
Abstract
In the present manuscript, an adaptive evolutionary multiple sequence alignment algorithm is proposed that uses a combination of consensus and SP-score methods. The algorithm searches intermediate pairwise consensus strings that are used to identify the final consensus string for a given set of DNA/RNA/protein sequences. The proposed algorithm is an extension of MPSAGA algorithm that uses positional matrix representation of sequences. An empirical study was performed in the present work to compare the proposed algorithm with the other three contemporary ClustalW, TCOFFEE, and MUSCLE algorithms on the four datasets. The overall observations from the various experiments revealed that the proposed algorithm outperforms than the other algorithms tested in aligning multiple sequences with an average increase of 0.03% in alignment length by inserting 0.02% increased number of gaps.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
A multiple sequence alignment is an alignment of three or more DNA or RNA or protein sequences that can organize data in such a way that similar sequence features are aligned together [1, 2]. The goal of the multiple sequence alignment is to reveal features that may be shared by many sequences and to identify alterations that further elucidate functional and phenotypic variability [2]. The main applications of sequence alignment include secondary or tertiary structure prediction, phylogenetic tree construction, function prediction, hidden Markov modeling, PCR primer design, and data validation [2]. The computation of an exact multiple sequence alignment (MSA) of a large set of sequences is extremely complex and is classified as an NP-complete problem [3]. Multiple sequence alignment provides more information than pairwise sequence alignment because it reveals regions which are conserved within a protein family that have structural and functional importance [1]. Multiple sequence alignment is used for distinctive objectives such as performing similarity search of sequences. The approach is used in classification problems (e.g., to classify members in the protein family, to identify close and distant relationship to infer phylogeny).

To explain MSA, let us consider a set of three or more DNA/RNA/protein sequences as depicted above. MSA will aim to align these sequences by introducing gaps in each sequence. For example, if there are k number of sequences of N length, then Si, i = 1, 2,…, K and:
Consequently, MSA of S will be obtained by inserting gaps (‘-‘) into the sequences in such a way that all resulting sequences \({S}_{i}^{ *}\) will have equal length N and \({S}_{i}^{*} = {S}_{i}\) after removal of all gaps from \({S}_{i}^{*}\), and no column will consist of gaps. Consider another MSA \({S}^{*}\) that consists of two sequences \({s}_{p}^{*}\) and \({s}_{q}^{*}\) in the alignment. Let us consider two nucleotides a and b in the aligned sequence here the score of the sequence alignment will be defined as:
To find the divergence d of a given set of aligned sequences, the following three methods are used. The divergence between sequences can also be called as the total distance between sequences or the alignment score.
Consensus Method: In the consensus method, a common character from each column is searched and the string created in this way is called the consensus string. The total distance between two sequences is calculated by adding a penalty for each character in its column that differs in the sequence from the consensus string. Let us consider S as a set of sequence wherein S = {S1, S2, ..Sk} and:
Here, S* is the consensus string of S and disti is the distance of i-th sequence from S*.
Evolutionary Tree Method: A weighted evolutionary tree is created using sequences where adjacent nodes correspond to the sequence pair. The weight of the tree is defined as the summation of the number of changes between two adjacent nodes in the tree.
Sum of Pairs (SP score): The sum of pairs score is the pairwise distance between all sequence pairs. SP score is widely used similarity function. SP score for the two protein sequences is given as predefined BLOSUM or PAM matrix but for more than two sequences, and since the number of possible combinations is too large, SP score needs to be calculated. Let us consider S as a given set of sequence:
Here, the align score is the alignment score between Si and Sj sequences. The align_score is equal to the sum of the similarity score of every pair minus gap penalties [4]. The problem of finding a multiple sequence alignment that maximizes the SP score (or minimizes the SP distance) is known to be NP hard [5, 6].
2 Literature Review
To perform multiple sequence alignment, four distinctive approaches have been discussed in the literature namely global optimization, approximation, heuristic, and probabilistic methods. The probabilistic approach finds the probability of mutation and indels that leads to generating information related to the probability of evolution. Probabilistic methods work efficiently for phylogenetic analysis [7,8,9]. The global optimization, approximation, and heuristic methods find the optimized score for multiple sequence alignment and are suitable for classification problems. Dynamic programming is a global optimization approach, but the limitation of dynamic programming is that it takes exponential time. Simulated annealing and genetic algorithms have been also used by some researchers to get optimal results [10, 11]. Another approach to overcome the limitation of dynamic programming is to use different search methodologies and improve the efficiency of the global optimization [12,13,14]. These methods work efficiently with the small datasets but for the large datasets, approximation method is highly useful [15, 16]. Heuristics-based algorithms for multiple sequence alignment can be classified into two groups that are progressive heuristics and iterative heuristics-based algorithms. ClustalW [17, 18] and MUSCLE [19, 20] are well known examples of progressive heuristics and iterative heuristic algorithms, respectively. A combination of heuristic and probabilistic methods has been also suggested by few researchers [19, 21,22,23,24,25]. Other heuristics-based multiple-sequence alignment methods include simulated annealing [26], branch and bound [27], genetic algorithms [28, 29], Tabu search [30], hidden Markov modeling [31], countless agglomerative and progressive alignment [32], etc. Moreover, some other publically available tools for multiple sequence alignment are Clustal-Omega [33], KAlign [34], MAFET [35], Prank [36,37,38], TCOFFEE [39,40,41], ContraAlign [42], Prime [43], and DiAlign [44,45,46].
3 The Proposed Algorithm
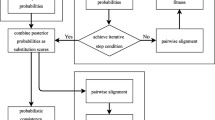
The proposed algorithm is dynamic programming-based multiple-sequence alignment algorithm which is an improved version of the already proposed adaptive evolutionary clustering algorithm MPSAGA [47]. The proposed method was executed with a set of sequences \({S} = \left\{ {{s}_{1} ,{s}_{2} ,{s}_{3} , \ldots {s}_{n} } \right\}\). The pair of sequences was identified from the sequence set such as paired_sequences \(\left( {\text{PS}} \right) = \left\{ {\left\{ {{s}_{1} ,{s}_{2} } \right\},\left\{ {{s}_{3} ,{s}_{4} } \right\},..\left\{ {{s}_{{{n} - 1}} ,{s}_{n} } \right\}} \right\}\). Using MPSAGA algorithm, these sequences were aligned pairwise. The alignment of these paired sequences was denoted as Aij where i and j denoted the index of the aligned sequences (si and sj). The set of all resultant alignments was denoted as \({A}^{*} = \left\{ {{A}_{12} ,{A}_{34} , \ldots ,{A}_{pq} } \right\}\), where p = (n − 1)/2 and q = (n − 1), when there was even number of sequences in the alignment. However, if the odd number of sequences were provided, then one sequence remained unpaired and got paired with the first sequence. For example, if there are 6 (even) sequences to be aligned, then the sequence pairs will be denoted as \({\text{PS}} = \left\{ {\left\{ {{s}_{1} ,{s}_{2} } \right\},\left\{ {{s}_{3} ,{s}_{4} } \right\},\left\{ {{s}_{5} ,{s}_{6} } \right\}} \right\}\). But if provided 7 (odd) sequences, then the sequence pairs will be denoted as \({\text{PS}} = \left\{ {\left\{ {{s}_{1} ,{s}_{2} } \right\},\left\{ {{s}_{3} ,{s}_{4} } \right\},\left\{ {{s}_{5} ,{s}_{6} } \right\},\left\{ {{s}_{7} ,{s}_{1} } \right\}} \right\}\) wherein the last unpaired sequence will be paired with the first sequence. The distance between these sequences will be reflected as match_score. The match_score of alignment can be calculated by the following formula: Match_Score = matches reward–mismatches penalty - gap opening penalty - gap extension penalty—indels penalty (7).
The default values used for the parameters in this algorithm are Match_Reward = +2, Gap_Opening = −1, Gap_Extension = −2, Mismatch = −2, and Indel = −2. In the next step, to group similar data items, the resultant pairs were clustered with an adaptive evolutionary clustering algorithm [48]. The step is helpful for the large datasets and can be skipped if the method is applied to the small datasets. The fitness of the clusters is calculated based on the fitness score of the individual clusters, i.e., match_score, and the clusters are sorted based on their average health [48]. Intracluster sorting is performed with each cluster based on their fitness. Finally, all the clusters are merged and sorted. These aligned sequences resulting from the multiple sequence alignment are sorted according to their match score. The flowchart of the proposed algorithm has been provided in Fig. 1.

The proposed MSA-MPSAGA (MPS) algorithm
4 Materials and Methods
The present research study was performed on a Windows-based system having an intel i5 processor with 8 GB RAM and 1 TB hard disk. The algorithm was implemented in Java 8 and executed for multiple sequence alignment on nine randomly chosen sequences downloaded from NCBI (https://www.ncbi.nlm.nih.gov/). The NCBI data are publically available for research use, and one can retrieve it by simply submitting the sequence ID.
4.1 Datasets Used
To check the performance and accuracy of the proposed multiple sequence aligner, an empirical study was performed in which the following four datasets were used: BAliBASE [49], MattBench [50], Homstrad [51], and Sisyphus [52]. These datasets contained 4–25 sequences. Random sampling was performed on the datasets to create an artificial dataset of 115 sequences as dataset 1, dataset 2, dataset 3, and dataset 4.
4.2 Evaluation Criteria
To check the accuracy of the alignments, FastSP v. 1.6.0 [53, 54] was used. FastSP calculates the alignment accuracy with respect to SP score. The accuracy measures provide a value between 0.0 and 1.0. The value of SP score 1.0 indicates the perfect accuracy, and the value of SP score 0.0 indicative of the sequence alignment is incorrect. FastSP also indicates an expansion ratio which is the ratio between the number of matches in the estimated alignment and the observed alignment. The value of expansion ratio less than 1.0 is an indication of over alignment, and value more than 1.0 corresponds to under alignment.
5 Results
The proposed algorithm was executed in a single run to perform multiple sequence alignment for the nine sequences downloaded from NCBI (Table 1). The results of multiple sequence alignment have been shown in a similarity matrix. The percent similarity of each sequence with the other sequence is called conservancy, and in the present study, it was calculated using MSA-MPSAGA (MPS) (Table 2). MSA was also performed using ClustalW (CW) [55], TCOFFEE (TC) [56], and MUSCLE (ML) aligners [57, 58]. To compare the results of MSA obtained using all the algorithms tested, visualization method was used. Consequently, the overall results of the multiple sequence alignment were used to construct phylogenetic trees using Phylogenetic Tree Viewer—ETE Toolkit (Table 3).
The empirical study was performed on the four data subsets. The summary of the results is shown in Table 4. The average number of the aligned sequences was observed to be 13, 7, 8, and 10 for the dataset 1, dataset 2, dataset 3, and dataset 4, respectively. The average length of the sequences in the dataset 1 was found to be 765 in which 38 gaps were inserted by the proposed algorithm to align the sequences. The average gap length in the aligned sequences in the dataset 1 was observed to be 9. In dataset 2, the average length of sequences was 260. To align these sequences, average 17 gaps were inserted with an average gap length of 4. A total of 8 sequences of average 421 lengths were aligned by inserting a total of 47 gaps and with an average of 3 basepair long gap length. While ten sequences with average 185 lengths were aligned by inserting 25 gaps with an average of 6 gap length.
The comparison of modeler score and SP score for the four tested algorithms is given in Table 5. It indicated that the MPS algorithm provides the SP score similar to the expected score. The modeler score and SP score of CW, TC, ML, and MPS were observed to be (0.70 and 0.50), (0.78 and 0.77), (0.678 and 0.69), and (0.72 and 0.72), respectively. Each dataset used in these experiments had at most 25 sequences. A total of 115 sequences from a subset of four datasets were used (46 from dataset 1, 36 from dataset 2, 18 from dataset 3, and 15 from dataset 4) (Fig. 2).

The comparison of modeler score and SP score between four benchmarking datasets
In the other experiment, ten sequence sets of different protein categories were aligned using CW, ML, TC, and MPS algorithms. The consolidated results of the aligned sequence such as average aligned sequence length, the average number of matches in the aligned sequences, number of gaps inserted to align the sequences, and the average gap length inserted in the aligned sequences have been detailed out in Table 6. A comparison of the average length of the aligned sequences for each category of proteins is given in Fig. 3.

Comparison of alignment length of MSA-MPSAGA with ClustalW, MUSCLE, and TCOFFEE multiple sequence aligners
Multiple sequence alignment using CW, TC, ML, and MPS aligners provided the average alignment length to be 224.8, 216.8, 229.5, and 230.1, respectively. The proposed algorithm MPS aligned sequences with an increased length of 0.004%, 0.067%, and 0.027% than the CW, TC, and ML algorithms, respectively. The number of matches in the aligned sequences is shown in Fig. 4. The average number of matches in the aligned sequences was observed to be 29.1, 25.2, 27.7, and 29.2 for CW, TC, ML, and MPS algorithms, respectively. MPS algorithm was found to align the sequences with an increased match of 0.025%, 0.181%, and 0.088% than the CW, TC, and ML algorithms. Comparison based on the number of gaps inserted in the aligned sequences by four multiple sequence alignment algorithms is shown in Fig. 5. The numbers of gaps inserted in the aligned sequences by CW, TC, ML, and MPS aligners were observed to be 271, 280, 280, and 279, respectively. The proposed MPS aligner inserted an increased number of gapes in the aligned sequences than the CW (0.03%), TC (0.014%), and ML (0.014%) algorithms.

Comparison of the number of matches occurred in the aligned sequences using ClustalW, TCOFFEE, and MUSCLE aligners with MSA-MPSAGA

Comparison of gaps inserted in the aligned sequences by MSA-MPSAGA with ClustalW, TCOFFEE, and MUSCLE aligners
A comparative study based on the match scores of the multiple alignments was also performed using CW, TC, ML, and MPS algorithms. The match score was calculated for the multiple sequence alignments performed by CW, TC, ML, and MPS algorithms that were observed to be 4484.6, 4084.7, 4589.6, and 4682.3, respectively (Fig. 6).

Comparison of Match Score for MSA performed by MSA-MPSAGA with ClustalW, TCOFFEE, and MUSCLE aligners
6 Conclusion
Conclusively, it can be stated that the proposed multiple sequence aligner based on adaptive evolutionary clustering algorithm (MSAMPSAGA or MPS) accurately identifies the sequence alignments. Furthermore, an average increase in sequence alignment length using the proposed aligner was observed to be 0.03% than the other tested algorithms ClustalW, TCOFFEE, and MUSCLE. The phylogenetic trees constructed from the MSA obtained from the aligners also indicated that the MPS provides more accurate results. The overall comparison of MPS with the other three tested algorithms showed that the qualitative and quantitative performance of the proposed algorithm is at par as compared to the other aligners. The only limitation of the proposed MPS algorithm is that the algorithm is more useful in doing MSA of biological sequences. The implementation of the proposed algorithm in aligning other types of sequences in the varied dataset is a scope of future study.
References
Wiltgen M (2018) Algorithms for structure comparison and analysis: homology modelling of proteins. Encyclopedia Bioinform Comput Biol: ABC Bioinform 21:38
Carsten K, Notredame C (2009) Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics 25:2455–2465 (Oxford, England)
Wang L, Jiang T (1994) On the complexity of multiple sequence alignment. J Comput Biol 1(4):337–348
Sung WK. Algorithms in bioinformatics: a practical introduction by (CHAPMAN & HALL/CRC mathematical and computational biology series) ISBN 978-1-4200-7033-0
Just W (2001) Computational complexity of multiple sequence alignment with SP-score. J Comput Biol 8(6):615–623
Wang L, Jiang T (1994) On the complexity of multiple sequence alignment. J Comput Biol 1(4):337–348
Holmes I (2003) Using guide trees to construct multiple-sequence evolutionary HMMs. Bioinformatics 19(Suppl 1):i147–i157
Holmes I, Bruno WJ (2001) Evolutionary HMMs: a Bayesian approach to multiple alignment. Bioinformatics 17(9):803–820
Sonnhammer EL, Eddy SR, Birney E, Bateman A, Durbin R (1998) Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res 26(1):320–322
Kim J, Pramanik S, Chung MJ (1994) Multiple sequence alignment using simulated annealing. Comput Appl Biosci 10(4):419–426
Notredame C, Higgins DG (1996) SAGA: sequence alignment by genetic algorithm. Nucleic Acids Res 24(8):1515–1524
Gupta SK, Kececioglu JD, Schaffer AA (1995) Improving the practical space and time efficiency of the shortest-paths approach to sum-of-pairs multiple sequence alignment. J Comput Mol Cell Biol 2(3):459–472
Lipman DJ, Altschul SF, Kececioglu JD (1989) A tool for multiple sequence alignment. Proc. Natl Acad Sci USA 86(12):4412–4415
Stoye J, Moulton V, Dress AW (1997) DCA: An efficient implementation of the divide-and-conquer approach to simultaneous multiple sequence alignment. CABIOS 13(6):625–626
Gusfield D (1993) Efficient methods for multiple sequence alignment with guaranteed error bounds. Bull Math Biol 5(1):141–154
Pevzner P (1992) Multiple alignment, communication cost, and graph matching. SIAM J Appl Math 52(6):1763–1779
Higgins DG, Sharp PM (1988) CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene 3(1):237–244
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Loytynoja A, Milinkovitch MC (2003) A hidden Markov model for progressive multiple alignment. Bioinformatics 19(12):1505–1513
Edgar RC, Sjöander K (2004) COACH: profile-profile alignment of protein families using hidden markov models. Bioinformatics 20(8):1309–1318
Edgar RC, Sjölander K (2003) SATCHMO: sequence alignment and tree construction using hidden Markov models. Bioinformatics 19(11):1404–1411
Loytynoja A, Goldman N (2005) An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci USA 102(30):10557–10562
Do CB, Mahabhashyam MSP, Brudno M, Batzoglou S (2005) ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res 15(2):330–340
Abhiman S, Daub CO, Sonnhammer EL (2006) Prediction of function divergence in protein families using the substitution rate variation parameter alpha. Mol Biol Evol 23(7):1406–1413
Reinert K et al (1997) A branch-and-cut algorithm for multiple sequence alignment. In: Santa Fe NM (ed) Recomb97. ACM Press, pp 241–249
Gondro C, Kinghorn BP (2007) A simple genetic algorithm for multiple sequence alignment. Genet Mol Res 6(4):964–982
Notredame C, Higgins DG (1996) SAGA: sequence alignment by genetic algorithm. Nucleic Acids Res 24(8):1515–1524
Riaz T, Yi W, Li KB (2005) A tabu search algorithm for post-processing multiple sequence alignment. J Bioinformatics Comput Biol 3(01):145–156
Rawlings CJ (1995) ISMB-95: Proceedings, third international conference on intelligent systems for molecular biology. AAAI Press
Hogeweg P, Hesper B (1984) The alignment of sets of sequences and the construction of phyletic trees: an integrated method. J Mol Evol 20(2):175–186
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7(1):539
Lassmann T, Sonnhammer ELL (2005) Automatic assessment of alignment quality. Nucleic Acids Res 33(22):7120–7128
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30(14):3059–3066
Loytynoja A, Goldman N (2008) Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320(5883):1632–1635
Löytynoja A, Goldman N (2005) An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci USA 102(30):10557–10562
Notredame C, Higgins DG, Heringa J (2000) T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302(1):205–217
Notredame C (2007) Recent evolutions of multiple sequence alignment algorithms. PLoS Comput Biol 3(8):1405–1408
O’Sullivan O, Suhre K, Abergel C, Higgins DG, Notredame C (2004) 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J Mol Biol 340(2):385–395
Do CB, Gross SS, Batzoglou S (2006) Contralign: discriminative training for protein sequence alignment. In: Research in computational molecular biology: 10th annual international conference, RECOMB 2006, Venice, Italy. Springer, Heidelberg, pp 160–174
Yamada S, Gotoh O, Yamana H (2006) Improvement in accuracy of multiple sequence alignment using novel group-to-group sequence alignment algorithm with piecewise linear gap cost. BMC Bioinformatics 7:524
Golubchik T, Wise MJ, Easteal S, Jermiin LS (2007) Mind the gaps: evidence of bias in estimates of multiple sequence alignments. Mol Biol Evol 24(11):2433–2442
Morgenstern B (1999) DIALIGN 2: improvement of the segmentto-segment approach to multiple sequence alignment. Bioinformatics 15(3):211–218
Pei J, Grishin NV (2006) MUMMALS: multiple sequence alignment improved by using hidden markov models with local structural information. Nucleic Acids Res 34(16):4364–4374
Mirarab S, Warnow T (2011) FASTSP: Linear time calculation of alignment accuracy. Bioinformatics 27(23):3250–3258
Lakhani J, Khunteta A, Choudhary A, Harwani D (2019) MPSAGA: a matrix-based pairwise sequence alignment algorithm for global alignment with position based sequence representation. Sādhanā 44(7):171
Lakhani J, Khunteta A, Chowdhary A, Harwani D (2016) Auto-evolving clusters based on rejection and migration. In: Bishnoi SK, Kuri M, Goar V (eds) Proceedings of the International Conference on Advances in Information Communication Technology & Computing (AICTC ‘16). ACM, New York, NY, USA, Article 98
Thomson JD, Plewniak F, Poch O (1999) BAliBASE: a benchmark alignment database for the evaluation of multiple alignment programs. Bioinformatics 15(1):87–88
Daniels NM, Kumar A, Cowen LJ, Menke M (2012) Touring protein space with Matt. IEEE/ACM Trans Comput Biol Bioinform 9:286–293
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539
Andreeva A, Prlić A, Hubbard TJP, Alexey GM (2007) SISYPHUS—structural alignments for proteins with non-trivial relationships. Nucleic Acids Res 35:D253–D259
Tang X, Wong DF (2001) FAST-SP: a fast algorithm for block placement based on sequence pair. In: Proceedings of the 2001 Asia and South Pacific design automation conference. ACM, pp 521–526
Mirarab S, Warnow T (2011) FastSP: linear time calculation of alignment accuracy. Bioinformatics 27(23):3250–3258
Thompson JD, Gibson TJ, Higgins DG (2003) Multiple sequence alignment using ClustalW and ClustalX. Curr Proto Bioinfo 1:2–3
Notredame C, Higgins DG, Heringa J (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302(1):205–217
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5(1):113
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Lakhani, J., Khunteta, A., Chowdhary, A., Harwani, D. (2021). Multiple Sequence Alignment Algorithm Using Adaptive Evolutionary Clustering. In: Goar, V., Kuri, M., Kumar, R., Senjyu, T. (eds) Advances in Information Communication Technology and Computing. Lecture Notes in Networks and Systems, vol 135. Springer, Singapore. https://doi.org/10.1007/978-981-15-5421-6_36
Download citation
DOI: https://doi.org/10.1007/978-981-15-5421-6_36
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-5420-9
Online ISBN: 978-981-15-5421-6
eBook Packages: EngineeringEngineering (R0)