
Abstract
Congenital pulmonary airway malformation (CPAM) type 1, CPAM type 2, bronchial atresia, and intralobar and extralobar pulmonary sequestration (IPS/EPS) are congenital cystic lung diseases (CCLD) that are most frequently submitted for pathological examination, whereas lobar emphysema, CPAM type 4, and bronchogenic cyst are less common. CPAM type 0, CPAM type 3, and alveolar capillary dysplasia are extremely rare. Pulmonary interstitial emphysema is not actually congenital, but usually occurs in the neonatal period, and a differential diagnosis with CCLD is often required. Fetal lung interstitial tumor (FLIT) is a tumorous lesion that usually occurs in the neonatal period, and sometimes in the fetal period.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

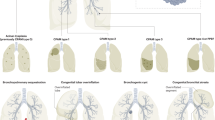

Keywords
- Congenital cystic lung disease
- Bronchogenic cyst
- Bronchial atresia
- Infantile lobar emphysema
- Congenital pulmonary airway malformation
- Intra/extralobar pulmonary sequestration
- Pulmonary interstitial emphysema
- Alveolar capillary dysplasia
- Fetal lung interstitial tumor
14.1 Bronchogenic Cyst
Bronchogenic cyst (BC) is assumed to originate from supernumerary lung buds. It is most often found in the middle mediastinum and less frequently adjacent to the pulmonary hilus. Macroscopically, it is a unilocular mass containing mucinous substances (Fig. 14.1a, b), and it usually has no communication with the preexisting trachea, bronchus, or pulmonary parenchyma. The diameter of the cyst is approximately 4–5 cm, but sometimes exceeds 10 cm in older patients. Microscopically, the cystic wall is lined with ciliated columnar or pseudostratified columnar epithelium, and the walls contain cartilaginous islands, bronchial glands, or smooth muscle bundles, mimicking the structure of the normal bronchus (Fig. 14.1c). The epithelium sometimes shows squamous metaplasia. A differential diagnosis may be required from CPAM type 1 and esophageal duplication. However, BC can be distinguished from these diseases by its epithelial type on histology.

Bronchogenic cyst. (a, b) Macroscopic findings (a) and semi-macroscopic findings (b). A unilocular mass containing mucinous substances is present at the hilus. (c) Microscopic findings. The cystic wall is lined with ciliated columnar or pseudostratified columnar epithelium, and cartilaginous islands, bronchial glands, or smooth muscle bundles occur in the wall, mimicking the structure of the normal bronchus
14.2 Bronchial Atresia/Infantile Lobar Emphysema
14.2.1 Bronchial Atresia
Bronchial atresia (BA) is a relatively common CCLD, and is defined as a malformation of the pulmonary parenchyma resulting from atresia of the bronchus or bronchiole. Atresia of the bronchus is often detected in imaging studies before resection. On macroscopic examination, a mucus plug is seen at the atretic bronchus and cystic lesions around the atretic bronchus (Fig. 14.2a). The distal pulmonary parenchyma of the lesion is often emphysematous. Microscopically, a mucus plug is seen in the dilated bronchus and in the peripheral smaller bronchiole, which is diagnostic for BA (Fig. 14.2b, c). Fibrous tissues are seen at the atretic site, and the bronchial cartilage is often malformed. Variously sized cystic lesions lined with ciliated columnar epithelium, some of which have back-to-back bronchiole-like structures and are occasionally surrounded by smooth muscle bundles, are observed in the peripheral pulmonary parenchyma of the atretic bronchus (Fig. 14.2d). These findings are considered to indicate microcystic parenchymal maldevelopment of the bronchial atresia sequence [1]. The cystic lesions are similar to those of CPAM type 2. If distinct atresia of the bronchus is identified on macroscopic or microscopic examination, the lesion should be diagnosed solely as BA, and the term hybrid lesion of BA and CPAM type 2 should be avoided [2, 3]. Emphysematous changes are often observed around the lesions (Fig. 14.2e).

Bronchial atresia. (a) Macroscopic findings. A mucus plug is present at the atretic bronchus (asterisk), and small cystic lesions occur around the atretic bronchus. The distal pulmonary parenchyma is emphysematous. (b–e) Microscopic findings. A mucus plug is present in the dilated bronchus (asterisk, b). The cartilage around the atretic bronchus is often malformed (c). Variously sized cystic lesions lined with ciliated columnar epithelium, some with back-to-back bronchiole-like structures, known as microcystic parenchymal maldevelopment, occur around the atretic bronchus (d). The peripheral pulmonary parenchyma is emphysematous (e)
14.2.2 Infantile (Congenital) Lobar Emphysema (Overinflation)
Infantile lobar emphysema (ILE) is defined as the overinflation or hyperplasia of the pulmonary parenchyma associated with bronchial atresia or stenosis. It is attributed to a bronchial structural abnormality or extrinsic or intrinsic obstruction, although the cause of the atresia/stenosis is unclear in a considerable number of cases, even on histological examination. The lesion may involve the entire lobe or part of the lobe. On macroscopic examination, the affected lobe is hyperexpanded, and individual alveoli are sometimes visible (Fig. 14.3a). Microscopically, two patterns can be identified: the classic pattern and the hyperplastic (polyalveolar) pattern. In the classic pattern, the alveolar ducts and sacs are distended, often by as much as 3–10 times their normal size, and the alveolar walls are often focally disrupted (Fig. 14.3b, c). In the hyperplastic (polyalveolar) pattern, the lung parenchyma is more complicated, with an increased number of alveoli, and the radial alveolar counts are often 2–3 times that expected for the patient’s age. The alveolar ducts and sacs may or may not be distended.

Lobar emphysema. (a) Macroscopic findings. The affected lobe is hyperexpanded. (b, c) Microscopic findings of the classic pattern. The alveolar ducts and sacs of the affected pulmonary parenchyma are distended (upper part of figure b) by up to 3–10 times the normal size. Lower part of (b) shows unaffected pulmonary parenchyma. The alveolar walls are often focally disrupted (c)
14.3 Congenital Pulmonary Airway Malformation
Congenital pulmonary airway malformation (CPAM) was originally described by Ch’In et al. in 1949 as a congenial cystic adenomatoid malformation (CCAM) [4]. In 1977, Stocker classified CCAM from type 1 to type 3, depending on the size of the cysts [5], and extended the classification to five types (types 0–4) based on the histological similarity of the cyst and airway structure of the trachea to alveoli, and introduced the term CPAM. CPAM is currently more commonly used than CCAM. In 2003, Langston established revised criteria for congenital cystic disease [1]. Further consideration of the definition of CPAM, including the clarification of its genetic background, is warranted.
14.3.1 CPAM Type 0
CPAM type 0, also described as congenital acinar dysplasia (CAD), is considered to arise from maturation arrest at the pseudoglandular stage, and is extremely rare and absolutely lethal. Macroscopically, the lesions are firm masses, and in cut section, show tiny scattered cystic lesions. Microscopically, the lesion is composed of bronchus-like structures without alveolar development, i.e., irregularly shaped tubules lined with simple or pseudostratified columnar epithelium, surrounded by mesenchymal tissues containing scattered muscle fibers, immature cartilaginous tissues, and large thin-walled vascular channels. Disruption of the TBX4–FGF10 pathway is reported to be a cause of CAD [6].
14.3.2 CPAM Type 1
CPAM type 1 was originally defined as a congenital cystic lesion composed of one or more large cysts, measuring more than 2 cm, with smaller cysts around the large cysts (Fig. 14.4a, b). It is reported to account for approximately 65% of all CPAMs [7]. Microscopically, the cysts are lined with ciliated columnar epithelium, often showing a sawtooth configuration. Clusters of mucogenic cells are present in the linings, surrounded by fibromuscular tissues, and in some cases, by malformed cartilage plates (Fig. 14.4c–e).

Congenital pulmonary airway malformation, type 1. (a, b) Macroscopic (a) and semi-macroscopic findings (b). The lesion is composed of one or more large cysts, with smaller cysts around the large cysts (c, d). Microscopic findings. The cysts are lined with ciliated columnar epithelium, often showing a sawtooth configuration (c), and clusters of mucogenic cells are present in the linings (d)
CPAM type 1 that undergoes surgical resection in the neonatal period may show smaller cysts (<2 cm). A rare type of CPAM type 1 composed only of small cysts, and the epithelium that is characteristic of CPAM type 1 (solid variant) is present (Fig. 14.5). Histological evidence of epithelium, particularly mucogenic cells, in addition to the size of the cyst are important for the diagnosis of CPAM type 1.

Congenital pulmonary airway malformation, type 1, solid variant. (a) Macroscopic findings. The cut surface is firm, with no large cyst, but scattered small cystic lesions are present. (b) Microscopic findings. The epithelium of the cysts are similar to those of classical CPAM type 1. Clusters of mucogenic cells are seen quite frequently in solid variants. Inset: high-power view of the cluster of mucogenic cells
Some cases of CPAM type 1 were reported to develop into adenocarcinoma, usually in the form of bronchioloalveolar carcinoma, which is associated with the genetic background, including KRAS and EGFR mutations [8, 9].
14.3.3 CPAM Type 2
CPAM type 2 was originally defined as a congenital cystic lesion predominantly composed of uniformly sized cysts of <2 cm (Fig. 14.6a, b). It is reported to account for approximately 10–15% of CPAMs [7]. Microscopically, the cysts are lined with ciliated columnar epithelium, with back-to-back bronchiole-like structures, known as microcystic parenchymal maldevelopment, and are occasionally surrounded by smooth muscle bundles (Fig. 14.6c). Normal alveolar structures and bronchioles are involved in the lesions. Mucogenic cells are not found in CPAM type 2. Rhabdomyoblasts are occasionally observed around the cysts (rhabdomyomatous dysplasia).

Congenital pulmonary airway malformation, type 2 (a, b). Macroscopic (a) and semi-macroscopic findings (b). The lesion is composed of predominantly uniformly sized cysts of <2 cm (b). Microscopic findings. The cysts are lined with ciliated columnar epithelium, and back-to-back bronchiole-like structures, occasionally surrounded by smooth muscle bundles, are observed (microcystic parenchymal maldevelopment). Normal alveolar structures and bronchioles are present in the lesions. No mucous plug is present (Courtesy of Dr. Yoshida, Hyogo Prefectural Kobe Children’s Hospital)
The histological findings are similar to those in the pulmonary parenchyma of bronchial atresia and pulmonary sequestration. Therefore, a diagnosis of CPAM type 2 should be avoided in patients in whom atresia of the bronchus or the presence of an aberrant artery is confirmed.
14.3.4 CPAM Type 3
CPAM type 3 is composed predominantly of uniformly sized cysts that rarely exceed 0.2 cm, and are firm solid lesions on macroscopic examination (Fig. 14.7a). CPAM type 3 involves the entire lobe, or even the entire lung, and reportedly accounts for approximately 5% of all CPAMs [7]. Microscopically, the cysts resemble the fetal lung tissue of the pseudoglandular period, i.e., contain evenly distributed small cysts lined by cuboidal epithelium, with no bronchial structure (Fig. 14.7b). The septa are wider than in the normal fetal lung, and the vessels are sparse. Mucogenic cells and rhabdomyoblasts are not found in CPAM type 3.

Congenital pulmonary airway malformation, type 3. (a) Macroscopic findings. The affected lung appears as a firm solid lesion. The entire lung of the presenting patient is affected. (b) Microscopic findings. The cysts resemble fetal lung tissue of the pseudoglandular period; evenly distributed small cysts lined with cuboidal epithelium, with no bronchial structure. Vessels are sparse. Inset: high-power view
14.3.5 CPAM Type 4
CPAM type 4 is usually located at the periphery of the lung, often beneath the pleura, and occasionally the lesion protrudes from the lung tissue (Fig. 14.8a). CPAM type 4 reportedly accounts for 10–15% of CPAMs [7]. On gross examination, the lesion is often composed of large cysts with thin septa. Microscopically, the cysts are lined with single flattened epithelium, resembling alveolar epithelium (Fig. 14.8b, c). CPAM type 4 and pleuropulmonary blastoma (PPB) type I (purely cystic PPB) are barely distinguishable from one another on imaging studies or gross examination. These lesions are known to be related by their background, as both involve an abnormality of the DICER1 gene [10]. At the diagnosis of CPAM type 4, careful examination is required to detect the histological findings of PPB, such as a cluster of blastemal cells beneath the epithelium, described as cambium layer.

Congenital pulmonary airway malformation, type 4. (a) Macroscopic findings. The lesion is located at the periphery of the lung (arrow), and protrudes from the lung tissue (asterisk). (Courtesy of Dr. Matsuoka and Dr. Takeuchi, Osaka Women’s and Children’s Hospital). (b, c) Microscopic findings. The lesion is located beneath the pleura. Normal pulmonary parenchyma is seen in the lower part of (b). The lesion is composed of large cysts with thin septa, lined with single flattened epithelium resembling alveolar epithelium. No blastemal cells are present in the lesion
14.4 Sequestration
Pulmonary sequestration (PS) is defined as abnormal pulmonary tissue lacking any connection to the tracheobronchial tree, and has an anomalous systemic arterial supply. PS is considered to be a congenital malformation, possibly derived from a supernumerary lung bud. It is classified into two categories, intralobar and extralobar PS, depending upon its location in relation to the adjacent normal lung and its visceral pleural covering. Abnormal communication with the alimentary tract is observed in extremely rare cases.
14.4.1 Intralobar PS
Intralobar PS (IPS) is located within the lung tissue and lacks its own visceral pleura. It typically affects the lower lobes, most commonly in the left posterior basal segment. On gross inspection, an aberrant artery is present at the lower border of the pulmonary ligament, and the lymph nodes are often visible. On the cut surface, the sequestered segment of the lung shows variably sized cystic lesions or overinflation (Fig. 14.9a). Chronic inflammation is prominent in older children, but indistinct in infant patients. A dilated and tortuous aberrant artery is prominent in the sequestered segment (Fig. 14.9b). Microscopically, a large elastic artery occurs at the pulmonary ligament, and lymph nodes and a bronchus-like structure with bronchial cartilage and/or bronchial glands are seen adjacent to the artery (Fig. 14.9c–e).

Intralobar pulmonary sequestration. (a, b) Macroscopic findings. Cystically dilated blind-ended bronchus and small cystic lesions with mucous retained in the parenchyma are present (a). At the cut surface, many anomalous vascular structures are present in the lower base segment (b, arrow). (c–e) Microscopic findings of aberrant hilus-like structures. Dilated bronchus with cartilage and bronchial glands (c), and Lymph node and aberrant elastic-type artery are present (d, e, e: Elastica Masson stain). (f, g) Microscopic findings of pulmonary parenchyma. The peripheral parenchyma shows microcystic maldevelopment, similar to CPAM type 2 (f), or hyperplasia-type maldevelopment (g)
The structure of the bronchial tree in a sequestered lung was investigated in a case series by Ishida et al., who demonstrated that “congenital” IPS showed the “peripheral type” structure, consisting of blind-ended bronchial trees running toward the insertion site of the aberrant elastic artery of the pulmonary ligament, and often accompanied by lymph nodes adjacent to the aberrant artery [11]. These three components, an aberrant elastic artery, blind-ended bronchus with cartilage and/or bronchial glands, and lymph nodes, are regarded as ectopic hilus-like structures, and are the histological findings specific for congenital IPS.
IPS is considered to be a variant of bronchial atresia occurring in the posterior basal segment [1]. A parenchymal cystic lesion resembling CPAM type 2 is occasionally observed histologically (Fig. 14.9f, g), but it should not be diagnosed as a hybrid lesion of IPS and CPAM. The detection of an aberrant hilus-like structure is essential for a diagnosis of congenital IPS, and the aberrant artery must be an elastic artery. If the aberrant artery is a muscular artery, it should be regarded as a systemic–pulmonary shunt, which is usually associated with inflammatory changes.
14.4.2 Extralobar PS Without Foregut Communication
Extralobar PS (EPS) without foregut communication is a congenital anomaly, with its own pleural investment and separated from the normal lung parenchyma. On gross examination, EPS is an ovoid or pyramidal mass with a systemic arterial supply from the distal thoracic or proximal abdominal aorta, and venous outflow to the systemic veins. The cut surface reveals homogeneous lung parenchyma or a cluster of small cysts. Microscopically, a hilus-like structure is often seen near the cut surface (Fig. 14.10a, b). The parenchyma consists of dilated bronchioles, alveolar ducts, and alveoli, often containing mucous material and macrophages. An adenomatoid cystic lesion similar to that of CPAM type 2 is seen in about 50% of cases of EPS (Fig. 14.10c). Like IPS, these findings are considered to represent microcystic parenchymal maldevelopment of the bronchial atresia sequence, and EPS should not be diagnosed as a hybrid lesion of EPS and CPAM. In rare cases, striated muscle fibers in the cyst wall or around the alveoli are seen, described as rhabdomyomatous dysplasia (Fig. 14.10d).

Extralobar sequestration. (a–d) Microscopic findings. Aberrant artery and vein (arrow), lymph node (arrowhead), blind-ended bronchus (asterisk), and small cysts with mucous retention are noted. (a, b, b: Elastica Masson stein). The parenchyma shows microcystic parenchymal maldevelopment (c). Desmin-positive striated muscle fibers are present in the cystic septum (d)
14.4.3 Extralobar PS with Foregut Communication
Extralobar PS with foregut communication is an extremely rare congenital malformation, characterized by a communicating fistula between an isolated part of the respiratory system and the esophagus or stomach. The lining of the communicating fistula is ciliated or squamous epithelium.
14.5 Pulmonary Interstitial Emphysema
Pulmonary interstitial emphysema (PIE) is defined as the dissection of bronchovascular structures and interlobular septa by air, and is usually associated with mechanical ventilation. PIE is an acquired lesion, but it is one of the most important differential diagnoses involving congenital cystic lung lesions in the neonatal period. PIE is classified as acute or persistent. On gross examination, PIE presents as air blebs of several millimeters on the pleural surface. Microscopically, the cysts are located in the interlobular septal and peribronchial regions. In persistent PIE, multinucleated foreign-body giant cells and many histiocytes line the abnormal space (Fig. 14.11a, b). Acute PIE lacks a histiocytic reaction and often communicates with the lymphatics of the interlobular area. Alveolar microcystic changes are occasionally seen at adjacent abnormal air spaces. The epithelial lining in the abnormal air space may be continuous with the alveolar cystic lesion (Fig. 14.11c, d).

Pulmonary interstitial emphysema. (a–d) Microscopic findings. Large abnormal air spaces are present around the bronchovascular structure (a). Multinucleated foreign-body giant cells occur on the inner lumen of the air space, indicated with an arrow in figure a (b). Alveolar microcystic changes occur adjacent to the air spaces, and communication between the air spaces and the surrounding alveolar sacs can be seen (arrowhead). (c) TTF1-positive lining cells are continuous with the abnormal air space (d)
14.6 Other Rare Diseases
14.6.1 Alveolar Capillary Dysplasia
Alveolar capillary dysplasia (ACD) is a rare and uniformly lethal disease, characterized by the maldevelopment of the alveolar capillaries. On macroscopic examination, the vessels may be prominent, but the macroscopic findings are otherwise unremarkable. Microscopically, the alveolar capillaries are relatively large, and are located in the center of the wide alveolar septa (Fig. 14.12a, b). Consequently, gas exchange at the alveoli is difficult. Misalignment of the pulmonary veins is commonly seen in ACD, and the pulmonary veins share the same adventia as the arteries (Fig. 14.12c, d).


Alveolar capillary dysplasia. Microscopic findings. (a, b) Relatively large alveolar capillaries are located in the center of the wide alveolar septa. (c, d) Pulmonary vein shares the adventitia of the artery. (b, d) Elastica Masson stain
The patient’s condition is often complicated with other anomalies, such as congenital heart disease, urogenital anomalies, and gastrointestinal anomalies, and familial occurrence is also seen [12]. Deletions in the FOX gene and mutations in the FOXF1 gene have been described as the causes of ACD [13].
14.6.2 Fetal Lung Interstitial Tumor
Fetal lung interstitial tumor (FLIT) is a fetal intrapulmonary lesion newly characterized by Dishop et al. in 2010 [14], that usually develops in neonates or early infants. On macroscopic examination, FLIT presents as an intrapulmonary well-circumscribed solid mass (Fig. 14.13a). On microscopic examination, the lesion shows an immature alveola-like structure, resembling the fetal lung of the canalicular period (Fig. 14.13b). The alveola-like structure is lined with flattened or cuboidal epithelium without cilia. Scattered immature mesenchymal cells with round nuclei and abundant glycogen-rich cytoplasm within edematous stroma are present in the septa. No overt atypia is observed in either the epithelial or stromal cells. Small bronchus-like structures with smooth muscle bundles, immature cartilaginous tissues, and extramedullary hematopoiesis may occur in the lesion. The lesion has an incomplete fibrous capsule (Fig. 14.13c).

Fetal lung interstitial tumor. (a) Macroscopic findings. The lesion appears as an intrapulmonary well-circumscribed solid mass. (b, c) Microscopic findings. The lesion (upper part of the photo) has an incomplete fibrous capsule, containing immature cartilaginous tissue (asterisk, b). The lesion is composed of an immature alveola-like structure lined with flattened or cuboidal epithelium, without cilia. The septa contain scattered immature mesenchymal cells with round nuclei and abundant glycogen-rich cytoplasm within the edematous stroma (c)
Chromosome 8 trisomy [15] and ALK rearrangement [16] are reported in cases of FLIT. Patients with ALK rearrangement may require a differential diagnosis with inflammatory myofibroblastic tumor [17]. The histological similarity of FLIT and CPAM type 3 has also been reported [18]. Further clarification of whether FLIT is developmental disorder or a true neoplastic lesion is required.
References
Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12:17–37.
Imai Y, Mark EJ. Cystic adenomatoid change is common to various forms of cystic lung diseases of children: a clinicopathologic analysis of 10 cases with emphasis on tracing the bronchial tree. Arch Pathol Lab Med. 2002;126:934–40.
Riedlinger WF, Vargas SO, Jennings RW, et al. Bronchial atresia is common to extralobar sequestration, intralobar sequestration, congenital cystic adenomatoid malformation, and lobar emphysema. Pediatr Dev Pathol. 2006;9:361–73.
Ch’In KY, Tang MY. Congenital adenomatoid malformation of one lobe of a lung with general anasarca. Arch Pathol. 1949;48:221–9.
Stocker JT, Madewell JE, Drake RM. Congenital cystic adenomatoid malformation of the lung. Classification and morphologic spectrum. Hum Pathol. 1977;8:155–71.
Karolak JA, Vincent M, Deutsch G, et al. Complex compound inheritance of lethal lung developmental disorders due to disruption of the TBX-FGF pathway. Am J Hum Genet. 2019;104:213–28.
Dehner LP, Stocker JT, Mani H, et al. Congenital pulmonary airway malformation in the respiratory tract. Philadelphia: Wolters Kluwer; 2016.
Guo H, Cajaiba MM, Borys D, et al. Expression of epidermal growth factor receptor, but not K-RAS mutations, is present in congenital cystic airway malformation/congenital pulmonary airway malformation. Hum Pathol. 2007;38:1772–8.
Lantuejoul S, Nicholson AG, Sartori G, et al. Mucinous cells in type 1 pulmonary congenital cystic adenomatoid malformation as mucinous bronchioloalveolar carcinoma precursors. Am J Surg Pathol. 2007;31:961–9.
Feinberg A, Hall NJ, Williams GM, et al. Can congenital pulmonary airway malformation be distinguished from type I pleuropulmonary blastoma based on clinical and radiological features? J Pediatr Surg. 2016;51:33–7.
Ishida H, Hajikano H, Hayashi A. Intralobal sequestration in children—a new concept from the form of bronchial tree in sequestrated lung. Nihon Kyobu Geka Gakkai Zasshi. 1992;40:957–68.
Slot E, Edel G, Cutz E, et al. Alveolar capillary dysplasia with misalignment of the pulmonary veins: clinical, histological, and genetic aspects. Pulm Circ. 2018;8:2045894018795143.
Stankiewicz P, Sen P, Bhatt SS, et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009;84:780–91.
Dishop MK, McKay EM, Kreiger PA, et al. Fetal lung interstitial tumor (FLIT): a proposed newly recognized lung tumor of infancy to be differentiated from cystic pleuropulmonary blastoma and other developmental pulmonary lesions. Am J Surg Pathol. 2010;34:1762–72.
de Chadarevian JP, Liu J, Pezanowski D, et al. Diagnosis of “fetal lung interstitial tumor” requires a FISH negative for trisomies 8 and 2. Am J Surg Pathol. 2011;35:1085. author reply 1086–1087
Onoda T, Kanno M, Sato H, et al. Identification of novel ALK rearrangement A2M-ALK in a neonate with fetal lung interstitial tumor. Genes Chromosomes Cancer. 2014;53:865–74.
Tanaka M, Kohashi K, Kushitani K, et al. Inflammatory myofibroblastic tumors of the lung carrying a chimeric A2M-ALK gene: report of 2 infantile cases and review of the differential diagnosis of infantile pulmonary lesions. Hum Pathol. 2017;66:177–82.
Yoshida M, Tanaka M, Gomi K, et al. Fetal lung interstitial tumor: the first Japanese case report and a comparison with fetal lung tissue and congenital cystic adenomatoid malformation/congenital pulmonary airway malformation type 3. Pathol Int. 2013;63:506–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Tanaka, M., Takakuwa, E. (2020). Pathology of Congenital Cystic Lung Diseases. In: Sago, H., Okuyama, H., Kanamori, Y. (eds) Congenital Cystic Lung Disease. Springer, Singapore. https://doi.org/10.1007/978-981-15-5175-8_14
Download citation
DOI: https://doi.org/10.1007/978-981-15-5175-8_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-5174-1
Online ISBN: 978-981-15-5175-8
eBook Packages: MedicineMedicine (R0)