
Abstract
The DNA bases in the genome are susceptible to oxidation by reactive oxygen species (ROS). The oxidized DNA base lesions such as 8-oxoguanine (8-oxoG) and thymine glycol are primarily repaired by the base excision repair (BER) pathway. Increasing pieces of evidence suggest that oxidative stress-induced base damages in the gene promoter serve as epigenetics marks to regulate gene expression by recruitment of BER proteins. To shed light on this novel role of oxidative DNA base modifications and BER proteins, in this chapter, we focus on how controlled guanine oxidation in gene promoters and BER proteins 8-oxoguanine DNA glycosylase (OGG1) and AP-endonuclease 1 (APE1) regulate expression of multiple genes that drive cancer progression and metastases. Further, we highlight the current studies suggesting a novel role of 8-oxoG and BER in regulating the formation of noncanonical DNA secondary structures such as G-quadruplexes (G4s) to regulate gene expression. Since high oxidative stress in tumor cells creates selective pressure on DNA damage repair pathways to maintain sustained growth, invasion, and metastases via modulating gene expression, such efforts to address the intertwined roles of DNA base modification, BER pathway, and gene expression could broaden cancer-therapeutic strategies.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Oxidized DNA base
- 8-Oxoguanine
- Base excision repair
- OGG1
- APE1
- AP site
- Histone demethylation
- G-quadruplex structure
- Gene expression
Introduction
The genome is inherently unstable, and chemical modifications and damaging agents continuously challenge its integrity. Persistent exposure to exogenous and endogenous reactive oxygen species (ROS) generated by metabolic and inflammatory processes can cause DNA base oxidation, one of the most frequent insults to the genome (Winterbourn 2008). ROS comprise of a group of highly reactive molecules such as superoxide anion (O2˙−), and hydroxyl radical (OH•) – some potent oxidants which can directly react with DNA and produce oxidized bases (Schieber and Chandel 2014). Oxidation of purines and pyrimidines can generate multiple modified DNA bases (Bauer et al. 2015). ROS can also directly generate single-strand breaks (SSBs) with nonligatable termini. Deoxyribose in DNA is also prone to oxidation, which usually results in base hydrolysis to form an abasic site and/or SSB (Bauer et al. 2015). Among the four canonical nucleotides in DNA, G base has the lowest redox potential, resulting in G being the most susceptible to oxidative modification (Cadet et al. 2014; Fleming and Burrows 2017). Products of G oxidation by ROS include two-electron oxidation product 8-oxo-7-8-dihydroguanine (8-oxoG), which can pair with A during replication and leading to G to T transversion mutation (Grollman and Moriya 1993). Thus, oxidative damage to DNA may lead to mutations accumulation in the genome, and this eventually results in pathological consequences such as cancer (Cooke et al. 2003).
The base excision repair (BER) pathway is the primary mechanism for repairing oxidative, alkylated base lesions, and SSBs in the genome (Lindahl and Barnes 2000; Mitra et al. 2002). BER is an evolutionarily conserved DNA damage repair pathway that is involved in maintaining genomic integrity by removing mutagenic lesions induced due to oxidative stress. BER relies on sequential recruitment and coordinated actions of multiple enzymes, which include DNA glycosylases (such as OGG1), AP-endonucleases (APE1), DNA polymerases, and DNA ligases (Scott et al. 2014). Several studies have demonstrated that in the absence of BER enzymes, cells accumulate mutations and are susceptible to various DNA-damaging agents. Thus, one can envision that BER plays a crucial role in cancer prevention. Overexpression of BER proteins confers protection of the cell genome from oxidative stress and toxicity to DNA-damaging drugs. Studies have shown elevated levels of APE1 in diverse cancer tissues and its association with chemo and radiation resistance (Abbotts and Madhusudan 2010; Sengupta et al. 2016).
Given the unavoidable exposure to ROS, cells have evolved strategies to utilize ROS as a biological stimulus or signaling molecule to regulate multiple cellular processes, including cell signaling and transcription. An emerging view in transcriptional regulation is that base oxidation in DNA, which is conventionally thought to be mutagenic and detrimental for cells, has an essential regulatory role in controlling transcription and gene expression. Despite high mutagenic potential of 8-oxoG , GC content in the promoter regions of genes in the vertebrate genome is high (Deaton and Bird 2011). This evolutionarily conserved high GC content has been linked with gene expression (Ba and Boldogh 2018). Indeed, low-resolution mapping of 8-oxoG in mammalian cells by several laboratories found that 8-oxoG is enriched in gene promoters and within potential G-quadruplex-forming sequences (PQSs) (Amente et al. 2019; Fang and Zou 2020). The promoter region of many proto-oncogenes, including c-MYC, KRAS, and VEGF, contains PQS that can fold to noncanonical DNA G-quadruplex (G4) structures to regulate their expression (Fleming et al. 2017a; Pastukh et al. 2015). Over the years, the intertwined roles of the 8-oxoG, BER pathway, and transcriptional regulation have merged as a novel epigenetic mechanism for regulating gene expression. The initial cellular observation of controlled G oxidation in VEGF gene promoter upon hypoxia driving the production of Hif-1 protein was reported by Gillespie’s laboratory (Pastukh et al. 2015, 2007). Later, several other laboratories, including ours, have provided evidence that oxidation of bases in these G-rich sequences may serve as critical sensors through which the cells sense the ROS signals, and BER proteins OGG1 and APE1 regulate the transcription of oxidative stress-responsive genes (Fleming et al. 2017a; Pan et al. 2016; Roychoudhury et al. 2020). In this chapter, the roles of oxidative base 8-oxoG and BER enzymes – OGG1 and APE1 in transcriptional regulation – are discussed in detail with an emphasis on how they facilitate the assembly of the transcriptional activators at gene promoters to promote gene expression. Further, we shed light on our current understanding of how oxidative base modification and BER proteins regulate the formation of higher-order DNA secondary structures such as G4s to regulate expression of many oncogenes and promote tumorigenesis.
Oxidative DNA Base Damage Shaping the Mutation Burden in Cancer
Cancer cells are characterized by persistent oxidative stress and high levels of ROS. Out of the several different DNA base derivatives that can arise due to ROS, 8-oxoG is one of the major oxidized bases in DNA that pairs with adenine (A) as well as cytosine (C) during DNA replication, potentially leading to G:C to T:A transversion mutations (Grollman and Moriya 1993) (Fig. 1). Left unrepaired, 8-oxoG can compromise transcription, DNA replication, and telomere maintenance, and being a highly mutagenic lesion, it has been associated with cellular transformation and cancer initiation. Elevated levels of 8-oxoG and other oxidative lesions have been found in the urine and tumor tissue DNA from patients with various malignancies (Guo et al. 2017). The levels of 8-oxoG are considered as a factor in risk assessment in cancer (Loft et al. 2012). Apart from 8-oxoG, oxidation of G can also result in the formation of 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG), a major oxidative lesion (Cadet and Wagner 2013).

A schematic representation of how unrepaired oxidative base damage (8-oxoG) in the DNA can lead to mutation in the genome
8-Oxo-7,8-dihydro-2′-deoxyadenosine (8-oxoA) and 4,6-diamino-5-formamidopyrimidine (FapyA) are the two major products that can arise from the oxidation of A. C can also be oxidized to form 5-hydroxy-2′-deoxycytidine (OH5C), spontaneously on the DNA, or can arise after exposure to ROS generating chemicals (Bjelland and Seeberg 2003; Cadet and Wagner 2013). T can be attacked by free radicals resulting in the generation of different oxidative products. Thymine glycol (Tg) can inhibit replicative polymerases and is highly mutagenic (McCulloch and Kunkel 2008). The 5-methyl group of T can be oxidized to form 5-hydroxymethyluracil (5hmU), which induces transition mutations (Maiti and Drohat 2011).
All these mutagenic events pose a serious threat to the DNA and might lead to genetic instability and malignant transformation. Defective DNA maintenance pathways have been associated with distinct signatures that arise due to these mutations (Alexandrov and Stratton 2014). Though the entire course of malignancy is associated with different mutational processes, there can be specific mutational types that are dominant in a particular type of cancer. For instance, a high proportion of T to G and C to T base mutations was found in the immunoglobulin genes of chronic lymphocytic leukemia and melanoma patients (Alexandrov et al. 2013; Puente et al. 2015; Hodis et al. 2012).
Oxidative DNA Base Damage Repair
Considering the high frequency at which DNA damaging events occur, and the broad spectrum of damages incurred by the cells, it is remarkable that a majority of these DNA lesions are repaired with impressive precision and efficiency by a host of DNA damage repair pathways.
As mentioned before, the BER pathway is the primary mechanism that protects the genome from the deleterious effects of exposure to ROS. Being the most versatile among the excision repair pathways, BER repairs multiple types of modified (oxidized or alkylated) DNA bases as well as DNA single-strand breaks (SSBs) (Hegde et al. 2008; Lindahl and Barnes 2000; Robertson et al. 2009). BER is a highly coordinated multistep cellular process that includes (i) damaged base removal, (ii) 3′- or 5-end cleaning, (iii) DNA synthesis, and (iv) DNA nick-sealing (Scott et al. 2014). It is generally initiated by a lesion-specific DNA glycosylase, which removes the damaged base. Among 11 human DNA glycosylases, 8-oxoguanine DNA glycosylase 1 (OGG1), NTH1, endonuclease VII-like (NEIL) 1, NEIL2, and NEIL3 are primarily involved in reaping the oxidized base damages from DNA. Once the base lesions are removed, the product apurinic/apyrimidinic (AP) sites or SSBs with nonligatable termini are a substrate for the next enzyme, AP endonuclease 1 (APE1). APE1 cleaves AP site generating 3′-hydroxyl and 5′-deoxyribose-phosphate (5′-dRP) end which is recognized by DNA polymerase β (pol β). Pol β fills the single nucleotide gap by incorporating a complementary base, and finally, DNA ligase III (Lig III) seals the nick (Fig. 2). Of note is that each of these reactions consequently leaves a new intermediate lesion in the DNA, which is harmful if the entire repair process is not completed through nick-sealing by the DNA ligases (Wilson and Kunkel 2000). Thus, BER is dependent on sequential recruitment and coordinated activities of multiple proteins via the formation of a series of transient repair complexes that assemble at the site of the DNA lesion (Hegde et al. 2012).

A schematic representation of the steps involved in the BER pathway
Apart from the core proteins mentioned above, poly (ADP-ribose) polymerase (PARP) and X-ray repair complementing defective repair in Chinese hamster cells 1 (XRCC1) do not directly participate in the DNA processing but form a scaffold and interact with BER proteins such as APE1 and Lig III which are critical for BER efficiency (El-Khamisy et al. 2003). Furthermore, several other non-BER proteins that are known to be involved in transcription or RNA metabolism have been demonstrated to regulate the activity of BER enzymes (Das et al. 2007). Such auxiliary proteins interact with BER proteins to enhance the overall DNA repair efficiency and coordinate repair and transcription.
The Interplay Between Posttranslational Modification of BER Proteins and Transcriptional Regulation
Reversible acetylation of histones plays a profound role in epigenetic regulation of gene expression. Many studies have shown the role of acetylation of BER proteins in the regulation of the BER pathway and gene expression (Bhakat et al. 2009; Carter and Parsons 2016). We discovered that the major human glycosylases, OGG1, NEIL1, and NEIL2, which cleave most oxidatively damaged bases, are acetylated at specific lysine (K) residues in cells (Bhakat et al. 2004, 2006; Sengupta et al. 2018). Acetylation of OGG1 at K338 and K341 increased the activity of OGG1 (Bhakat et al. 2006). OGG1 acetylation was enhanced in cells after oxidative stress, suggesting a DNA damage-dependent activation of OGG1 acetylation due to its acetylation. We also showed that NEIL1 is acetylated at multiple K residues (K296, K297, and K298) located in the intrinsically unstructured C-terminal domain, and acetylation of NEIL1 enhanced its glycosylase activity in vitro (Sengupta et al. 2018). We discovered that human APE1 is also acetylated at multiple K residues located in the unstructured N-terminal region (Bhakat et al. 2003; Lirussi et al. 2012). APE1 is acetylated in chromatin, and acetylation of APE1 enhances its AP-endonuclease activity in vitro and modulates its transcriptional coregulatory functions (Roychoudhury et al. 2017). Notably, while unmodified OGG1 and APE1 are present in cytosol and nucleus, acetylated OGG1 and APE1 are exclusively bound to chromatin (Bhakat et al. 2006; Roychoudhury et al. 2017).
Using unbiased genome-wide mapping of AP sites, a common intermediate in the BER pathway, and binding of acetylated APE1 and acetylated OGG1, we have documented that endogenous oxidative base damages are not randomly located (Roychoudhury et al. 2020). There is a distinct distribution of oxidative damage and enrichment of acetylated OGG1 or acetylated APE1 predominately in the promoter/enhancer regions of the transcribed genes suggesting a connection of DNA base damage or assembly of repair complexes with the regulation of gene expression. Acetylated APE1-bound regions significantly overlap the regions that bear active enhancer and promoter histone marks (Roychoudhury et al. 2020). Studies have shown the interaction of APE1 with transcription activator or repressor factors such as STAT, HIF-1α, AP-1, NF-κB, and HDAC1 in multiple gene promoters (Bhakat et al. 2009; Evans et al. 2000; Xanthoudakis and Curran 1992). Acetylated APE1 was found to be present in the protein complexes that bind to negative calcium response element in gene promoter (Bhakat et al. 2003). Binding of Y-box-binding protein 1 (YB-1) to multidrug resistance gene MDR1 was shown to be dependent on APE1’s acetylation (Sengupta et al. 2011, Chattopadhyay et al. 2008). Further, we demonstrated elevated levels of AcAPE1 in diverse cancer types. The loss of APE1 acetylation resulted in altering the expression of hundreds of genes and enhanced tumor cells’ sensitivity to chemotherapeutic agents (Sengupta et al. 2016).
8-OxoG and OGG1 as a Novel Epigenetic Regulator for Controlling Gene Expression
Both transcription and DNA repair have intimate transactions with the DNA, and these two processes are often coupled and are perhaps also interdependent and cross-functional. A growing list of DNA damage repair proteins that were thought to function in repair pathways solely is increasingly becoming important in transcriptional regulation as well. 8-oxoG is the most prevalent oxidized DNA base found in the genome. Studies have shown that a controlled or localized oxidation of G in a specific region in the genome, such as promoter/enhancer, can occur in a signal-dependent manner through localized histone demethylation by LSD1, a flavin-dependent monoamine oxidase that generates H2O2 (Shi et al. 2005). The locally formed H2O2 drives G oxidation in the discrete regions leading to the recruitment of OGG1 and activation BER pathway to promote induction of transcription (Perillo et al. 2008). Furthermore, supporting this, several laboratories found that 8-oxoG is enriched in gene promoters and within G-rich PQS or G4 sequences (see details below). Lastly, the genome-wide mapping of OGG1 (the primary enzyme responsible for removing 8-oxoG from DNA) further supports that oxidative base damage is not random; they are enriched in promoter regions in the mammalian genome (Ba and Boldogh 2018; Roychoudhury et al. 2020).
BER in Hypoxia-Induced VEGF mRNA Expression
The first evidence for a direct connection between oxidized DNA bases, BER pathway, and gene regulation came from the studies by Gillespie’s group. A study by Pastukh et al. demonstrated the hypoxia-mediated formation of 8-oxoG in hypoxic response elements (HREs) in the VEGF gene promoter (Pastukh et al. 2007). Hypoxia-induced oxidative stress was shown to promote the assembly of hypoxia-inducible factor-1 (Hif-1α) and BER enzymes OGG1 and APE1. This recruitment of BER machinery was essential for the assembly of transcription complex on the VEGF HRE and for driving VEGF mRNA expression. Inhibition of the BER pathway resulted in hypoxia-induced accumulation of 8-oxoG and attenuated Hif-1α and APE1 binding to VEGF HRE sequences and blunted VEGF mRNA expression. Moreover, compared to wild-type HRE luciferase reporter, luciferase activity increased with AP-site containing HRE element in reporter plasmid (Pastukh et al. 2015, 2007). As described above, the AP site is a common intermediate generated after the removal of oxidized bases in the BER pathway. As the BER pathway repairs AP sites, these results suggested that oxidative base lesions in promoters of hypoxia-inducible genes are linked with transcriptional activation. More, recent studies have demonstrated that the signal-induced formation of localized oxidized bases in the promoter regions in many oncogene promoters such as KRAS, c-MYC, etc., modulates their expression (Fleming et al. 2017b); however, the molecular basis of G oxidation, AP sites formation, and modulation of their gene expression is different in each case (see below).
Role of 8-OxoG and OGG1 in the Regulation of Inflammatory Response Genes
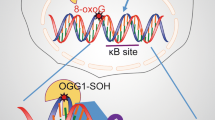
It is well-known that inflammation induces ROS production and that ROS-mediated signaling regulates transcription proinflammatory genes. Previous studies have shown ROS’s role in posttranslational modification of TFs such as NF- κB, and AP-1(Ba and Boldogh 2018). In a study by Ba et al., it was shown that TNF-α-induced ROS production results in elevating 8-oxoG levels in the genome, including the promoter regions (Ba and Boldogh 2018). Simultaneously, the binding of OGG1 also increased Cxcl2 expression by facilitating the loading of TFIID, NF- κB/RelA, Sp1, and p-RNA Pol II (Fig. 3). Depletion of OGG1 reduced transcription factor recruitment and the subsequent TNF-α-mediated innate immune response (Ba et al. 2014).

Binding of OGG1 to 8-oxoG in promoter sequences bends the DNA and augments proinflammatory gene transcription by facilitating the recruitment of site-specific transcription factors
A master regulator of gene expression, nuclear factor κB (NF- κB) , is a transcription factor (TF) necessary for the inflammatory response, cell proliferation, and differentiation (Oeckinghaus and Ghosh 2009). Constitutive NF- κB activity due to the inflammatory microenvironment and the various oncogenic mutations is found in several human cancers. DNA sequences that contain three or more Gs (5’-GGGRNYYYCC-3′ where R is a purine, Y is pyrimidine, and N is any nucleotide) are signatures where NF- κB classically binds. As mentioned before, oxidation of these Gs can lead to the formation of 8-oxoG lesions, which can either change the sequence context of the TF-binding site due to its mutagenicity or directly modulate DNA-protein interactions and thus gene expression. A study by Pan et al. demonstrated that NF- κB-driven gene expression requires 8-oxoG and OGG1 enrichment in the promoter sequences after TNF-α exposure to the cells. OGG1 bound to 8-oxoG facilitates NF- κB’s binding to the DNA (Pan et al. 2016). Hence, binding of OGG1 to its substrate was shown to function in epigenetic regulation of gene expression in cells that are under oxidative stress. Additionally, the Tell laboratory proposed that oxidative stress-induced 8-oxoG in the negative calcium responsive elements (nCaRE) in the promoter of sirtuin-1 (SIRT-1) gene recruits OGG1 (Antoniali et al. 2014).
The Ras pathway is often activated in human cancers. Boldogh et al. demonstrated that 8-oxoG is bound by OGG1 at a nonsubstrate site with high affinity (Boldogh et al. 2012). The study was one of the first to document the DNA repair-independent function of OGG1 in modulating cellular signaling.
Histone Demethylation, Oxidation of DNA, BER, and Gene Expression
Unlike histone acetylation, methylation of histone was considered a permanent and irreversible modification. In 2004, Shi et al. demonstrated that LSD1-mediated removal of the methyl group from both H3K4me2 and H3K4me1 in vitro via Flavin adenine dinucleotide oxidation reaction, generates H2O2 during demethylation (Shi et al. 2004). This demethylation resulted in local oxidation of DNA base around promoter or gene body regions and produced 8-oxoG , which is recognized by OGG1. OGG1 initiates the BER pathway, which can regulate transcription and gene expression (Fig. 4). The first direct evidence of histone demethylation and BER’s involvement in transcriptional regulation came from the study by Perillo et al. (Perillo et al. 2008). The authors showed that treatment with 17-beta-estradiol induced estrogen receptor recruitment to the estrogen-responsive elements in the promoter and enhancer of Bcl-2 and pS2 genes and formed a chromatin loop between the promoter and enhancer to activate the gene expression. Demethylation of H3K9me2 occurred at the promoter and enhancer sites of these genes, and 8-oxoG could be detected after treatment with estradiol. Importantly, siRNA-mediated knockdown of LSD1 abolished the accumulation of 8-oxoG lesions. Removal of 8-oxoG lesions by OGG1 followed by recruitment of APE1 generates an SSB facilitating chromatin looping, which provides a local signal for the assembly of transcriptional initiation complex to activate gene expression. OGG1 appeared to be required to remove 8-oxoG, and APE1 was necessary to complete the repair process. However, no additional downstream BER proteins were evaluated in this initial study. Downregulation of OGG1 and APE1 was used to confirm the functional role of BER in transcriptional control, and it dramatically reduced the promoter-enhancer loop formation and gene expression.

LSD1-mediated histone demethylation in gene promoters gives rise to H2O2, which causes local oxidation of DNA base (8-oxoG), leading to the initiation of BER and assembly of transcription factors (TFs) to drive target gene expression
The same group also demonstrated that DNA oxidation drives Myc-mediated transcription (Amente et al. 2010b). Myc, a transcription factor that is activated in many cancers, contributes to a wide variety of gene regulation involved in cell proliferation, differentiation, and apoptosis (Amente et al. 2010b). They provided a mechanistic framework that links histone demethylation to oxidation of DNA and repairs underlying Myc-activated transcription. They demonstrated that local demethylation of H3K4me2 promotes the recruitment of the OGG1 and APE1 following Myc binding, which produces nicks on the Myc target gene. This subsequently facilitates chromatin loops that bring together RNA polymerase II and Myc. Silencing of OGG1 or APE1 inhibits Myc-mediated transcription (Amente et al. 2010a).
Regulatory Roles of G Oxidation and BER Proteins in G4 Structure Formation in the Genome to Regulate Gene Expression
G-quadruplexes (G4s) are DNA secondary structures that can form when four Gs associate through Hoogsteen hydrogen bonds to form a planar tetrad. Stacking of two or more tetrads forms a G4 structure (Bochman et al. 2012; Burge et al. 2006). G4 DNA structures in key regulatory regions in the genome, such as promoters, replication origins, etc., regulate multiple biological processes, including transcription, replication, and telomere maintenance (Hansel-Hertsch et al. 2016, 2017). There is a growing consensus among Cynthia Burrows’ laboratory, ours suggesting that 8-oxoG and BER pathway plays a pivotal role in G4 structure formation in the genome to regulate gene expression (Fleming et al. 2017a, b; Roychoudhury et al. 2020). G4s are overrepresented in regulatory regions such as gene promoters, 5′ and 3′ untranslated regions, replication initiation sites, and telomeres (Fleming et al. 2019; Hansel-Hertsch et al. 2016). The promoter region of many proto-oncogenes, including c-MYC, KRAS, and VEGF, which are activated by oxidative stress, contains PQS that can fold to G4 to regulate their expression. Through biochemical, cellular, and genetic approaches, Burrows’ laboratory showed that 8-oxoG or AP sites in the PQS of VEGF and endonuclease III-like protein 1(NTHL1) promoter activate transcription of a reporter gene expression (Fleming et al. 2017a). OGG1 and APE1 are required for the induction of transcription. Further, it was found that oxidative modified PQS close to the transcription start site (TSS) can activate transcription when the damaged base and repair chemistry occur in the coding strand (Fleming et al. 2019). On the other hand, modification of PQS in the template strand turned off transcription. Thus formation of 8-oxoG under oxidative stress conditions functions as a signaling or epigenetic mark to promote G4 fold, thus leading to transcriptional activation. This also implies that G oxidation is an epigenetic modification, and G4-forming sequences serve as sensors for oxidative stress.
Similarly, in a separate study, Redstone et al. showed that oxidative modification of PQS in the proliferating cell nuclear antigen (PCNA) gene promoter could turn on its transcription (Redstone et al. 2019). PCNA overexpression has been detected in malignancies ranging from colorectal cancer to breast cancer. The study showed the possible mechanism of PCNA gene activation during oxidative stress conditions. Briefly, the G-rich PQS promoter element of the PCNA gene can undergo oxidation to produce 8-oxoG . This event directs DNA repair via the BER pathway toward the promoter for initiating events that ultimately induce transcription of the gene.
In an independent study by Cogoi et al., it was found that 8-oxoG formation in the PQS of the KRAS oncogene promoter positioned from 148 to 116 relative to the TSS, turned on the transcription (Cogoi et al. 2018). In this study, the authors demonstrated that 8-oxoG in the G4 region of KRAS promoter recruits OGG1. OGG1 removes 8-oxoG from the G4 motif in duplex, but when folded, it binds to the G4 in a nonproductive way. Recruitment of MAZ and hnRNP A1, the two nuclear factors that are essential for KRAS transcription to the KRAS promoter, is enhanced by 8-oxoG (Cogoi et al. 2018). Thus, this oxidative lesion in the G4 region was demonstrated to be a novel player in oncogene expression regulation, adding to other pieces of evidence of the epigenetic potential of oxidative base damage and its associated repair pathway in controlling gene expression.
Although these studies mentioned above have established that an oxidized G or AP site in PQSs of many oncogene promoters such as KRAS, MYC, and VEGF modulates their expression, the requirement of G oxidation, AP sites, and APE1 in the spatiotemporal regulation of gene expression in cells was mostly unknown. We have recently demonstrated a mechanistic framework for the role of oxidized DNA-base-derived AP sites and APE1 in the formation and stability of G4 structures (Roychoudhury et al. 2020). We showed that endogenous oxidized DNA-base-derived AP sites in G4 sequences and subsequent recruitment of APE1 drive the spatiotemporal formation and stability of G4 structures to regulate KRAS and MYC oncogene expression. Mapping of the genome-wide APE1 binding and G4 structures revealed that the binding of APE1 is predominant in PQS sequences and is nonrandom. Furthermore, it was shown that APE1 supports the formation of G4 structures and G4-mediated gene expression. Binding of APE1 to G4 sequences promotes G4 folding, and the acetylation of APE1 stabilizes G4 structures by enhancing its residence time. APE1 subsequently facilitates transcription factor loading to the promoter, leading to G4-mediated gene expression. We postulated that cellular oxidants could oxidize G bases within a PQS into 8-oxoG , which is excised by OGG1 to generate an AP site. Unlike 8-oxoG paired with C, an AP site significantly impacts the thermal stability of duplex DNA (Fleming et al. 2017a). We proposed that the generation of an AP site in a PQS destabilizes the duplex and shifts the equilibrium to form an unstable G4. Subsequent binding of APE1 results in the stabilization of a G4 structure, and acetylation of APE1 by p300 enhances its affinity/interaction to stabilize G4 structure and facilitates the loading of TFs to activate gene expression (Fig. 5). This study highlighted a novel role of oxidized DNA bases and APE1 in controlling the formation of G4 structures to regulate transcription (Fleming et al. 2017a; Roychoudhury et al. 2020).

Schematic representation linking the role of endogenous oxidized DNA base (8-oxoG) in gene regulatory elements and APE1 in controlling the formation of G4 structures to modulate transcription of target genes
Thus, all these studies collectively demonstrated that oxidative base modifications such as 8-oxoG in the gene promoters that contain PQS might serve as epigenetic marks and serve as a common ground to coordinate the initial steps of DNA repair and the assembly of transcriptional machinery to launch adequate alteration of expression of the gene.
Conclusion
Over the years, studies from several laboratories have supported the view that oxidative DNA modifications such as 8-oxoG in the promoter/enhancer regions could serve as an epigenetic mark to regulate gene expression. Additionally, the BER pathway has been deemed necessary for modulating DNA G4 structures and histone demethylation-mediated epigenetic regulation, which are distinct from its well-established role in maintaining genome stability. Defects in BER may affect the formation of G4 structures and reduce promoter-enhancer formation affecting many oncogene gene expressions. Although several studies discussed above support the concept that action of BER in gene promoters guided by oxidative modification of G regulates transcription of several genes, some obvious questions warranting answers are (a) How a nonselective oxidant such as H2O2 generated during LSD1-mediated histone demethylation induces specific G oxidation in promoter/enhancer sequences to regulate gene expression? (b) Do oxidative base modifications other than 8-oxoG also function as an epigenetic regulator, and are they often required for transcription of oxidative stress-dependent genes? In the context of G4s, future studies are required to understand whether promoter PQSs serve as sensors of oxidative stress to modulate transcription of downstream genes through direct oxidation of DNA bases in the promoters under stressed conditions. Given the fact that tumor cells are highly transcriptionally active and have elevated levels of oxidative stress and BER enzymes, addressing these questions would help illuminate the complex interplay between oxidative DNA damage, BER, and transcriptional regulation in cancer.
References
Abbotts R, Madhusudan S (2010) Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Treat Rev 36:425–435. https://doi.org/10.1016/j.ctrv.2009.12.006
Alexandrov LB, Stratton MR (2014) Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev 24:52–60. https://doi.org/10.1016/j.gde.2013.11.014
Alexandrov LB, Nik-Zainal S, Wedge DC et al (2013) Signatures of mutational processes in human cancer. Nature 500:415–421. https://doi.org/10.1038/nature12477
Amente S, Bertoni A, Morano A et al (2010a) LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene 29:3691–3702. https://doi.org/10.1038/onc.2010.120
Amente S, Lania L, Avvedimento EV et al (2010b) DNA oxidation drives Myc mediated transcription. Cell Cycle 9:3002–3004. https://doi.org/10.4161/cc.9.15.12499
Amente S, Di Palo G, Scala G et al (2019) Genome-wide mapping of 8-oxo-7,8-dihydro-2′-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res 47:221–236. https://doi.org/10.1093/nar/gky1152
Antoniali G, Lirussi L, D’Ambrosio C et al (2014) SIRT1 gene expression upon genotoxic damage is regulated by APE1 through nCaRE-promoter elements. Mol Biol Cell 25:532–547. https://doi.org/10.1091/mbc.E13-05-0286
Ba X, Boldogh I (2018) 8-Oxoguanine DNA glycosylase 1: beyond repair of the oxidatively modified base lesions. Redox Biol 14:669–678. https://doi.org/10.1016/j.redox.2017.11.008
Ba X, Bacsi A, Luo J et al (2014) 8-oxoguanine DNA glycosylase-1 augments proinflammatoryproinflammatory gene expression by facilitating the recruitment of site-specific transcription factors. J Immunol 192:2384–2394. https://doi.org/10.4049/jimmunol.1302472
Bauer NC, Corbett AH, Doetsch PW (2015) The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res 43:10083–10101. https://doi.org/10.1093/nar/gkv1136
Bhakat KK, Izumi T, Yang SH et al (2003) Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J 22:6299–6309. https://doi.org/10.1093/emboj/cdg595
Bhakat KK, Hazra TK, Mitra S (2004) Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res 32:3033–3039. https://doi.org/10.1093/nar/gkh632
Bhakat KK, Mokkapati SK, Boldogh I et al (2006) Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol Cell Biol 26:1654–1665. https://doi.org/10.1128/MCB.26.5.1654-1665.2006
Bhakat KK, Mantha AK, Mitra S (2009) Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multifunctional protein. Antioxid Redox Signal 11:621–638. https://doi.org/10.1089/ARS.2008.2198
Bjelland S, Seeberg E (2003) Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res 531:37–80. https://doi.org/10.1016/j.mrfmmm.2003.07.002
Bochman ML, Paeschke K, Zakian VA (2012) DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet 13:770–780. https://doi.org/10.1038/nrg3296
Boldogh I, Hajas G, Aguilera-Aguirre L et al (2012) Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J Biol Chem 287:20769–20773. https://doi.org/10.1074/jbc.C112.364620
Burge S, Parkinson GN, Hazel P et al (2006) Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res 34:5402–5415. https://doi.org/10.1093/nar/gkl655
Cadet J, Wagner JR (2013) DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol 5. https://doi.org/10.1101/cshperspect.a012559
Cadet J, Wagner JR, Shafirovich V et al (2014) One-electron oxidation reactions of purine and pyrimidine bases in cellular DNA. Int J Radiat Biol 90:423–432. https://doi.org/10.3109/09553002.2013.877176
Carter RJ, Parsons JL (2016) Base excision repair, a pathway regulated by post-translational modifications. Mol Cell Biol 36:1426–1437. https://doi.org/10.1128/MCB.00030-16
Chattopadhyay R, Das S, Maiti AK et al (2008) Regulatory role of human AP-endonuclease (APE1/Ref-1) in YB-1-mediated activation of the multidrug resistance gene MDR1. Mol Cell Biol 28:7066–7080. https://doi.org/10.1128/MCB.00244-08
Cogoi S, Ferino A, Miglietta G et al (2018) The regulatory G4 motif of the Kirsten ras (KRAS) gene is sensitive to guanine oxidation: implications on transcription. Nucleic Acids Res 46:661–676. https://doi.org/10.1093/nar/gkx1142
Cooke MS, Evans MD, Dizdaroglu M et al (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17:1195–1214. https://doi.org/10.1096/fj.02-0752rev
Das S, Chattopadhyay R, Bhakat KK et al (2007) Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J Biol Chem 282:28474–28484. https://doi.org/10.1074/jbc.M704672200
Deaton AM, Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25:1010–1022. https://doi.org/10.1101/gad.2037511
El-Khamisy SF, Masutani M, Suzuki H et al (2003) A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res 31:5526–5533. https://doi.org/10.1093/nar/gkg761
Evans AR, Limp-Foster M, Kelley MR (2000) Going APE over ref-1. Mutat Res 461:83–108. https://doi.org/10.1016/s0921-8777(00)00046-x
Fang Y, Zou P (2020) Genome-wide mapping of oxidative DNA damage via engineering of 8-Oxoguanine DNA. Glycosyl Biochem 59:85–89. https://doi.org/10.1021/acs.biochem.9b00782
Fleming AM, Burrows CJ (2017) 8-Oxo-7,8-dihydro-2′-deoxyguanosine and abasic site tandem lesions are oxidation prone yielding hydantoin products that strongly destabilize duplex DNA. Org Biomol Chem 15:8341–8353. https://doi.org/10.1039/c7ob02096a
Fleming AM, Ding Y, Burrows CJ (2017a) Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc Natl Acad Sci U S A 114:2604–2609. https://doi.org/10.1073/pnas.1619809114
Fleming AM, Zhu J, Ding Y, Burrows CJ (2017b) 8-Oxo-7,8-dihydroguanine in the context of a gene promoter G-Quadruplex is an on-off switch for transcription ACS. Chem Biol 12:2417–2426. https://doi.org/10.1021/acschembio.7b00636
Fleming AM, Zhu J, Ding Y, Burrows CJ (2019) Location dependence of the transcriptional response of a potential G-quadruplex in gene promoters under oxidative stress. Nucleic Acids Res 47:5049–5060. https://doi.org/10.1093/nar/gkz207
Grollman AP, Moriya M (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet 9:246–249. https://doi.org/10.1016/0168-9525(93)90089-z
Guo C, Li X, Ye M et al (2017) Discriminating patients with early-stage breast cancer from benign lesions by detection of oxidative DNA damage biomarker in urine. Oncotarget 8:53100–53109. https://doi.org/10.18632/oncotarget.17831
Hansel-Hertsch R, Beraldi D, Lensing SV et al (2016) G-quadruplex structures mark human regulatory chromatin. Nat Genet 48:1267–1272. https://doi.org/10.1038/ng.3662
Hansel-Hertsch R, Di Antonio M, Balasubramanian S (2017) DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat Rev Mol Cell Biol 18:279–284. https://doi.org/10.1038/nrm.2017.3
Hegde ML, Hazra TK, Mitra S (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res 18:27–47. https://doi.org/10.1038/cr.2008.8
Hegde ML, Banerjee S, Hegde PM et al (2012) Enhancement of NEIL1 protein-initiated oxidized DNA base excision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via direct interaction. J Biol Chem 287:34202–34211. https://doi.org/10.1074/jbc.M112.384032
Hodis E, Watson IR, Kryukov GV et al (2012) A landscape of driver mutations in melanoma. Cell 150:251–263. https://doi.org/10.1016/j.cell.2012.06.024
Lindahl T, Barnes DE (2000) Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 65:127–133. https://doi.org/10.1101/sqb.2000.65.127
Lirussi L, Antoniali G, Vascotto C et al (2012) Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol Biol Cell 23:4079–4096. https://doi.org/10.1091/mbc.E12-04-0299
Loft S, Svoboda P, Kawai K et al (2012) Association between 8-oxo-7,8-dihydroguanine excretion and risk of lung cancer in a prospective study. Free Radic Biol Med 52:167–172. https://doi.org/10.1016/j.freeradbiomed.2011.10.439
Maiti A, Drohat AC (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem 286:35334–35338. https://doi.org/10.1074/jbc.C111.284620
McCulloch SD, Kunkel TA (2008) The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res 18:148–161. https://doi.org/10.1038/cr.2008.4
Mitra S, Izumi T, Boldogh I et al (2002) Choreography of oxidative damage repair in mammalian genomes. Free Radic Biol Med 33:15–28. https://doi.org/10.1016/s0891-5849(02)00819-5
Oeckinghaus A, Ghosh S (2009) The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1:a000034. https://doi.org/10.1101/cshperspect.a000034
Pan L, Zhu B, Hao W et al (2016) Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase-1-mediated epigenetic regulation of nuclear factor kappaB-driven gene expression. J Biol Chem 291:25553–25566. https://doi.org/10.1074/jbc.M116.751453
Pastukh V, Ruchko M, Gorodnya O et al (2007) Sequence-specific oxidative base modifications in hypoxia-inducible genes. Free Radic Biol Med 43:1616–1626. https://doi.org/10.1016/j.freeradbiomed.2007.08.027
Pastukh V, Roberts JT, Clark DW et al (2015) An oxidative DNA “damage” and repair mechanism localized in the VEGF promoter is important for hypoxia-induced VEGF mRNA expression. Am J Physiol Lung Cell Mol Physiol 309:L1367–L1375. https://doi.org/10.1152/ajplung.00236.2015
Perillo B, Ombra MN, Bertoni A et al (2008) DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319:202–206. https://doi.org/10.1126/science.1147674
Puente XS, Bea S, Valdes-Mas R et al (2015) Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 526:519–524. https://doi.org/10.1038/nature14666
Redstone SCJ, Fleming AM, Burrows CJ (2019) Oxidative modification of the potential g-quadruplex sequence in the pcna gene promoter can turn on transcription. Chem Res Toxicol 32:437–446. https://doi.org/10.1021/acs.chemrestox.8b00332
Robertson AB, Klungland A, Rognes T, Leiros I (2009) DNA repair in mammalian cells: Base excision repair: the long and short of it. Cell Mol Life Sci 66:981–993. https://doi.org/10.1007/s00018-009-8736-z
Roychoudhury S, Nath S, Song H et al (2017) Human apurinic/apyrimidinic endonuclease (APE1) is acetylated at DNA damage sites in chromatin, and acetylation modulates its DNA repair activity. Mol Cell Biol 37. https://doi.org/10.1128/MCB.00401-16
Roychoudhury S, Pramanik S, Harris HL et al (2020) Endogenous oxidized DNA bases and APE1 regulate the formation of G-quadruplex structures in the genome. Proc Natl Acad Sci U S A 117:11409–11420. https://doi.org/10.1073/pnas.1912355117
Schieber M, Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24:R453–R462. https://doi.org/10.1016/j.cub.2014.03.034
Scott TL, Rangaswamy S, Wicker CA et al (2014) Repair of oxidative DNA damage and cancer: recent progress in DNA base excision repair. Antioxid Redox Signal 20:708–726. https://doi.org/10.1089/ars.2013.5529
Sengupta S, Mantha AK, Mitra S et al (2011) Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene 30:482–493. https://doi.org/10.1038/onc.2010.435
Sengupta S, Mantha AK, Song H et al (2016) Elevated level of acetylation of APE1 in tumor cells modulates DNA damage repair. Oncotarget 7:75197–75209. https://doi.org/10.18632/oncotarget.12113
Sengupta S, Yang C, Hegde ML et al (2018) Acetylation of oxidized base repair-initiating NEIL1 DNA glycosylase required for chromatin-bound repair complex formation in the human genome increases cellular resistance to oxidative stress. DNA Repair (Amst) 66–67:1–10. https://doi.org/10.1016/j.dnarep.2018.04.001
Shi Y, Matson C, Lan F et al (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119:941–953. https://doi.org/10.1016/j.cell.2004.12.012
Shi YJ, Matson C, Lan F et al (2005) Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 19:857–864. https://doi.org/10.1016/j.molcel.2005.08.027
Wilson SH, Kunkel TA (2000) Passing the baton in base excision repair. Nat Struct Biol 7:176–178. https://doi.org/10.1038/73260
Winterbourn CC (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 4:278–286. https://doi.org/10.1038/nchembio.85
Xanthoudakis S, Curran T (1992) Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J 11:653–665
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Singapore Pte Ltd.
About this entry

Cite this entry
Pramanik, S., Roychoudhury, S., Bhakat, K.K. (2022). Oxidized DNA Base Damage Repair and Transcription. In: Chakraborti, S., Ray, B.K., Roychoudhury, S. (eds) Handbook of Oxidative Stress in Cancer: Mechanistic Aspects. Springer, Singapore. https://doi.org/10.1007/978-981-15-9411-3_156
Download citation
DOI: https://doi.org/10.1007/978-981-15-9411-3_156
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-9410-6
Online ISBN: 978-981-15-9411-3
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences