
Abstract
Lynch syndrome is a cancer-predisposing syndrome inherited in an autosomal dominant manner, wherein colon cancer and endometrial cancer develop frequently in the family, it results from a loss of function of one of four different protein (MLH1, MSH2, MSH6, and PMS2), which are the products of mismatch repair genes. An abnormal EPCAM gene at the position adjacent to the MSH2 gene also inhibits MSH2 expression and causes Lynch syndrome.
Mismatch repair proteins are involved in repairing of incorrect pairing, including point mutations and deletion/insertion of simple repetitive sequences, so-called microsatellites, that can arise during DNA replication. MSH2 forms heterodimers with MSH6 or MSH3 (MutSα, MutSβ, respectively) and is involved in mismatch-pair recognition and initiation of repair. MLH1 forms a complex with PMS2 and functions as an endonuclease. If the mismatch repair system is thoroughly working, genome integrity is maintained at a high level. Lynch syndrome is a state of mismatch repair deficiency (MMRd) due to a monoallelic abnormality of the mismatch repair genes. The phenotype indicating the mismatch repair deficiency can be frequently observed as a microsatellite instability (MSI) in tumors.
Generally, Lynch syndrome develops in adulthood, but MMR gene abnormalities are observed in children with different genotypes and phenotypes. Children with germline biallelic mismatch repair gene abnormalities were reported to develop conditions such as gastrointestinal polyposis, colorectal cancer, brain cancer, leukemia, and so on. This condition is called constitutional mismatch repair deficiency (CMMRD).
In addition, for promoting cancer genome medicine in a new era, such as by utilizing immune checkpoints, it is important to understand the genetic and genomic molecular background, including the status of mismatch repair deficiency.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Mismatch repair gene
- Lynch syndrome
- Microsatellite instability
- Constitutional mismatch repair deficiency
- Immune checkpoint inhibitor
1.1 Introduction
Cancer is fundamentally a genetic disease, and pathogenic variants, also called “mutation,” are pivotal to its etiology and progression. Carcinogenesis develops by the accumulation of numerous genetic and epigenetic abnormalities [1,2,3,4]. Therefore, cancer has the following six characteristics: sustained proliferative signaling, evasion of growth suppressors, resistance cell death, replicative immortality, angiogenesis induction, and activation of invasion and metastasis [5]. Therefore, elucidation of carcinogenesis is essential for therapeutic development [6]. Although rare, hereditary cancer syndromes are observed in cancers derived from any organ. In individuals with hereditary cancer syndrome, the initial cancer-causing pathogenic variant is inherited through the germ cell and therefore, is already present in all 37 trillion cells that make up the body. Lynch syndrome (MIM# 120435) is an autosomal dominant syndrome with a penetration rate of about 80% characterized by several individuals in the family affected with colorectal cancer (CRC) or extracolonic tumors of the endometrium, stomach, small bowel, ureter, renal pelvis, ovary, and hepatobiliary tract [7].
Lynch syndrome occurs due to loss of function of the mismatch repair mechanism for genomic replication errors. This article outlines the basis of molecular genetics involved in Lynch syndrome.
1.2 DNA Repair System
The frequency of replication errors is 10−10 per base of DNA per cell division, and in an estimated 1015 cell divisions during an individual’s lifetime replication errors cause thousands of new DNA variants in the genome in every cell. Eukaryotes have multiple repair systems to avoid replication errors (Table 1.1) [8]. Maintaining DNA-integrity through genome repair suppresses cancer development and progression by genomic abnormalities. Genes encoding molecules involved in genome repair are referred to as DNA repair genes and “caretaker tumor suppressor genes.”
The mismatch repair system was recognized in 1961, with proposal that the correction of DNA base pair mismatches within recombination intermediates is the basis for gene conversion [9]. Elucidation of the mismatch repair system has been advanced by fundamental research based on Escherichia coli, developed four E. coli mutator genes: mutH, mutL, mutS, and uvrD [10,11,12,13]. Inactivation of any of these genes increases the generation of variants in the E. coli cell by 50-to 100-fold, indicating the importance of this pathway in variant avoidance and genetic stability. The reduction in mutability afforded by the E. coli methyl-directed system has been attributed to its role in the strand-specific elimination of DNA errors (Table 1.2) [6, 8, 14,15,16,17,18,19]. Research on the mismatch repair system has advanced extensively and has clarified its mechanism and role as an essential mechanism for maintaining genome integrity in organisms and involved in predisposition to cancer development.
1.3 Genes Responsible for Lynch Syndrome
Lynch syndrome, is called hereditary nonpolyposis colorectal cancer: HNPCC in the past, is an autosomal dominant inherited disorder caused by germline pathogenic variants in DNA mismatch repair (MMR) genes. Patients with Lynch syndrome are at an increased risk of developing tumors from a young age and throughout their lifetime. Most of them suffer from multiple synchronous and/or metachronous primary tumors. Colorectal cancer and endometrial cancer (female) are well known in the tumor spectrum of Lynch syndrome. In addition, patients with Lynch syndrome have high potential for developing cancer of the urinary tract, the stomach, the small intestine, the biliary tract, the skin, the brain, and others.
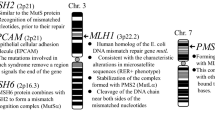
Multiple types of human mismatch repair (MMR) proteins have been discovered and several encoding genes have been isolated so far. Currently, four types of MMR genes, MLH1 (MIM# 120436), MSH2 (MIM# 609309), MSH6 (MIM# 600678), and PMS2 (MIM# 600259), are used in the clinical applications related to Lynch syndrome. An outline of the responsible genes is shown in Fig. 1.1. The EPCAM, which encodes a cell adhesion molecule, is not an MMR gene, but its structural abnormality causes Lynch syndrome, because it is adjacent to the MSH2 gene [20]. This content will be described later.

The genes responsible for Lynch syndrome [8]
In 1993, MSH2 gene was isolated at chromosome 2p22–p21 in 1993 and has high homology with the mutator phenotype gene, mutS of E. coli [21,22,23,24]. In 1994, as the second responsible gene of Lynch syndrome, MLH1, the E. coli mutL homologue, was isolated from 3p22.2 [25,26,27]. In 1995, mismatch binding factors were found as the 100 kD MSH2 or as heterodimers of the 160 kD polypeptide called GTBP/MSH6 (for G/T binding protein), which was recognized as a new member of the MutS homologue [28, 29]. MSH6 gene was first reported by Japanese researchers as a gene responsible for Lynch syndrome [30, 31]. In 1994, a germline deletion of the PMS2 was also identified in families with Lynch syndrome. Moreover, additional deletions in tumor samples with microsatellite instability high (MSI-high) showed the presence of two-hits [32, 33].
1.4 Structure and Function of MMR Proteins
Each MMR protein encoded by the corresponding MMR gene has a unique function in repairing replication errors. Therefore, MMR proteins possess unique functional domains. When pathogenic variants of MMR genes occur in the DNA site corresponding to the functional domain, DNA repair function may be impaired. Schematic representations of MLH1, MSH2, MSH6, and PMS2 proteins are shown in Fig. 1.2 [8, 34,35,36,37,38]. Both MLH1 and PMS2 have an ATP binding domain and require ATP molecules for the endonuclease function.


Structure of mismatch repair proteins (a) MLH1, (b) MSH2, (c) MSH6, (d) PMS2 [8]
Many human MMR related proteins have been identified as homologues of E. coli MMR proteins [8, 22,23,24,25,26,27,28,29, 39,40,41,42,43,44,45,46,47,48,49]. These include human homologues of MutS, MutL, ExoI, DNA polymerase δ (pol δ), proliferating cellular nuclear antigen (PCNA), replication factor (RFC), DNA ligase I, and so on. MSH2 heterodimerizes with MSH6 or MSH3 to form MutSα or MutSβ, respectively. These are involved in the mismatch-pair recognition and initiation of repair [50,51,52,53,54]. In addition, various kinds of complexes such as MutLα, MutLβ, and MutLγ are formed and involved in the mismatch repair system [37, 38, 40, 51, 52, 54,55,56,57,58,59,60,61,62,63].
1.5 Mechanisms of Mismatch Repair
The mismatch repair (MMR) system consists of sequential steps for the recognition, removal, and resynthesis of the mismatch site in DNA. This system that maintains DNA fidelity is well conserved from E. coli to eukaryotes. A schematic diagram of the pathway is shown in Fig. 1.3 [8, 53, 58, 60, 62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82]. Base–base mismatches in double-strand DNA are recognized by MutSα (heterodimer of MSH2-MSH6). MutSα binds as a sliding clamp around the double-strand DNA. MutSα and MutLα form a tetrameric complex and then initiate the process of mismatch repair. The tetrameric complex recruits proliferating cell nuclear antigen (PCNA), replication factor C (RFC), exonuclease 1 (Exo 1) to remove the nascent (daughter) strand, and resynthesize the correct strand. Then, exonuclease 1 (Exo 1) is recruited and removes the nascent (daughter) strand around the error region. The resynthesis step is accomplished by DNA polymerase (Polδ or Polε) and Ligase 1.

Mechanistic model of mismatch repair [8]
1.6 Relationship Between MMR System and DNA Damages
Depending on the DNA damage pattern, specific mismatch repair molecules, and complexes are involved. The outline is shown in Fig. 1.4 [8, 50, 64, 66, 83,84,85,86]. The MutSα (heterodimer of MSH2-MSH6) contributes to mismatch recognition by single nucleotide substitution (e.g., G:T mismatch pair) and recognition of small insertion-deletion loops (IDL, e.g., error of the repeat number in adenine clusters), whereas MutSβ (heterodimer of MSH2-MSH3) contributes to the repair of small loops and relatively large damages up to about ten nucleotide loops. Recently, the function of MutSβ has attracted attention for its biological characteristics and as a prognostic factor of elevated microsatellite instability at selected tetranucleotide (EMAST) colorectal cancer, which shows instability in the repeat sequence of the tetranucleotides [87,88,89,90,91].

Schematic of DNA damage recognized by the mismatch repair pathway [8]
1.7 EPCAM as the Gene Responsible for Lynch Syndrome
EPCAM is located at 2p21 adjacent to the MSH2 on the 5′ upstream and encodes the EPCAM protein, expressed on the membrane of cells in epithelial tissues and plasma cells, and is involved in cell-cell adhesion function [92, 93]. Although EPCAM is not the direct responsible gene of Lynch syndrome, but it is located just 17 kb upstream of MSH2. The deletion of EPCAM affects MSH2 gene expression, resulting in Lynch syndrome. The schema is shown in Fig. 1.5 [8, 20]. The cis-deleted alleles inhibit MSH2 expression and finally causes Lynch syndrome in 1.3% of the affected families [20, 94].

A cis-deletion of EPCAM gene causes an epimutation of the MSH2 gene [8]
In addition, biallelic inactivation of EPCAM is responsible for congenital tufting enteropathy (CTE, MIM# 613217) with an estimated incidence of one in 50,000–100,000 births in Western Europe [95,96,98]. CTE presents within the first months of life with severe chronic watery diarrhea and growth restriction. EPCAM abnormalities responsible for CTE are usually missense mutations, nonsense mutations, minute insertions/deletions, and splicing errors, unlike Lynch syndrome [98].
1.8 Constitutional Mismatch Repair Deficiency Syndrome
Constitutional mismatch repair deficiency syndrome (CMMR-D) is caused by biallelic homozygous or compound heterozygous pathogenic germline pathogenic variants of MMR genes and is a distinct childhood cancer preposition syndrome (MIM# 276300) with an autosomal recessive inheritance [98,99,101]. In biallelic germline pathogenic variant carriers of MMR genes, hematological malignancies, brain/central nervous system (CNS) tumors, and Lynch syndrome associated carcinomas develop frequently. In the gastrointestinal tract, bowel adenomatous polyposes are often observed as premalignant lesions that require differential diagnosis from FAP. By the way, the pathological condition classified as a subtype of FAP called Turcot’s syndrome is considered to be exactly CMRR-D [102, 103].
The median age at diagnosis of hematological malignancies and brain/CNS tumors was respectively, 6.6 (age range: 1.2–30.8) and 10.3 (age range: 3.3–40) years. However, Lynch syndrome-associated tumors developed later (median age at diagnosis: 21.4 years (age range: 11.4–36.6)). Moreover, the spectrum of Lynch syndrome is mostly colorectal cancer and/or endometrial cancer [104]. Various nonneoplastic features are related to CMMR-D including Cafe au lait spots (NF1 like), skin hypopigmentation, mild defects in immunoglobulin class switching recombination, agenesis of the corpus callosum, cavernous brain hemangioma, capillary hemangioma of the skin, combination of various congenital malformations, and lupus erythematosus.
1.9 Genetic Testing for Lynch Syndrome
In order to select high-risk individuals with Lynch syndrome from among patients with colorectal cancer and to increase the efficiency of detecting germline pathogenic variants, microsatellite instability (MSI) testing and/or immunohistochemical staining (IHC) of MMR proteins is recommended as universal tumor screening and is recommended to do first [102, 105, 106]. The MSI testing is a method to easily identify events in which genetic integrity has been damaged due to repair failures of DNA replication errors using simple repeated microsatellite sequences [106,107,108,109,111]. Five types of repeat markers including mononucleotide and dinucleotide repeats have been used, but recently mononucleotide repeat markers have been preferred. Cases with different numbers of repeats between normal tissue-derived DNA and cancer-derived DNA are considered as positive [112]. If two or more of the five markers show instability, the tumor is evaluated as MSI-high (MSI-H). The results of MSI-H colorectal cancer are shown in Fig. 1.6. If one of the markers shows instability, the tumor is considered as MSI-low (MSI-L). If positive markers are not observed, the mismatch repair system is evaluated to be proficient and is called MS-stable (MSS).

Analytic image of MSI testing: four out of five markers show microsatellite instability [8]
Immunohistochemical staining of MMR proteins can reveal damaged molecules using specific antibodies. Staining with four antibodies: MLH1, MSH2, MSH6, and PMS2 can predict the gene causing Lynch syndrome (Table 1.3) [112,113,114,115,116,117,118,120].
For MSI testing, sensitivity ranged from 66.7 to 100.0% and specificity ranged from 61.1 to 92.5%, whereas for IHC staining, sensitivity ranged from 80.8 to 100.0%, and specificity ranged from 80.5 to 91.9% [121].
Approximately 10–15% of sporadic colorectal cancers show MSI-H findings. The cause is mostly the loss of MSH1 protein due to methylation of the MLH1 gene promoter region. About half of MSI-H sporadic colorectal cancers show BRAF V600E mutation, which is rarely detected in colorectal cancers from patients with Lynch syndrome. MLH1 methylation analysis and BRAF V600E mutation testing in colorectal cancers can improve the efficiency of the diagnosis for Lynch syndrome [36, 122].
Final genetic testing for Lynch syndrome is performed using DNA sequencing in selected cases excluding sporadic colon cancer from all colorectal cancers. For a long time, genetic testing has mainly been performed using Sanger sequencing, and multiplex ligation-dependent probe amplification (MLPA) has been adopted for a wide range of abnormalities such as large deletions/insertions [123]. Clinical genetics is currently transitioning from phenotype-directed single gene testing to multigene panels [124]. Multigene panel testing using next generation sequencing for hereditary colorectal cancer has been evaluated as a feasible, timely, and cost-effective approach compared to single gene testing [125]. Previously, the distribution of germline pathogenic variants in MMR and EPCAM genes in Lynch syndrome was thought to predominantly occur in MSH2 and MLH1 and less frequently in MSH6 and PMS2. As a result of multigene panel testing without universal tumor screening, Espenschied et al. reported that MSH6 pathogenic variants were the most frequent, followed by PMS2, MSH2, MLH1, and EPCAM (Table 1.4) [8, 123, 125,126,128]. About 12% of individuals carrying MMR gene pathogenic variants have breast cancer alone. Furthermore, even MMR gene pathogenic variant carriers do not always meet the criteria for Lynch syndrome or the BRCA1/BRCA2 testing criteria. However, MSH6 and PMS2 germline pathogenic variants are associated with an increased risk for breast cancer [126, 129]. Table 1.4 shows the gene-specific distributions of germline variants by the types of abnormalities in mismatch repair genes. Most MSH2, MLH1, and MSH6 pathogenic variants were truncated types such as nonsense mutations or frameshift mutations [8, 130]. Knowledge of choice of analysis method is important. A wide range of rearrangements were detected at 10%, 7%, and 10% for MSH2, MLH1, and PMS2, respectively. Therefore, the selection of an appropriate analysis method is required for genetic testing.
1.10 Effectiveness of Immune Check Point Blockades and a Hypermutable State (High Tumor Mutational Burden)
As cancer cells escape the host immune system by suppressing T cell activation, thus exert an immunosuppressive function due to immune checkpoint molecules. The immune checkpoint molecules include cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1, CD279), and so on [131, 132], which were found to negatively control the immune system [133, 134]. In human cancer treatment, anti-PD-1 antibody was found to be effective for non-small cell lung cancer, malignant melanoma, and renal cell cancer and was also clinically applicable in safety [135]. The clinical efficacy of PD-1 inhibitor was found to be higher in mismatch repair-defective colorectal and non-colorectal cancers compared to proficient-mismatch repair cancers [136]. According to recent survey results, as shown in Fig.1.7, high tumor mutational burden (TMB) is an excellent biomarker for predicting the efficacy of immune checkpoint inhibitors (ICIs) [136,137,139], and the group of colorectal cancer patients with the biological characteristics of mismatch repair deficient (MMRd) has a significantly better response to ICIs than those with mismatch repair proficient (MMRp) [136, 137]. In gastrointestinal cancer, because the state of microsatellite instability high (MSI-H) state has been shown to correlate well with high TMB based on an analysis of many cancer genomes, the microsatellite instability (MSI) testing is used as a standard biomarker to predict the response of ICIs [139,140,142].

Correlation between tumor mutational burden (TMB) and objective response rate with immune checkpoint inhibitors [137]
1.11 Future Directions
The cancer-accumulating family reported by Warthin AS more than 100 years ago led to the establishment of Lynch syndrome by the vigorous genetic epidemiological approach of Lynch HT et al. On the other hand, mismatch repair genes have been elucidated as part of the genome integrity system of Escherichia coli and yeast. These basic researchers worked together to understand the clinical, genetic, and molecular biological aspects of Lynch syndrome. With its natural history and molecular biological characteristics clarified, presymptomatic diagnosis by genetic testing for at-risk persons in the family, and appropriate medically actionable interventions, such as early diagnosis, are becoming possible.
The development of ICIs is a major milestone in the treatment of patients with Lynch Syndrome. Most malignant tumors in patients with LS have MSI-H status and are expected to respond to ICIs. These studies have shown new possibilities for the treatment of hereditary tumor syndrome. In future, we hope that advances in the integrated understanding of the clinical and molecular biology of Lynch syndrome will lead to the development of novel diagnostic methods and effective treatments.
Abbreviations
- CMMR-D:
-
Constitutional mismatch repair deficiency
- CNS:
-
Central nervous system
- CTE:
-
Congenital tufting enteropathy
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- IHC:
-
Immunohistochemical staining
- ISI:
-
Immune checkpoint inhibitor
- MLPA:
-
Multiple ligation-dependent probe amplification
- MMR:
-
Mismatch repair
- MSI:
-
Microsatellite instability
- PCNA:
-
Proliferating cellular nuclear antigen
- PD-1:
-
Programmed cell death protein 1
- RFC:
-
Replication factor
- TMB:
-
Tumor mutational burden
References
Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32.
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67.
Bodmer W, Bishop T, Karran P. Genetic steps in colorectal cancer. Nat Genet. 1994;6:217–9.
Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Tamura K, Utsunomiya J, Iwama T, et al. Mechanism of carcinogenesis in familial tumors. Int J Clin Oncol. 2004;9:232–45.
Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–32.
Tamura K, Kaneda M, Futagawa M, et al. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int J Clin Oncol. 2019;24:999–1011.
Holliday R. A mechanism for gene conversion in fungi. Genet Res. 1964;5:282–304.
Modrich P. Mechanisms in E. coli and mismatch repair. Angew Chem Int Ed Engl. 2016;55:8490–501.
Nevers P, Spats HC. Escherichia coli mutants uvr D and uvr E deficient in gene conversion of lambda-heteroduplexes. Mol Gen Genet. 1975;139:233–43.
Rydberg B. Bromouracil mutagenesis and mismatch repair in mutator strains of Esherichia coli. Mutat Res. 1978;52:11–24.
Glickman BW, Radman M. Esherichia coli mutator mutants deficient in methylation-instructed DNA mismatch correction. Proc Natl Acad Sci U S A. 1980;77:1063–7.
Lauhe RS, Su SS, Morich P. Requirement for d(GATC) sequences in Esherichia coli mutHLS mismatch correction. Proc Natl Acad Sci U S A. 1987;84:1482–6.
Su SS, Morrich P. Esherichia coli mutS-encoded protein binds to mismatched DNA base pairs. Proc Natl Acad Sci USA. 1986;83:5057–61.
Meselson M. Methyl-directed repair of DNA mismatches. In: Low KB, editor. Recombination of the genetic material. San Diego, CA: Academic Press; 1988. p. 91–113.
Modrich P. Methyl-directed DNA mismatch correction. J Biol Chem. 1989;264:6597–600.
Grilley M, Holmes J, Yashar B, et al. Mechanisms of DNA-mismatch correction. Mutat Res. 1990;236:253–67.
Modrich P. Mechanisms and biological effects of mismatch repair. Annu Rev Genet. 1991;25:229–53.
Ligtenberg MJL, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3-prime exons of TACSTD1. Nat Genet. 2009;41:112–7.
Fishel R, Lescoe MK, Rao MRS, et al. The human mutator gene homolog MSH2 and its association. Cell. 1993;75:1027–38.
Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–25.
Kolodner RD, Hall NR, Lipford J, et al. Structure of the human MSH2 locus and analysis of two Muir-Torre kindreds for msh2 mutations. Genomics. 1994;24:516–26.
Fishel R, Ewel A, Lee S, et al. Binding of mismatched microsatellite DNA sequences by the human MSH2 protein. Science. 1993;266:1403–5.
Papadopoulos N, Nicolaides NC, Wei Y-F, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–9.
Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–61.
Han H-J, Maruyama M, Baba S, et al. Genomic structure of human mismatch repair gene, hMLH1, and its mutation analysis in patients with hereditary non-polyposis colorectal cancer (HNPCC). Hum Mol Genet. 1995;4:237–42.
Drummond JT, Li G-M, Longley MJ, et al. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. 1995;268:1909–12.
Palombo F, Gallinari P, Iaccarino I, et al. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science. 1995;268:1912–4.
Miyaki M, Konishi M, Tanaka K, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17:271–2.
Akiyama Y, Sato H, Yamada T, et al. Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res. 1997;57:3920–3.
Nicolaides NC, Papadopoulos N, Liu B, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371:75–80.
De Vos M, Hayward BE, Picton S, et al. Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. Am J Hum Genet. 2004;74:954–64.
Ban C, Juno M, Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell. 1999;97:85–97.
Tran PT, Liskay RM. Functional studies on the candidate ATPase domains of Saccharomyces cerevisiae MutLalpha. Mol Cell Biol. 2000;20:6390–8.
Räschle M, Dufner P, Marra G, et al. Mutations within the hMLH1 and hPMS2 subunits of the human MutLalpha mismatch repair factor affect its ATPase activity, but not its ability to interact with hMutSalpha. J Biol Chem. 2002;277:21810–20.
Guerrette S, Acharya S, Fishel R. The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer. J Biol Chem. 1999;274:6336–41.
Kondo E, Horii A, Fukushige S. The interaction domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS2. Nucleic Acids Res. 2001;29:1695–708.
Reenan RA, Kolodner RD. Isolation and characterization of two Saccharomyces cerevisiae genes encoding homologs of the bacterial HexA and MutS mismatch repair proteins. Genetics. 1992;132:963–73.
Li GM, Modrich P. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc Natl Acad Sci U S A. 1995;92:1950–4.
Johnson RE, Kovvali GK, Guzder SN, et al. Evidence for involvement of yeast proliferating cell nuclear antigen in DNA mismatch repair. J Biol Chem. 1996;271:27987–90.
Umar A, Buermeyer AB, Simon JA, et al. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell. 1996;87:65–73.
Tishkoff DX, Boerger AL, Bertrand P, et al. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc Natl Acad Sci U S A. 1997;94:7487–92.
Longley MJ, Pierce AJ, Modrich P. DNA polymerase delta is required for human mismatch repair in vitro. J Biol Chem. 1997;272:10917–21.
Schmutte C, Marinescu RC, Sadoff MM, et al. Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res. 1998;58:4537–42.
Tishkoff DX, Amin NS, Viars CS, et al. Identification of a human gene encoding a homologue of Saccharomyces cerevisiae EXO1, an exonuclease implicated in mismatch repair and recombination. Cancer Res. 1998;58:5027–31.
Lin YL, Shivji MK, Chen C, et al. The evolutionarily conserved zinc finger motif in the largest subunit of human replication protein A is required for DNA replication and mismatch repair but not for nucleotide excision repair. J Biol Chem. 1998;273:1453–61.
Gu L, Hong Y, McCulloch S, et al. ATP-dependent interaction of human mismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Res. 1998;26:1173–8.
Zhang Y, Yuan F, Presnell SR, et al. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705.
Genschel J, Littman SJ, Drummond JT, et al. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem. 1998;273(31):19895–901.
Iyer RR, Pluciennik A, Genschel J, et al. MutLalpha and proliferating cell nuclear antigen share binding sites on MutSbeta. J Biol Chem. 2010;285(15):11730–9.
Plotz G, Raedle J, Brieger A, et al. N-terminus of hMLH1 confers interaction of hMutLalpha and hMutLbeta with hMutSalpha. Nucleic Acids Res. 2003;31(12):3217–26.
Dahal BK, Kadyrova LY, Delfino KR, et al. Involvement of DNA mismatch repair in the maintenance of heterochromatic DNA stability in Saccharomyces cerevisiae. PLoS Genet. 2017;13(10):e1007074.
Villahermosa D, Christensen O, Knapp K, et al. Schizosaccharomyces pombe MutSα and MutLα maintain stability of tetra-nucleotide repeats and Msh3 of hepta-nucleotide repeats. G3 (Bethesda). 2017;7(5):1463–73.
Prolla TA, Baker SM, Harris AC, et al. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet. 1998;18(3):276–9.
Jäger AC, Rasmussen M, Bisgaard HC, et al. HNPCC mutations in the human DNA mismatch repair gene hMLH1 influence assembly of hMutLalpha and hMLH1-hEXO1 complexes. Oncogene. 2001;20(27):3590–5.
Cannavo E, Marra G, Sabates-Bellver J, et al. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Res. 2005;65(23):10759–66.
Kadyrov FA, Dzantiev L, Constantin N, et al. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126(2):297–308.
Peng M, Litman R, Xie J, Sharma S, Brosh RM Jr, Cantor SB. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007;26(13):3238–49.
Pluciennik A, Dzantiev L, Iyer RR, et al. PCNA function in the activation and strand direction of MutLα endonuclease in mismatch repair. Proc Natl Acad Sci U S A. 2010;107(37):16066–71.
Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710.
Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98.
Kunkel TA, Erie DA. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet. 2015;49:291–313.
Marti TM, Kunz C, Fleck O. DNA mismatch repair and mutation avoidance pathways. J Cell Physiol. 2002;191(1):28–41.
Friedberg EC. DNA damage and repair. Nature. 2003;421(6921):436–40.
Martin SA, Lord CJ, Ashworth A. Therapeutic targeting of the DNA mismatch repair pathway. Clin Cancer Res. 2010;16(21):5107–13.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073–87.
Fishel R. Mismatch repair. J Biol Chem. 2015;290(44):26395–403.
Groothuizen FS, Sixma TK. The conserved molecular machinery in DNA mismatch repair enzyme structures. DNA Repair (Amst). 2016;38:14–23.
Hingorani MM. Mismatch binding, ADP-ATP exchange and intramolecular signaling during mismatch repair. DNA Repair (Amst). 2016;38:24–31.
Kadyrova LY, Kadyrov FA. Endonuclease activities of MutLα and its homologs in DNA mismatch repair. DNA Repair (Amst). 2016;38:42–9.
Peltomäki P. update on Lynch syndrome genomics. Familial Cancer. 2016;15:385–93.
Liu D, Keijzers G, Rasmussen LJ. DNA mismatch repair and its many roles in eukaryotic cells. Mutat Res. 2017;773:174–87.
Lee JB, Cho WK, Park J, et al. Single-molecule views of MutS on mismatched DNA. DNA Repair (Amst). 2014;20:82–93.
Plotz G, Raedle J, Brieger A, et al. hMutSalpha forms an ATP-dependent complex with hMutLalpha and hMutLbeta on DNA. Nucleic Acids Res. 2002;30(3):711–8.
Plotz G, Piiper A, Wormek M, et al. Analysis of the human MutLalpha.MutSalpha complex. Biochem Biophys Res Commun. 2006;340(3):852–9.
Friedhoff P, Li P, Gotthardt J. Protein-protein interactions in DNA mismatch repair. DNA Repair (Amst). 2016;38:50–7.
Jeon Y, Kim D, Martin-Lopez JV, et al. Dynamic control of strand excision during human DNA mismatch repair. Proc Natl Acad Sci U S A. 2016;113(12):3281–6.
Fishel R. Mismatch repair, molecular switches, and signal transduction. Genes Dev. 1998;12(14):2096–101.
Ban C, Junop M, Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell. 1999;97(1):85–97.
Spampinato C, Modrich P. The MutL ATPase is required for mismatch repair. J Biol Chem. 2000;275(13):9863–9.
Lamers MH, Winterwerp HH, Sixma TK. The alternating ATPase domains of MutS control DNA mismatch repair. ENBO J. 2003;22(3):746–56.
Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999;9(1):89–96.
Peltomäki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum Mol Genet. 2001;10(7):735–40.
Bellacosa A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death Differ. 2001;8(11):1076–92.
Scmidt MHM, Pearson CE. Disease associated repeat instability and mismatch repair. DNA Repair (Amst). 2016;38:117–26.
Campregher C, Schmid G, Ferk F, et al. MSH3-deficiency initiates EMAST without oncogenic transformation of human colon epithelial cells. PLoS One. 2012;7(11):e50541. https://doi.org/10.1371/journal.pone.0050541.
Tseng-Rogenski SS, Chung H, Wilk M, et al. Oxidative stress induces nuclear-to-cytosol shift of hMSH3, a potential mechanism for EMAST in colorectal cancer cells. PLoS One. 2012;7(11):e50616. https://doi.org/10.1371/journal.pone.0050616.
Watson MMC, Berg M, Søreide K. Prevalence and implications of elevated microsatellite alterations at selected tetranucleotides in cancer. Br J Cancer. 2014;111(5):823–7.
Carethers JM, Koi M, Tseng-Rogenski SS. EMAST is a form of microsatellite instability that is initiated by inflammation and modulates colorectal cancer progression. Genes (Basel). 2015;6(2):185–205.
Venderbosch S, van Lent-van Vliet S, de Haan AF, et al. EMAST is associated with a poor prognosis in microsatellite instable metastatic colorectal cancer. PLoS One. 2015;10(4):e0124538. https://doi.org/10.1371/journal.pone.0124538.
Dollé E, Theise ND, Schmelzer E, et al. EpCAM and the biology of hepatic stem/progenitor cells. Am J Physiol Gastrointest Liver Physiol. 2015;308:G233–50.
Huang L, Yang Y, Yang F, et al. Functions of EpCAM in physiological processes and diseases. Int J Mol Med. 2018;42(4):1771–85.
Kovacs ME, Papp J, Szentirmay Z, et al. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat. 2009;30(2):197–203.
Reifen RM, Cutz E, Griffiths AM, et al. Tufting enteropathy: a newly recognized clinicopathological entity associated with refractory diarrhea in infants. J Pediatr Gastroenterol Nutr. 1994;18(3):379–85.
Goulet O, Salomon J, Ruemmele F, et al. Intestinal epithelial dysplasia (tufting enteropathy). Orphanet J Rare Dis. 2007;2(1):20.
Sivagnanam M, Mueller JL, Lee H, et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 2008;135(2):429–37.
Pathak SJ, Mueller JL, Okamoto K, et al. EPCAM mutation update: variants associated with congenital tufting enteropathy and Lynch syndrome. Hum Mutat. 2019;40(2):142–61.
Wimmer K, Kratz CP, Vasen HFA, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet. 2014;51(6):355–65.
Ricciardone MD, Ozçelik T, Cevher B, et al. Human MLH1 deficiency predisposes to hematological malignancy and neurofibromatosis type 1. Cancer Res. 1999;59(2):290–3.
Wang Q, Lasset C, Desseigne F, et al. Neurofibromatosis and early onset of cancers in hMLH1-deficient children. Cancer Res. 1999;59(2):294–7.
Turcot J, Despres JP, St Pierre F, et al. Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases. Dis Colon Rectum. 1959;2:465–8.
Hamilton SR, Liu B, Parsons RE, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332(13):839–47.
Lavoine N, Colas C, Muleris M, et al. Constitutional mismatch repair deficiency syndrome: clinical description in a French cohort. J Med Genet. 2015;52(11):770–8.
Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11(1):35–41.
Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multisociety Task Force on colorectal cancer. Am J Gastroenterol. 2014;109(8):1159–79.
Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223–62.
Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–57.
Ionov Y, Peinado MA, Malkhosyan S, et al. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363(6429):558–61.
Peltomäki P, Aaltonen LA, Sistonen P, et al. Genetic mapping of a locus predisposing to human colorectal cancer. Nature. 1993;260:810–2.
Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Nature. 1993;260(5109):816–9.
Rodriguez-Bigas MA, Boland CR, Hamilton SR, et al. A National Cancer Institute Workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997;89(23):1758–62.
Leach FS, Polyak K, Burrell M, et al. Expression of the human mismatch repair gene hMSH2 in normal and neoplastic tissues. Cancer Res. 1996;56(2):235–40.
Thibodeau SN, French AJ, Roche PC, et al. Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res. 1996;56(21):4836–40.
Hendriks Y, Franken P, Dierssen JW, et al. Conventional and tissue microarray immunohistochemical expression analysis of mismatch repair in hereditary colorectal tumors. Am J Pathol. 2003;162(2):469–77.
de Jong AE, van Puijenbroek M, Hendriks Y, et al. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10(39):972–80.
Lipkin SM, Wang V, Jacoby R, et al. MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet. 2000;24(1):27–35.
Rigau V, Sebbagh N, Olschwang S, et al. Microsatellite instability in colorectal carcinoma. The comparison of immunohistochemistry and molecular biology suggests a role for hMSH6 [correction of hMLH6] immunostaining. Arch Pathol Lab Med. 2003;127(6):694–700.
Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352(18):1851–60.
Hendriks YMC, de Jong AE, Morreau H, et al. Diagnostic approach and management of Lynch syndrome (hereditary nonpolyposis colorectal carcinoma): a guide for clinicians. CA Cancer J Clin. 2006;56(4):213–25.
Snowsill T, Coelho H, Huxley N, et al. Molecular testing for Lynch syndrome in people with colorectal cancer: systematic reviews and economic evaluation. Health Technol Assess. 2017;21(51):1–238.
Jin M, Hampel H, Zhou X, et al. BRAF V600E mutation analysis simplifies the testing algorithm for Lynch Syndrome. Am J Clin Pathol. 2013;140(2):177–83.
Lagerstedt-Robinson K, Rohlin A, Aravidis C, et al. Mismatch repair gene mutation spectrum in the Swedish Lynch syndrome population. Oncol Rep. 2016;36(5):2823–35.
Lorans M, Dow E, Macrae FA, et al. Update on hereditary colorectal cancer: improving the clinical utility of multigene panel testing. Clin Colorectal Cancer. 2018;17(2):e293–305. https://doi.org/10.1016/j.clcc.2018.01.001.
Gallego CJ, Shirts BH, Bennette CS, et al. Next-generations sequencing panels for the diagnosis of colorectal cancer and polyposis syndromes: a cost-effectiveness analysis. J Clin Oncol. 2015;33(18):2084–91.
Espenschied CR, LaDuca H, Li S, et al. Multigene panel testing provides a new perspective on Lynch syndrome. J Clin Oncol. 2017;35(22):2568–75.
Yurgelun MB, Kulke MH, Fuchs CS, et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J Clin Oncol. 2017;35(10):1086–95.
Thompson BA, Spurdle AB, Plazzer JP, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46(12):107–15.
Roberts ME, Jackson SA, Susswein LR, et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet Med. 2018;20(10):1167–74.
Plazzer JP, Sijmons RH, Woods MO, et al. The InSiGHT database: utilizing 100 years of insights into Lynch syndrome. Familial Cancer. 2013;12(2):175–80.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6.
Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–95.
Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–7.
Nishimura H, Nose M, Hiai H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11(2):141–51.
Topalian SL, Hodi S, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Yarchoan M, Hopkins A, Jaffee EM, et al. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2018;377(25):2500–1.
Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99.
Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Charmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. https://doi.org/10.1186/s13073-017-0424-2.
Dudley JC, Lin MT, Le DT, et al. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res. 2016;22(4):813–20.
Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–13.
Acknowledgments
This work was supported in part by a grant from the Japanese Ministry of Education, Science, Sports and Culture of Japan (19K07763).
Disclosure Statement of COI
The author declares no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Tamura, K. (2020). Molecular Mechanism of Lynch Syndrome. In: Tomita, N. (eds) Lynch Syndrome. Springer, Singapore. https://doi.org/10.1007/978-981-15-6891-6_1
Download citation
DOI: https://doi.org/10.1007/978-981-15-6891-6_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-6890-9
Online ISBN: 978-981-15-6891-6
eBook Packages: MedicineMedicine (R0)