
Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease with multiple etiologies. If remains untreated, it leads to high rate of morbidity and mortality, and a median survival of only 5–7 years. Clinically, PAH is defined as mean pulmonary arterial pressure >20 mmHg at rest with normal left atrial pressure. PAH is characterized by remodeling of the pulmonary vasculature due to pulmonary vascular endothelial and smooth cell proliferation. These events eventually reduce the pulmonary arterial compliance and thus causing increased pulmonary vascular resistance. If remains unmanaged, it will ultimately end up in right ventricular failure. However, the Food and Drug Administration (FDA) has approved drugs that target the endothelial pathway, the nitric oxide pathway, and the prostacyclin pathway. These therapeutic strategies, mainly inducing pulmonary vasodilation, however, have poor effect on the signaling pathways activated during the pathogenesis of PAH. This might be one of the reasons why they are unable to reverse the pathology of the disease, and eventually patients stop responding to them. If medications fail, the only viable alternative will be lung transplantation to save a patient’s life. Evidences show the contribution of heredity, inflammation, drugs, toxins, HIV infection, and hepatic diseases in the progression of PAH. Therefore, there is a need for potential therapeutics, which, via affecting the pathological signaling pathways, halt the disease process and eventually reverse the pulmonary remodeling in the patients. In this chapter, we discuss the emerging novel therapeutics and their mechanism of actions, which have advanced beyond preclinical research and are currently being investigated in the clinical cases of PAH.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Pulmonary arterial hypertension
- Vascular smooth muscle proliferation
- Clinical trials
- Lung inflammation
- Bone morphogenetic protein receptor 2 (BMPR2)
- Hypoxia
- Endothelial cells
- Pulmonary artery
1.1 Introduction
The pulmonary hypertension is defined as elevated pulmonary arterial pressure (PAP) [1,2,3,4]. It is characterized by pulmonary vascular remodeling in the small pulmonary arteries, increased PAP and elevated pulmonary vascular resistance (PVR) [5]. The etiology of pulmonary hypertension (PH) is multifactorial [1, 6]. Other noticeable changes during the development of PH include intimal proliferation, vascular smooth muscle proliferation or hypertrophy, and adventitial thickening [7, 8]. The symptoms of the disease are progressive such as shortness of breath, fatigue, chest discomfort, abdominal fullness, and light-headedness or even syncope with more advanced and severe form of the disease [9, 10]. If remain untreated, or uncontrolled by the currently available medications, patients of PH will eventually need heart and lung transplantation. Otherwise, it leads to increased rate of mortality [5, 11]. Thus, PH is emerging as an urgent medical need, which requires a thorough understating of its pathogenesis at cellular and molecular levels, and thereby development of potential novel therapeutics to treat the PH [1, 2, 6].
Several confounding factors, including aberrations in the genes, environmental toxins, drugs, hepatic disease, and right heart dysfunction, to mention but a few, are involved in the pathogenesis of PH [3, 12]. Clinically, PH is a mean pulmonary arterial pressure more than 25 mmHg determined by resting supine right heart catheterization (RHC) [9, 11, 13]. According to the current classification and criteria [13], PH may be categorized into (1) idiopathic, which is defined by the absence of underlying risk factor, (2) heritable/genetic, for instance, the presence of germline mutation in bone morphogenetic protein receptor 2 (BMPR2), (3) PH induced by drugs or toxins, or (4) it may be associated with other pathological conditions such as connective tissue disease, portal hypertension, HIV infections, and congenital heart disease. The most updated PH classification was defined at the 5th World Symposium on Pulmonary Hypertension in 2013 [3, 13]. This classification is based on a shared disease histology and pathophysiology, clinical presentation and therapeutic interventions. Thus, this put the PH in five differed groups: (1) pulmonary arterial hypertension (PAH), (2) PH due to left heart disease or pulmonary venous hypertension, (3) PH due to chronic lung disease and/or hypoxia, (4) chronic thromboembolic PH (CTEPH), and (5) PH due to undefined or unknown mechanism [9, 12].
1.2 Basic Review of Pulmonary Circulation
The basic understanding of physiology and pathophysiology of pulmonary circulation will be helpful to better understand and define the pathogenesis, diagnosis, prognosis, and therapeutic aspects of the PH. Briefly, the major function of the pulmonary circulation is to carry deoxygenated blood from the right side of the heart to the lungs via pulmonary artery to get it oxygenated [13, 14] and return oxygen-rich blood back to the left side of the heart via pulmonary vain. Therefore, maintenance of pulmonary circulation function is vital to provide oxygenated blood to the systemic circulation [12].
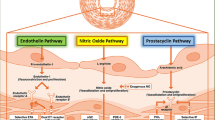
The pulmonary arterial hypertension (PAH) is a progressive process that leads to an increase in pulmonary vascular resistance, and is a most common type of PH in clinical practice. This is due to the multiple pathophysiologic outcomes such as pulmonary vascular remodeling and sustained vasoconstriction. Since pulmonary circulation is getting the entire cardiac output, low pressure and PVR in the pulmonary artery are maintained through the high cross-sectional area containing network of small pulmonary arteries and capillaries. One may predict that any change or fluctuation in the PVR will be reflected in the increase in pulmonary arterial pressure and leads to PH. Anatomically, the pulmonary artery is comprised of three distinct types of tissue layers. The innermost intima is comprised of pulmonary artery endothelial cells, followed by the middle layer of media containing pulmonary artery smooth muscle cells. The outermost layer or lining is made of the fibrous tissue called adventitia. In the pathogenesis of PH, the above cell types play a fundamental role in causing narrowing of the pulmonary artery and increasing PVR. The current line of therapeutics mostly targets proliferation of intimal and medial cells proliferation but is unable to reverse the disease progression (Fig. 1.1) [6, 12, 14].

Pathways targeted in current therapies for pulmonary arterial hypertension. Newly approved therapies are listed in blue. cAMP cyclic adenosine monophosphate, cGMP cyclic guanylate monophosphate, ERA endothelin receptor antagonist, FDA US Food and Drug Administration, INH inhaled, IP2 prostacyclin receptor 2, IV intravenous, NO nitric oxide, PAH pulmonary arterial hypertension, PDE-5 phosphodiesterase-5, PDE-5i phosphodiesterase-5 inhibitor, PGI2 prostaglandin I2, sGC soluble guanylate cyclase, SQ subcutaneous. (Reproduced with permission from Tsai H, Sung YK and de Jesus Perez V. Recent advances in the management of pulmonary arterial hypertension [version 1]. F1000Research 2016, 5:2755 [6])
1.3 Pulmonary Hypertension: Clinical Classification
Under clinical setting, PH is more of comorbid condition, which is most of the time associated with other types of clinical conditions. This makes a poor prognosis and ends up in high rate of morbidity and mortality [2, 12, 14]. This puts a great challenge on clinicians and pulmonary physician to correctly diagnose the disease. Over the past several years, a great stride has been done to improve the understanding of the pathophysiology of the PH. One of the challenging tasks is to correctly classify the disease based on the pathophysiological origin of the disease. Here, we will expand the classification of PH so that we may be able to better understand the disease process. In 1970, the efforts led by the World Health Organization (WHO) resulted in the understanding of the underlying causes of PH. This further led to putting PH as primary or secondary PH. This was the beginning of the understanding of the tip of the iceberg basically, which opened doors for the further discoveries in the field of PH. The most current and updated PH classifications were defined at the 5th World Symposium on PH in 2013 and put the pH cases in five separate types, as defined above [12, 14]. Now the question is: What was the outcome on the patients’ overall health and survival in PH? The above classification system impacted significantly on the patients’ diagnosis and treatment process due to more focused identification of the factors affecting morbidity and mortality in PH [12, 13].
The question which is still vaguely answered is why the diagnosis of PH is so challenging. There is no single and clear answer to this question. One of the most important challenges with the diagnosis of PH is that the early symptoms of the disease are nonspecific and that may lead to misdiagnosis [6, 15]. This may require more intensive physical examination and may be more invasive clinical procedures to delineate the disease in patients at early stage [8, 16].
Patients with PH displayed varying symptoms starting from unexplained dyspnea upon exertion, chest pain, fatigue, hemoptysis, syncope, and Raynaud’s phenomena. These symptoms most of the time are poor oxygen transport and impaired cardiac output due to the increased pulmonary vascular resistance. Therefore, only a thorough clinical investigation may reveal the underlying pathophysiology pertinent to PH. Other signs, which were revealed upon clinical examination, include jugular venous distensions, hepatomegaly, cyanosis, the presence of hepatojugular reflex, mottled extremities, diminished peripheral pulses, peripheral edema, and ascites [12, 16]. Cardiac catheterization is a reliable mode to delineate and differentiate multiple abnormal sounds associated with pulmonary artery hypertension such as RV, S3 and S4 sounds, accentuated pulmonic valve component (P2) of the second heart sound, systolic murmur suggesting pulmonary regurgitation, and a parasternal lift may be detectable [12]. This indicates how rigorously patient should be examined to reach at a conclusion, from where the medications will be initiated [12, 16].
Besides what is explained above, other invasive and noninvasive techniques are further required at the final stage for the explicit diagnostic purpose of PH. These include electrocardiography, pulmonary function testing, chest radiography, echocardiography, serologic testing, and right heart catheterization [12]. With such an advancement of the diagnostic technology in PH, it is a fact that the current therapeutics, which is being used at the clinic, is merely a supportive care that targets pulmonary vasocontraction. At this point, we may ask a question why at the verge of advancement of medical technology, we are feeling so helpless therapeutically and relying on conjecture rather than really treating or reversing the disease. The answer is maybe either we are still lacking the correct understanding of pathological process that takes place at the cellular and molecular level or we need to develop rationale drugs that target the disease at the cellular and molecular level. We will discuss the advancement in these areas in forthcoming paragraphs. First, we will briefly review the different types of PH [1, 16].
1.3.1 Group 1: Pulmonary Arterial Hypertension (PAH)
Group 1 pulmonary hypertension or pulmonary arterial hypertension (PAH) is a progressively increased pressure in the pulmonary artery or increase in mean pulmonary arterial pressure (Fig. 1.2) [6, 8, 12]. Primarily, it is due to the proliferation of vascular smooth muscle in the pulmonary artery, which leads to the obstruction in the blood flow in pulmonary vasculature. If it remains untreated, it will eventually end up in right ventricular failure and often cause premature death. In terms of the etiology, there are multiple factors involved in the progression of the disease and few of them are well understood. For instance, in idiopathic pulmonary hypertension, we do not have any clear understanding of the etiology of the disease. Other important factors which are involved include heredity, drug-induced PAH, various connective tissue-related abnormalities, human immunodeficiency virus, or HIV infection, and congenital abnormities in the heart. PAH may also arise following other vasculo-occlusive disorders including portal hypertension, veno-occlusive diseases and persistent pulmonary hypertension of the newborns. It was a time when there was a lack of therapeutics and the median life span oftentimes was less than 3 years [12, 17]. With the advent of more targeted therapies, along with the early diagnosis of the disease, the clinical outcome is much improved. Recent research demonstrates involvement of gene mutation and development of PAH. For instance, mutation in the bone morphogenetic protein receptor 2 (BMPR2) gene has been involved in more than 75% cases of clinically diagnosed PAH. Intriguingly, these cases mostly have a known family history of PAH. There are up to 25% of sporadic cases with more than 300 known independent mutations in the gene alone [12, 18]. This suggests that mutated BMPR2 is the most commonly involved denominator in the pathogenesis of PAH. The mutated gene product binds and activates members of the transforming growth factor (TGF)-β family, including activin-like receptor kinase-1 (ALK-1), endoglin (ENG), and mothers against decapentaplegic homolog 9 (SMAd9) have also been identified in the etiology of the PAH. This suggests a significant role of this signaling cascade in the development of PAH [19]. Besides the clear central role of TGF-β family, recent studies indicate mutations in caveolin-1 (CAV-1) and potassium two pore domain channel subfamily K member 3 (KCNK3) has a significant role in the etiology and pathogenesis of PAH [19, 20].

Treatment algorithm from the 2015 European Society of Cardiology/European Respiratory Society guidelines for the diagnosis and treatment of pulmonary hypertension. (Reproduced with permission from Tsai H, Sung YK and de Jesus Perez V. Recent advances in the management of pulmonary arterial hypertension [version 1]. F1000Research 2016, 5:2755 [6])
Research studies demonstrate the potential role of drugs and toxins in the pathology of PAH [19, 21, 22]. However, exactly how these chemical compounds lead to PAH is still in obscurity. Very few drugs and chemicals have been known so far, which cause PAH with some defined understanding. Under these categories, for instance, certain anorexigens (e.g., aminorex, fenfluramine, and dexfenfluramine), toxic rapeseed oil, selective serotonin inhibitors (SSRIs) and benfluorex cause PAH in the patients. Although the precise mechanism of anorexigens in inducing PAH is unclear, studies show that they basically block the uptake of serotonin via blocking the serotonin transporter [1, 12, 15]. This allows serotonin to induce proliferation of pulmonary vascular smooth cells and induction of PAH. Benflurex shares an active metabolite with fenfluramine, and is a drug used in Europe to treat hyperglycemia and other metabolic disorders, causing PAH. Moreover, SSRIs are used in pregnancy, and increase the risk of PAH. It also increases the risk of mortality and worsening the PAH if used in patients who are already diagnosed with PAH [12, 21, 22].
Other drugs that have strong link to induce PAH include amphetamine, methamphetamine, l-tryptophan, dasatinib, cocaine, phenylpropanolamine, St. John’s wort, chemotherapeutic agents, and interferon α and β [23]. However, there is no strong relation between cigarette smoking, estrogens, or oral contraceptives in the induction of PAH [12, 23].
1.3.2 Group 2: Pulmonary Hypertension (PH) Due to Left Heart Disease
Group 2 PH due to left heart disease includes left ventricular (LV) diastolic dysfunction, left ventricular (LV) systolic dysfunction, valvular disease, congenital and or acquired left heart inflow/outflow tract obstruction, and congenital cardiomyopathies (25 and current) [24]. Most common presence of group 2 PH has been observed in the patients suffering heart failure with reduced (HFeEF) or preserved ejection fraction (HFpEF). Hemodynamically, the disease is defined as mean pulmonary arterial pressure (mPAP) ≥ 25 mmHg, a pulmonary artery wedge pressure (PAWP) or left ventricular end-diastolic pressure (LVEDP) > 15 mmHg, and a normal or reduced cardiac output [12]. The pathophysiological manifestation involved the passive backward transmission of filling pressures that increases mPAP (e.g., loss of left atrial compliance, diastolic dysfunction, mitral valve regurgitation), further increase in mPAP (e.g., endothelial dysfunction, vasoconstriction), and eventually ends up in worsening of pulmonary vascular remodeling, right ventricular dysfunction and, if remain untreatable, ultimately leads to high rate of mortality [12, 24].
Group 2 PH is further categorized into two sub categories. This subclassification is based on the diastolic pressure difference (DPD, defined as [diastolic PAP − mean PAWP]) during right heart catheterization under resting conditions. These categories are isolated post capillary PH (PAWP > 15 mmHg and DPD < 15 mmHg) and combined post and precapillary PH (PAWP > 15 mmHg and DPD ≥ 7 mmHg) [12, 24].
The clinical features of Group 2 PH are different compared to Group 1 PAH. Group 2 PH patients with HFpEF were older in age, superimposed with other cardiovascular comorbidities such as hypertension, coronary artery disease, worse exercise capacity, and renal dysfunction [25]. They were also suffering from high frequency of left atrial enlargement, lower frequency of right atrial enlargement and less severe PH. Therapeutically, there is more focus to treat the underlying heart disease present in Group 2 PH to achieve desirable effects [12, 24].
1.3.3 Group 3: Pulmonary Hypertension (PH) Due to Lung Diseases and Hypoxia
The pathological conditions associated with the underlying cause of Group 3 PH include chronic obstructive pulmonary disease (COPD), interstitial lung disease (ILD) (e.g., idiopathic pulmonary fibrosis, sarcoidosis), and other lung diseases such as with the involvement of sleep disorder-induced breathing problems, alveolar hyperventilation disorders, chronic high-altitude exposure, and developmental lung diseases [12, 26]. Since there is an increased propensity of above associated pulmonary disorders, sometimes, it is very difficult to distinguish the differences between idiopathic PAH and Group 3 PH. At the diagnostic end, findings suggest that the Group 3 PH is associated with moderate-to-severe impairment in ventilation function, reduced breathing reserve, and characteristic airway and/or parenchymal abnormalities [12].
Two significant pathophysiological features underlying Group 3 PH include hypoxic vasoconstriction and obliteration of the pulmonary vascular bed. Hypoxia leads to a significant damage to the vasculature and release of vasoconstrictor substance such as endothelin from the endothelium. Upon release, endothelin causes underlying vascular smooth muscle cell contraction and leads to vasoconstriction and proliferation. Initial hypoxia-induced vasoconstriction is reversible; however, once there is pulmonary remodeling due to the chronic hypoxic condition, it is largely irreversible. Like Group 2 PH, Group 3 PH management also involves identification and treatment of underlying pathophysiologic cause rather than only focusing on the increased in mPAP [12].
1.3.4 Group 4: Chronic Thromboembolic PH (CTEPH)
Group 4 pulmonary hypertension is due to the chronic thromboembolic disease that leads to prolonged occlusion of the pulmonary vasculature [27]. This disease arises due to the abnormal activation of blood coagulation cascade in the vasculature. Pathologically, there is an abnormality at the level of fibrinolysis or any underlying hematological or autoimmune disorders ultimately leading to the hypercoagulable state and poor resolution of thrombosis. Group 4 PH is manageable through surgical intervention along with drug therapy. In this unique situation, through pulmonary thromboendarterectomy, a surgeon removes chronic blood clots from the pulmonary artery. Long-term anticoagulation medicines are recommended [27, 28].
1.3.5 Group 5: PH with Unclear Multifactorial Mechanism
Group 5 PH includes all the clinical conditions of PH where there are unclear multifactorial mechanisms of the disease origin. This category includes hematologic disorders (sickle cell disease), β-thalassemia, chronic hemolytic anemia, myeloproliferative disorders, splenectomy, metabolic disorders (glycogen storage disease, Gaucher disease, and thyroid disorders), and systemic disorders (sarcoidosis, lymphangioleiomyomatosis, and pulmonary histiocytosis). Post capillary PH in sickle cell disease is secondary to left ventricular dysfunction, whereas precapillary PH may cause by vasculopathy from the intravascular hemolysis, chronic pulmonary thromboembolism, or enhanced activity of pulmonary hypoxic response [29]. This category is so far the most poorly studied type of PH. The reason is its multimechanism activation etiology. Therefore, to well characterize this class, further research is warranted. So far, there is a serious lack of underrating of its pathology [29].
1.4 Mechanisms of Pathogenesis and Pathophysiology of Pulmonary Hypertension
As we consistently noted in the previous sections of this chapter, the main vascular changes in all forms of the pulmonary hypertension are vasoconstriction, smooth muscle cell proliferation, and even thrombosis. These pathological findings suggest an imbalance between vasodilation and vasoconstriction mechanisms [6, 15]. They also indicate perturbations even at the level of growth inhibitors and mitogenic factors and cause more inclination toward the prothrombotic state and alteration in the antithrombotic milieu. Hence, these off-balance homeostatic processes may ultimately lead to pulmonary endothelial dysfunction and injury [6, 15, 30].
1.4.1 Prostacyclin and Thromboxane A2
Prostacyclin and thromboxane A2 are potent vasodilator and vasoconstrictor, respectively [31]. They are generated in the vascular tissue during arachidonic acid metabolism. In contrast to the thromboxane A2, prostacyclin inhibits platelets aggregation and prevents smooth muscle proliferation [31]. In the pulmonary hypertension, there is abundance in the generation of thromboxane A2 compared to prostacyclin, and henceforth, pulmonary vasoconstriction, smooth muscle proliferation, and initiation of the process of thrombosis [32]. Moreover, it was observed that the generation of prostacyclin has been reduced in the small and medium-sized pulmonary arteries of the patients diagnosed with pulmonary hypertension, especially cases of the idiopathic pulmonary arterial hypertension [30].
1.4.2 Endothelin-1
Endothelin-1 is produced by the endothelial cells and is a powerful vasoconstrictor. It stimulates the pulmonary artery smooth muscle proliferation. Plasma levels of the endothelin-1 have been found to be increased in the patients of pulmonary hypertension [33]. The levels of the endothelin-1 are found to be inversely proportional to the degree of the pulmonary vascular blood flow and the cardiac output [34]. These important observations suggest a significant role of the endothelin-1 in the pathophysiologic hemodynamic changes that occur in patients of pulmonary hypertension [9, 30].
1.4.3 Nitric Oxide
Nitric Oxide (NO) is similar to Endothelin-1 produced by the endothelial cells; however, it induces a vasodilation via relaxing the vascular smooth muscle of the pulmonary vasculature [35]. It also inhibits pulmonary smooth muscle proliferation and prevents the platelets aggregation. The synthesis of the NO in the endothelial cell is regulated by the family of the enzymes, nitric oxide synthase (NOS). Studies show the decreased levels of the NOS in the pulmonary vessel of patients of pulmonary hypertension [36]. Thus, this sets the stage for pulmonary endothelial dysfunction, smooth muscle proliferation, and prothrombotic state [30, 36].
1.4.4 Serotonin
Serotonin, a 5-hydroxythryptamine, is vasoconstrictor and induces a smooth muscle cell proliferation [37, 38]. Due to the defect in the platelets that leads to hampered uptake of serotonin in platelets, it has been found to be involved in the pathogenesis of pulmonary hypertension. Research studies have described the genetic mutation in the serotonin transporter (5-HTT) and 5-hydroxytryptamine 2B receptor (5HT2B) in the platelets, which is associated with pulmonary arterial hypertension [37, 38]. However, other studies have observed that selective serotonin-reuptake inhibitors (SSRIs) increase the serotonin via inhibiting the serotonin transport, are rather protective in the setting of hypoxia, and not increasing the pulmonary hypertension. Thus, it may suggest that the levels of the serotonin itself may not be the only determinant of pulmonary hypertension. Therefore, more research is required to delineate the exact role of serotonin in the pathogenies of pulmonary hypertension [30].
1.4.5 Vasoactive Intestinal Peptide
Vasoactive intestinal peptide (VIP) is a powerful vasodilator and decreases pulmonary artery pressure and vascular resistance [10, 39]. It also has inhibitory effects on the platelet aggregation and suppresses smooth muscle proliferation. Clinical study showed the decreased levels of VIP in the serum and in the lungs of the patients of pulmonary arterial hypertension [10, 39]. However, providing inhalation therapy of VIP to the patients improved the symptoms and the hemodynamic alterations in the patients of pulmonary hypertension [6, 30].
1.4.6 Vascular Endothelial Growth Factors
Both acute hypoxia and chronic hypoxia lead to increased secretions of vascular endothelial growth factors (VEGF) along with increased expression of VEGF receptors, VEGF receptor-1 (KDR/flk) and VEGF receptor-2 (flt) in the lungs. Increased VEGF signaling in pulmonary arterial hypertension may be involved in abrupt angiogenesis, an underlying cause of the formation of the plexiform lesions and the clonal expression of the endothelial cells within the lesions [30, 40].
The above observations show that there is a clear inclination toward an increased vascular constriction response, enhanced vascular smooth muscle cell proliferation, and thrombosis [41]. Current treatment strategies are focused on bringing back the vascular and coagulation imbalances toward normalcy [34]. For instance, the therapeutic strategies developed based upon above observations, such as vasodilator prostaglandin analogue, epiprotenol, endothelial-derived relaxing factor, nitric oxide (NO), and endothelial-derived vasoconstrictor, and endothelin receptor antagonists, have been in the clinic to improve hemodynamic responses in pulmonary vascular [42]. Presently, there are important therapeutic approaches available to manage pulmonary arterial and idiopathic pulmonary hypertension in patients. They significantly increase the life expectancy of patients [42]. However, on the flip side, these medications have their own cons such as they have strong side effects profile, and most of the time patients stop to respond to these medications. And eventually, they need heart or lung transplantation [41]. Therefore, without any ambiguity, pulmonary hypertension is an unmet medical need, which warrants the development of newer therapeutics to target the disease. Recent studies suggest the emergence of the newer and improved therapeutic approaches to manage the pulmonary arterial hypertension [30, 40]. In the next sections, we will discuss the newer medications and their mechanisms of action in detail.
1.5 Emerging Therapeutics in the Management of Pulmonary Arterial Hypertension
The prevalence of all forms of the pulmonary hypertension (PH) is approximately about 15–50 per million individuals, with a prevalence of 2.4 per million per year [2, 9]. Studies further show that for the different forms of pulmonary arterial hypertension (PAH), such as idiopathic PH or heritable PH, female sex ratio is emerging as a risk factor. Out of the total cases of PAH, 80% are women. PH causes approximately about 15% risk of mortality within 1 year and 30% risk of death within 3 years in poorly controlled patients. One of the reasons for the poor prognosis of the disease may be incomplete understanding of the underlying pathophysiology of the disease. Newer and emerging therapies targeting the novel disease pathways discovered in the pathogenies of the disease will be discussed hereunder [1, 9].
For the last two decades, there is significant improvement in the treatment strategies of PH. US Food and Drug Administration (FDA) has approved 14 newer therapies to be delivered via oral, inhaled, subcutaneous, and intravenous routes to target the three potential established pathological pathways involved in the progression of pulmonary arterial hypertension. These pathways are endothelin, prostacyclin, and nitric oxide-mediated pulmonary vascular homeostasis (Fig. 1.1) [6]. Besides, investigators have pursued and identified new mechanistic pathways with novel interventions to halt the disease progression in difficult-to-manage conditions (Fig. 1.1) [1, 4, 6, 43].
1.5.1 Sex Hormones
Since women are more affected with PH than man, multicentered, international, preclinical, and clinical studies took the initiative to delineate the involvement of female sex per se and the female sex hormone, estrogen, in the pathogenesis of pulmonary arterial hypertension (PAH) [1, 38]. For instance, estrogen treatment shows the downregulation of expression of BMPR2, the gene that causes the expression of bone morphogenetic protein receptor II. Mutation in the BMPR2 leads to heritable pulmonary arterial hypertension in the patients [44]. Genetic variants of the cytochrome P450 and CYP1B1 increase the risk of development of pulmonary arterial hypertension in BMPR2 mutation carrier [1, 45]. This is due to the increased breakdown of estrogen into metabolites, which causes proliferation and prevents apoptosis in the vascular smooth muscle, thus narrowing the narrowing of the pulmonary vasculature. Administration of anastrozole and fulvestrant, which are estrogen receptor blockers, reversed the vascular remodeling in the BMPR2 transgenic mice [44, 46].
Clinical studies show that those women who are devoid of any cardiovascular disease are having better right ventricular ejection fraction (RVEF) compared to men [47]. This may be due to higher levels of estrogen in women. Thus, even though women are on a higher risk of getting pulmonary hypertension, and if they get the disease, their survival rate is much higher due to their better right ventricular (RV) function compared with men [48]. Research findings show that both men and post-menopausal women with pulmonary arterial hypertension have higher levels of estrogen and lower levels of dehydroepiandrosterone sulfate (DHEA-S) compared with control male and females [49]. Higher levels of estrogen are associated with shorter 6-min walk distance (6MWD), whereas a higher level of DHEA-S was linked with the lower pulmonary vascular resentence (PVR) and right atrial pressure [49].
Clinical trials were initiated to target the female sex hormone, estrogen, to manage pulmonary hypertension [50]. Findings from the small placebo-controlled randomized clinical trials (RCT) show that treatment of anastrozole reduced the circulating levels of estrogen, but no beneficial effect was observed in tricuspid annular plane systolic excursion (a marker of RV function and primary endpoint) [1]. Anastrozole significantly increased 6MWD over 12 weeks’ treatment [51]. Currently, several NIH-funded trials are ongoing, targeting estrogen or androgen using fulvestrant, tamoxifen, and DHEA in the management of pulmonary artery hypertension [1, 52].
1.5.2 Genes, Epigenetics, and MicroRNAs
Research studies demonstrate that more than 70% of patients diagnosed with familial or heritable pulmonary arterial hypertension and about 20% of patients with idiopathic pulmonary arterial hypertension have heterozygous mutations in BMPR2 [1, 53]. It is almost a conclusive argument that there is a significant involvement of altered signaling of BMP in pulmonary arterial hypertension, thus targeting this pathway should be an ideal scientific acuity [53]. Multiple experimental approaches were utilized to restore BMPR2 signaling using exogenous BMPR2 delivery via gene therapy, correction of mutation through medications, improvement of trafficking of the BMPR2 to the cell membrane by chemical chaperons, inhibition of the lysosomal degradation process, and increase in BMPR2 gene expression [1].
Recent advances in the molecular and cellular biology reveal a fascinating process of the regulation of biological functions including gene expressions and pulmonary vascular physiology using small noncoding RNA molecules, termed as micro RNAs or miRNAs or miRs [54]. Dysregulation of miRs has been shown to be associated with the development and progression of PAH [54]. This suggests a potential need for a thorough investigation of the role of miRs in the pathogenesis of PAH. The possible miR therapy in the management of PAH includes the inhibitors of miR-17, miR-130/301, miR-143/145, miR-20a, and miR-10, and the mimics of miR-204, miR-424/503, and miR-96 [55]. Currently, our understanding is in infancy about the specific targets of the above miRs, and their effective delivery to the patients’ body [1]. For instance, current challenges that need to be addressed include, but not limited to, the route of delivery, the mode of delivery, such as via vectors or naked oligonucleotides packaged in nanoparticles or liposomal delivery. Despite the initial hurdles, the future of the miRs therapy looks promising. miRs, via regulating the cellular gene expressions, may improve the pulmonary vascular homeostasis and thus emerge as better therapeutics tools in the management of PAH in patients [1]. One of the lead mechanisms, which cause the pulmonary vascular smooth muscle proliferation and ultimately narrowing the artery, is improper repair of the damaged DNA. This may also give rise to cancer-like phenotype and abnormal multiplication of the vascular smooth cells in PAH. Thus, DNA damage is the first step, which follows the development of PAH [56]. DNA is evident in the setting of the generation of reactive oxygen species, hypoxia, inflammation, reduced BMPR2 expression, and anorexigen drugs [56]. Consistently, the DNA damage in peripheral blood mononuclear cells, pulmonary artery smooth muscle and endothelial cells is a prior event that leads to clinical PAH [1, 56].
Downstream of DNA damages causes activation of poly (ADP-ribose) polymerase (PARP-1), which induces DNA repair process. However, research studies show overactivation of the PARP-1 pathway in clinical PAH [57]. This overactive PARP-1 signaling in PAH leads to cell dysfunction and further activation of inflammatory cell signaling response. Genetic deletion or therapeutic inhibition of PARP-1 prevents endothelial cell dysfunction, vascular remodeling, increases RV pressure and thereby attenuation of RV hypertrophy [1, 57].
In clinical studies, FK506 has been shown to significantly improve BMPR2 signaling. FK506 was tested in three advanced PAH patients who were found be resistant on the current line of treatments and were the candidates of lung transplantation [58]. FK506 improved and stabilized their conditions after 1 year of treatment. Significant improvement was observed in 6MWD, NT-ProBNP, and RV functions. Olaparib, an oral PARP-1 inhibitor, an approved drug by the FDA to treat ovarian cancer, is being investigated in an open-labeled single-arm study to investigate its effect on PVR over a 16-week duration (NCT03251872) [59]. The study is currently going on and outcomes have yet to be released [1].
1.5.3 Elastase Inhibition
Preclinical studies show a consistent finding of the disintegration of pulmonary vascular internal elastic lamina [1, 60]. This is associated with smooth muscle cell proliferation and neointima formation [60], which occurs 4 days after the monocrotaline (MCT) injection in the rats. Preclinical studies further showed that elastases, proteolytic enzymes, and target elastin can release growth factors from the extracellular matrix [60]. Treatment with the oral serine elastase inhibitor revered pulmonary vascular remodeling in MCT-exposed rats by enhancing apoptosis of smooth muscle cells. Elafin, an endogenous elastase inhibitor, is accompanied by a potent antiinflammatory activity. Elafin-overexpressed transgenic mice were found to be protected against hypoxia-induced pulmonary hypertension [46, 47]. Elafin reduced neointima formation via increasing the apoptosis in smooth muscle cells in lung in patients with PAH [61]. Elafin also demonstrated protective effects on vascular endothelium through BMPR2 and caveolin-mediated signaling mechanism [61].
Clinical studies have been initiated by the financial support of NIH to develop elafin, a potential drug to treat PAH (HL108797) [1]. These clinical investigations will assess the effectiveness, safety, and tolerability of elafin, and the effects of elafin on inflammatory markers [1].
1.5.4 Inflammation and Immunity
Inflammation induced by the generation of proinflammatory cytokines, such as IL-6, has been involved in inflammatory lung disease [62]. Mice overexpressed with IL-6 displayed an increased RV systolic pressure, hypertrophy, and severe form of occlusive angioproliferative lesions in the small distal pulmonary vessels. Besides, increased infiltration of lymphocytes was also found [48]. Deletion of IL-6 protected the mice from hypoxia-induced pulmonary hypertension [49]. Similarly, transgenic mice devoid of IL-6 receptor on the smooth muscle cells were protected against hypoxia-induced pulmonary hypertension [63]. Treatment with IL-6 receptor antagonist reversed the experiential hypertension in rats [63].
Dysregulated B-cell function, regulatory T-cell (T reg) deficiency, and pathological antibodies generation against endothelial cells are found to be associated with the pathogenesis of PAH [1, 64]. Research findings show that exposing the athymic nude rats (lacking T reg) to sugen-hypoxia resulted in increased number of B cells and generation of antiendothelial cell antibodies in pulmonary vasculature [64] and led to severe PAH [64]. Immune reconstitution using healthy T reg cells prevented accumulation of B cell and generation of antibodies against endothelial cells, resulting in attenuation of PAH [1].
Dimethyl fumarate (DMF) is NRF2 pathway-activating agent in various cell types and has been demonstrated to exert a potent antiinflammatory role in pulmonary inflammatory diseases [65]. Part of the antiinflammatory property of the DMF is through inhibition of NFkB signaling pathway. DMF is an FDA-approved medication to manage multiple sclerosis. Treatment of DMF in the sugen-hypoxia model reversed pathological hemodynamic changes, and reduced inflammation and oxidative stress [1, 65].
Clinical studies show increased levels of IL-6 in the serum of the patients diagnosed with idiopathic PAH and PAH associated with autoimmune disorders. Increased IL-6 levels are associated with increased incidents of mortality in PAH patients [66]. Increased levels of expression of IL-6 receptor have been reported in the smooth muscle cells from the patients of PAH [66].
Tocilizumab, a humanized monoclonal antibody, is approved for autoimmune disorders, such as rheumatoid arthritis [67]. Being aware of a clear role of IL-6 and its downstream singling in the pathogenesis and poor prognosis of PAH, currently, a phase II clinical study, the Therapeutic Open Label Study of Tocilizumab in the Treatment of Pulmonary Artery Hypertension (TRANSFORM-UK, NCT02676947), is underway in which Tocilizumab is administered once a month for 6 months to the patients of PAH [67]. The primary endpoints of the study are safety and change in PVR.
Based upon a potential preclinical data showing the role of Treg signaling in PAH, human trials are going on using rituximab, a chimeric monoclonal antibody against CD20, that targets B cells [1]. Rituximab has anecdotally been demonstrated to be effective with connective tissue disease (CTD)-related PAH and systemic sclerosis-associated PAH, thus requiring detailed scientific investigations of its safety and efficacy. The ASC01 study, a NIH-funded, double-blind, placebo-controlled phase II trial (NCT01086540), was initiated to evaluate the safety and efficacy of rituximab on systemic sclerosis-associated PAH and its progression. The primary endpoint is the change in PVR at 24 weeks. Secondary endpoint includes RV function measured by MRI [1].
Studies have also been initiated to target NRF-2 pathway, including the role of bardoxolone and DMF in the patients associated with systemic sclerosis-induced PAH (NCT02657356, NCT02981082) [1].
1.5.5 Mitochondrial Dysfunction
The metabolic theory of the induction of PAH is based on multiple molecular abnormalities in PAH-caused mitochondrial suppression in both pulmonary vascular cells and extrapulmonary tissue [68]. This ultimately inhibits the oxidation of the glucose metabolism and upregulation of glycolysis [68]. It is not clear yet if mitochondrial alteration is the cause or an outcome of PAH. Mitochondrial dysfunction displays an inhibition of glucose oxidation due to the abnormality at the level of the enzyme, pyruvate dehydrogenase (PVD) [68]. PVD is inhibited following the phosphorylation induced by a pyruvate dehydrogenase kinase (PDK) because of the hypoxia, inflammation, endoplasmic reticulum (ER) stress, and tyrosine kinase activation [68]. PDH inhibition leads to attenuation of apoptotic pathway, cell proliferation and inflammation—salient features of the pathogenesis of the PAH. In line with the above findings, a small molecule inhibitor, dichloroacetate (DCA), activates PDH and thereby reverses the above pathological changes in several animal models of PAH [69].
A clinical study, a 4-month, open-label trial of DCA (3–6 mg/kg b.i.d) was conducted in 20 stable WHO functional class II–III idiopathic PAH patients [70]. The results of the study showed a reduction in mPAP and PVR, and improvement in functional capacity in patients with SIRT3 and UCP2 variants that decreased PDH [70]. Based on the outcomes, further clinical studies are required to determine the drug’s safety and efficacy in a large set of population.
1.5.6 Other Metabolic Pathways
Metformin, an antidiabetic drug, demonstrated the improvement in the endothelial cell function by increasing NO synthase activity and thereby reduced pulmonary artery smooth muscle cell proliferation [71]. Further studies showed that metformin also inhibits aromatase and estrogen production [72]. Metformin provided protection in MCT and hypoxia-induced PAH in rats via inhibition of smooth muscle cell proliferation [73]. Clinical studies of metformin are currently going on at Vanderbilt University for PAH (NCT01884051, NCT03617458) [1].
The mechanistic target of rapamycin complex (mTORC) pathway signals the proliferation of vascular smooth muscle cells [74]. mTORC1 and mTORC2 have been linked to PAH as their expressions are increased in pulmonary smooth muscle cells in PAH patients [75]. Studies show that mTORC2 is required for adenosine triphosphate (ATP) generation that leads to the survival of pulmonary artery smooth muscle proliferation in PAH [75]. The mTOR kinase inhibitor PP242 has a potent antiproliferative activity on mTORC2. It also induces smooth muscle cell apoptosis in small pulmonary arteries and reverses hypoxia-induced pulmonary vascular remodeling in rat model [1].
A phase I trial is initiated to investigate the effect of ABI-009, (a nanoparticle-bound rapamycin targeted to the lungs) in PAH (NCT02587325) [1].
1.5.7 Nervous System
Sympathetic nervous system activation and para-sympathetic nervous system inhibition lead to the pulmonary vascular constriction, and thereby contribute in the pathophysiology of PAH [76]. Catecholamines, upon release from sympathetic nervous system, activate α1 receptor and thereby induce pulmonary vasoconstriction and an increase in pulmonary artery pressure [76]. However, pharmacological inhibition of α1-receptors or activation of the parasympathetic activation causes vasorelaxation in pulmonary artery [76]. Antagonism of adrenergic receptors prevents MCT-induced pulmonary hypertension and decreases the RV size [1]. Carvedilol treatment demonstrated the improvement in survival of the animals treated with MCT [77].
Based upon animal research, several pharmacological agents targeting autonomic nervous system were investigated in clinical PAH. Small pilot studies used β-blocker such as carvedilol or bisoprolol to PAH patients [43]. The outcome of the studies showed decreased heart rate, improvement in cardiac indices and in 6MWD. A placebo-controlled crossover trial of carvedilol is current going on (NCT02507011) [1].
1.5.8 Renin-Angiotensin-Aldosterone System (RAAS)
The role of the renin-angiotensin-aldosterone system (RAAS) in the pathogenesis of PAH is recently being investigated [78]. Angiotensin II (Ang II) increases pulmonary vascular fibrosis and RV collagen deposition. Angiotensin-converting enzyme (ACE) inhibitors have a cornerstone role in the treatment of cardiovascular diseases. An increased expression of angiotensin II type 1 (AT1) receptor is observed in the pulmonary vascular of PAH patients, which may cause vasoconstriction, oxidative stress, inflammation, and smooth muscle cell proliferation. Losartan, an AT1 receptor blocker, prevented MCT-induced diseases progression, restored right ventricular (RV)-pulmonary artery (PA) coupling, and improved RV diastolic function in animals [78, 79].
Increased levels of aldosterone are reported in the blood of in animal models of PAH, as well in in some human studies [79, 80]. Administration of aldosterone receptor antagonist, spironolactone, improved RV morphology, function, and pulmonary vascular remodeling, and reduced pulmonary smooth muscle proliferation in MCT and in sugen-hypoxia rats and in hypoxia mouse model [81].
Based on the potential experimental data [82], clinical trials have been initiated and are currently going on to investigate the clinical efficacy and safety of spironolactone in patients with PAH (NCT02253394, NCT01712620, NCT01468571) [62].
1.6 Summary
Pulmonary arterial hypertension is contributing high rate of morbidity, mortality, and economic burden even in developed countries. Pulmonary vascular remodeling due to enhanced endothelial and smooth muscle cell proliferation causes the narrowing of the pulmonary artery and increases pulmonary arterial pressure. Despite recent advances in the treatment of PAH, current medications failed to halt or reverse the progression of the disease in the patients (Fig. 1.2) [6, 8]. This requires a better understanding of the disease at cellular and molecular levels. There is also a need to enhance the drug discovery process in the laboratory. Therefore, a rational drug design will yield lesser failures at preclinical and clinical stages of drug testing and may expedite the approval process of the medications to treat PAH. We hope that the medications which are currently at various stages of clinical trials will be able to change the current treatment paradigm in near future, and thus hold a promise to improve the treatment of PAH.
References
Spiekerkoetter E, Kawut SM, de Jesus Perez VA (2019) New and emerging therapies for pulmonary arterial hypertension. Annu Rev Med 70:45–59
Humbert M, Lau EM, Montani D, Jais X, Sitbon O, Simonneau G (2014) Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 130(24):2189–2208
Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A et al (2009) Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 54(1 Suppl):S55–S66
Thenappan T, Ormiston ML, Ryan JJ, Archer SL (2018) Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ 360:j5492
Thenappan T, Khoruts A, Chen Y, Weir EK (2019) Can intestinal microbiota and circulating microbial products contribute to pulmonary arterial hypertension? Am J Physiol Heart Circ Physiol 317(5):H1093–HH101
Tsai H, Sung YK, de Jesus Perez V (2016) Recent advances in the management of pulmonary arterial hypertension. F1000Res 5:2755
Lau EMT, Giannoulatou E, Celermajer DS, Humbert M (2017) Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol 14(10):603–614
Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A et al (2015) 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 46(4):903–975
Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V et al (2006) Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 173(9):1023–1030
Mathioudakis A, Chatzimavridou-Grigoriadou V, Evangelopoulou E, Mathioudakis G (2013) Vasoactive intestinal peptide inhaled agonists: potential role in respiratory therapeutics. Hippokratia 17(1):12–16
Hemnes AR, Champion HC (2008) Right heart function and haemodynamics in pulmonary hypertension. Int J Clin Pract Suppl 160:11–19
Sysol JR, Machado RF (2018) Classification and pathophysiology of pulmonary hypertension. Continuing Cardiol Educ 4(1):2–12
Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A et al (2013) Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62(25 Suppl):D34–D41
Godbey PS, Graham JA, Presson RG Jr, Wagner WW Jr, Lloyd TC Jr (1995) Effect of capillary pressure and lung distension on capillary recruitment. J Appl Physiol (1985) 79(4):1142–1147
Wilkins MR, Aman J, Harbaum L, Ulrich A, Wharton J, Rhodes CJ (2018) Recent advances in pulmonary arterial hypertension. F1000Res 7. https://doi.org/10.12688/f1000research.14984.1
Rich JD, Rich S (2014) Clinical diagnosis of pulmonary hypertension. Circulation 130(20):1820–1830
Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM et al (1987) Primary pulmonary hypertension. A national prospective study. Ann Intern Med 107(2):216–223
Soubrier F, Chung WK, Machado R, Grunig E, Aldred M, Geraci M et al (2013) Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 62(25 Suppl):D13–D21
Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G et al (2000) Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67(3):737–744
International PPHC, Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA III et al (2000) Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 26(1):81–84
Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X et al (1996) Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med 335(9):609–616
Sadoughi A, Roberts KE, Preston IR, Lai GP, McCollister DH, Farber HW et al (2013) Use of selective serotonin reuptake inhibitors and outcomes in pulmonary arterial hypertension. Chest 144(2):531–541
Montani D, Bergot E, Gunther S, Savale L, Bergeron A, Bourdin A et al (2012) Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 125(17):2128–2137
Melenovsky V, Kotrc M, Borlaug BA, Marek T, Kovar J, Malek I et al (2013) Relationships between right ventricular function, body composition, and prognosis in advanced heart failure. J Am Coll Cardiol 62(18):1660–1670
Guazzi M, Vicenzi M, Arena R, Guazzi MD (2011) Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 124(2):164–174
Seeger W, Adir Y, Barbera JA, Champion H, Coghlan JG, Cottin V et al (2013) Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol 62(25 Suppl):D109–D116
Kim NH, Delcroix M, Jenkins DP, Channick R, Dartevelle P, Jansa P et al (2013) Chronic thromboembolic pulmonary hypertension. J Am Coll Cardiol 62(25 Suppl):D92–D99
Mahmud E, Madani MM, Kim NH, Poch D, Ang L, Behnamfar O et al (2018) Chronic thromboembolic pulmonary hypertension: evolving therapeutic approaches for operable and inoperable disease. J Am Coll Cardiol 71(21):2468–2486
Gordeuk VR, Castro OL, Machado RF (2016) Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood 127(7):820–828
Farber HW, Loscalzo J (2004) Pulmonary arterial hypertension. N Engl J Med 351(16):1655–1665
Gerber JG, Voelkel N, Nies AS, McMurtry IF, Reeves JT (1980) Moderation of hypoxic vasoconstriction by infused arachidonic acid: role of PGI2. J Appl Physiol Respir Environ Exerc Physiol 49(1):107–112
Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM et al (1992) An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 327(2):70–75
Allen SW, Chatfield BA, Koppenhafer SA, Schaffer MS, Wolfe RR, Abman SH (1993) Circulating immunoreactive endothelin-1 in children with pulmonary hypertension. Association with acute hypoxic pulmonary vasoreactivity. Am Rev Respir Dis 148(2):519–522
Komai H, Adatia IT, Elliott MJ, de Leval MR, Haworth SG (1993) Increased plasma levels of endothelin-1 after cardiopulmonary bypass in patients with pulmonary hypertension and congenital heart disease. J Thorac Cardiovasc Surg 106(3):473–478
Giaid A, Saleh D (1995) Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 333(4):214–221
Xue C, Johns RA (1995) Endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 333(24):1642–1644
Jiao YR, Wang W, Lei PC, Jia HP, Dong J, Gou YQ et al (2019) 5-HTT, BMPR2, EDN1, ENG, KCNA5 gene polymorphisms and susceptibility to pulmonary arterial hypertension: a meta-analysis. Gene 680:34–42
Marcos E, Fadel E, Sanchez O, Humbert M, Dartevelle P, Simonneau G et al (2004) Serotonin-induced smooth muscle hyperplasia in various forms of human pulmonary hypertension. Circ Res 94(9):1263–1270
Baliga RS, Macallister RJ, Hobbs AJ (2013) Vasoactive peptides and the pathogenesis of pulmonary hypertension: role and potential therapeutic application. Handb Exp Pharmacol 218:477–511
Voelkel NF, Gomez-Arroyo J (2014) The role of vascular endothelial growth factor in pulmonary arterial hypertension. The angiogenesis paradox. Am J Respir Cell Mol Biol 51(4):474–484
Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F (2011) Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 8(8):443–455
Guignabert C, Tu L, Girerd B, Ricard N, Huertas A, Montani D et al (2015) New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: importance of endothelial communication. Chest 147(2):529–537
da Silva Goncalves Bos D, Van Der Bruggen CEE, Kurakula K, Sun XQ, Casali KR, Casali AG et al (2018) Contribution of impaired parasympathetic activity to right ventricular dysfunction and pulmonary vascular remodeling in pulmonary arterial hypertension. Circulation 137(9):910–924
Lahm T, Tuder RM, Petrache I (2014) Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307(1):L7–L26
Austin ED, Hamid R, Hemnes AR, Loyd JE, Blackwell T, Yu C et al (2012) BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ 3(1):6
Chen X, Austin ED, Talati M, Fessel JP, Farber-Eger EH, Brittain EL et al (2017) Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur Respir J 50(2):1602337
Kawut SM, Lima JA, Barr RG, Chahal H, Jain A, Tandri H et al (2011) Sex and race differences in right ventricular structure and function: the multi-ethnic study of atherosclerosis-right ventricle study. Circulation 123(22):2542–2551
Jacobs W, van de Veerdonk MC, Trip P, de Man F, Heymans MW, Marcus JT et al (2014) The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 145(6):1230–1236
Hemnes AR, Maynard KB, Champion HC, Gleaves L, Penner N, West J et al (2012) Testosterone negatively regulates right ventricular load stress responses in mice. Pulm Circ 2(3):352–358
Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP et al (2009) Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir J 34(5):1093–1099
Kawut SM, Archer-Chicko CL, DeMichele A, Fritz JS, Klinger JR, Ky B et al (2017) Anastrozole in pulmonary arterial hypertension. A randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med 195(3):360–368
Baird GL, Archer-Chicko C, Barr RG, Bluemke DA, Foderaro AE, Fritz JS et al (2018) Lower DHEA-S levels predict disease and worse outcomes in post-menopausal women with idiopathic, connective tissue disease- and congenital heart disease-associated pulmonary arterial hypertension. Eur Respir J 51(6):1800467
Orriols M, Gomez-Puerto MC, Ten Dijke P (2017) BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell Mol Life Sci 74(16):2979–2995
Boucherat O, Bonnet S (2016) MicroRNA signature of end-stage idiopathic pulmonary arterial hypertension: clinical correlations and regulation of WNT signaling. J Mol Med (Berl) 94(8):849–851
Chun HJ, Bonnet S, Chan SY (2017) Translational advances in the field of pulmonary hypertension. Translating microRNA biology in pulmonary hypertension. It will take more than “miR” words. Am J Respir Crit Care Med 195(2):167–178
Ranchoux B, Meloche J, Paulin R, Boucherat O, Provencher S, Bonnet S (2016) DNA damage and pulmonary hypertension. Int J Mol Sci 17(6)
Kaur G, Singh N, Lingeshwar P, Siddiqui HH, Hanif K (2015) Poly (ADP-ribose) polymerase-1: an emerging target in right ventricle dysfunction associated with pulmonary hypertension. Pulm Pharmacol Ther 30:66–79
Lallouet M, Sornin G (1985) [Craniomandibular joint dysfunction. Report of a clinical case]. Inf Dent 67(36):3811–3812
Spiekerkoetter E, Sung YK, Sudheendra D, Scott V, Del Rosario P, Bill M et al (2017) Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur Respir J 50(3)
Maruyama K, Ye CL, Woo M, Venkatacharya H, Lines LD, Silver MM et al (1991) Chronic hypoxic pulmonary hypertension in rats and increased elastolytic activity. Am J Phys 261(6 Pt 2):H1716–H1726
Zaidi SH, You XM, Ciura S, Husain M, Rabinovitch M (2002) Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation 105(4):516–521
Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB (2009) Interleukin-6 overexpression induces pulmonary hypertension. Circ Res 104(2):236–44, 28p following 44
Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM et al (2018) Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest 128(5):1956–1970
Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C et al (2007) Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med 175(12):1280–1289
Grzegorzewska AP, Seta F, Han R, Czajka CA, Makino K, Stawski L et al (2017) Dimethyl fumarate ameliorates pulmonary arterial hypertension and lung fibrosis by targeting multiple pathways. Sci Rep 7:41605
Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD et al (2010) Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 122(9):920–927
Hernandez-Sanchez J, Harlow L, Church C, Gaine S, Knightbridge E, Bunclark K et al (2018) Clinical trial protocol for TRANSFORM-UK: a therapeutic open-label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm Circ 8(1):2045893217735820
Paulin R, Michelakis ED (2014) The metabolic theory of pulmonary arterial hypertension. Circ Res 115(1):148–164
McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K et al (2004) Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 95(8):830–840
Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L et al (2017) Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med 9(413)
Sartoretto JL, Melo GA, Carvalho MH, Nigro D, Passaglia RT, Scavone C et al (2005) Metformin treatment restores the altered microvascular reactivity in neonatal streptozotocin-induced diabetic rats increasing NOS activity, but not NOS expression. Life Sci 77(21):2676–2689
Dean A, Nilsen M, Loughlin L, Salt IP, MacLean MR (2016) Metformin reverses development of pulmonary hypertension via aromatase inhibition. Hypertension 68(2):446–454
Agard C, Rolli-Derkinderen M, Dumas-de-La-Roque E, Rio M, Sagan C, Savineau JP et al (2009) Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br J Pharmacol 158(5):1285–1294
Pena A, Kobir A, Goncharov D, Goda A, Kudryashova TV, Ray A et al (2017) Pharmacological inhibition of mTOR kinase reverses right ventricle remodeling and improves right ventricle structure and function in rats. Am J Respir Cell Mol Biol 57(5):615–625
Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP et al (2014) Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129(8):864–874
Vaillancourt M, Chia P, Sarji S, Nguyen J, Hoftman N, Ruffenach G et al (2017) Autonomic nervous system involvement in pulmonary arterial hypertension. Respir Res 18(1):201
Drake JI, Gomez-Arroyo J, Dumur CI, Kraskauskas D, Natarajan R, Bogaard HJ et al (2013) Chronic carvedilol treatment partially reverses the right ventricular failure transcriptional profile in experimental pulmonary hypertension. Physiol Genomics 45(12):449–461
de Man FS, Tu L, Handoko ML, Rain S, Ruiter G, Francois C et al (2012) Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med 186(8):780–789
Shenoy V, Kwon KC, Rathinasabapathy A, Lin S, Jin G, Song C et al (2014) Oral delivery of Angiotensin-converting enzyme 2 and Angiotensin-(1-7) bioencapsulated in plant cells attenuates pulmonary hypertension. Hypertension 64(6):1248–1259
Maron BA, Opotowsky AR, Landzberg MJ, Loscalzo J, Waxman AB, Leopold JA (2013) Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail 15(3):277–283
Maron BA, Oldham WM, Chan SY, Vargas SO, Arons E, Zhang YY et al (2014) Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation 130(2):168–179
Hemnes AR, Rathinasabapathy A, Austin EA, Brittain EL, Carrier EJ, Chen X et al (2018) A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur Respir J 51(6)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Heo, S. et al. (2020). Therapeutic Advances in the Management of Pulmonary Arterial Hypertension. In: Rayees, S., Din, I., Singh, G., Malik, F. (eds) Chronic Lung Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-15-3734-9_1
Download citation
DOI: https://doi.org/10.1007/978-981-15-3734-9_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-3733-2
Online ISBN: 978-981-15-3734-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)