
Abstract
Generation of induced photoreceptors (PRs) holds promise as a tool for in vitro modeling of inherited retinal diseases. Direct reprogramming, direct conversion or redirect differentiation of somatic cells by overexpression of transcription factors is a promising, simple, and low-cost approach to generate target cells from somatic cells without using induced pluripotent stem cells. My research group has successfully generated PR-like cells from human somatic cells; iris cells, dermal fibroblasts, and peripheral blood mononuclear cells (PBMCs) using this redirect differentiation technique. In this chapter, I introduce this method and demonstrate its application as a cellular model of inherited retinal diseases.
First, we tried to define the transcription factor combinations that can induce PR-like cells. A mixture of these genes was then transduced into iris cells, which were examined for inducible expression of PR-specific phenotypes. Expression patterns were dependent on combinations of transcription factors: A combination of CRX and NeuroD induced rhodopsin and blue opsin, but not green opsin; a combination of CRX and RAX induced blue opsin and green/red opsin, but not rhodopsin. After transduction with CRX, RAX, and NeuroD, rhodopsin-positive, blue opsin-positive, or green/red opsin-positive cells were found in induced PR-like cells by immunostaining, and these cells were determined to be photo-responsive by functional analysis using whole cell patch-clamp recordings. However, the response was an inward current instead of the typical outward current. Next, we tested whether human dermal fibroblasts could be converted into PRs. Transduction with the same combination of genes, CRX, RAX, and NeuroD, upregulated expression of the PR-specific genes. Additional OTX2 gene transduction increased the upregulation of these genes. Both the NRL gene and the NR2E3 gene were also endogenously upregulated in these cells. Global gene expression data by microarray analysis showed that phototransduction-related genes were significantly increased in these cells, where a photo-response, i.e., outward or inward current, was detected using the whole cell patch-clamp recordings. We then examined whether human PBMCs could be converted into PRs. Retinal disease-related genes, most of which are crucial to PR functions, were detected in CRX-transduced PBMCs. Functional studies showed that a light-induced inward current was detected in some CRX-transduced PBMCs.
Retinitis pigmentosa (RP) is an inherited retinal dystrophy that leads to visual impairment. The EYS gene was reported as the most common gene responsible for autosomal recessive (ar) RP. arRP with EYS gene defects is denoted by “EYS-RP.” We produced PR-directed fibroblasts from EYS-RP patients using redirect differentiation as a replacement for degenerative retinas. A combination of four transcription factors, CRX, RAX, OTX2, and NeuroD, was transduced into dermal fibroblasts from three EYS-RP patients with homozygous or heterozygous mutations. We analyzed the defective transcripts of the EYS gene in these cells to elucidate the phenotypes of the EYS-RP patients, as the decay of the transcripts may be involved in the phenotypic variation associated with the disease. As a result, expression levels of defective transcripts were markedly different depending on the type of mutation. In conclusion, we suggest that the redirect differentiation method may be a valuable tool for disease modeling, despite some limitations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Redirect differentiation
- Iris
- Dermal fibroblast
- Peripheral blood mononuclear cells (PBMC)
- Photoreceptor (PR)
- Disease modeling
- Retinitis pigmentosa
- EYS
- Truncating mutation
- Nonsense-mediated mRNA decay (NMD)
- Phenotypic variation
- Genotype-phenotype relationship
13.1 Introduction
Retinitis pigmentosa (RP) is an inherited retinal dystrophy that leads to visual impairment. Generation of induced photoreceptors (PRs) holds promise for in vitro modeling of inherited retinal diseases such as RP. The ideal tool for analysis of transcripts of the pathogenic genes is a retina from a patient, but for research purposes, cellular models are available as a substitute for human retinas. Induced PRs generated from disease-specific iPSCs of RP patients were reported to reproduce pathogenic phenotypes [1,2,3,4]. Although methods to generate PRs from induced pluripotent stem cells (iPSCs) have been established [5, 6], they are expensive and time-consuming. We established an alternative method, “redirect differentiation,” wherein photosensitive PR-like cells are generated more easily and rapidly [7,8,9]. Direct reprogramming, direct conversion or redirect differentiation of somatic cells by overexpression of transcription factors is a promising, simple, low-cost approach to generate target cells from somatic cells without using iPSCs. My research group successfully generated PR-like cells from human somatic cells; iris cells, dermal fibroblasts, and peripheral blood mononuclear cells (PBMCs), using a redirect differentiation technique. Because we determined that continuous expression of exogenous transgenes is necessary to maintain the properties of PRs, we call this method “redirect differentiation.”
We further generated and analyzed induced PR-like cells from human somatic cells derived from healthy volunteers and RP patients by redirect differentiation to examine the possibility of using these cells for disease modeling of RP.
13.2 What Is “Redirect Differentiation ”?
The possibility of redirecting cell differentiation by overexpression of genes was first suggested by Weintraub with the identification of the “master gene,” MyoD [10]. The process of “direct reprogamming” or “direct conversion” is thought to be direct lineage switching [11] rather than lineage switching back to a branch point and out again in a different direction. Examples of “direct conversion” has been shown in beta-cells, cardiomyocytes, and neurons: A specific combination of three transcription factors (Ngn3, Pdx1, and MafA) reprogram differentiated pancreatic exocrine cells in adult mice into cells that closely resemble beta-cells [12]; a combination of three factors (Gata4, Tbx5, and Baf60c) induces non-cardiac mesoderm to differentiate directly into contractile cardiomyocytes [13]; and a combination of three factors (Ascl1, Brn2, and Myt1l) converts mouse fibroblasts into functional neurons [14]. We tried to generate PR-cells by this “direct reprogramming” or “direct conversion.”
13.3 Methods of Differentiation and Assessment of Induced PR-Like Cells
At first, we defined the transcription factor combinations that can determine photoreceptor cell fate using human iris cells. Detailed methods are available in our previous paper [7]. In brief, we selected Six3, Pax6, Rax, Crx, Nrl, and NeuroD, genes that were expected to contribute to the induction of PR-specific phenotypes. Full length of transcription factors SIX3 [15], PAX6 [16], RAX [17], CRX [18], NRL [19, 20] and NeuroD [21], were amplified from cDNAs prepared from total RNA of adult human retina (Clontech, CA, USA) by PCR, and cloned into the XmnI-EcoRV sites of pENTR11 (Invitrogen). The resulting pENTR11-transcription factors were recombined with pMXs-DEST (modified pMXs (gift from T Kitamura to A Umezawa) by Y Miyagawa) by use of LR recombination reactions (Invitrogen). The retroviral DNAs were then transfected into 293FT cells and after 3 days, the media was collected and concentrated. The iris-derived cells were plated onto laminin-coated dishes and maintained for 1 day. The cells were transduced with media containing retroviral vector particles with 8 μg/ml of polybrene for 5 h at 37 °C. After retroviral transduction, the media was replaced with the DMEM/F12/B27 medium supplemented with 20 ng/ml bFGF, 40 ng/ml EGF, fibronectin, and 1% FBS. The retrovirus-transduced cells were cultured for up to 21 days. In order to measure the efficiency of transduction, we transduced retroviral eGFP under the same conditions. The frequency of eGFP-positive cells was 90–94% of all cells 48 h after transduction. Each vector contained one transcription factor and a mixture of vectors was used. Transduced cells were examined for inducible expression of PR-specific phenotypes using RT-PCR and immunocytochemistry. In addition, photo-responsiveness of induced PR-like cells was investigated using patch-clamp recordings.
13.4 Combinations of Transcription Factors Determining Photoreceptor Cell Fate
Transduction of a single gene for SIX3, PAX6, RAX, CRX, NRL, or NeuroD did not induce rod- or cone-specific phenotypes in iris cells, but the six genes together upregulated blue opsin and rhodopsin as shown previously [7]. To determine which of the six candidates were critical, we tested the effect of withdrawal of individual factors from the pool of transduced candidate genes on the expression of the opsin genes. We identified two genes, NeuroD and CRX , which were essential for PR-induction; withdrawal of NeuroD resulted in the loss of expression of rhodopsin, and withdrawal of CRX resulted in the loss of blue opsin. Expression patterns were dependent on combinations of transcription factors: A combination of CRX and NeuroD induced rod-specific genes, but did not induce the red opsin gene. Additional RAX gene transduction significantly upregulated blue opsin gene expression. A combination of CRX and RAX induced blue opsin and green/red opsin, but did not induce rhodopsin. NeuroD significantly decreased expression of the cone-specific genes, i.e., genes for green opsin and cone channel B3 (CNGB3) in human iris cells (p < 0.005). It was clearly demonstrated that the expression of rhodopsin and S-antigen, which are specifically expressed in rod-PRs, were much higher in CRX, RAX, and NeuroD-transduced cells than in CRX and RAX-transduced cells (rhodopsin, p < 0.05; S-antigen, p < 0.005, Welch’s t-test). From these results, it was speculated that the combination of CRX and RAX generated immature PRs: and additional NeuroD promoted maturation.
We then tested whether human dermal fibroblasts could be converted into PRs [8]. Human dermal fibroblasts can be differentiated to PR-like cells by the same transcription factor combination as human iris cells. Transduction of a combination of the CRX, RAX, and NeuroD genes upregulated expression of PR-specific genes, recoverin, blue opsin, and PDE6C. Additional OTX2 gene transduction increased up-regulation of these genes. Both the NRL gene and the NR2E3 gene, which were reported to determine photoreceptor cell fate, were endogenously upregulated in PR-directed fibroblasts by four transcription factors, CRX, RAX, OTX2, and NeuroD, by microarray analysis and endpoint RT-PCR, implying that exogenous CRX, RAX, OTX2, and NeuroD, but not NRL, are sufficient to generate PR-like cells with expression of rod-specific genes.
13.5 Endogenous and Exogenous Expression of Transcription Factors
We performed RT-PCR to investigate whether endogenous expression of transcription factors was induced in the PR-like cells that we generated [7, 8]. Both transgenic and endogenous CRX, RAX, and NeuroD were expressed. This indicates that human somatic cells, such as iris cells and dermal fibroblasts, were reprogrammed into PRs, at least to some extent. We then suppressed the CRX and NeuroD genes by siRNA. Expression of the PR-specific genes such as blue opsin, s-antigen, and recoverin decreased significantly in siCRX and siNeuroD-transfected cells, suggesting that continuous expression of CRX and NeuroD is necessary to maintain the properties of PRs. This is why we call our method “differentiation,” not “reprogramming.”
13.6 Photo-Responsiveness of Induced PR-Like Cells
For functional assessment of transduced cells, electrical recordings were made using the whole cell patch-clamp technique. The membrane current before and after light stimulation was recorded and analyzed. The PR-like cells derived from iris cells, induced by CRX, RAX, and NeuroD, responded to light. However, the response was an inward current instead of the typical outward current [7]. Since the light-induced inward current seemed to be mediated by melanopsin-associated phototransduction, we investigated the expression of melanopsin by RT-PCR and immunocytochemistry. CRX, RX, and NEUROD-transduced iris cells expressed melanopsin, suggesting that melanopsin expression was associated with inward current.
PR-directed fibroblasts, transduced by CRX, RAX, NeuroD, and OTX2, clearly responded to light. Global gene expression data by microarray analysis showed that phototransduction-related genes were significantly increased in induced PR-like cells. We also demonstrated that physiological responses to light differed between two different commercially available cell lines [22]. Under light stimulation, Ishii et al. found that an outward current (photoreceptor-like responses) was observed in both cell lines, while an inward current (intrinsically photosensitive retinal ganglion cell-like responses) was observed only in one cell line. Although cell age (passage number) may have differed, our data suggest that properties of the photosensitive cells produced by redirect differentiation may be controlled by the origin of the cell source.
However, some CRX-transduced PBMCs exhibited a light-induced inward current [9], instead of the typical outward current. Since the light-induced inward current seemed to be mediated by melanopsin-associated phototransduction as observed in iris-derived PR-like cells [7, 23], Komuta et al. investigated the expression of melanopsin by RT-PCR. The expression of melanopsin was not detected in PR-directed PBMCs. The reason an inward current was detected in CRX-transduced cells expressing PR-related genes was not determined; however, it might be possible that signals passed from blue, red, or green opsin to a downstream point in the melanopsin signaling cascade in CRX-transduced cells, leading to depolarization by light stimulation.
13.7 Variation of Cell Types of Sources for Induced PR-Like Cells (Fig. 13.1)
13.7.1 Iris
My research group defined the transcription factor combinations that can induce PR-like cells from human infantile iris cells [7]. Expression patterns were dependent on combinations of transcription factors: A combination of CRX and NeuroD induced rhodopsin and blue opsin, but not green opsin; a combination of CRX and RAX induced blue opsin and green/red opsin, but not rhodopsin. Expression levels of rhodopsin genes and blue opsin genes reached maximum levels 1 week after gene transduction of transcription factors and remained unchanged for up to 3 weeks. Expression of green/red opsin reached maximum levels 3 days after gene transduction. After transduction with CRX, RAX, and NeuroD, rhodopsin-positive, blue opsin-positive, and green/red opsin-positive cells were 29% (per 954 cells), 37% (per 235 cells), and 25% (per 193 cells) of total cells, respectively, by immunostaining. Hybrid PRs were also detected by double-staining immunocytochemistry. Ultrastructural analysis revealed a cilia-associated structure, i.e., centriole, surrounded by mitochondria [7].

Variation of cell types of sources for induced photoreceptor-like cells
Although it has been shown that retinal stem cells are not present in the human iris [24, 25], our previous study demonstrated that human iris cells expressed stem cell markers such as nestin, N-cadherin, Sox2, Musashi-1, and Pax6 [7]. Expression of stem cell markers in iris cells may be attributed to the cell source, i.e., cells from infants. However, PR cell differentiation with exogenously added chemicals and growth factors was limited [7]. Other experimental evidence has also suggested the limitation in mammals without genetic manipulation. Progenitor cells from the mammalian iris, pars plana, and ciliary body do not show a convincing immunoreactivity for rhodopsin, phosducin, recoverin, PKC, or RPE65 [26], but are induced into PR progeny with retinal transcription factors [27, 28]. Derivation of PR-like cells can be attributed to transgene-dependent differentiation of retinal progenitors that exist in the iris. We also indicated that human iris stromal (IS) cells that originate from neural crest [7], as well as IPE cells, differentiate into PR-like cells.
13.7.2 Dermal Fibroblasts
The induced pluripotent stem cells (iPS) developed by Takahashi and Yamanaka was the first model for “direct reprogramming,” in which mouse adult fibroblasts were reprogrammed by transduction of four transcription factor genes, Oct3/4, Sox2, c-Myc, and Klf4 [29]. Additionally, functional neurons were generated from mouse fibroblasts by a combination of three factors (Ascl1, Brn2, and Myt1l) [14], and functional platelets were generated from mouse and human fibroblasts by a combination of three factors (p45NF-E2, MafG, and MafK) [30]. Because human dermal fibroblasts are less specialized than iris cells, we tested whether human dermal fibroblasts could be converted into PRs by the same defined combination of genes used successfully for human iris cells, CRX, RAX, and NeuroD, to generalize and establish our technology for generating PRs [8]. In human dermal fibroblasts, recoverin, blue opsin, PDE6c were upregulated by a combination of CRX, RAX, and NeuroD. Additional OTX2 gene transduction increased up-regulation of the PR-specific genes; that is blue opsin, recoverin, S-antigen, CNGB3, and PDE6C. These results suggest that OTX2 may work as an amplifier [8].
For functional assessment of transduced cells, electrical recordings were made using the whole cell patch-clamp technique. The membrane current before and after light stimulation was recorded and analyzed. Induced PR-like cells derived from human dermal fibroblasts, induced by CRX, RAX, and NeuroD, responded to light. A typical outward current was detected [8, 22].
Dermal fibroblasts are of mesodermal origin and immunogenic, while iris-pigmented epithelial cells (IPE cells) are of neural ectoderm origin and show immune tolerance. Iris cells studied here include not only IPE cells but also iris stromal cells, which are of neural crest origin. We have previously shown that iris cells, IPE cells, and iris stromal cells are differentiated into photoreceptor cells in the same way [7]. However, dermal fibroblasts are harvested easily and safely, and iris cells are obtained surgically. To find a more suitable cell source than the iris cells for reprogramming into photoreceptor cells, we compared signal ratios between PR-direct fibroblasts and PR-directed iris cells by a microarray analysis. The results show that there was an increase in both the expression levels and the variety of upregulated PR-specific genes in PR-directed iris cells when compared with PR-direct fibroblasts [8]. The difference in induced endogenous expression of transcription factors CRX, RAX, and NeuroD between CRN-Fib and CRN-Iris as well as the difference in upregulated photoreceptor-specific genes may suggest a difference in reprogramming potential between the two types of cells. From the standpoint of regenerative medicine, iris cells may be more suitable than dermal fibroblasts based on their characteristics of immune tolerance and higher expression of retina-specific genes in differentiated cells. It may be possible to improve dermal fibroblasts as a source by use of other transcription factors or by manipulating the histone methylation signature [31]. Dermal fibroblasts have an important advantage in that these cells are obtained safely and easily from patients. Because the direct reprogramming method may be suitable to provide the small numbers of cells required for individualized drug screening and disease modeling, dermal fibroblasts may be useful for such purposes despite their limitations.
13.8 Peripheral Blood Mononuclear Cells (PBMCs)
We further investigated another cell type, peripheral blood mononuclear cells, or PBMCs. Though dermal fibroblasts are often utilized for reprogramming, sampling by dermal biopsies requires surgical intervention and expertise. Therefore, we tested whether human PBMCs could be converted into PRs. Based on our previous studies of the generation of photosensitive PR-like cells from human iris cells and human dermal fibroblasts, we transduced the same transcription factors into PBMCs via Sendai virus vectors. PBMCs expressed cone-related genes after the transduction of CRX alone using SeV vectors. Blue opsin and red/green opsin were more efficiently and intensely expressed in CRX-transduced PBMCs prepared using SeV vectors than in those using retrovirus vectors because transduction by SeV vectors is efficient. However, the expression levels of the blue opsin gene increased in CRX-transduced PBMCs but not in fibroblasts, although transduction was performed by SeV vectors in both cell types. Endogenous CRX expression was detected in dermal fibroblasts transduced with CRX by both retrovirus and SeV vectors, but was detected in PBMCs transduced only by SeV vectors. These differences might be attributed to variable reprogramming efficiencies based on different methylation signatures dependent on cell types, as previously reported [31, 32].
We found that some PR-related genes, blue opsin, PDE6H, and SAG, were efficiently detected in CRX-transduced cells. Expression levels of blue opsin and PDE6H peaked at 1 week and that of SAG peaked earlier, at 3 days. By immunocytochemistry, on the third day after transduction, blue opsin-positive cells constituted about 20% of the CRX-transduced cells. Surprisingly, in functional studies, patch-clamp recordings showed that a light-induced inward current was detected in some CRX-transduced cells. Photostimulation of the rod or cone pathway produces hyperpolarizing responses, while activation of the melanopsin pathway produces depolarizing responses [23]. Since the light-induced inward current seemed to be mediated by melanopsin-associated phototransduction as observed in iris-derived photoreceptor-like cells [7], Komuta et al. investigated the expression of melanopsin by RT-PCR. However, the expression of melanopsin was not detected in photoreceptor-directed PBMCs. We therefore examined photoreceptor-related and melanopsin-related genes that function in phototransduction. Strong expression of downstream genes of the melanopsin cascade, such as TRPC and Gqα, was detected. Abundant CNGB3 expression was detected in CRX-transduced cells, but CNGA3, which coordinates with CNGB3, was not sufficiently expressed. This might be the reason why the phototransduction cascade could not mediate the light stimuli as the typical outward current of photoreceptors. Proteins involved in the signal transduction cascade of melanopsin, such as TRPC and Gqα proteins, which induce depolarization, were abundantly expressed, while Gαt and CNG proteins, which induce hyperpolarization, were not sufficiently expressed. The reason why an inward current was detected in CRX-transduced cells expressing photoreceptor-related genes was unknown at this time; however, it might be possible that signals passed from blue, red, or green opsin to a downstream point in the melanopsin signaling cascade in CRX-transduced cells, leading to producing the depolarization by light stimuli.
Furthermore, numerous retinal disease-related genes were efficiently detected in CRX-transduced cells, most of which are crucial to the photoreceptor function. In order to increase differentiation efficiency, Komuta et al. modified the culture conditions. By adding transduction of RAX1 and NEUROD1, additional conditioned medium of cultured retinal pigment epithelial cells and Activin A, DKK, and Lefty2, they saw expression of a greater variety of retinal disease-related genes than that observed in CRX-transduced PBMCs. Polycistronic vectors, with four transcription factors, CRX, RAX, NeuroD, and OTX2, were inserted in a cistronic manner via Sendai virus, were employed with the aim to improve the differentiation efficiency of PBMC to PRs. However, expression levels of rhodopsin were higher in PR-directed fibroblasts with a mixture of mono-cistronic retrovirus vectors than in PR-directed PBMC by polycistronic vectors via Sendai virus vectors (unpublished data).
PBMC proliferation is induced by IL-2, are easily collected, and are safer to use compared to dermal fibroblasts; these cells have the potential for use as a cell source for differentiation into PRs. CRX transduced by SeV acts as a master control gene for reprogramming of PBMCs into PRs, specifically, cone PR-like cells. In PR-directed PBMCs, expression of rod-photoreceptor specific genes was very low; the differentiation needs to be improved.
13.9 Application of Induced PR-Like Cells to RP Research
We examined the possibility of using our induced PR-like cells derived from dermal fibroblasts of RP patients as disease modeling for RP [33] because of the shortcomings of our differentiation methods.
RP displays degeneration of PR/RPE via gene defects, leading to the deterioration of nyctalopia and narrowing of the visual field. RP is progressive and incurable, leading to a major causative disease of juvenile blindness. It is speculated that gene defects lead to cellular dysfunction of PRs in patients. Disease modeling of RP should be useful for disease diagnosis, elucidation of pathogenesis, and drug screening.
Defects in the EYS gene on chromosome 6q12 were found to be a major cause of autosomal recessive (ar) RP in several populations [34,35,36,37,38,39]. In Japan, c.4957dupA (p.Ser1653Lysfs*2) and c.8805C > A (p.Tyr2935*) were identified as pathogenic mutations in about 20%–30% of arRP patients [40, 41]. To date, many EYS variants have been reported as causative defects of RP [42]. Hereafter arRP caused by defects in the EYS gene is denoted as “EYS-RP.” RP is a highly heterogeneous disease, and accordingly, EYS-RP exhibits heterogeneous phenotypes with a wide range in severity. In order to clarify the genotype-phenotype correlation in EYS-RP, the analysis of transcripts may be helpful. EYS (OMIM 612424) is currently the largest gene expressed in the human eye, spanning over 2 Mb within the RP25 locus (6q12) [34, 37]. The ideal tool for analysis of the EYS gene transcripts is a retina from an EYS-RP patient. For research purposes, cellular models are available as an alternative for human retinas.
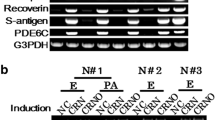
We collected fibroblasts of patients with “EYS-RP.” Dermal fibroblasts were harvested from three healthy donors: N#1, N#2, N#3, and three EYS-RP patients with homozygous or heterozygous mutations (Table 13.1) under the approval of the Ethics Committee of the National Rehabilitation Center for Persons with Disabilities (NRCPD). Using “redirect differentiation” by CRX, RAX, NeuroD, and OTX2, we generated PR-directed fibroblasts derived from these subjects. We tested the inducible expression of the PR-specific genes (blue opsin, rhodopsin, recoverin, S-antigen, PDE6C, EYS) in these cells. PR-specific genes were upregulated in all the PR-directed fibroblasts tested. However, expression levels of defective transcripts of the EYS gene were markedly different, depending on the type of mutation. To analyze transcripts derived from three different types of the defective EYS gene, c.1211dupA, c.4957dupA, and c.8805C > A, we performed RT-PCR and analyzed DNA sequences of amplified products for exon 6–11, exon 26–27, and exon 42–43 that carry c.1211dupA, c.4957dupA and c.8805C > A, respectively, using total RNAs extracted from PR-directed fibroblasts of Pt#1, Pt#2 and Pt#3 10 days after gene transduction. Transcripts derived from these three defective genes were barely detectable, expressed at a lower level, or expressed at almost the same level as in normal volunteers, respectively.
Generally, faulty transcripts are immediately triaged for destruction by nonsense-mediated mRNA decay (NMD) [43]. All three EYS-RP donors had the frameshift mutation c.4957dupA, in at least one allele of exon 26 (Table 13.1). Therefore, we expected that NMD would cause the loss of the EYS gene transcripts corresponding to exon 26–27. However, the transcripts with c.4957dupA were clearly detected in PR-directed fibroblasts from Pt#1, carrying homozygous mutations and Pt#2, carrying compound heterozygous mutations. However, the expression levels of the transcript with c.4957dupA in Pt#1 and Pt#2 were lower than the mean of N#1, N#2, and N#3. To explain this phenomenon, we referred to a previous study [44] where it was reported that a cis element that inhibits NMD is located within the first 200 nt when positioned in the downstream proximal region of the premature termination codon (PTC) [44]. The authors showed several examples with significant enrichment of A/U nucleotides (63%–71%) and hypothesized that this may be a condition for NMD evasion. To determine whether our data was consistent with their hypothesis, we analyzed sequences in the proximal downstream region of the PTC in the transcripts derived from defected alleles of the EYS gene and calculated the A/U nucleotide content. For c.4957dupA, A/U content was 67%, which is in the range previously reported (63%–71%). This result supports our hypothesis that transcripts having the frameshift mutation, c.4957dupA, may partially escape from NMD.
Pt#2 has the nonsense mutation c.8805C > A, on an allele of exon 43 (Table 13.1). Because this mutation produces a PTC in the last exon, the transcript with this mutation may escape degradation by NMD [45]. These transcripts, corresponding to exon 42–43, were clearly detected in PR-directed dermal fibroblasts derived from Pt#2. The peak amplitudes of normal and mutated bases on the electropherogram were nearly the same. The expression levels from Pt#2 were similar to those from normal volunteers, suggesting that escape from NMD occurred in transcripts with c.8805C > A. Interestingly, the exon 42–43 region of the EYS gene was expressed in human dermal fibroblasts without PR-induction. The expression level of the exon 42–43 fragment in default state fibroblasts was higher than in PR-directed fibroblasts. Because the EYS is reported to be expressed exclusively in the retina, our research group intensively studied on this exon 42–43 fragment that is expressed in human dermal fibroblasts. As a result, Takita et al. identified a new variant, transcribed from exon 37, which is specifically expressed in dermal fibroblasts [46].
Pt#3 has the frameshift mutation c.1211dupA (p.Asn404Lysfs*3) in exon 8, which has previously been reported in an Israeli arRP patient [38]. By endpoint RT-PCR and sequencing, only the transcript derived from the normal allele was detected, suggesting that the transcript derived from the mutant allele was degraded by NMD, as expected.
To pursue the relationship between phenotypic variations of EYS-RP patients, large samples are needed. The present study also suggests that the redirect differentiation method could be a valuable tool for disease modeling, despite some limitations. Our induced PR-like cells may contribute to individualized drug screening and disease modeling of inherited retinal degeneration.
13.10 Conclusion
My research group has successfully generated PR -like cells from human somatic cells; iris cells, dermal fibroblasts, and peripheral blood mononuclear cells (PBMCs) using a redirect differentiation technique. Expression patterns of PR-specific genes were dependent on combinations of transcription factors in PR-like cells that we generated.
By the redirect differentiation technique, an in vitro EYS-RP model was created by transduction of a combination of transcription factor genes, CRX, RAX, NeuroD, and OTX2, into dermal fibroblasts derived from EYS-RP patients with homozygous or heterozygous mutations. The expression of the defective EYS transcripts was markedly different, depending on the type of mutation. Nonsense mutations of the EYS gene transcripts, which are the same as in the genome, were detected. These results suggest that nonsense-mediated mRNA decay, NMD, is inhibited, in part, by a cis-acting mechanism. Molecular changes in the in vitro model of RP mimic the pathological condition of RP, in part.
In conclusion, we believe that our redirect differentiation method may be a valuable tool for disease modeling, despite some limitations.
References
Jin ZB, Okamoto S, et al. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PLoS One. 2011;6:e17084.
Jin ZB, Okamoto S, et al. Integration-free induced pluripotent stem cells derived from retinitis pigmentosa patient for disease modeling. Stem Cells Transl Med. 2012;1:503–9.
Phillips MJ, Perez ET, et al. Modeling human retinal development with patient-specific induced pluripotent stem cells reveals multiple roles for visual system homeobox 2. Stem Cells. 2014;32:1480–92.
Yoshida T, Ozawa Y, et al. The use of induced pluripotent stem cells to reveal pathogenic gene mutations and explore treatments for retinitis pigmentosa. Mol Brain. 2014;7:45.
Osakada F, Jin ZB, et al. In vitro differentiation of retinal cells from human pluripotent stem cells by small-molecule induction. J Cell Sci. 2009;122:3169–79.
Ohlemacher SK, Iglesias CL, et al. Generation of highly enriched populations of optic vesicle-like retinal cells from human pluripotent stem cells. Curr Protoc Stem Cell Biol. 2015;32:1H 8 1–1H 8 20.
Seko Y, Azuma N, et al. Derivation of human differential photoreceptor-like cells from the iris by defined combinations of CRX, RX and NEUROD. PLoS One. 2012;7:e35611.
Seko Y, Azuma N, et al. Derivation of human differential photoreceptor cells from adult human dermal fibroblasts by defined combinations of CRX, RAX, OTX2 and NEUROD. Genes Cells. 2014;19:198–208.
Komuta Y, Ishii T, et al. In vitro transdifferentiation of human peripheral blood mononuclear cells to photoreceptor-like cells. Biol Open. 2016;5:709–19.
Weintraub H, Tapscott SJ, et al. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci USA. 1989;86:5434–8.
Gurdon JB, Melton DA. Nuclear reprogramming in cells. Science. 2008;322:1811–5.
Zhou Q, Brown J, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–32.
Takeuchi JK, Bruneau BG. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009;459:708–11.
Vierbuchen T, Ostermeier A, et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–41.
Manavathi B, Peng S, et al. Repression of Six3 by a corepressor regulates rhodopsin expression. Proc Natl Acad Sci USA. 2007;104:13128–33.
Glaser T, Walton DS, et al. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 1992;2:232–9.
Mathers PH, Grinberg A, et al. The Rx homeobox gene is essential for vertebrate eye development. Nature. 1997;387:603–7.
Furukawa T, Morrow EM, et al. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell. 1997;91:531–41.
Rehemtulla A, Warwar R, et al. The basic motif-leucine zipper transcription factor Nrl can positively regulate rhodopsin gene expression. Proc Natl Acad Sci USA. 1996;93:191–5.
Mears AJ, Kondo M, et al. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29:447–52.
Morrow EM, Furukawa T, et al. NeuroD regulates multiple functions in the developing neural retina in rodent. Development. 1999;126:23–36.
Ishii T, Yin C, et al. Variation in the phenotype of photosensitive cells produced from human fibroblast cell lines. J Nippon Med Sch. 2018;85:110–6.
Hattar S, Liao HW, et al. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002;295:1065–70.
Coles BL, Angenieux B, et al. Facile isolation and the characterization of human retinal stem cells. Proc Natl Acad Sci USA. 2004;101:15772–7.
Froen RC, Johnsen EO, et al. Pigment epithelial cells isolated from human peripheral iridectomies have limited properties of retinal stem cells. Acta Ophthalmol. 2011;89:e635–44.
MacNeil A, Pearson RA, et al. Comparative analysis of progenitor cells isolated from the Iris, Pars Plana, and ciliary body of the adult porcine eye. Stem Cells. 2007;25:2430–8.
Akagi T, Akita J, et al. Iris-derived cells from adult rodents and primates adopt photoreceptor-specific phenotypes. Invest Ophthalmol Vis Sci. 2005;46:3411–9.
Inoue T, Coles BL, et al. Maximizing functional photoreceptor differentiation from adult human retinal stem cells. Stem Cells. 2010;28:489–500.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Ono Y, Wang Y, et al. Induction of functional platelets from mouse and human fibroblasts by p45NF-E2/Maf. Blood. 2012;120:3812–21.
Bramswig NC, Everett LJ, et al. Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J Clin Invest. 2013;123:1275–84.
Nishino K, Toyoda M, et al. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011;7:e1002085.
Seko Y, Iwanami M, et al. The manner of decay of genetically defective EYS gene transcripts in photoreceptor-directed fibroblasts derived from retinitis pigmentosa patients depends on the type of mutation. Stem Cell Res Ther. 2018;9:279.
Abd El-Aziz MM, Barragan I, et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet. 2008;40:1285–7.
Abd El-Aziz MM, O'Driscoll CA, et al. Identification of novel mutations in the ortholog of Drosophila eyes shut gene (EYS) causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2010;51:4266–72.
Barragan I, Borrego S, et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum Mutat. 2010;31:E1772–800.
Collin RW, Littink KW, et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am J Hum Genet. 2008;83:594–603.
Bandah-Rozenfeld D, Littink KW, et al. Novel null mutations in the EYS gene are a frequent cause of autosomal recessive retinitis pigmentosa in the Israeli population. Invest Ophthalmol Vis Sci. 2010;51:4387–94.
Littink KW, van den Born LI, et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology. 2010;117:2026–33.
Iwanami M, Oshikawa M, et al. High prevalence of mutations in the EYS gene in Japanese patients with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2012;53:1033–40.
Hosono K, Ishigami C, et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS One. 2012;7:e31036.
Messchaert M, Haer-Wigman L, et al. EYS mutation update: in silico assessment of 271 reported and 26 novel variants in patients with retinitis pigmentosa. Hum Mutat. 2018;39:177–86.
Trcek T, Sato H, et al. Temporal and spatial characterization of nonsense-mediated mRNA decay. Genes Dev. 2013;27:541–51.
Toma KG, Rebbapragada I, et al. Identification of elements in human long 3' UTRs that inhibit nonsense-mediated decay. RNA. 2015;21:887–97.
Holbrook JA, Neu-Yilik G, et al. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–8.
Takita S, Miyamoto-Matsui K, Seko Y. Intra- and inter-species comparison of EYS transcripts highlights its characteristics in the eye. FASEB J. 2019;33:9422–33.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Seko, Y. (2021). Generation and Analysis of Induced Photoreceptor-Like Cells from Fibroblasts of Patients with Retinitis Pigmentosa. In: Prakash, G., Iwata, T. (eds) Advances in Vision Research, Volume III. Essentials in Ophthalmology. Springer, Singapore. https://doi.org/10.1007/978-981-15-9184-6_13
Download citation
DOI: https://doi.org/10.1007/978-981-15-9184-6_13
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-9183-9
Online ISBN: 978-981-15-9184-6
eBook Packages: MedicineMedicine (R0)