryavtseva AV, Kardymon OL et al (2019) ROS generation and antioxidant defense systems in normal and malignant cells. Oxidative Med Cell Longev 2019:6175804
Srinivas S, Tan B, Vellayappan A, Anand D (2018) Author’ s accepted manuscript. Redox Biol:101084. https://doi.org/10.1016/j.redox.2018.101084
The Lancet (2018) GLOBOCAN 2018: counting the toll of cancer. Lancet 392:985. https://doi.org/10.1016/S0140-6736(18)32252-9
Trachootham D, Alexandre J, Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8:579–591. https://doi.org/10.1038/nrd2803
Ushio-Fukai M, Nakamura Y (2008) Reactive oxygen species and angiogenesis NADPH oxidase as. Cancer Lett 266:37–52. https://doi.org/10.1016/j.canlet.2008.02.044.Reactive
Walser T, Cui X, Yanagawa J et al (2008) Smoking and lung cancer: the role of inflammation. Proc Am Thorac Soc 5:811–815. https://doi.org/10.1513/pats.200809-100TH
Wanders RJA, Waterham HR (2006) Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem 75:295–332. https://doi.org/10.1146/annurev.biochem.74.082803.133329
Wang M, Zhao J, Zhang L et al (2017) Role of tumor microenvironment in tumorigenesis. J Cancer:8. https://doi.org/10.7150/jca.17648
Waris G, Ahsan H (2006) Reactive oxygen species: role in the development of cancer and various chronic conditions. 8:1–8. https://doi.org/10.1186/1477-3163-5-14
Weinberg F, Ramnath N, Nagrath D (2019) Reactive oxygen species in the tumor. Cancers (Basel) 11(8):1191
Whiteside TL (2008) The tumor microenvironment and its role in promoting tumor growth. Oncogene 27:5904–5912. https://doi.org/10.1038/onc.2008.271
Wu Y, Antony S, Meitzler JL, Doroshow JH (2014) Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett 345:164–173. https://doi.org/10.1016/j.canlet.2013.08.014
Xia C, Meng Q, Liu LZ et al (2007) Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res 67:10823–10830. https://doi.org/10.1158/0008-5472.CAN-07-0783
Yang H, Villani RM, Wang H et al (2018) The role of cellular reactive oxygen species in cancer chemotherapy. J Exp Clin Cancer Res 37:1–10. https://doi.org/10.1186/s13046-018-0909-x
Zhang J, Wang X, Vikash V et al (2016) ROS and ROS-mediated cellular Signaling. Oxidative Med Cell Longev 2016. https://doi.org/10.1155/2016/4350965
Acknowledgment
The authors express their gratitude to SASTRA-Deemed-to-be-University, Tamil Nadu, India, for infrastructure and financial support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Singapore Pte Ltd.
About this entry

Cite this entry
Muralidar, S., Gopal, G., Ambi, S.V. (2022). ROS-Mediated Inflammatory Response in Cancer. In: Chakraborti, S., Ray, B.K., Roychoudhury, S. (eds) Handbook of Oxidative Stress in Cancer: Mechanistic Aspects. Springer, Singapore. https://doi.org/10.1007/978-981-15-9411-3_181
Download citation
DOI: https://doi.org/10.1007/978-981-15-9411-3_181
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-9410-6
Online ISBN: 978-981-15-9411-3
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences