
Abstract
Voltage-gated sodium ion channels are essential to maintain the excitability and activity of neurons and neuronal network. Several studies have been done to explore the basic properties of ion channels, their existence and physiological characteristics. Till date we know that 11 genes are responsible for encoding of 9 families of sodium channels (Nav 1.1 to Nav 1.9) and are classified according to varying degrees of sensitivity to Tetrodotoxin (TTX). These are localized in various sites such as skeletal muscles, central nervous system (CNS), cardiac muscles, and peripheral sensory neurons. Any aberration in its structure and function leads to various clinical pathologies such as channelopathies (where dysregulation in receptors are directly responsible for initiation and progression) and diseases contributing to dysregulation of expression of sodium channels cause various neurological disorders. In this chapter, we emphasize the composition, function, and regulation of sodium channels at the molecular level and the crucial role of sodium channels in the development and progression of various disease pathologies such as epilepsy, schizophrenia, familiar hemiplegic migraine and neuropathic pain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Voltage-gated sodium channels
- Composition
- Regulation
- Channelopathies
- Neurological disorders
- Neuropathic pain
18.1 Introduction
Voltage-gated sodium ion channels are required for excitation of cells. Their history can be traced back to 1941, when Kenneth S. Cole and Richard F. Baker confirmed the existence of voltage-gated membrane pores in giant squid axon (Cole and Curtis 1939). Alan Hodgkin and Andrew Huxley won the Nobel prize for their work on basic properties of ion channels and their mechanisms of excitation and inhibition in 1952. Later in 1970 and 1981, the existence and physiological characteristics of ion channels were explored by Bernard Katz, Ricardo, and Miledi et al. and Erwin Neher and Bert Sakmann et al., using noise analysis and patch clamp techniques, respectively (Cole and Baker 1941; Roger et al. 2015).
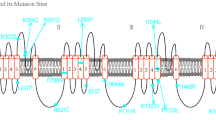
So far, 11 genes are responsible for the encoding of 9 families of sodium channels (Nav 1.1 to Nav 1.9). These genes are classified according to varying degrees of sensitivity to Tetrodotoxin (TTX) and can be traced to four paralogous locations in the chromosome segment (Ambrose et al. 1992; Wang et al. 1992; Burgess et al. 1995; Beckers et al. 1996; Kozak and Sangameswaran 1996; Plummer et al. 1998; Catterall et al. 2005; Bagal et al. 2015) (Table 18.1). More than 20 exons encode for 9 sodium channel α-subunits. Nav channels (1.1, 1.2, 1.3, and 1.7) are situated on the second chromosome in both humans as well as in the mouse whereas Nav channels 1.5 are situated at 2q24 and in case of mouse, present on chromosome 2. And the Nav 1.8 and 1.9 channels are present on 3p21–24 in case of humans and chromosome 9 in the mouse. Skeletal muscle contains the Nav 1.4 and the CNS contains the Nav 1.6. The genes of Nav 1.4 are situated on the 17th chromosome (humans) and 11th chromosome (mouse) whereas genes of Nav 1.6 can be traced to the 12th chromosome (humans) and the 15th chromosome (mouse) (Catterall et al. 2005; Mantegazza and Catterall 2012). All the sodium channels structures are analogous to each other, but few amino acid replacements confer this resistance, for example, in Nav 1.5 which is predominantly localized in cardiac muscles, substitution of phenylalanine to cystine in pore area provides 200 times reduction in sensitivity to TTX (Yamagishi et al. 2001). A similar replacement is also observed in Nav 1.8 and Nav 1.9, which are localized in peripheral sensory neurons where phenylalanine is replaced by serine, which provides even greater resistance (Mantegazza and Catterall 2012). Channelopathies are the group of diseases characterized by any aberration in the structure or function arising from a mutation in the genes encoding for sodium channels. In this chapter, we will study the composition, function, and regulation at the molecular level of sodium channels and the crucial role of sodium channels in the development and progression of various disease pathologies (Roger et al. 2015).
18.1.1 Composition of Sodium Channels
Sodium channels are heteromeric integral glycoproteins composed of two types of subunits, α (initiator) and β (regulatory). Each α subunit contains six segments, which houses four domains (DI-DIV) around the central pore. Arginine-rich segment 4 (S4) is also known as the “voltage sensor,” and the hairpin segment (P loop) forms the “ion filter” between segment 5 (S5) and segment 6 (S6) responsible for ion permeability (King and Vetter 2014). There is a group of amino acids called IFMT, segment “I” Isoleucine, “F” Phenylalanine, “M” Methionine, “T” Tryptophan, present between segment 6 of DIII and Segment 1 of DIV (Fig.18.1). This IFMT segment has an affinity to bind to hydrophobic amino acids (like alanine and asparagine) between segment 4 and segment 5 of DIII and DIV; this binding is responsible for the refractive nature of the ion channel (Tata 2013). Pore consists of tubular vestibule that forms a cavity, ion-selective filter, and intracellular gate activator. There are two models that explain the organization of voltage-gated sodium channels: (1) Sliding helix and (2) Helical screw. Both models suggest the presence of positively charged residues in segment 4 that serves as gating (Frank and Catterall 2003). These positively charged residues are held together by corresponding residues having negative charge, of segments 1, 2, and/or 3 in active state. But in the inactive state, these residues are held inside due to the Coulomb force of negatively charged resting membrane potential. When depolarization takes place, changes in membrane polarity occur, which results in relieving of this electrostatic force. As a result, these positive, rich residues move out opening the pore, causing an influx of sodium ions, thus initiating the cell activation process. The outward movement of segment 4 is “voltage dependent.” On the other hand, all the downstream mechanisms are “voltage independent.”

Composition of sodium channel: α subunit (Domains- I, II, III & IV); β subunit and IFMT segment (Isoleucine, Phenylalanine, Methionine, and Tryptophan)
β subunits are integral regulatory proteins found to be associated with the α subunit. It consists of three domains: extracellular, transmembrane, and intracellular domains. β subunits are served as cell adhesion molecules that are responsible for control of the surface expression of voltage-gated sodium channels, cell-to-cell communication and cellular migration. β1 subunit in association with Neurofascin 186 and Contactin provides increase in surface expression of voltage-gated sodium channels. β1 subunit knockout leads to disruption of neuronal–glial interaction and reduction in the number of Nodes of Ranvier in myelinated neurons which results in disrupted saltatory conduction (Marban et al. 1998).
18.1.2 Molecular Functioning of Sodium Channels
Voltage-gated sodium channels (VGSCs) occur in three forms: (1) active (2) refractory, (3) closed. From closed state to go on to open state requires depolarization, changes in membrane polarity to positive, which usually happens during an action potential, and within a few milli-seconds VGSCs progress to refractory state, also known as the inactive state, is a mechanism that protects cells from excessive stimulation. This is followed by the closed state when the cell membrane attains the normal repolarized potential. During closed state, the “voltage sensor” segment 4 is held inside due to the overall negative resting membrane potential of the cell. The depolarization relieves the electrostatic force on segment 4 pushing it to move out, thus opening the intracellular ion gate. This allows sodium ion conductions, leading to increased sodium concentration inside the cell. When the intracellular gate is opened, the IFMT segment binds to hydrophobic amino acids (like alanine and asparagine) between segment 4 and segments 5 of DIII and DIV, as there is no steric hindrance caused by intracellular ion gate due to conformational change. This state is called a refractory state/inactive state. In the inactive state, sodium ions cannot pass through causing prevention of the cell from overstimulation. From a refractory state to go to closed state again requires a change in membrane potential. Whenever cell resting membrane potential is restored, segment 4 domain is attracted toward the cell, augmenting its electrostatic influence on segment 4 resulting in a conformational change to the closed ion channel (Catterall 1992; Marban et al. 1998) (Fig. 18.2).

Molecular functioning of sodium channels: Closed, opened and inactivated state; IFMT segment (Isoleucine, Phenylalanine, Methionine, and Tryptophan), RMP; Resting Membrane Potential
18.2 Regulation of Voltage-Gated Sodium Channels
Channel Interacting Proteins (ChIPs) are endogenous proteins that interact with voltage-gated Na+ channels. Some examples of ChIPs are connexion 43, Caveolin 3, Calcium-calmodulin kinase 2, ankyrins, telethonin, plakophilin, neuronal precursor cell-expressed developmentally downregulated 4 (nedd4), fibroblast growth factor (FGF) and its homologous factors (FHFs). These endogenous proteins regulate the sodium channel expression and its functioning (Savio-Galimberti et al. 2012).
18.2.1 Cellular Trafficking and Surface Expression of Voltage-Gated Sodium Channels: Caveolae/Caveolin Interaction-Kinesin-VGSCs Interaction
Caveolae are membrane folding proteins predominantly found in muscle tissues, implicated for surface expression of number of receptors and Caveolins are linker proteins responsible for the rapid surface expression of sodium channels. These Caveolin proteins act as linker proteins and cause recruitment of voltage-gated ion channels from the intracellular pool following the cell stimulation. Kinesins are the molecular motor proteins responsible for the delivery of newly synthesized receptors (like VGSCs) vesicles from the nucleus, rough endoplasmic reticulum and Golgi body to the synapse, creating the intracellular pool of vesicles containing receptors. This intracellular pool of vesicles containing receptors is dynamic in nature. When a cell is stimulated, these caveolin proteins act as linker proteins causing mobilization of these vesicles and with the help of caveolae proteins, cause surface expression of voltage-gated ion channels receptors. Some recent studies show that kif 1B and kif 5A, kif 5B are kinesin members of kinesin 3 and kinesin 1 families respectively which are implicated in the cellular trafficking of voltage-gated ion channels like Nav 1.8. Mutation or overexpression of these genes and proteins leads to neuropathic conditions like Charcot and Marie tooth disorder and neuropathic pain. Another study proved that β cell stimulation leads to increased surface expression of VGSCs. This is due to intracellular VGSCs pool mobilization by caveolin 3 and caveolae membrane proteins, VGSCs are surface expressed (Savio-Galimberti et al. 2012; Roger et al. 2015).
18.3 Sodium Channel and Related Clinical Pathologies
Sodium channel as discussed above is crucial for the functioning of any excitable cells, and any aberration in its structure or functioning leads to various clinical pathologies. There are two types of disorders that are classified based on the role played by the sodium channels on pathogenesis of diseases: (1) channelopathies (where dysfunctional receptors are directly responsible for initiation and progression) and (2) disease contributing to dysregulated expression of sodium channels (like cancer, neurological disorders) (Kaplan et al. 2016).
18.3.1 Function of Sodium Channels in the PNS and CNS
Sodium channels are important to maintain the excitable character as well as the activity of neurons and the neuronal network. They are ubiquitously expressed throughout the neurocyton and axons in neurons. Denseness of sodium channel is more at Nodes of Ranvier and axon initial segment (AIS), which is responsible for speeding nerve impulse conduction. This phenomenon is often referred to as saltatory conduction (Kaplan et al. 2016). The action potential is initiated from AIS and this characteristic is attributed to relatively high sodium channel receptor density at this site. Various IHC studies have validated this claim of determining distribution of sodium channel expression along AIS. In such studies Nav1.6 was densely expressed at distal AIS of retinal ganglion cell of rat, whereas Nav1.1 was aggregated at proximal AIS. Similar trend in expression was observed in cortical pyramidal neurons of rat showing increased Nav1.2 and Nav 1.6 at AIS. Hyperpolarization of NaV1.6 that depend on NaV 1.2 activity results in differential expression of sodium channel. When depolarlizing signals enter AIS, activation threshold of Nav 1.6 crosses more readily as compared to Nav1.2, which consequently generates action potential. This causes two types of currents: the first one propagates down the axon and the other results in backpropagating currents, which leads to activation of Nav1.2. This action potential is propagated from soma to axons and dendrites. The backpropagating currents are crucial for relating the neuronal output and regulating neuronal platicity. Dendritic VGSCs also contributes to action potential by amplifying the depolarlization response. Collectively, this data suggests the complex relationship of sodium channels distribution and neuronal activity. The data also hints at the susceptibility of brain networks to any aberrations in sodium channels function and its number by mutations, disorders, or action of drug (Kaplan et al. 2016).
18.3.2 Dysfunctional and Dysregulated Expressions of Sodium Channels as a Cause for Neurological Disorders
Channelopathies are the group of disorders arising from a mutation in genes encoding for voltage-sensitive ion channel or dysfunctional receptors arising from an autoimmune attack. This condition has been indicated for several diseases including epilepsy, Dravet Syndrome, complications associated with pain like congenital pain insensitivity, paroxysmal extreme pain disorder, primary erythromelalgia and cardiac arrhythmia (characterized by long QT syndrome), slow ventricular conduction and atrial standstill. Aggressive nature of breast (Nav1.5) and prostate cancer (Nav1.7) has been related with change in epigenetic regulation. Dysregulation of sodium channels has also been over-served in diabetes and chemical-induced neuropathies, neuromuscular disorders (like Eaton Lambert syndrome, Charcot Marie Tooth disorder) and various neurodegenerative disorders (like Alzheimer’s, Parkinson’s, and Schizophrenia). Sodium channels have been an important target due to their major role in these diseases.
18.3.3 Role of Sodium Channels in Epilepsy
Analysis of mutation in VGSCs in epilepsy has shown a complex data that results in a specific pattern showing connection between channel mutation and epilepsy. In VGSCs of interneuron, there are alterations in channel biological organization that are cell dependent in nature; for example, there is a loss of function in Nav1.1 or Nav1.6 due to depolarization in shifts in the voltage dependence of activation, hyperpolarization in shifts in the voltage dependence of inactivation, and haploinsufficiency (Hargus et al. 2013). Contrastingly, alteration in pyramidal cell channels in NaV1.2 or NaV1.6 leads to depolarization of shifts in the voltage dependence of inactivation and augmentation in persistent current (Ye et al. 2018). Haploinsufficiency in SCNIA triggers Dravet Syndrome (Bechi et al. 2012) and functional dropping in SCN1B (Patino et al. 2009). The function in SCN2A is associated with BFNIS (Misra et al. 2008). The functional gaining in SCN8A is related to epileptic encephalopathy and functional loss is related with its nonoccurrence (Martin et al. 2007). In patients of epilepsy, additional potential pathological mechanisms have been suggested, but not yet specifically identified. Mutations in the voltage sensor in votage-gated sodium channel result in exchange of the amino, acid, i.e., arginines (charged) with residues (neutral). As a result, voltage sensor leads to development of a leak current (omega/gating pore current) (Sokolov et al. 2005). In spite of the fact that it is not seen in epilepsy patients till now, in hypokalemic periodic paralysis, Nav1.4 mutation is associated with an increase in omega current, proposing involvement in channelopathies (Sokolov et al. 2007). Resurgence of sodium current is proposed to be involved in epileptical conditions. In some subtypes of voltage-gated sodium channels, by membrane repolarization followed by prolonged depolarization pulse, a minor transient ingoing current can be attained (Raman and Bean 1997). Augmented resurgent current amplitude has been related to augmentation in action potential firing frequency, proposing it as a possible pathological mechanism in epileptical conditions (Jarecki et al. 2010).
Some important advancement has been made in psychiatric disorders treatment and diagnosis. From data it has been supported that these disorders occur due to interplay of environmental and genetic risk factors. These disorders are caused by structural and functional damage in several brain areas such as the thalamus, amygdala, midbrain, and the prefrontal cortex. Since the early 1990s, gene mapping of ion channels have been done on human chromosomes, and identification of mutations in these genes has been performed. Many neurological disorders like epilepsy, migraine, and episodic ataxia are caused by dysfunction of brain electrical circuits, which has been attributed to ion channel mutations. Hence, it has been suggested that any change in activity of ion channel in the respective brain regions has been associated with psychiatric disorders etiopathology.
18.3.4 Sodium Channels Involved in Schizophrenia
Schizophrenia is a psychotic disorder with 1% prevalence worldwide. Symptoms involve negative symptoms (decrease in social interaction, anhedonia), positive symptoms (hallucination and delusion), and cognitive impairment. Many evidences have increasingly proposed that schizophrenia is a brain development and plasticity disorder, which involves strong alteration in activity and excitability of substantia nigra, hippocampus, ventral tegmental area, and prefrontal cortex. Recently, genetic linkage and association studies suggested risk genes for schizophrenia, including some ion channels genes (Imbrici et al. 2013).
By targeting the sequence of 10,198 samples, Rees et al. 2019 confirmed irregularities of activity of neuron and associated voltage-gated sodium channels in pathogenesis of schizophrenia. This is a sequence-based study that includes the schizophrenia of rare coding variants in neuronally expressed genes, including N-methyl-D-aspartate receptor (NMDAR) complexes and activity-regulated cytoskeleton-associated protein (ARC); but, bigger sample number is essential to disclose novel genes and particular biological mechanisms. In this study, they have sequenced 187 genes in a new dataset of 5207 cases and 4991 controls, which are selected on the basis of previous information related to schizophrenia. These genes were involved as members of ARC and NMDAR postsynaptic protein complexes, including voltage-gated sodium and calcium channels. A data for a total of 11,319 cases, 15,854 controls, and 1136 trios has been published sequentially and on the basis of this data, a rare variant meta-analysis was conducted. From this data, it was found that there is no single gene significantly involved in schizophrenia but exonic variants in the ARC (p = 4.0 × 10−4) and NMDAR (p = 1.7 × 10−5) synaptic complexes are a significantly involved risk factor for schizophrenia. Other than this, in this study it was also found that loss-of-function variants and missense variants at paralog-conserved sites were enhanced in voltage-gated sodium channels, specially the alpha subunits (p = 8.6 × 10−4) involved in schizophrenia. From this study it was evidenced that multiple voltage-gated sodium channels are involved in schizophrenia pathogenesis and verify the involvement of ARC and NMDAR postsynaptic complexes (Rees et al. 2019).
18.3.5 Sodium Channels Involved in Familial Hemiplegic Migraine (FHM)
Familial hemiplegic migraine (FHM) can be autosomal-dominant in nature although its occurrence is rare (Watanabe et al. 1971). Manifestation of illness includes visual and speech hampering, aura and short duration of motor lethargy to some extent. Mutations of genes like CACNA1A, ATP1A2, and SCN1A through encoding of various ion transporting proteins are involved in occurrence of FHM. When compared with migraine arising due to periodic seizures of diseases associated with nervous system occurrence of FHM is atypical in nature caused due to alteration in a typical gene. Mutation of gene present in channel generated by calcium ion was the prime gene to be recognized and termed as CACNA1 α1-subunit and was primarily responsible for FHM1, which is most affected by the mutation where encoding gene of catalytic α2 subunit, present in Na+/K+-ATPase and termed as ATP1A2 gets mutated and around one-fifth of FHM2 families gets affected. Alteration of SCIN1A gene (Q1489K and L1649Q) has been found to cause FHM3. Formation of pores in α subunit of neuronal voltage-dependent gated sodium ion channel occurs through this gene (Dichgans et al. 2005; Carreño et al. 2013). As per current reports, atypical occurence of gene alteration leading to FHM3 was observed in a Chinese household on a 62-year-old woman (Shao et al. 2018). Earlier manifestation was transient ischemic attack which were ultimately identified as FHM having c.4495T>C alteration observed in SCN1A gene. The case study report ultimately deduced about role of SCN1A gene in FHM pathophysiology. Another study also reported occurrence of FHM3 through alteration in voltage-dependent sodium channel gates Nav 1.1 which is being encoded by SCNIA gene. This study was conducted to determine molecular flaws that are inheritable in nature and occur due to alteration related to FHM3 (L263V, Q1489K, and L1649Q). L1649Q is one of the mutations that failed to produce measurable current because it significantly reduced cell surface expression. Production of pronounced currents by two mutations occurs through co-expression of human beta1 and beta2 accessory subunits by tsA201 cells. Mutations when compared with WT-Nav1.1 showed principal depletion of functional phenotype that was additionally expressed by depletion of channel accessibility during consecutive stimulation. Data suggested that Q1489K mutation causes high current in continuation and decelerates the recovery from fast and slow inactivation as well as increased entry into slow inactivation. L263V, on the other hand, showed augmentation in functional characteristics which included detained entrance to slow inactivation, retarded entry but accelerated recovery in case of fast inactivation as well as high current in continuation. The two alternations (Q1489K and L1649Q) associated with typical FHM generated either full or partial loss in producing the outcome. On the other hand, L236V, which is an alteration, leads to attainment of function and was associated with FHM as well as increased occurrence of generalized epilepsy (Kahlig et al. 2008).
18.3.6 Sodium Channels Involved in Neuropathic Pain
From the generation of action potential to its dissemination, voltage-dependent sodium ion channels perform a pivotal role. These channels are further identified as the genes that codes them from Nav1.1 to Nav1.9 with further subclassification based on function as genes that are sensitive to tetrodotoxin- (TTX-S) or genes that are resistant to tetrodotoxin (TTX-R). These are present on primary afferent sensory neurons and recent pharmacological and genetic data has provided evidence of the involvement of Nav1.3, 1.7, 1.8, and 1.9 in nociceptive transmission. Out of these, Nav1.8 is a TTX-R sodium channel that is highly localized on primary sensory afferent neurons.
18.3.6.1 Nav 1.3 Channel
Nav1.3 can be termed as sodium channel sensitive to tetrodotoxin and is found in nerve cells of embryo but is also present in axotomized sensory nerve cells (Waxman et al. 2017). Nav1.3 channel produces fast stimulating and nonstimulating kinetics and quick retrieval from nonstimulating phase. Its presence has also been confirmed in distal tips of axon in experimental neuromas in rats and in human neuromas. Hyperresponsiveness and unpremeditated firing of neurons was observed with an increase in pain and altered electrophysiologal properties as manifestation when Na 1.3 was upregulated, whereas down-regulation of channel showed pain in trigeminal nerves in trigeminal ganglionic regions in ferret (Nassar et al. 2006). Protein and mRNA level of Nav1.3 was found to be diminished following administration of antisense oligodeoxynucleotides through intrathecal route resulting in reduction of hyperexcitability of DRG neurons and reduced pain in nerves after injury of spinal cord and sciatic nerve (Lindia et al. 2005).
18.3.6.2 Nav1.7 Channel
Nav1.7 channel is sodium channel sensitive to tetrodotoxin and present largely in sensory neurons and sympathetic ganglia providing fast stimulating and nonstimulating kinetics but in turn causes considerably slower recovery period from fast inactivation making it different from other tetrodotoxin sensitive channels. Down-regulation of mRNA and protein in Nav1.7 channels was done through tying of spinal nerve of neuropathic pain. Mutations that cause gain of function of SCN9A coding for sodium channel Nav1.7 are present in patients suffering from critical pain syndrome inherited erythromelalgia (IE) (Cregg et al. 2013) and paroxysmal extreme pain disorder (PEPD) (Dabby et al. 2011), whereas mutations that cause loss of function of SCN9A gene have been found in patients suffering from inherited insensitivity to pain and disabled sense of smell. Inherited erythromelalgia is specified by burning pain and hot skin flashes, which causes change in hyperpolarization of voltage, which in turn effects the activation. Paroxysmal extreme pain disorder is manifested by extreme pain and burning sensation in rectal, ocular and submandibular region, which causes change in depolarizing voltage, which in turn effects steady-state inactivation.
18.3.6.3 Nav1.8 Channel
Nav1.8 channel is present mainly in sensory neurons of small diameter and trigeminal ganglionic neuron. This channel is tetrodotoxin resistant in nature and produces lower time period of depolarized stimulation and steady-state voltage-dependent nonstimulating kinetics but has high rate of retrieval from nonstimulation state. Depolarized stimulation of Nav1.8 sodium channel leads to upstroke in action potential by 80–90% of inward channels of sensory neurons from Nav1.8 null mice (Harty and Waxman 2007). Ongoing nociceptive unit can regulate both biophysical properties of Nav1.8 as well as its expression. Patients suffering from chronic pain due to dysfunction of nervous system or with increased sensitivity to chronic pain in local areas had increased content of Nav1.8 channels in regions having proximity to peripheral injury site. Manifestation of inflammatory mediators leads to altered level of tetrodotoxin channel with stimulating threshold shifted toward more negative potential through stimulation of protein kinase A. Increase in magnitude of Nav1.8 mediated channel is caused by prostaglandin E2, adenosine, and serotonin, which results in shift of relation between conductance of voltage toward negative spectrum and increasing time period of stimulating and nonstimulating of sodium channels in sensory neurons having small diameter (Black et al. 2004; Wood et al. 2004).
18.3.6.4 Nav1.9 Channel
Nav1.9 channel is sodium channel resistant to tetrodotoxin and is present mainly in sensory nerve cells having smaller diameter. Nav1.9 channel provides voltage-dependent activation close to the potential of membrane present in the resting state (-70 mV). Steady-state nonstimulation occurs at comparatively positive potential (-45 mV) (Shah et al. 2010). These kinetic properties suggest that activation of Nav1.9 may prolong response to subthreshold depolarization and generate a persistence channel. Experiments on electrophysiology on sensory neurons of Nav1.9 null mice results in lowering of threshold for action potential electrogenesis and produces persistent channels leading to hyperexcitability. Inflammation leads to increased expression of Nav1.9 gene along with Nav1.9 mRNA, which ultimately results in substantial down-regulation of proteins in axotomized afferent neurons and several in vivo models that are suffering from pain in the somatosensory nervous system (Lolignier et al. 2011).
18.4 Conclusion
Normal functional and number of sodium channels are crucial for the functioning of any excitable cells. This, when mutations or disorder arise, which disturbs this balance, leads to various clinical pathologies. Channelopathies are disarrays arising due to alternations in sodium channels. These disorders include manifestations like epilepsy, Dravet Syndrome, complications associated with pain, like loss of sensitivity to pain from birth, primary erythromelalgia and manifestation of paroxysmal extreme pain disorder, irregular heartbeat (signified by extreme QT syndrome), slow ventricular conduction, and atrial standstill. Aggressive nature of prostate (Nav1.7) and breast cancer (Nav1.5) has been associated with alteration in epigenetic regulation. Sodium channel upregulation has also been involved in diabetes and chemical induced neuropathies, presence of neuromuscular anomalies like Eaton Lambert Syndrome, Charcot Marie tooth disorder along with several disorders that are neurodegenerative in nature like Alzheimer’s, Schizophrenia, and most importantly Parkinson’s. Sodium channels are an important target due to their main role in these diseases. Dravet Syndrome (DS) also leads to haploinsufficiency of SCN1A along with functional loss of SCN1B. On the other hand, functional gain of SCN2A is associated with BFNIS. Likewise, functional gain of SCN8A is associated with epileptic encephalopathy (EE). Substitution of arginine I by uncharged amino acid residues will lead to alteration in VGSCs voltage sensor and hence lead to outflow in dispensing of current or alteration in sodium channel function resulting in resurgence of inward current, leading to increase in neuronal firing frequency. All these changes are some responsible factors for epilepsy. Sodium channels are also associated with development of psychotic diseases such as Schizophrenia, alteration in activity of hippocampus, substantia-nigra, ventral tegmental area, and prefrontal cortex, which in turn are manifestations associated with the development of Schizophrenia. Dysregulation in activity of neuron and voltage-gated sodium channels (VGSCs) was found to be involved in the development of Schizophrenia through targeted sequencing of 10,198 samples. Development of familial hemiplegic migraine (FHM) was also found to be related to sodium channels. FHM, which manifests by transient motor weakness to some extent and difficulty in having conversation along with optical disturbances is an autosomal dominant type disease although its occurrence is rare. Alterations in ATP1A2 that encodes for Na+/K + -ATPase dependent alpha2 subunit, which is catalytic in nature, and SCN1A (encodes for pore region of Nav1.1) are some of the etiological factors involved in the pathogenesis of FHM. Dysregulation of sodium channels (predominantly Nav1.5, Nav1.7, Nav1.8, and Nav1.9) leads to hypersensitivity of nervous system, which leads to hyperalgesia and allodynia observed during neuropathic pain. In another study, alternations that lead to functional gain of SCN9A gene, which in turn encrypts sodium channel Nav1.7, was found to be expressed in patients suffering from diseases associated with pain manifestations such as inherited erythromelalgia (IE) and paroxysmal extreme pain disorder (PEPD). On the other hand, functional loss of SCN9A is observed in patients with inborn insensitivity to pain and impaired sense of smell. Sodium channels targeting is an emerging concept that provides help in mitigation of various clinical disorders. However, there has been more than 70 years of research in understanding molecular dynamics and the functioning of sodium channels. The drugs that effectively target specific Nav channels and that are able to modulate its expression are yet to be discovered. Kinesins and Caveolae/Caveolin interaction plays an essential role in maintaining required number of functional VGSCs. Drugs that are able to target these markers can be developed or repurposed as an alternative safe therapy for treatment of channelopathies.
Abbreviations
- AIS:
-
Axon initial segment
- ARC:
-
Activity-regulated cytoskeleton-associated protein
- BFNIS:
-
Benign familial neonatal-infantile seizures
- ChIPs:
-
Channel Interacting Proteins
- CNS:
-
Central nervous system
- DRG:
-
Dorsal root ganglia
- EE:
-
Epileptic encephalopathy
- FGF:
-
Fibroblast growth factor
- FHF:
-
Fibroblast homologous factor
- FHM:
-
Familiar hemiplegic migraine
- IE:
-
Erythromelalgia
- IFMT:
-
Isoleucine, Phenylalanine, Methionine, and Tryptophan
- IHC:
-
Immunohistochemistry
- PEPD:
-
Paroxymal Extreme Pain Disorder
- PNS:
-
Peripheral Nervous System
- Nav:
-
Voltage-gated sodium channel
- NMDAR:
-
N-methyl-D-aspartate receptor
- TTX:
-
Tetrodotoxin
- VGSCs:
-
Voltage-Gated Sodium Channels
References
Ambrose C et al (1992) The α-subunit of the skeletal muscle sodium channel is encoded proximal to Tk-1 on mouse chromosome 11. Mamm Genome 3:151–155. https://doi.org/10.1007/BF00352459
Bagal SK et al (2015) Voltage gated sodium channels as drug discovery targets. Channels 9:360–366. https://doi.org/10.1080/19336950.2015.1079674
Bechi G et al (2012) Pure haploinsufficiency for Dravet syndrome Na V1.1 (SCN1A) sodium channel truncating mutations. Epilepsia 53:87–100. https://doi.org/10.1111/j.1528-1167.2011.03346.x
Beckers MC et al (1996) A new sodium channel α-subunit gene (Scn9a) from Schwann cells maps to the Scn1a, Scn2a, Scn3a cluster of mouse chromosome 2. Genomics 36:202–205. https://doi.org/10.1006/geno.1996.0447
Black JA et al (2004) Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108:237–247. https://doi.org/10.1016/j.pain.2003.12.035
Burgess DL et al (1995) Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease”. Nat Genet 10:461–465. https://doi.org/10.1038/ng0895-461
Carreño O et al (2013) Screening of CACNA1A and ATP1A2 genes in hemiplegic migraine: clinical, genetic, and functional studies. Mol Genet Genomic Med 1:206–222. https://doi.org/10.1002/mgg3.24
Catterall WA (1992) Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev 72:S15–S48. https://doi.org/10.1152/physrev.1992.72.suppl_4.S15
Catterall WA et al (2005) Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. https://doi.org/10.1124/pr.57.4.5.units
Cole KS, Baker RF (1941) Longitudinal impedance of the squid giant axon. J Gen Physiol. https://doi.org/10.1085/jgp.24.6.771
Cole KS, Curtis HJ (1939) Electric impedance of the squid giant axon during activity. J Gen Physiol. https://doi.org/10.1085/jgp.22.5.649
Cregg R et al (2013) Novel mutations mapping to the fourth sodium channel domain of Nav1. 7 result in variable clinical manifestations of primary erythromelalgia. Neuromol Med 15(2):265–278. Springer
Dabby R et al (2011) Chronic non-paroxysmal neuropathic pain—novel phenotype of mutation in the sodium channel SCN9A gene. J Neurol Sci 301(1–2):90–92. Elsevier
Dichgans M et al (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366:371–377. https://doi.org/10.1016/S0140-6736(05)66786-4
Frank HY, Catterall WA (2003) Overview of the voltage-gated sodium channel family. Genome Biol 4(3):207. BioMed Central
Hargus NJ et al (2013) Evidence for a role of Na v 1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J Neurophysiol 110:1144–1157. https://doi.org/10.1152/jn.00383.2013
Harty TP, Waxman SG (2007) Inactivation properties of sodium channel Nav1.8 maintain action potential amplitude in small DRG neurons in the context of depolarization. Mol Pain. https://doi.org/10.1186/1744-8069-3-12
Imbrici P, Camerino DC, Tricarico D (2013) Major channels involved in neuropsychiatric disorders and therapeutic perspectives. Front Genet 4. https://doi.org/10.3389/fgene.2013.00076
Jarecki BW et al (2010) Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Investig 120:369–378. https://doi.org/10.1172/JCI40801
Kahlig KM et al (2008) Divergent sodium channel defects in familial hemiplegic migraine. Proc Natl Acad Sci 105:9799–9804. https://doi.org/10.1073/pnas.0711717105
Kaplan DI, Isom LL, Petrou S (2016) Role of sodium channels in epilepsy. Cold Spring Harb Perspect Med 6. https://doi.org/10.1101/cshperspect.a022814
King GF, Vetter I (2014) No gain, no pain: NaV1. 7 as an analgesic target. ACS Chem Neurosci 5(9):749–751. ACS Publications
Kozak CA, Sangameswaran L (1996) Genetic mapping of the peripheral sodium channel genes, Scn9a and Scn10a, in the mouse. Mamm Genome 7:787–788. https://doi.org/10.1007/s003359900235
Lindia JA et al (2005) Relationship between sodium channel NaV1.3 expression and neuropathic pain behavior in rats. Pain 117:145–153. https://doi.org/10.1016/j.pain.2005.05.027
Lolignier S et al (2011) Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS One 6:e23083. https://doi.org/10.1371/journal.pone.0023083
Mantegazza M, Catterall WA (2012) Voltage-gated Na+ channels: structure, function, and pathophysiology. In: Jasper’s basic mechanisms of the epilepsies. National Center for Biotechnology Information (US), Bethesda, MD
Marban E, Yamagishi T, Tomaselli GF (1998) Structure and function of voltage-gated sodium channels. J Physiol 508:647–657. https://doi.org/10.1111/j.1469-7793.1998.647bp.x
Martin MS et al (2007) The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet 16:2892–2899. https://doi.org/10.1093/hmg/ddm248
Misra SN, Kahlig KM, George AL (2008) Impaired NaV1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia 49:1535–1545. https://doi.org/10.1111/j.1528-1167.2008.01619.x
Nassar MA et al (2006) Nerve injury induces robust allodynia and ectopic discharges in Nav 1.3 null mutant mice. Mol Pain 2:1744-8069-2-33. https://doi.org/10.1186/1744-8069-2-33
Patino GA et al (2009) A functional null mutation of SCN1B in a patient with Dravet syndrome. J Neurosci 29:10764–10778. https://doi.org/10.1523/JNEUROSCI.2475-09.2009
Plummer NW et al (1998) Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene SCN8A. Genomics 54:287–296. https://doi.org/10.1006/geno.1998.5550
Raman IM, Bean BP (1997) Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci 17(5):4517–4526
Rees E et al (2019) Targeted sequencing of 10,198 samples confirms abnormalities in neuronal activity and implicates voltage-gated sodium channels in schizophrenia pathogenesis. Biol Psychiatry 85:554–562. https://doi.org/10.1016/j.biopsych.2018.08.022
Roger S et al (2015) Voltage-gated sodium channels and cancer: is excitability their primary role? Front Pharmacol 6. https://doi.org/10.3389/fphar.2015.00152
Savio-Galimberti E, Gollob MH, Darbar D (2012) Voltage-gated sodium channels: biophysics, pharmacology, and related channelopathies. Front Pharmacol 3. https://doi.org/10.3389/fphar.2012.00124
Shah KU et al (2010) Voltage gated sodium channel blockers: potential treatment for neuropathic pain. Curr Res Inform Pharm Sci 11:11–16
Shao N et al (2018) Familial hemiplegic migraine type 3 (FHM3) with an SCN1A Mutation in a Chinese family: a case report. Front Neurol 9. https://doi.org/10.3389/fneur.2018.00976
Sokolov S, Scheuer T, Catterall WA (2005) Ion permeation through a voltage- sensitive gating pore in brain sodium channels having voltage sensor mutations. Neuron 47:183–189. https://doi.org/10.1016/j.neuron.2005.06.012
Sokolov S, Scheuer T, Catterall WA (2007) Gating pore current in an inherited ion channelopathy. Nature 446:76–78. https://doi.org/10.1038/nature05598
Tata A-G (2013) Effects of cysteine mutations in the S4-S5 linkers from domains D1 to D4 on the fast inactivation of the voltage-gated sodium channel Nav1. 4. Universität Ulm. Medizinische Fakultät, Ulm
Wang J et al (1992) Sequence and genomic structure of the human adult skeletal muscle sodium channel α subunit gene on 17q. Biochem Biophys Res Commun 182:794–801. https://doi.org/10.1016/0006-291X(92)91802-W
Watanabe K, Sato I, Kosaki T (1971) Familial hemiplegic migraine. No to Hattatsu. https://doi.org/10.11251/ojjscn1969.3.14
Waxman SG, Kocsis JD, Black JA (2017) Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol 72:466–470. https://doi.org/10.1152/jn.1994.72.1.466
Wood JN et al (2004) Voltage-gated sodium channels and pain pathways. J Neurobiol 61:55–71. https://doi.org/10.1002/neu.20094
Yamagishi T et al (2001) Molecular architecture of the voltage-dependent Na channel: functional evidence for alpha helices in the pore. J Gen Physiol 118:171–182. https://doi.org/10.1085/jgp.118.2.171
Ye M et al (2018) Differential roles of Na V 1.2 and Na V 1.6 in regulating neuronal excitability at febrile temperature and distinct contributions to febrile seizures. Sci Rep 8:753. https://doi.org/10.1038/s41598-017-17344-8
Acknowledgment
This work is supported by the Department of Pharmaceutical Engineering and Technology, Indian Institute of Technology (BHU). Varanasi.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Tiwari, V., Uniyal, A., Akhilesh, Gadepalli, A., Tiwari, V., Agrawal, S. (2020). Sodium Channels: As an Eye of the Storm in Various Clinical Pathologies. In: Kumar, P., Deb, P.K. (eds) Frontiers in Pharmacology of Neurotransmitters. Springer, Singapore. https://doi.org/10.1007/978-981-15-3556-7_18
Download citation
DOI: https://doi.org/10.1007/978-981-15-3556-7_18
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-3555-0
Online ISBN: 978-981-15-3556-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)