
Abstract
Besides COPD, pulmonary fibrosis, and asthma, autophagy also participates in the development of many other respiratory diseases. Cystic fibrosis is an innate lung disease. Unlike idiopathic pulmonary fibrosis, cystic fibrosis has unique pathogenesis. Autophagy is an essential biological mechanism for the removal of misfolded proteins and damaged organelles in cells. Abnormal autophagy activity is involved in the pathogenesis of cystic fibrosis. Various studies have demonstrated that abnormalities or impaired autophagy are associated with cardiovascular diseases including pulmonary vascular disease. Autophagy plays a key role in maintaining normal vascular biological functions and vascular cell tissue homeostasis, and also plays an important role in the pathogenesis of various vascular diseases. For example, recent studies have found that autophagy participates in the occurrence and development of pulmonary hypertension. In addition, autophagy plays a central role in both innate and adaptive immune responses in immune cells or other cells with immune function. Thus, autophagy is the important cellular biological mechanism which causes cell fighting against pathogenic microorganisms including viruses, bacteria, and parasites. In this chapter, we discuss the work related to autophagy and other lung diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Autophagy and Cystic Fibrosis
1.1 Cystic Fibrosis is a Hereditary Lung Disease
Cystic pulmonary fibrosis (CF), also known as the viscous obstructive disease, is a congenital disease with a family autosomal recessive inherited disease. This disease is more common in the lungs but can also occur in the pancreas, liver, kidneys, and intestines; the main symptoms of the lungs are difficulty in breathing and coughing. CF has a high incidence in North America, with less frequency in Asia and Africa. According to pathological changes, CF belongs to the exocrine gland disease, and its primary cause is the CFTR gene mutation. CFTR mainly regulates the body to produce sweat, digestive fluid, and mucus. When the CFTR function is disordered, the airway surface fluid is decreased, the acid glycoprotein content in the mucus gland secretion is also increased. In addition, the secondary infection is the primary pathological basis of the patients with CF. Recurrent episodes of atelectasis and secondary infections eventually lead to extensive fibrosis and obstructive emphysema in the lungs.
1.2 Glutamine Transaminase 2 Regulates Autophagy in Cystic Fibrosis
The most common mutant site of CFTR is the 508 site (phenylalanine) deletion. This CFTR mutant has a tendency to aggregate and therefore tends to be prematurely degraded in protein processing in the endoplasmic reticulum. Autophagy is an important pathway for cells to clear misfolded proteins and damaged organelles. It has also been proved to be one of the important biological mechanisms of the pathogenesis of cystic fibrosis.
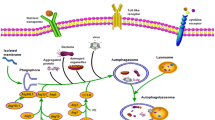
Glutamine transaminase 2 (TG2) is a direct cross-linking enzyme that catalyzes acyl transfer reactions in the cell and induces cross-linking within or between proteins by inducing a series of biochemical reactions. TG2 in airway epithelial cells which deficient CFTR occurs SUMO modification. SUMO modification is one of the fundamental post-transcriptional modification mechanisms of proteins, which can regulate the transcriptional activity, nuclear translocation, and biological functions of proteins. TG2 has three SUMOylated lysine sites, which can inhibit TG2 from ubiquitination and inhibit TG2 degradation after SUMOylation. Increased ROS content in CF epithelial cells leads to increased expression of the E3 ligase PIASy, which mediates TG2 SUMOylation, ultimately leading to an increase in intracellular TG2 protein levels. There is evidence that CFTR mutants can induce large amounts of ROS in the lung epithelial cells and trigger endoplasmic reticulum stress, and the autophagy pathway is activated (Fig. 42.1). Moreover, the interaction between beclin1 and Bcl-2 is weakened in CF epithelial cells, indicating that the microenvironment in CF cells is conducive to autophagy activation. However, high expression and sustained activation of TG2 can promote the crosstalk between beclin1, which leads to the decreasing stability of autophagy core complex and inhibition of autophagy activity. After autophagy was inhibited, the damaged mitochondria and p62 appeared to be accumulated. At the same time, the CFTR mutant could not be cleared by the autophagy pathway, which caused more ROS in the cells to induce a vicious circle. Overexpression of beclin1 in CF epithelial cells restores autophagy activity in cells. Similarly, intratracheal instillation of beclin1 lentivirus in CFTR mutant mice increases the number of LC3 puncta in cells and reduces the level of p62 in epithelial cells. It can be seen that the dysfunction of autophagic induces the onset of CF which depends on the formation of autophagy core complex.

The CFTR mutant inhibits autophagy in cells. The production of intracellular ROS is increased in CFTR mutated cells, and ROS can induce an increase in the expression of E3 ubiquitin ligase PISy. PISy inhibits TG2 degradation through the ubiquitin-proteasome pathway by causing SUMOylation of TG2. The large amount of TG2 accumulated in the cells crosstalk with beclin 1, which ultimately affects the formation of autophagy core complex and inhibits autophagy activity. After inhibition of autophagy, CFTR mutants cannot be degraded by autophagy pathway
At present, impaired autophagy in epithelial cells has become a new target for CF therapeutic research (Junkins et al. 2014). Cysteine stimulates CF epithelial cells to increase the maturation of CFTR mutants, promotes the mutant entry into the Golgi apparatus to exert their original biological activities, and increases the stability of CFTR on the cell membrane surface. After cysteine treatment, autophagy activity in CF epithelial cells is increased, which may be a result secondary to the recovery of CFTR function. After restoring CFTR function, the level of SUMOylation of TG2 and intracellular ROS is decreased, the level of cross-linking of beclin1 is reduced, and the stability of autophagy core complex is increased, thus leading to the recovery of autophagy activity. In the normal bronchial epithelial inhibition of CFTR function, the mature glycosylated CFTR on the cell membrane is reduced by 2/3, which also leads to an increase in intracellular ROS content, which induces a decrease in the stability of the TG2-mediated autophagy core complex, and this phenomenon disappears after the cysteine treatment (Luciani et al. 2010). It has been shown that cystine does not directly activate autophagy. If cystine-induced cell surface CFTR stability persists, the ROS/TG2 pathway in CF epithelial cells cannot inhibit autophagy. At the same time, cystine-treated CF epithelial cells can also attenuate CFTR by activating the autophagosome lysosomal pathway to await the degradation of protein aggregates, further reducing intracellular ROS levels.
1.3 P62 and Cystic Fibrosis
CF is an intracellular aggregate-prone disease, and there are a large number of misfolded or modified protein aggregates in CF epithelial cells, such as CFTR, PPAR, and beclin1. P62 is a classical receptor for autophagy, and its protein structure determines its easy formation of aggregates. When the autophagy activity of the cells is impaired, p62 will be accumulated in cells and promotes the onset of CF. After the accumulation of p62, the autophagy activity was inhibited, which led to the dysfunction of the proteasome pathway. A large number of proteins to be degraded in the cells accumulated and could not be removed. Thus, after the autophagy activity of CF epithelial cells was inhibited, the accumulation of p62 also affected the degradation of CFTR mutants to some extent. It has been shown that inhibition of CFTR function in normal bronchial epithelial cells leads to its own ubiquitination, and polyubiquitinated CFTR can interact with the p62 and form aggregates within the cells (Luciani et al. 2010). Moreover, it has been found that internalized CFTR and p62 colocalization in endosomal vesicles. Knockdown of p62 in CF epithelial cells with impaired autophagy activity not only reduces the accumulation of protein aggregates but also promotes the maturation and transport of CFTR proteins. This suggests that restoring cellular autophagy can promote sufficient functional CFTR (Luciani et al. 2011).
2 Pulmonary Hypertension and Autophagy
2.1 Drugs that Reduce the Death of Pulmonary Hypertension Remain to be Discovered
Pulmonary hypertension is a fatal pulmonary vascular disease characterized by a continuous increase in pulmonary vascular pressure (quiescent pulmonary artery pressure >25 mm Hg) leading to right heart failure even death. A large number of studies have shown that pulmonary hypertension is a reconstituted disease of pulmonary vascular tissue caused by a combination of increased proliferation of vascular smooth muscle cells and decreased apoptosis. Abnormal pulmonary artery remodeling causes vascular obstruction leading to cardiac hypertrophy and cardiac fibrosis; persistent cardiac tissue remodeling leads to right heart failure, left heart failure, and death. Therefore, continuous heart failure is the main cause of disability and death in patients with pulmonary hypertension. Vascular tissue hypoxia caused by various causes is a key mechanism that stimulates vascular smooth muscle cell proliferation and inhibits cell death, leading to pulmonary hypertension. Although there are currently relatively specific therapeutic drugs for pulmonary hypertension, including endothelial cell receptor antagonists, phosphodiesterase inhibitors, and prostacyclin and analogs, the disease is progressive even in the presence of these specific treatments, because these treatments can’t reduce the mortality of patients. Therefore, it is still important to study the pathogenesis of pulmonary hypertension and discover a new strategy for pulmonary hypertension treatment. Recent studies have shown that lung tissue from patients with pulmonary hypertension have increased autophagy activity, suggesting that autophagy may be involved in the development of pulmonary hypertension.
2.2 Pulmonary Hypertension and Autophagy
Studies on the pathogenesis of autophagy involved in pulmonary hypertension have just started. Evidence currently shows that activated autophagy may provide protection against the occurrence of pulmonary hypertension, and there is evidence to suggest that inhibition of autophagy may prevent the development of pulmonary hypertension (Nussenzweig et al. 2015). The number of autophagosomes formed in the lung cells is increased in pulmonary hypertension mice induced by chronic hypoxia, and the increase in high pressure occurs in chronic hypoxia which leads to the decreasing of autophagy activity in LC3B-deficient mouse (Rawat et al. 2014). In addition, inhibiting mTORC1, an autophagy negative regulator, activates autophagy to prevent pulmonary vascular smooth muscle cell proliferation. These studies demonstrate that activation of autophagy or inhibition of the mTOR signaling pathway can prohibit the development of pulmonary hypertension. Studies have found that knockout of autophagy signaling molecule beclin1 leads to a lack of autophagy activity, which inhibits the proliferation of pulmonary artery endothelial cells. It has also been found that hypoxia causes an increase in angiogenesis in the beclin1−/+ mice. These results demonstrate that beclin1 mediated autophagy is involved in the development of pulmonary hypertension. On the other hand, studies have also provided evidence that autophagy activity promotes the development of pulmonary hypertension (Chen et al. 2018). Indeed, the autophagy-lysosomal inhibitor chloroquine prevents the progression of lung hypertension in experimental animals. Interestingly, recent studies have shown that inhibition of glucose-6-phosphate dehydrogenase in animals with pulmonary hypertension and reduction of nicotinamide adenine dinucleotide phosphate production can inhibit the development of lung hypertension in animals, and improve the heart function. Obviously, studies on the pathogenesis of autophagy and pulmonary hypertension have just begun. The contradictory findings shown above indicate that the role and mechanism of autophagy in the pathogenesis of pulmonary hypertension is unclear.
3 Autophagy and Lung Infectious Diseases
3.1 Tuberculosis is a Chronic Infectious Lung Disease
Pulmonary tuberculosis, commonly known as pulmonary consumption, is a chronic infectious disease caused by Mycobacterium tuberculosis infection in human lungs and is the most common type of tuberculosis in human body. Typical pulmonary tuberculosis is slow, and the course of the disease is long. Common symptoms include low fever, fatigue, loss of appetite, cough, and hemoptysis. However, most patients who suffer from pulmonary tuberculosis often have no obvious symptoms, so that only when examined with X-ray or checked for hemoptysis, patients realize that they got tuberculosis. If tuberculosis can be discovered in time and treated through convincing strategy, most of the patients can be cured. The extent of tuberculosis symptoms is related to the extent of the lesion, progression, and body reactivity. M. tuberculosis belongs to the actinomycete family, and its pathogenic effect depends on the inflammatory reaction induced by the proliferation of bacteria in the cells. It also can induce the damage due to promoting the immunoreaction. The infection rate of M. tuberculosis is high, but the incidence of tuberculosis is very low, which indicates that the human body has certain immunity against tuberculosis infection. Autophagy is one of the important biological mechanisms of human cells. In addition to regulating cell energy balance, autophagy has also been shown to be involved in pathogen clearance in vivo. To date, studies on the pathogenesis of tuberculosis have focused on autophagy for the elimination of M. tuberculosis.
3.2 Immune Signal and Autophagy are Effectors of M. tuberculosis
In addition to being induced by starvation, autophagy is also regulated by innate immune signals and inflammatory cytokines during the immune response. It has been shown that activation of many pattern recognition receptors (PRRs) can induce autophagy. For example, ligands of TLR can induce autophagy by stimulating TRAF6 or enhance the stability of autophagy-related proteins such as Beclin1 and ULK1, and NOD receptors signaling can activate autophagy by increasing the protein stability of autophagy-associated proteins RIPK2 and ULK1 also. In addition, cGAMP produced by stimulation with viruses, mitochondria or bacterial DNA, and 3′-5′ circular double GMP or circular double AMP produced by secreted bacteria can also induce autophagy of cells. There is evidence that IL-1β induced autophagy activation is closely related to its anti-tuberculous mycobacterial effect. MyD88 is an important linker regulated by the signaling of different immune receptors. Interestingly, the MyD88 signaling pathway has been shown to play a key role in early anti-tuberculosis processes, but activation of MyD88 signaling via multiple TLRs does not induce significant resistance to tuberculosis. However, the MyD88-dependent IL-1 signaling pathway plays a key role in the early stages of anti-tuberculosis. Similar to the biological effects induced by IL-1 Th1-type cytokines can also act as intracellular anti-tuberculosis by activating autophagy (Moraco and Kornfeld 2014). IFN-γ is a typical Th1-type cytokine that has been shown to activate autophagy, whereas the Th2-type cytokines IL-4 and IL-13 are capable of inhibiting IFN-γ induced autophagy by STAT6. In addition, Th2-type cytokines can also inhibit starvation-induced autophagy through the Akt signaling pathway.
Calcitriol, a metabolite of Vitamin D, is an important synergist for IFN-γ activation of autophagy. There is a synergistic effect between calcitriol and IFN-γ in macrophages to induce autophagy activation, which effectively promotes intracellular M. tuberculosis clearance. Calcitriol can activate the AMPK signaling pathway through Ca2+, activate the ULK1 phosphorylation cascade, and ultimately induce autophagy by forming autophagy core complexes. IFN-γ induced autophagy in macrophages is dependent on calcitriol, but starvation or rapamycin-induced autophagy does not require the involvement of calcitriol. Thus, the decrease of serum calcitriol and its precursor calcifediol in patients with pulmonary tuberculosis limits the anti-tuberculosis effect of IFN-γ.
3.3 The Mechanism of the Anti-tuberculosis Effects of Autophagy in Cells
A large body of evidence indicates that autophagy removes intracellular M. tuberculosis by the unique antibacterial properties of autophagosomes (Deretic 2014). Through the analysis of autophagy-related factors, it was found that the autophagy cargo protein p62 is a key initiator of the entire autophagy pathway against M. tuberculosis. The p62 protein can deliver specific ribosomes (rpS30) and a large number of ubiquitinated cytoplasmic proteins into autophagosomes, and then a series of acidification decomposition reactions occur, resulting in anti-tuberculosis effects. NDP52, another autophagy receptor protein, is also thought to be involved in macrophage-induced autophagy against tuberculosis in mice. Autophagy activation promotes acidification and maturation of M. tuberculosis phagosomes, which in turn become the organelles that kill mycobacteria. The classic anti-tuberculosis strategy is to inhibit phagosome-lysosomal fusion, thereby inhibiting autophagy. However, current evidence suggests that induction of autophagy activation is far superior to the previous classical strategy. The results show that the main component of anti-tuberculosis antibacterial peptide can stimulate the fusion of lysosomes in tuberculosis host cells. Autophagy can also induce antibacterial effects by capturing and digesting components within cells. Host cells infected with M. tuberculosis can form autophagosome with antibacterial properties in the cells after induction of autophagy activation. Such autophagosomes can mediate anti-tuberculosis action after fusion with lysosomes. In some cases, intracellular autophagy degradation products contain antimicrobial peptides and derivatives thereof that promote the clearance of M. tuberculosis. In addition, some M. tuberculosis escapes from autophagosomes into the cytoplasm, and autophagy can mediate the clearance of escaped M. tuberculosis in host cells (Liu et al. 2018).
There is also a close relationship between autophagy and first-line drugs against tuberculosis (Ryter and Choi 2015). For example, isoniazid and pyrazinamide can exert therapeutic effects by activating autophagy. In the treatment of tuberculosis with isoniazid or pyrazinamide, the M. tuberculosis product and the oxidative stress products produced by mitochondria and NADPH can synergistically induce activation of autophagy. In addition, several compounds that induce autophagy activation can also act as anti-tuberculosis, suggesting that inducing autophagy is a promising strategy to treat tuberculosis. When the autophagy activity is inhibited by small molecule inhibitors in cells infected with M. tuberculosis, the cells no longer have antibacterial activity.
3.4 Autophagy and Pulmonary Inflammation Caused by Acute Infection
Pulmonary inflammation is caused by continuous exposure of the respiratory tract to microorganisms, atmospheric particulate matter, irritants, pollutants, allergens, and pathogens. When stimulated, the lungs produce a range of defense reactions. The first is the epithelial barrier, which secretes a large number of substances including mucin, lysozyme, defensin, and nitric oxide; the second is mucociliary, which play an important role in innate defense mechanism. It will remove harmful particles from the mucus of the respiratory tract. In addition, the cells of the natural immune system have the function of aggregating, encapsulating, and killing microorganisms, including macrophages, dendritic cells, monocytes, neutrophils, eosinophils, Natural killer cells, and mast cells. These cells produce a variety of inflammatory mediators such as ROS and various cytokines such as IL6, TNF-γ, and IL1β.
The pathogen recognition receptors expressed on alveolar macrophages and dendritic cells can recognize pathogen-associated molecular patterns and molecular patterns of damage, and respond to infection or injury of the respiratory tract. Subsequent inflammatory cascades and cytokines further recruit immune cells such as neutrophils and monocytes. In addition, dendritic cells also acted as an antigen-presenting cell to associate innate immunity with acquired immunity by phagocytizing microorganisms and migrating to local lymph nodes to activate lymphocytes including T cells and B cells. During pulmonary infection, alveolar macrophages can regulate inflammation and damage repair processes by killing microbes and clearing apoptotic cells. During the infection, neutrophils are located in the pulmonary capillaries and interstitial spaces, and are recruited into the alveolar cavity during infection by the chemokines. Neutrophils can secrete antibacterial proteins and reactive oxygen species to kill the ingested microorganisms, and simultaneously release chemokines to recruit more monocytes to the site of infection.
The lymphocytes in the lung tissue are distributed throughout the airway and lung parenchyma and consist of thymus-dependent T cells and bone marrow-dependent B cells. The activated Th1 cells can produce pro-inflammatory cytokines including TNF-α and IFN-γ while Th2 cells secrete IL4, IL13 that stimulate B cells to produce immunoglobulin E concentration, Serum (IGES) to activate mast cells. In addition, IL5, another important Th2 cytokine, could stimulate eosinophils and further exacerbate local tissue inflammation. During inflammation, mast cells also produce cytokines, leukotrienes, and proteases by activating IGES receptor (FCER1).
In order to remove pathogens and harmful particles from the lung tissues, the body needs a rapid, powerful, and highly regulated inflammatory defense reaction. Furthermore, the steady state in the respiratory system depends on the collaboration between the innate immune system and the adaptive immune system. Autophagy plays an important role in the inflammatory response of the lungs suffered from infection or stress. At homeostasis, autophagy in alveolar macrophages is essential for the inhibition of spontaneous inflammation, and autophagy is also the basic mechanism for airway goblet cells to secrete airway mucus. Atg5−/− or Atg7−/− deficient mice develop spontaneous aseptic pulmonary inflammation characterized by a marked increase in the number of inflammatory cells, submucosal thickening, goblet cellification, and collagen increase. Knocking out ATG 5 in mouse ITGAX/CD11c+ cells results in spontaneous airway hyperresponsiveness and severe neutrophilic lung inflammation.
In acute lung injury, autophagy activation is a mechanism of host protection following bacterial and viral infection. Atg7 knockout mice reduce the ability of pathogen clearance, increase neutrophil-mediated inflammation and IL1β production, leading to severe lung damage and reduced survival after infected with P. aeruginosa. Similarly, map1l3b knockout mice infected with respiratory syncytial virus present severe IL17A-dependent lung pathology change and increase Th2 cytokine levels, mucus secretion, and infiltrating of eosinophils and dendritic cells in the lungs. At the same time, it has been suggested that autophagy appears to be a protective response in infected lungs, and autophagy may play an adverse role in the late stages of sepsis due to excessive autophagosome accumulation, leading to acute lung injury. In addition, in LPS-induced pulmonary inflammation, knockdown of PI3KC3 in macrophages results in loss of autophagy, which reduces the infiltration of immune cells in the bronchi and alveoli of the lungs, and the concentration of cytokines in the lungs. These evidences suggest that autophagy plays a key role in acute lung infections, including clearance of pathogenic microorganisms, the development of inflammatory responses, and the maintenance of lung tissue homeostasis.
3.5 Sepsis and Autophagy
Although the immune response is critical for the host to fight against sepsis, excessive immune responses and inflammation often cause tissue/organ damage and subsequent secondary infection. The role of autophagy in sepsis is investigated based on transmission electron microscopy studies of liver specimens from patients with sepsis. These studies have shown an increase in the number of autophagic vacuoles in the liver of patients with sepsis compared with non-sepsis patients. However, it is unclear whether the increase in autophagic vacuoles in patients with sepsis means an increase in autophagy activity in patients with sepsis, or an inhibition of autophagy leading to excessive accumulation of autophagic vacuoles. However, these data indicate that autophagy plays an important role in the pathogenesis of sepsis. Autophagy has been shown to play an important role in preclinical models of sepsis such as sepsis caused by cecal ligation and perforation (CLP) and endotoxin-induced septic shock. The level of LC3B is up-regulated in target organs of sepsis such as lung, liver, and kidney of mice receiving CLP or endotoxin treatment. The study of autophagy in the sepsis models is based on various transgenic mice or autophagy modulators. Deletion of autophagy genes such as MAP1LC3B, BECN 1, or Vps 34 increases inflammation, bacterial burden, organ damage, and mortality in mice with sepsis caused by CLP or LPS. In contrast, autophagy activators such as rapamycin or overexpressing LC3 inhibit inflammatory and apoptotic activities and increase survival in CLP mice. Genetic studies have further shown that autophagy has important biological functions in sepsis. The immune-related GTPase acts as an important molecule regulating autophagy induction and elimination of intracellular mycobacteria, and its genetic polymorphism is associated with mortality in patients with severe sepsis. Mutations in the ATG 16 allele are also associated with sepsis severity and ventilator-associated pneumonia.
Although modulating inflammation is an important method of preventing multiple organ dysfunction in sepsis, proper immune response is crucial for eliminating microorganisms during infection. In this regard, autophagy can upregulate immune function through allogeneic autophagy and mitochondrial autophagy, increasing bacterial killing and inhibiting inflammation. The killing effect of allogeneic autophagy on virulence pathogens can reduce the number of pathogens, leading to immune reactions and inflammation during sepsis. In addition to allogeneic autophagy, autophagy can also regulate immune responses and inflammation by controlling mitochondrial quality. Increased plasma mitochondrial DNA levels in patients with sepsis are associated with disease severity and mortality. In contrast, a recent study showed that the copy number of mitochondrial DNA in monocytes and lymphocytes in patients with sepsis is reduced and inversely associated with the severity of the disease, which suggested that mitochondrial DNA can be released from these cells. The release of mitochondrial DNA is associated with mitochondrial DNA-mediated activation of NLRP 3 inflammatory bodies. Therefore, these results indicate that mitochondrial integrity may be impaired in patients with sepsis.
Mitochondrial dysfunction promotes ROS production in mitochondrial and activation of NLRP 3 inflammatory bodies, leading to cell death and secretion of pro-inflammatory cytokines. Mitochondrial dysfunction caused by infection or oxidative stress during sepsis may be one of the leading causes of immune response. Therefore, the elimination of dysfunctional mitochondria by mitochondrial autophagy is critical for regulating immune responses and inflammation. LC3B is increased and co-localized with mitochondria in the lungs of mice with sepsis caused by S. aureus infection, indicating that mitochondrial autophagy has changed after infection. Indeed, the deletion of LC3B and Beclin 1 further increased the production of IL-1β and IL-18 in septic mice and mortality. In addition, the lack of Parkin gene impaired myocardial contractility recovery in LPS-treated septic mice. Defects in mitochondrial autophagy caused by kinase JNK 2 deficient mice also cause excessive activation of inflammasome and increase mortality due to endotoxic shock.
The immunomodulatory effects of autophagy may be related to changes in cellular metabolism. Recent studies have shown that downregulation of cellular metabolic pathways, including glycolysis, lipid synthesis, and fatty acid synthesis, is critical for the activation of inflammasome. Hexokinase-1 (HK-1), uncoupling protein-2 (UCP2), and NADPH oxidase 4 (NOX4) are known key molecules for this process. Although the important role of autophagy in immune cells such as macrophages has been confirmed, it is unclear whether other types of cells are involved in the pathogenesis of sepsis. Given the complexity of sepsis pathology, further studies using genetic and other approaches are needed to determine the role of autophagy in various cell types/tissues.
4 Obstructive Sleep Apnea (OSA) and Autophagy
Obstructive sleep apnea (OSA) is a respiratory disorder that occurs during sleep, with recurrent upper airway obstruction leading to apnea, hypopnea, and/or respiratory arousal. These respiratory disorders often lead to sleep disruption, hypoxemia, hypercapnia, and increased sympathetic activity, while increasing the risk of hypertension, congestive heart failure, type 2 diabetes, stroke, and premature death. This is an area of increasing public health that is of concern. The latest data show that the prevalence of moderate to severe OSA in men is 10–17%, and in women it is 3–9%. Compared with the past 20 years, the prevalence rate has increased by more than 25%. The underlying pathological mechanisms leading to systemic dysfunction in OSA patients are complex and not fully understood, but it has been illustrated that this effect is associated with intermittent hypoxia (IH) in patients. The relationship between intermittent hypoxia and abnormal glucose metabolism and obesity has been established by in vivo experiments. However, there is increasing evidence that IH plays an important role in causing inflammation, which induces OSA-associated cardiac metabolic diseases. IH is a known activator of the NF-κB 1/2 pathway, which acts as a potent inflammatory activating factor leading to the release of TNF-α, IL6, IL8, and CCL 2/MCP-1. Patient data indicate that increased circulating ROS in OSA patients, but the evidence is limited. Although this issue needs further study, systemic inflammation does have an important role in the pathogenesis of OSA-related diseases.
In recent years, the role of autophagy in the pathogenesis of OSA has been questioned. In vivo studies have linked autophagy to chronic intermittent hypoxia. Using a chronic intermittent hypoxic rat model, activating autophagy by injecting melatonin protects the body from heart damage, which is a common cause of OSA disease. Interestingly, another study tested the role of autophagy in insulin resistance caused by chronic intermittent hypoxia, and there is no relationship between these processes. These data suggest that autophagy may play a role in the processes of chronic intermittent hypoxia in OSA patients. However, more research is needed to determine which processes are dependent on the regulation of autophagy. As an important research field, using autophagy activators/inhibitors may be an important approach to modulate systemic complications of OSA disease.
5 Conclusion
Respiratory diseases are usually chronic disorders that have long threat to public health and life. The constant exploration of the pathogenesis of these diseases will provide new information to discover new therapy strategies and drugs. Autophagy, as one of the important biological regulatory mechanisms of cells, is involved in the occurrence and development of many respiratory diseases, especially considering that the basal autophagy activity in cells is a necessary condition for the resolution of various respiratory diseases. Indeed, autophagy is an important mechanism for repairing lung tissue damage, regardless of the cause of lung tissue damage. However, chronic inflammation in the damaged lung tissue will block the autophagy process and decreases autophagy activity. Neither the over-activated autophagy nor the blocked autophagic flux can trigger tissue repair and lung regeneration, and both of these processes cause lung injury by inducing pro-inflammatory and pro-apoptotic effects. Therefore, understanding the role of autophagy in the development of various respiratory diseases is important for the therapy of a variety of lung diseases, especially chronic respiratory diseases.
References
Chen R, Jiang M, Li B et al (2018) The role of autophagy in pulmonary hypertension: a double-edge sword. Apoptosis 23(9–10):459–469
Deretic V (2014) Autophagy in tuberculosis. Cold Spring Harbor Perspect Med 4:a018481
Junkins RD, McCormick C, Lin TJ (2014) The emerging potential of autophagy-based therapies in the treatment of cystic fibrosis lung infections. Autophagy 10(3):538–547
Liu F, Chen J, Wang P et al (2018) MicroRNA-27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium-associated autophagy. Nat Commun 9(1):4295
Luciani A, Villella VR, Esposito S et al (2010) Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 12(9):863–875
Luciani A, Villella VR, Esposito S et al (2011) Cystic fibrosis: a disorder with defective autophagy. Autophagy 7(1):104–106
Moraco AH, Kornfeld H (2014) Cell death and autophagy in tuberculosis. Semin Immunol 26(6):497–511
Nussenzweig SC, Verma S, Finkel T (2015) The role of autophagy in vascular biology. Cir Res 116(3):480–488
Rawat DK, Alzoubi A, Gupte R et al (2014) Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertension 64(6):1266–1274
Ryter SW, Choi AM (2015) Autophagy in lung disease pathogenesis and therapeutics. Redox Biol 4:215–225
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Science Press and Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Lv, X., Li, K., Hu, Z. (2020). Autophagy and Others Respiratory Diseases. In: Le, W. (eds) Autophagy: Biology and Diseases. Advances in Experimental Medicine and Biology, vol 1207. Springer, Singapore. https://doi.org/10.1007/978-981-15-4272-5_42
Download citation
DOI: https://doi.org/10.1007/978-981-15-4272-5_42
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-4271-8
Online ISBN: 978-981-15-4272-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)