
Abstract
Originally treated as part of a cellular waste, extracellular vesicles (EVs) are being shown to possess a vast variety of functions, of which exosome is the most studied one. Most cells, such as tumor cells, immunocytes, and fibroblasts can secrete exosomes, especially under certain stresses the amount is much higher, and the contents of exosome represent the status of the donor cells and the tumor microenvironment. As crucial transporters for cells’ content exchange, much attention has been raised in the utilities of exosomes to suppress immune response, and to modify a microenvironment favorable for cancer progression. Exosomal immune checkpoints, such as programmed cell death ligand 1 (PD-L1), contribute to immunosuppression and are associated with anti-PD-1 response. Many forms of soluble immune checkpoint receptors have also been shown to influence efficacy mediated by their therapeutic antibodies. Therefore, targeting pro-tumorous exosomes may achieve antitumor effect supplementary to existing therapies. Exosome, itself natural liposome-like structure, allows it to be a potential drug delivery tool.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
19.1 The Introduction of Exosome in Cancer Immunology
Mammalian cells synthesize and release heterogeneous EVs which can be generally subclassified as exosomes (30–120 nm in diameter), microvesicles (MVs, or ectosomes or microparticles, 0.1–1.0 μm), and apoptotic bodies (0.8–5.0 μm), each differing in their biogenesis, composition, and biological functions from others (Han et al. 2019) (Fig. 19.1). Exosomes are considered to be endosomally derived, while MVs bud from the surface of plasma membrane. EVs are circular pieces of membranes that incorporate various bioactive molecules including membrane receptors, proteins, mRNAs, microRNAs (miRNAs), and organelles, and thus potentially affect target cells by transferring their cargo or by receptor-to-ligand interaction. Endosomally-derived vesicles was first reported in transferrin recycling Rat reticulocyte endocytoses and then releases transferrin through multivesicular endosomes or multivesicular bodies (MVBs), which were described as often in 250–300 nm diameter but range from 120 to 800 nm diameter and may be irregularly shaped, into the surrounding microenvironment via fusion with plasma membrane of the parental cell. The MVBs intracellular passage does not enter into lysosome (Pan and Johnstone 1983; Harding et al. 1983).

The biogenesis of exosomes
The biogenesis of exosomes includes three distinct steps, begin with the development of endocytic vesicles by inward budding of the plasma membrane, then the generation of MVBs by invagination of the endosomal membrane, and at last, the fusion of plasma membrane with MVBs and subsequent release of the vesicular components, named exosomes (Batista et al. 2011) (Fig. 19.1). Exosomes and other EVs differ in size, density, morphology, marker expression, and rely on specific enzymes for their biogenesis. Vital enzymes in their generation include budding of intravesicular vesicles promoter—NSMASE2 (aka SMPD3), and fusion of the MVB to the plasma membrane catalyzer—RAB27A (Ostrowski et al. 2010; Kosaka et al. 2010). Genetic manipulation of these enzymes provides an opportunity to dissect the role of exosomes in vivo. Various exosome biomarkers have been identified, for example, TSG101, Ras-related protein Rab-11B (RAB11B), CD63, charged multivesicular body protein 2a (CHMP2A), and CD81 proteins, as well as lipids, including cholesterol, ceramide, sphingomyelin, and phosphatidylserine (Dickens et al. 2017; Colombo et al. 2014; Raposo and Stoorvogel 2013).
The isolation of pure exosomes is vital to uncover their action mechanisms and for further application in biomedical sciences. Since they are tiny and the isolation is challenging, a variety of techniques have been developed to facilitate the isolation. Successful isolation of exosomes can be achieved in a variety of ways like ultracentrifugation, ultrafiltration, chromatography, affinity capture on antibody-coupled magnetic beads, and polymer-based precipitation (Peterson et al. 2015). Practically, the size difference between cells, subpopulations of EVs, and proteins allow them to be separated and isolated by centrifugation. Ultrafiltration is rapid but hard to get rid of contaminating proteins, which can be improved by combining with ultracentrifugation. Membranes are used to filtrate cells and large EVs, and then ultracentrifugation can further separate exosomes from proteins (Alvarez et al. 2012). Aiming to purify exosome isolations, immuno-affinity purification (IP) techniques have been adopted that can selectively capture desired exosomes from a complex population basing on certain surface markers. IP techniques are rapid, convenient, and compatible with regular laboratory equipment, during which process anti-CD63, anti-CD9, and anti-CD81 antibodies are typically used (Tauro et al. 2012).
Exosomes released from tumor cells actively lead to tumor progression and metastasis (Chiodoni et al. 2019). It has been shown by studies that exosome released from cancer cells can be stimulated under different stresses, which also cause the alteration of exosome’s content. Oxidative and heat stresses have been shown to induce the exosome generation from leukemia/lymphoma T and B cell lines (Hedlund et al. 2011) and hypoxic environment has been reported to effectively augment tumor-derived exosomes (TEX) shedding from breast cancer cells (Wang et al. 2014). Moreover, sublethal doses of a number of chemotherapeutic drugs, such as proteasome inhibitors and genotoxic drugs, enhance exosome release in various cancer models. As such, multiple myeloma (MM) cells can enhance the release of nanovesicles receiving bortezomib or melphalan treatment (Lehmann et al. 2008; Yu et al. 2006).
Tumor-suppressor gene p53 has been described in stress-induced exosome secretion. In this regard, irradiation of prostate cancer cells trigged an amplified secretion of exosome-like vesicles by p53 activation (Lehmann et al. 2008). Furthermore, a p53-inducible gene product, the tumor-suppressor-activated pathway 6 (TSAP6), was found to regulate exosome trafficking and secretion in cells undergoing DNA damage (Yu et al. 2006; Amzallag et al. 2004). The fact that exosome secretion is reported to be severely restrained in TSAP6-deficient mice can further support these observations (Lespagnol et al. 2008). Moreover, increased exosome secretion in tumor cells can be observed in a senescent phenotype (Borrelli et al. 2018), which can pose pro-tumorigenic effects in pre-malignant recipient cells. Senescence-associated secretory phenotype (SASP) can be enhanced either by inactivation of p53 or gain of oncogenic RAS (Coppe et al. 2008).
EVs are implicated in multiply processes, such as tumor cells proliferation, chemoresistance, immune escape, metabolic reprogramming, metastatic enhancement, and angiogenesis. Herein, we will mostly focus on how exosomes affect cancer immunology.
19.2 The Role of TEX in Cancer Immunology (Detailed in Fig. 19.2)
By generating an immunosuppressive microenvironment, TEX is beneficial for tumor growth and distant dissemination. Studies have been showing the mechanisms and targets of immune suppression by TEX are diverse (Graner et al. 2018), while some reporting TEX is immune activator. Both success of immune-based therapies and active endogenous antitumor immune responses rely on effector cells that directly target and kill cancer cells, such as cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells. We will herein discuss how TEX contributes to those cancer-related immunocytes’ function.

The roles of tumor-derived exosomes in cancer immunology
19.2.1 TEX Suppresses T Cell Functions
Tumor cells actively release immunosuppressive MVs into the microenvironment, disrupting T cell immunosurveillance. Tumor-derived MVs harvested in suspension of head and neck squamous cell carcinoma (SCCHN) cell line, PCI-13, with combination of size-exclusion chromatography and ultracentrifugation methods, were variably enriched in major histocompatibility complex (MHC) class I. And those MVs suppressed signaling and expansion, and induced apoptosis of activated CTLs, while enhancing the proliferation and suppressor activity of CD4+ CD25+ FOXP3+ Treg cells (Wieckowski et al. 2009), leading to immune suppression to allow tumor escape. Melanoma cell line-derived exosomes are able to alter the metabolic function of CTL cell line, CTLL-2, by transferring mRNA/miRNA contents. Notably, the delivery of mRNA/miRNA loaded exosome happened within a short time frame. Four of the top 20 mRNAs found within B16F0 exosomes (such as Cmtm4, Wsb2, Ptpn14, and Fam168b) were detected in CTLL-2 cells in 30 min. Another cluster of genes was upregulated at 4 h upon exosome treatment, which contained several gene targets involved with cellular metabolism. CMTM4 mRNA is upregulated in TEX exposed CTLL-2 cells, and this is potentially significant given its role in PD-L1 trafficking (Mezzadra et al. 2017).
Exosomes isolated from the ascites of ovarian cancer (OVCA) patients are identified, which can be internalized rapidly by T cells, and inhibited various T cell response endpoints like translocation of NFAT and NFκB into the nucleus, upregulation of CD69 and CD107a, release of cytokines, and elevated cell proliferation. However, T cell viability was not affected and T cell arrest was transient. After removal of the immunosuppressive exosomes, T cell regained their activation potential within 24 h (Shenoy et al. 2018).
However, TEX can activate CTL clones after processing by antigen-presenting cell (APC) expressing the correct MHC haplotype (Andre et al. 2002). TEX comprises cancer-related antigens that may initiate an immune response using dendritic cell (DC) as intermediaries. Melanoma-associated antigen (MAGE) was enriched in TEX from malignant ascites of patients with melanoma, and it can be recognized by T cells (MART-1). These TEX, once delivered to DCs, facilitated in vitro cross-presentation of the antigen, and response of a CTL clone, leading to an efficient in vitro antitumor cellular activation, as monitored by the quantity of interferon (IFN)-γ produced, and by the induction of specific cancer cell lysis (Andre et al. 2002; Wolfers et al. 2001).
19.2.2 Mechanisms of TEX Influencing NK Cells and DCs
TEX can regulate the functions of NK cells, by affecting the differentiation of precursors to mature antigen-presenting cells. NKG2D (activating natural killer group 2 member D) ligand on tumor cells triggers cytotoxic activity in NK cells through interacting with NKG2D (Bauer et al. 1999; Rincon-Orozco et al. 2005). TEX NKG2D is loaded with many other functional molecules including death receptor ligands, MHC class I/II, adhesion molecules, etc. (Whiteside 2013). Therefore, the alteration of the immune response by NKG2DL containing TEX is also impacted by other co-existing molecules within the exosome (Clayton et al. 2008). NKG2D interacts with several kinds of ligands. For instance, the NKG2DL in humans includes MICA\B and UL16-binding proteins 1–6 (ULBP1–6), exosomal MICA*008 decreased NKG2D expression on NK cell and suppressed its cytotoxicity (Ashiru et al. 2010). Exosomal ULBP3 was also reported to bring down NKG2D on primary NK cells and restrain NK cell‐mediated elimination of MICA‐expressing target cells (Fernandez-Messina et al. 2010). Nevertheless, human DCs can secrete exosomal NKG2DL to directly enhance NK cells’ function ex vivo (Viaud et al. 2009).
The other extracellular presence of NKG2DL is soluble form, which can downregulate NKG2D on NK cells or T cells, leading to suppressed cytotoxicity. Soluble MICA (sMICA) downregulated NKG2D by enhancing its endocytosis and then degradation, leading to a reduced NKG2D expression on tumor‐infiltrating T cells (Groh et al. 2002). Similarly, sULBP was found to downregulate NKG2D on NK cells (Fernandez-Messina et al. 2010).
Moreover, heat shock protein 70 (Hsp70) on tumor cell surfaces is a recognized ligand for the NK cell receptor CD94 and can enhance the cytotoxicity of NK cells against Hsp70-positive tumor target cells (Gross et al. 2003). By contrast, TEX surface Hsp70 will cause reduction of NK CD94, and in this case TEX functions as systemic decoy for NK cells (Hedlund et al. 2011). In addition, transforming growth factor β (TGF-β), acting as a component of TEX (Graner et al. 2009), has also been shown to be an immune suppressor of NK cells (Szczepanski et al. 2011). Fetal liver mesenchymal stem cell (mSC)-derived exosomes contain several immunomodulatory molecules—latency-associated peptide (LAP), TGFβ, and thrombospondin 1 (TSP1), through stimulating the downstream TGF-β/Smad2/3 signaling, inhibiting proliferation, activation, and cytotoxicity of NK cells (Fan et al. 2019).
In normal human cells, inhibition of exosome secretion reduces removal of harmful nuclear DNA, causes the stacking of nuclear DNA, and thus activates cGAS-STING pathway and causes type I IFN production from those cells (Takahashi et al. 2017), indicating exosome secretion is vital for maintaining parental cell’s homeostasis. Exosomal double-stranded (Ds) DNA secreted from irradiated mouse breast cancer cells could be transferred to DCs and upregulate DCs’ surface costimulatory molecules, leading to the STING-dependent activation of type I IFNs (Diamond et al. 2018). The same phenomenon has been observed under the antitumor agent topotecan, an inhibitor of topoisomerase I treatment (Kitai et al. 2017). Collectively, these findings indicate that dsDNA associated with TEXs induces type I IFN production directly from cancer cells or indirectly through the DCs stimulation. Type I IFNs are known to play a crucial role in cancer progression through the promotion of anti-cancer immune responses, (Medrano et al. 2017; Zitvogel et al. 2015) such as directly activate NK cell-mediated functions, increase perforin-dependent cytotoxicity (Nguyen et al. 2002), and induce TNF-related apoptosis-inducing ligand (TRAIL) expression. In addition, with the coordinated action of IL-12, type I IFNs greatly promote NK cell-mediated IFN-γ production.
19.2.3 Mechanisms of TEX Influencing Macrophages
Preclinical and clinical studies indicate that tumor-associated macrophages (TAMs) provide important pro-tumorigenic and survival factors (Noy and Pollard 2014). TEX stimulates the macrophage infiltration and polarization in remote site for establishment of premetastatic niche. After injection of breast TEXs into mice, the amount of macrophage is shown to be increased in axillary lymph nodes, with CD206 positive M2 macrophages much more detected than NOS2 positive M1 macrophages (Piao et al. 2018). Regarding macrophages, toll-like receptor (TLR) signaling is vital for pro-inflammatory cytokines, miRNAs, and other components secretion, greatly enhancing inflammation and favoring cancer development. Through TLR on macrophages, breast TEXs activate NF-кB and induce secretion of G-CSF, TNF-α, IL-6, and CCL2, while genetic depletion of TLR2 or MyD88, a vital signaling adaptor of the NF-кB pathway, completely abrogates this effect (Chow et al. 2014). TEX can also induce monocytes to release immune-modifying factors. TEX in chronic lymphocytic leukemia (CLL) can enhance monocytes to secrete cytokines IL-6, CCL2, and CCL4, and express PD-L1 (Haderk et al. 2017). HY4, a noncoding Y RNA enriched in exosomes of CLL patient plasma can achieve the same effects on monocytes as TEX, resulting in cancer-associated inflammation and potential immune evasion via PD-L1 upregulation.
Under hypoxic stress, TEX enhances oxidative phosphorylation in bone marrow-derived macrophages through transfer of let-7a miRNA and subsequent suppression of the insulin-Akt-mTOR signaling pathway, leading to M2 polarization (Park et al. 2019).
However, macrophage receiving TEX miRNAs also have antitumor effects, as in the case where murine breast cancer was treated with EGCG (epigallocatechin gallate), a component of green tea extract with known anti-cancer properties (Jang et al. 2013). EGCG enhances TEX-derived miR-16, which is an important regulator of CHUK/IKK α (inhibitor of nuclear factor kappa-B kinase subunit alpha) complex. Delivery of these TEX to macrophage is favorable for M1 phenotype cytokines secretion. MiR-16 causes reduction of CHUK/IKK α complex and subsequent accumulation of I-κB, thus preventing NF-кB activation and M2 phenotype cytokines production (Hagemann et al. 2009; Lawrence 2009).
19.2.4 Other Immune Cell Lineages
Other immune cells are also subject to the impact of EVs produced by tumor cells. B cells primed by hepatocellular carcinoma (HCC) cell line-derived exosome strongly expressed T cell immunoglobulin-1(TIM-1) protein and were endowed with suppressive activity against CD8+ T cells. A major portion of TIM-1 is expressed by B cells and serves as a marker for Breg cells (Jang et al. 2013). HCC-exosome has high level of HMGB1, which activates B cells and promotes TIM-1+Breg cell expansion through the TLR2/4 and mitogen-activated protein kinase (MAPK) signaling pathways (Yan et al. 2012, 2018). TIM-1+ Breg cells secrete the highest proportion of IL-10 among all types of B cells.
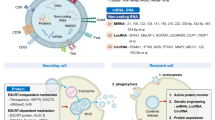
19.3 Exosome and Cancer Immune Checkpoints (Fig. 19.3)
19.3.1 Exosomal PD-L1
Tumor cells evade the immune surveillance through increasing surface expression of PD-L1, which interacts with PD-1 on T cells (Dong et al. 2002; Chen and Han 2015), contributing to dephosphorylation of the T cell receptor as well as its co-receptor CD28 through Shp2 phosphatase, thus inhibiting antigen-driven activation of T cells (Graner et al. 2018; Hui et al. 2017). Therapeutic antibodies of PD-L1 and PD-1 block this interaction, and therefore can reactivate the antitumor immune effect (Chen and Mellman 2017). Both anti-PD-1 and PD-L1 antibodies have shown remarkable promise in curing tumors, such as renal cell carcinoma and metastatic melanoma (Chen and Han 2015; Ribas et al. 2016; Topalian et al. 2016). Unfortunately, a major portion of patients have low response rate (Ribas et al. 2016; Zaretsky et al. 2016), requiring deep understanding of PD-L1-mediated immune evasion to predict patient response and promote treatment efficacy.

Cross talk among tumor cells, APCs and T cells
PD-L1 is found on the surface of EVs, and more interestingly EV PD-L1 levels have been related with cancer progression and response to immunotherapy (Yang et al. 2018; Theodoraki et al. 2018; Ricklefs et al. 2018; Chen et al. 2018). Chen et al. reported that metastatic melanomas released EVs, with exosomes being the major form, bearing PD-L1 on their surface. Treatment with IFN-γ can enhance PD-L1 level on these vesicles, leading to CTLs dysfunction and tumor growth acceleration.
The evolvement of systemic exosomal PD-L1 along the course of anti-PD-1 therapy has predictive value for cancer prognosis. The amplitudes of the increase in systemic exosomal PD-L1 during early stages of pembrolizumab treatment, indicating the adaptive response of the tumor cells to T cell re-invigoration, can stratify clinical responders from non-responders. However, circulating exosomal PD-L1 before and on treatment may reflect different states of antitumor immunity. High pretreatment level may signify the “exhaustion” of patient T cells to a turning point, by which they are unable to be re-invigorated by the anti-PD-1 treatment. For the on-treatment patients, on the contrary, a rise in the level of exosomal PD-L1, correlating proportionally with the T cell re-invigoration, would represent a strong antitumor immunity, thereby a favorable prognosis. Therefore, TEX PD-L1 may serve as a predictor for anti-PD-1 therapy efficacy (Chen et al. 2018).
PD-L1 from TEX imposes systemic immunosuppression through inhibiting T cell activation in the draining lymph node. Wild-type tumor cells grow slower with exposure to exosomal PD-L1-deficient tumor cells, which are injected simultaneously at a distant site. Inhibition of exosomal PD-L1 induces systemic antitumor immunity, even in models resistant to anti-PD-L1 antibodies (Poggio et al. 2019). Systemically introduced exosomal PD-L1 rescues growth of tumors unable to secrete their own. Anti-PD-L1 antibodies work additively, not redundantly, with exosomal PD-L1 blockade to suppress tumor growth.
Not only PD-L1 proteins, but also PD-L1 mRNA can be detected in exosomes. Researchers have found that PD-L1 mRNA contained exosome was more enriched in periodontitis patients than control subjects. Exosomal PD-L1 mRNA level in saliva correlates with the severity/stage of periodontitis, and can potentially be used to distinguish periodontitis from the healthy (Yu et al. 2019).
Yang et al. revealed in cancer cell lines, exosomal PD-L1 significantly suppressed CD3/CD28-driven ERK phosphorylation and NF-κB stimulation of T cells in a dose-dependent manner, as well as PHA-induced interleukin-2 (IL-2) secretion, leading to T cell dysfunction. Exosomes are capable of transferring functional PD-L1 to other cells. Exosomal PD-L1 detected on the surface of target cells is able to bind to PD-1 and mount an immunosuppressive effect. Suppression of exosome secretion from 4T1 tumor cells by either pharmacological inhibitor GW4869 treatment or Rab27a knocks down remarkably restrained tumor growth, and hence the antitumor effect is relatively superior than anti-PD-L1 therapeutic treatment. Combination of inhibition of exosome secretion and anti-PD-L1 antibody treatment achieved much stronger tumor suppression.
Researchers also reported exosomal release of PD-L1 occurs at the expense of surface PD-L1 levels. ALIX, as endosomal sorting complexes required for transporting (ESCRT)-associated protein, is the regulator of both PD-L1 surface presentation and EGFR activity in basal-like breast cancer (BLBC) cells. Besides locating on cell surface, PD-L1 also exits in the limiting membrane as well as intraluminal vesicle (ILVs) of CD63-positive MVBs in HCC1954 cells after IFNγ treatment. ILVs are the intracellular precursors of extracellular vesicles. Failure of PD-L1 incorporation in ILV leads to defective exosomal packaging, and following MVB-PM (plasma membrane) fusion thereby enhances cell surface PD-L1. In ALIXKD cells, a higher ratio of PD-L1 was observed at the limiting membrane of MVBs, compared within the endosomal lumen, resulting in prolonged and enhanced stimulation-induced EGFR activity as well as defective PD-L1 exosomal release, and its promoted redistribution to the cell surface, which implies an enhanced immunosuppressive phenotype (Monypenny et al. 2018).
19.3.2 Soluble Immune Checkpoint Receptors
The immunomodulatory interactions of cytotoxic T lymphocyte antigen-4 (CTLA-4), 4-1BB and PD-1 on T cells with their corresponding ligands on APCs or tumor cells have been extensively studied. Antibodies such as anti-CTLA-4 (ipilimumab) and anti-PD-1 (nivolumab) that counter these ligand–receptor interactions have shown clinical improvements in patients with solid tumors or autoimmune diseases. Still, a large portion of patient is unresponsive to these therapies and the full immunomodulatory mechanisms of these ligand–receptor interactions have not been resolved. More researchers are trying to uncover alternatively spliced soluble isoforms of these receptors to amplify efficacy mediated by their therapeutic antibodies.
19.3.2.1 Soluble PD-L1 as a Biomarker in Patients Treated with Checkpoint Inhibitors
Soluble PD-L1 (sPD-L1) may serve as a putative predictive biomarker for disease outcome and patient stratification under some circumstances. For instance, sPD-L1 may be a marker of systemic inflammation in pancreatic cancer (Kruger et al. 2017). sPD-L1 level in sera correlates with aggressiveness of renal cell carcinoma (RCC) and predicts survival in patients with MM or diffuse large B cell lymphoma (Wang et al. 2015; Rossille et al. 2014; Frigola et al. 2011).
SPD-L1 detected in the sera of RCC patients may cause systemic immunosuppression, facilitating tumor progression and resulting in poor prognosis (Frigola et al. 2011). Higher preoperative sPD-L1 levels were associated with poor clinical characteristics, including larger tumor volume, later stage, higher grade, and more tumor necrosis. Twice of sPD-L1 levels was related with a 41% increased risk of death. SPD-L1 was also detected in the cell supernatants of some PD-L1-positive RCC cell lines. Those sPD-L1s retained receptor-binding domain was indicated by protein sequencing and was able to trigger pro-apoptotic signals in T cells (Frigola et al. 2011).
Multiple splice variants of PD-L1 have been identified and elevated sPD-L1 was observed in sera of patients with metastatic melanoma compared with healthy. High pretreatment sPD-L1 levels are correlated with rapid deterioration under anti-CTLA-4 or anti-PD1-based therapy, perhaps due to enhancing aberrant splicing activities in tumor cells, large tumor burden, or a diminishing antitumor immune effect, which are not easy to treat with a checkpoint blockade. Rise in sPD-L1 after 5 months of treatment rather than early alterations of sPD-L1 levels correlated with partial responses when receiving anti-CTLA-4 or anti-PD-1 therapy (Zhou et al. 2017).
Soluble PD-L1 harbors potential utility for antitumor therapy by blocking PD-1/B7-H1 pathway in murine model. In preclinical models of hepatocarcinoma (HCC), intramuscular injection of a plasmid encoding sPD-1 was reported to enhance lysis of tumor cells and extend overall survival of tumor-bearing mice (He et al. 2005).
19.3.2.2 Soluble CTLA4
The transmembrane isoform of CTLA-4 (Tm-CTLA-4) receptor plays a critical role in downregulating the immune response and sustaining the immune homeostasis. Alternatively spliced mRNA of the CTLA-4 gene that lacks exon 3 is found in human, mouse, and rat immune cells (Magistrelli et al. 1999; Oaks et al. 2000). When sCTLA-4 was first described, it was considered as a product of resting T cells, with its manufacture being cut down following T cell activation (Oaks et al. 2000). However, recently researchers have shown human T cells release more sCTLA-4 under physiological stimuli such as peptide immunogens or other recall antigens, indicating that sCTLA-4 may have functions relevant to ongoing immune responses (Ward et al. 2013). Antagonism of sCTLA-4 by isoform-specific monoclonal antibody (mAb) could remarkably stimulate antigen-dependent immune effects in a range of experimental systems in vitro and in vivo, as is shown in enhanced cell proliferation and pro-inflammatory cytokine levels like IFN-γ. Furthermore, in a melanoma-bearing B16F10 mouse model, isoform-selective anti-sCTLA-4 mAb treatment can achieve similar effect as panCTLA-4 mAb on the reduction of lung metastases, demonstrating soluble isoform is capable of modulating the overall outcome (Ward et al. 2013).
The extracellular domain of sCTLA-4, similar to that of the integral membrane isoform, has the MYPPY motif that can bind to the CD28-shared CD80/CD86 ligands on APCs. In a mixed lymphocyte response, recombinant sCTLA-4 showed immunomodulatory effect on inhibiting cell proliferation in a dose-dependent manner (Oaks et al. 2000).
19.3.2.3 Other Soluble Immune Checkpoints
Soluble 4-1BB can restrain over-zealous immune responses by acting in a negative feedback loop, as shown in animal models to suppress development of type I diabetes (Kachapati et al. 2013). Moreover, human renal, lung, melanoma, and hepatocellular tumor cell lines can generate s4-1BB under hypoxic stress, causing 4-1BB ligand engagement, thus blocking its costimulatory effect (Labiano et al. 2016).
Lymphocyte-activation gene 3 (LAG-3) is an immune suppressive receptor, with major MHC-II as a canonical ligand. Wang et al. demonstrated that fibrinogen-like protein 1 (FGL1), secreted by liver, acted as the major LAG-3 ligand independent of MHC-II. Inhibition of antigen-specific T cell activation was observed by FGL1-LAG-3 interaction, ablation of which enhances T cell response in mice. Tumor cells generated excessive FGL1, and increased plasma FGL1 was correlated with stronger resistance to anti-PD-1/PD-L1 therapy and a poor outcome in patients (Wang et al. 2019).
19.4 Immunocytes-Derived Exosomes in Cancer Immunology
Not only tumor cells, but also a variety of immune cells are able to release exosomes, such as T cells, DCs, macrophages, B cells, and mast cells (Skokos et al. 2003). Immunocytes-derived exosomes are shown to modulate tumor microenvironment and affect cancer outcome.
19.4.1 T Cell-Derived Exosomes
T cell-derived exosomes contain heterogeneous components, targeting divergent cells, and perform distinct types of function. Using proteomic approach, researchers were able to show activated T cells can secrete exosomes that contain signaling components associated with RAS, such as ZAP70, RAP1, RASGRP1, and AKT, and these vesicles can lead to ERK phosphorylation in mast cells (Azoulay-Alfaguter and Mor 2018). Activated T cells can release EVs which promote proliferation of autologous resting CD8 T cells (Wahlgren et al. 2012). T cells were activated and released exosomes after interaction with antigen-bearing DCs. In return, T cells could enhance the protective roles of DCs via transfer of exosomal DNA, and may modulate the immune system when encountering threats (Torralba et al. 2018). On the other hand, T cell-derived exosomes can prevent autoimmune damage through bioactive FasL and TRAIL, which eliminated activated T cells (Monleon et al. 2001).
Fibroblastic tumor stroma consisting of mesenchymal stem cells (MSCs) and cancer-associated fibroblasts (CAFs) promotes the invasion and metastasis of cancer cells. EVs derived from activated CD8+ T cell could disrupt fibroblastic stroma-mediated tumor growth, as evidenced from activated CD8+ T cells in healthy mice transiently secreting cytotoxic EVs which causes significant inhibition of invasive and metastatic properties of tumor through apoptotic elimination of mesenchymal tumor stromal cells. EV-releasing CD8+ T cells infiltrate in neovascular areas with high mesenchymal cell density, and tumor MSCs preferentially engulf CD8+ T cell-derived EVs than other cell populations like tumor cells. Thereby, CD8+ T cells can prevent cancer progression via EV-mediated depletion of mesenchymal tumor stromal cells besides their conventional direct cytotoxicity (Seo et al. 2018).
Follicular helper T cells (Tfh cells) secreted exosomes correlate with occurrence and progression of antibody-mediated rejection (AMR) in renal transplantation. Tfh cell-derived exosomes could promote the proliferation and differentiation of B cells and may play a critical role in the development of AMR after renal transplantation. Analysis of the peripheral blood from 42 kidney transplant patients indicated that CTLA-4 level of CD4+ CXCR5+ exosomes was significantly lower in AMR group than that in non-AMR group (Yang et al. 2019).
19.4.2 NK Cell-Derived Exosomes
NK cells in tumor microenvironment could attenuate cancer progression through their exosomes. NK cell-derived exosomes performed cell-killing activity targeting cancer cells through the cytotoxicity factors, including perforin, granulysin, and granzymes A and B, which were able to activate caspase pathways in tumor cells, as well as blocking caspase inhibitors (Jong et al. 2017). NK cell-derived exosomes contain tumor-suppressive miR-186, which downregulates certain oncogenic proteins, for example, the mitotic kinase aurora kinase A (AUKRA) and N-myc proto-oncogene protein. TGF-β1 could inhibit the levels of miR-186 in NK cells, rendering NK cells inactive. Restoration of miR-186 levels increases the cell-killing capabilities of NK cells, resulting in decreased tumor burden and prolonged survival in neuroblastoma (Schmittgen 2019).
19.4.3 DC-Derived Exosomes
DC is the key player in antigen-presenting process, its EVs are in charge of intercellular communicators in adaptive immunity. DCs and DC-derived exosomes have several similarities. Like DCs, their exosomes express functional MHC-peptide complexes, T cell stimulatory factors, and other components that interact with other immune cells. After activated by T cells, DCs exhibited a capacity for antigen-specific T cell activation through exosomes (Lindenbergh et al. 2019). Accumulated evidence has shown that DC-derived exosomes could facilitate immune cell-dependent cancer therapy (Pitt et al. 2016). Given the component of DCs in modulating immune responses, a majority of studies focused on the effect of DC-derived exosomes against tumor progression, the potential immune-modifying function, and feasibility and safety of application (Chen et al. 2018a, b; Pitt et al. 2014). DC-derived exosomes can limit cancer cells via activation of naïve T cells and NK cells (Gao and Jiang 2018).
19.4.4 Macrophage-Derived Exosomes (MDE)
Clinical and experimental evidence has shown that tumor-associated macrophages induce cancer initiation and progression. MDE has been shown to accelerate colorectal cancer (CRC) cells’ migration and invasion through its miRNA contents including miR-21-5p and miR-155-5p. Both miR-21-5p and miR-155-5p downregulate expression of BRG1, which is crucial for the CRC metastasis (Lan et al. 2019). After activated by IL-4, macrophage can enhance invasiveness of breast cancer cells via the Mef2c-b-catenin pathway through transferring miR-233. MiR-223 antisense oligonucleotide reduced the expression of miR-223 in macrophages, thus depressing the invasiveness of the co-cultured breast cancer cells (Yang et al. 2011).
Hypoxic epithelial ovarian cancer (EOC) cells induced macrophages into a TAM-like phenotype, which then deliver exosomes to the co-cultivated EOC cells, enhancing the malignant phenotype and drug resistant of EOC cells via the PTEN-PI3K/AKT pathway (Zhu et al. 2019).
19.4.5 Other Immunocytes-Derived Exosomes
B cell-derived exosomes mediate part of B cell’s functions, including antigen-presenting capacities, MHC-restricted antigen recognition, and induction of different types of immune responses as well. In lymph nodes, B cell-derived exosomes were found to act reciprocally with CD169+ macrophages and further interrupted the spread of viruses or tumor cells (Saito et al. 2015). B cell-derived exosomes promoted T cell response, and those effects were independent of B cell presence or B cell-secreted antibody (Saunderson and McLellan 2017).
Myeloid-derived suppressor cells (MDSCs), known as an immune suppressor, are generated from immature myeloid cells under certain conditions. MDSC-derived exosomes contained different cargos in accordance with the immunosuppressive activity (Geis-Asteggiante et al. 2018). It was also demonstrated that MDSCs are present in cancer patients, inhibiting antitumor immunity and scrambling anti-cancer immunotherapies. MDSC-derived exosomes, which were primed by tumor milieu, could also promote oncogenesis (Burke et al. 2014).
19.5 Exosome as Biomarker and Vaccine for Cancer Progression
19.5.1 Liquid Biopsy
TEX delivers malignant signals in a variety of forms, including nucleic acids, such as messenger RNA (mRNA) and miRNAs, or proteins like chemokines, cytokines, growth factors, or angiogenic and immunomodulatory molecules (Chiodoni et al. 2019). Measuring exosomal contents would be noninvasive, however, a promising way to detect cancer occurrence and monitor tumor progression. The problem is exosomal-specific proteins are in very low abundance, and thus a large amount of serum or culture medium is needed to enrich sufficient exosome to conduct the proteomics or western blot measurement. Thereby, exosome protein profile is still in the starting stage. Genomic profile of the exosome, including miRNA, mRNA, long noncoding RNA (lncRNA), and mitochondrial RNA, being amplified by PCR to increase quantity seems to be excellent candidate biomarkers to subclassify tumor types.
19.5.2 Exosomal miRNA (Detailed in Table 19.1)
In recent years, much of the research on cancer blood biomarkers has shifted from protein-based to nucleic acid-based molecules (Chiodoni et al. 2019). For example, Fang et al. reported that hepatoma cells produced high levels of miR-103 and release it in exosomes to induce tumor metastasis, indicating exosome miR-103 can be used as a predictive marker for cancer progression (Fang et al. 2018). Shi et al. found exosome-derived miR-638 was significantly decreased in serum of HCC patients with advanced disease, such as at later TNM stage (III/IV) or with larger tumor size (>5 cm) (Shi et al. 2018). Moreover, several lncRNAs, like lnc-h19, lnc-sox2ot, and lncRNA-ARSR, have been investigated in circulating exosomes and closely related with tumor stage and overall survival of patients (Fang et al. 2018; Shi et al. 2018; Qu et al. 2016; Zhao et al. 2018; Lin et al. 2018; Conigliaro et al. 2015). Several studies have implicated exosome-derived miRNAs as potential biomarkers for detection of CRC occurrence and monitor recurrence. The serum levels of exosomal miRNAs, such as miR-1224-5p, miR-1229, miR-21, miR-223, miR-150, and let-7a, are much higher in CRC patients than healthy, dropping after tumor resection (Ruiz-Lopez et al. 2018). Exosomal miRNAs such as miR-19a, miR-18a, and miR-100 may be useful to detect the recurrence of CRC (Komatsu et al. 2014; Matsumura et al. 2015; Cha et al. 2015). These findings suggest that exosomal RNA molecules detected from circulation or other sources can serve as biomarkers to evaluate cancer occurrence and progression, with potentially high sensitivity and specificity.
19.5.2.1 TEX miRNAs and T Cells
Bland et al. found out that the tumor cell line B16F0 can deliver mRNA/miRNA loaded exosomes to CTLs and alter their metabolic function and IFN-γ production. TEX from nasopharyngeal carcinoma was reported to contain high levels of miR-106a-5p, miR-1908, miR-24-3p, miR-891a, and miR-20a-5p, yielding almost 20 targets linked to the MAPK1 pathway for potential downregulation. The net effect on T cells was a shift from Th1 and Th17 phenotypes to Th2 and Treg phenotypes, through suppression of ERK/STAT1/STAT3 phosphorylation (Ye et al. 2014). Evaluating overexpressed tumor miRNAs from patients with non-small cell lung cancer, breast cancer, pancreatic cancer or HCC, the ubiquitous miR-21 and miR-214 were shown to be consistently upregulated in tissue and in plasma exosomes/microvesicles. The same phenomenon was observed in murine sarcoma and lung cancer models, where miR-214 was enriched in MVs and downregulated PTEN in T cells, favoring Treg cell’s expansion (Yin et al. 2014; Walsh et al. 2006), but runs somewhat contradictory to other reports concerning the role of PTEN in Treg maintenance (Shrestha et al. 2015; Sharma et al. 2015).
19.5.2.2 TEX miRNAs and NK Cells
In lung cancer and leukemia, TEX-derived miR-23a was found to decrease the level of LAMP1 (lysosome-associated membrane glycoprotein 1)/CD107a (Berchem et al. 2016), which is an NK cell activation marker signifying lymphocytes degranulation (Cohnen et al. 2013). As the TEX also deliver TGFβ that inhibits NKG2D expression, they were generally considered as NK cell inhibitors. TEX-derived miR-362-5p has distinct effects depending on tumor cells themselves (Yang et al. 2015; Wu et al. 2015; Ni et al. 2016), but seems to be crucial in enhancing NK cell response via downregulation of CYLD, a suppressor of NF-κB signaling (Ni et al. 2015). However, the overabundance of the miRNA could lead to overstimulation resulting in hypo-responsiveness (Shifrin et al. 2014).
19.5.2.3 TEX MiRNAs and Monocytes
It was shown TEX-pulsed DCs could supply antigens to T cells and promote effector T cell’s response. The context in the immune system is likely a critical factor to determine TEX involvement of immune stimulation versus immune suppression (Kunigelis and Graner 2015). Researchers revealed an inhibition of stimulatory capacity in immature DCs when exposed to the human pancreatic cancer cell line-Panc-1 TEX (Zhou et al. 2014), which can deliver miR-203 to DCs and decrease TLR4 expression and downstream cytokines like TNFα and IL12. The changes of the cytokines in the tumor microenvironment can influence both T cells and B cells interacting with DCs.
A few years ago, Fabbri et al. revealed that TEX from non-small cell lung carcinoma (NSCLC) transfers miR-29a and miR-21 to macrophage existing in the tumor microenvironment (Fabbri et al. 2012). These miRNAs bound and enhanced TLR8 (murine TLR7) as ligands, activating the NF-κB pathway and resulting in production of IL6 and TFNα, creating a prometastatic inflammatory microenvironment. TLR7/8 belongs to intracellular TLRs subset, existing in endosomal and other vesicular membranes, and mediates innate immune reactions against multiple pathogens (Cervantes et al. 2012; Challagundla et al. 2015). MiR-21 from neuroblastoma TEX could also trigger TLR8 in monocytes, which led to upregulation of miR-155 in those cells, and the latter miRNA could then be sent back to the cancer cells via exosomes, resulting in downregulation of telomeric repeat binding factor 1 (TERF1). TERF1 is a telomerase inhibitor, downregulation of which increases cisplatin resistance in neuroblastoma cancer cells (Guo et al. 2009). Thus, the cross-interaction through exosomal miRNAs in the microenvironment is generally beneficial for the tumor.
19.5.3 Clinical Potential of Exosomes
The major role of TEX is to create an adaptive microenvironment for cancer cells to grow; however, several studies showed that EVs can inhibit cancer progression, either by direct effect of the EV-transported protein and nucleic acid contents or through antigen presentation to immunocytes. Tumor cells can deliver some of the same antigens through exosomes as the ones presenting on the surface (Chiodoni et al. 2018, 2019). For instance, DCs under the influence of rat glioblastoma cell-derived exosomes can trigger a strong antitumor reaction and dramatically prolong median survival in glioblastoma-bearing rats when used in combination with α-galactosylceramide (Liu et al. 2017). Given the relative longevity of EVs within the circulation, modification of those antitumor ones creates the potential to design new tools for cancer therapy.
The exosome liposome-like structure allows them to be loaded with various drugs. Exosomes are considered as a new generation of a natural nanoscale delivery system. Hemopurifier® is currently being accessed for its efficacy on seizing exosomes released by cancer cell lines or released in biofluids from cancer patients (Marleau et al. 2012). Researchers are studying a refined biomimetic nanostructure to deliver doxorubicin to breast cancer patient, by re-engineering immuno-exosome with a synthetic liposome (Rayamajhi et al. 2019).
Indeed, exosomes derived from different types of cells present different signaling molecules, and thereby have a great potential for targeted drug therapy (Xu et al. 2016). In a mouse breast cancer model, treatment with human-specific anti-CD9 or anti-CD63 antibodies inhibited metastasis to the lungs, lymph nodes, and thoracic cavity via the depletion of circulating EVs. EVs incubated with the targeted antibodies were preferentially internalized by macrophages and might be further eliminated by macrophages (Nishida-Aoki et al. 2017). Phase I clinical trial for advanced CRC has been performed using ascites-derived exosomes and granulocyte-macrophage colony-stimulating factor (GM-CSF). Combination of those two components efficiently enhances antitumor cytotoxic T cell response as a safe and feasible immunotherapy of advanced CRC (Dai et al. 2008).
19.6 CAF-Derived Exosome
The tumor microenvironment comprises tumor cells, nontumor cellular, and noncellular components such as fibroblasts, inflammatory cells, lymphocytes extracellular matrix, blood vessels, and signaling pathways. This dynamic context contributes to tumorigenesis through complex interactions of these elements. One of the main components of tumor microenvironment is CAFs. The interaction of tumor cells and CAFs has been reported to promote cancer progression (Alguacil-Nunez et al. 2018). Exosomes can induce normal fibroblast differentiation into CAFs through TGFβ signaling (Ringuette Goulet et al. 2018). CAFs then pose pro-tumor feedback to induce epithelial–mesenchymal transition of bladder cancer cells via paracrine IL-6 signaling (Goulet et al. 2019).
Next-generation sequencing and bioinformatics study on primary human normal and CAFs from nine paired normal colorectal mucosa and cancer tissues displayed significant differences between the ncRNA component and enrichment within exosomes of the normal and CAFs. NcRNA regulatory factors are specifically detected in CAF-generated exosomes, indicating a specific interaction between CAFs and CRC cells (Herrera et al. 2018).
Exosomal miRNAs were profiled from paired patient-derived normal fibroblasts and CAFs, from an ongoing prospective biomarker study. In vitro CAFs exosomes are delivered to CRC cells, with a subsequent increase in cellular miRNA levels, influencing tumor cell proliferation and chemoresistance. An exosomal CAF signature composed of miRNAs 21, 215, 181a, 329, 199b, and 382 was identified. Of these, miR‐21 showed highest abundance in CAF exosomes. In an orthotopic CRC murine model, co-injection with miR‐21‐overexpressing fibroblasts led to increased liver metastases than with control fibroblasts (Bhome et al. 2017).
CAFs constitute the majority of the tumor bulk of pancreatic ductal adenocarcinomas (PDACs). CAFs exposed to chemotherapy have an active role in regulating the survival and proliferation of cancer cells through exosome secretion. Gemcitabine increases the secretion of both miR-146a and Snail in pancreatic CAF exosomes. Blocking CAF exosome secretion inhibited PDAC tumor cell survival (Richards et al. 2017). Exosomal miR-196a derived from CAFs confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5 (Qin et al. 2019). In addition, loss of exosomal miR-3188 in CAFs leads to HNC progression (Wang et al. 2019).
Fibroblast growth factor 2(FGF2)-FGFR1 signaling regulates generation of leukemia-protective exosomes from bone marrow stromal cells. It was demonstrated that bone marrow stromal cells deliver exosome FGF2 to leukemia cells, protecting leukemia cells from tyrosine kinase inhibitors (TKIs). Expression of FGF2 and its receptor, FGFR1, are both enhanced in a subgroup of stromal cell lines and primary AML stroma. Activated FGF2/FGFR1 signaling can further enhance exosome secretion. Inhibiting FGFR cuts off stromal autocrine growth and remarkably suppresses secretion of FGF2-containing exosomes, contributing to compromised stromal guard of leukemia cells. In addition, Fgf2 −/− mice transplanted with retroviral BCR-ABL acute leukemia had prolonged survival compared with Fgf2 +/+ mice given TKI. Therefore, suppression of FGFR can downregulate stromal function, inhibit exosome secretion, and serve as a therapeutic target to conquer TKIs resistance (Javidi-Sharifi et al. 2019).
19.7 Conclusions
Tumor cells can develop a variety of mechanisms, including transferring TEX to evade and subvert the immune system for their survival. EVs represent a diverse category of cellular releasing products present in multiple types of biofluids and cell culture media. Exosomal contents directly reflect the metabolic state of the cells from which they originate. PD-L1 can be transferred to multiple cell types including tumor cells, macrophages, and DCs through PD-L1-containing exosomes in the tumor microenvironment, indicating a systemic regulatory role of exosomal PD-L1. Exosomal PD-L1 represents an unexplored therapeutic target, which could overcome resistance to current immune checkpoint inhibitors. The general pattern is that TEX deliver miRNAs to immune cells that ultimately lead to situations that benefit the cancer. Administration of therapeutic antibody effectively inhibits EV-induced tumor metastasis and that the removal of EVs could be a novel cancer treatment (Nishida-Aoki et al. 2017).
Though our understanding of EVs continues to grow, it is far from complete. Experimental data accumulated since decades ago evidently suggests that EVs play pivotal roles for some, if not all, cancer hallmarks. Until now, the field of EV research has drawn mounting interest from scientists and physicians, with growing number of investigators dissecting on the critical role of EVs in cancer biology, and thereby requires more transparent reporting and documenting to streamline interpretation and enable replication of experiments. EV-TRACK, a crowdsourcing knowledgebase (http://evtrack.org), is recently built to improve centralization of EV biology and relevant methodology to help reviewers, authors, editors, and funders to fulfill experimental guidelines and increase research reproducibility (Van Deun et al. 2017; Consortium et al. 2017). Vesiclepedia (http://www.microvesicles.org) is an established web-based compendium of components including RNA, lipids, proteins, and metabolites transported by EVs from both published and unpublished researches, with the input currently from 1254 EV investigations, consisting of 38,146 RNA entries, 349,988 protein entries, and 639 lipid/metabolite entries (Pathan et al. 2019). There are also alternative or supplementary initiatives to characterize EVs, for example, ExoCarta and EVpedia, two typical web domains that help researchers to promptly upload proteomic lists of identified proteins of the EVs being investigated (Mathivanan and Simpson 2009; Kim et al. 2015). Widespread implementation of those knowledgebases by the EV scientific community is believed to facilitate the success of the exosome research in the long run.
References
Alguacil-Nunez C, Ferrer-Ortiz I, Garcia-Verdu E, Lopez-Pirez P, Llorente-Cortijo IM, Sainz B Jr (2018) Current perspectives on the crosstalk between lung cancer stem cells and cancer-associated fibroblasts. Crit Rev Oncology/Hematology 125:102–110
Alvarez ML, Khosroheidari M, Kanchi Ravi R, DiStefano JK (2012) Comparison of protein, microrna, and mrna yields using different methods of urinary exosome isolation for the discovery of kidney disease biomarkers. Kidney Int 82:1024–1032
Amzallag N, Passer BJ, Allanic D, Segura E, Thery C, Goud B, Amson R, Telerman A (2004) Tsap6 facilitates the secretion of translationally controlled tumor protein/histamine-releasing factor via a nonclassical pathway. J Biol Chem 279:46104–46112
Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, Pomel C, Lhomme C, Escudier B, Le Chevalier T, Tursz T, Amigorena S, Raposo G, Angevin E, Zitvogel L (2002) Malignant effusions and immunogenic tumour-derived exosomes. Lancet 360:295–305
Ashiru O, Boutet P, Fernandez-Messina L, Aguera-Gonzalez S, Skepper JN, Vales-Gomez M, Reyburn HT (2010) Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Can Res 70:481–489
Azoulay-Alfaguter I, Mor A (2018) Proteomic analysis of human T cell-derived exosomes reveals differential RAS/MAPK signaling. Eur J Immunol 48:1915–1917
Batista BS, Eng WS, Pilobello KT, Hendricks-Munoz KD, Mahal LK (2011) Identification of a conserved glycan signature for microvesicles. J Proteome Res 10:4624–4633
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T (1999) Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285:727–729
Berchem G, Noman MZ, Bosseler M, Paggetti J, Baconnais S, Le Cam E, Nanbakhsh A, Moussay E, Mami-Chouaib F, Janji B, Chouaib S (2016) Hypoxic tumor-derived microvesicles negatively regulate nk cell function by a mechanism involving TGF-β and miR23a transfer. Oncoimmunology. 5:e1062968
Bhome R, Goh RW, Bullock MD, Pillar N, Thirdborough SM, Mellone M, Mirnezami R, Galea D, Veselkov K, Gu Q, Underwood TJ, Primrose JN, De Wever O, Shomron N, Sayan AE, Mirnezami AH (2017) Exosomal microRNAs derived from colorectal cancer-associated fibroblasts: role in driving cancer progression. Aging 9:2666–2694
Borrelli C, Ricci B, Vulpis E, Fionda C, Ricciardi MR, Petrucci MT, Masuelli L, Peri A, Cippitelli M, Zingoni A, Santoni A, Soriani A (2018) Drug-induced senescent multiple myeloma cells elicit nk cell proliferation by direct or exosome-mediated il15 trans-presentation. Cancer Immunol Res 6:860–869
Burke M, Choksawangkarn W, Edwards N, Ostrand-Rosenberg S, Fenselau C (2014) Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J Proteome Res 13:836–843
Cervantes JL, Weinerman B, Basole C, Salazar JC (2012) TLR8: The forgotten relative revindicated. Cell Mol Immunol 9:434–438
Cha DJ, Franklin JL, Dou Y, Liu Q, Higginbotham JN, Demory Beckler M, Weaver AM, Vickers K, Prasad N, Levy S, Zhang B, Coffey RJ, Patton JG (2015) KRAS-dependent sorting of mirna to exosomes. eLife 4:e07197
Challagundla KB, Wise PM, Neviani P, Chava H, Murtadha M, Xu T, Kennedy R, Ivan C, Zhang X, Vannini I, Fanini F, Amadori D, Calin GA, Hadjidaniel M, Shimada H, Jong A, Seeger RC, Asgharzadeh S, Goldkorn A, Fabbri M (2015) Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst 107
Chen L, Han X (2015) Anti-PD-1/PD-l1 therapy of human cancer: past, present, and future. J Clin Investig 125:3384–3391
Chen DS, Mellman I (2017) Elements of cancer immunity and the cancer–immune set point. Nature 541:321–330
Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, Xia H, Man Q, Zhong W, Antelo LF, Wu B, Xiong X, Liu X, Guan L, Li T, Liu S, Yang R, Lu Y, Dong L, McGettigan S, Somasundaram R, Radhakrishnan R, Mills G, Lu Y, Kim J, Chen YH, Dong H, Zhao Y, Karakousis GC, Mitchell TC, Schuchter LM, Herlyn M, Wherry EJ, Xu X, Guo W (2018a) Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 560:382–386
Chen S, Lv M, Fang S, Ye W, Gao Y, Xu Y (2018b) Poly (I:C) enhanced anti-cervical cancer immunities induced by dendritic cells-derived exosomes. Int J Biol Macromol 113:1182–1187
Cheng WC, Liao TT, Lin CC, Yuan LE, Lan HY, Lin HH, Teng HW, Chang HC, Lin CH, Yang CY, Huang SC, Jiang JK, Yang SH, Yang MH, Hwang WL (2019) RAB27b-activated secretion of stem-like tumor exosomes delivers the biomarker microRNA-146a-5p, which promotes tumorigenesis and associates with an immunosuppressive tumor microenvironment in colorectal cancer. Int J Cancer 145:2209–2224
Chiodoni C, Di Martino MT, Zazzeroni F, Caraglia M, Donadelli M, Meschini S, Leonetti C, Scotlandi K (2019) Cell communication and signaling: how to turn bad language into positive one. J Exper Clin Cancer Res CR 38:128
Chow A, Zhou W, Liu L, Fong MY, Champer J, Van Haute D, Chin AR, Ren X, Gugiu BG, Meng Z, Huang W, Ngo V, Kortylewski M, Wang SE (2014) Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-κB. Sci Rep 4:5750
Chulpanova DS, Kitaeva KV, James V, Rizvanov AA, Solovyeva VV (2018) Therapeutic prospects of extracellular vesicles in cancer treatment. Front Immunol 9:1534
Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z (2008) Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol 180:7249–7258
Cohnen A, Chiang SC, Stojanovic A, Schmidt H, Claus M, Saftig P, Janssen O, Cerwenka A, Bryceson YT, Watzl C (2013) Surface CD107a/LAMP-1 protects natural killer cells from degranulation-associated damage. Blood 122:1411–1418
Colombo M, Raposo G, Thery C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Ann Rev Cell Dev Biol 30:255–289
Conigliaro A, Costa V, Lo Dico A, Saieva L, Buccheri S, Dieli F, Manno M, Raccosta S, Mancone C, Tripodi M, De Leo G, Alessandro R (2015) CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol Cancer 14:155
Consortium E-T, Van Deun J, Mestdagh P, Agostinis P, Akay O, Anand S, Anckaert J, Martinez ZA, Baetens T, Beghein E, Bertier L, Berx G, Boere J, Boukouris S, Bremer M, Buschmann D, Byrd JB, Casert C, Cheng L, Cmoch A, Daveloose D, De Smedt E, Demirsoy S, Depoorter V, Dhondt B, Driedonks TA, Dudek A, Elsharawy A, Floris I, Foers AD, Gartner K, Garg AD, Geeurickx E, Gettemans J, Ghazavi F, Giebel B, Kormelink TG, Hancock G, Helsmoortel H, Hill AF, Hyenne V, Kalra H, Kim D, Kowal J, Kraemer S, Leidinger P, Leonelli C, Liang Y, Lippens L, Liu S, Lo Cicero A, Martin S, Mathivanan S, Mathiyalagan P, Matusek T, Milani G, Monguio-Tortajada M, Mus LM, Muth DC, Nemeth A, Nolte-’t Hoen EN, O’Driscoll L, Palmulli R, Pfaffl MW, Primdal-Bengtson B, Romano E, Rousseau Q, Sahoo S, Sampaio N, Samuel M, Scicluna B, Soen B, Steels A, Swinnen JV, Takatalo M, Thaminy S, Thery C, Tulkens J, Van Audenhove I, van der Grein S, Van Goethem A, van Herwijnen MJ, Van Niel G, Van Roy N, Van Vliet AR, Vandamme N, Vanhauwaert S, Vergauwen G, Verweij F, Wallaert A, Wauben M, Witwer KW, Zonneveld MI, De Wever O, Vandesompele J, Hendrix A (2017) Ev-track: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods 14:228–232
Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic ras and the p53 tumor suppressor. PLoS Biol 6:2853–2868
Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H, Li G (2008) Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther J Am Soc Gene Ther 16:782–790
Diamond JM, Vanpouille-Box C, Spada S, Rudqvist NP, Chapman JR, Ueberheide BM, Pilones KA, Sarfraz Y, Formenti SC, Demaria S (2018) Exosomes shuttle TREX1-sensitive ifn-stimulatory dsdna from irradiated cancer cells to dcs. Cancer Immunol Res 6:910–920
Dickens AM, Tovar YRLB, Yoo SW, Trout AL, Bae M, Kanmogne M, Megra B, Williams DW, Witwer KW, Gacias M, Tabatadze N, Cole RN, Casaccia P, Berman JW, Anthony DC, Haughey NJ (2017) Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal 10
Ding G, Zhou L, Qian Y, Fu M, Chen J, Chen J, Xiang J, Wu Z, Jiang G, Cao L (2015) Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget 6:29877–29888
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L (2002) Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8:793–800
Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ, Zanesi N, Crawford M, Ozer GH, Wernicke D, Alder H, Caligiuri MA, Nana-Sinkam P, Perrotti D, Croce CM (2012) Micrornas bind to toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA 109:2110–2116
Fan Y, Herr F, Vernochet A, Mennesson B, Oberlin E, Durrbach A (2019) Human fetal liver mesenchymal stem cell-derived exosomes impair natural killer cell function. Stem Cells Dev 28:44–55
Fang JH, Zhang ZJ, Shang LR, Luo YW, Lin YF, Yuan Y, Zhuang SM (2018) Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology 68:1459–1475
Fernandez-Messina L, Ashiru O, Boutet P, Aguera-Gonzalez S, Skepper JN, Reyburn HT, Vales-Gomez M (2010) Differential mechanisms of shedding of the glycosylphosphatidylinositol (GPI)-anchored NKG2D ligands. J Biol Chem 285:8543–8551
Frigola X, Inman BA, Lohse CM, Krco CJ, Cheville JC, Thompson RH, Leibovich B, Blute ML, Dong H, Kwon ED (2011) Identification of a soluble form of B7-H1 that retains immunosuppressive activity and is associated with aggressive renal cell carcinoma. Clin Cancer Res 17:1915–1923 An Official Journal of the American Association for Cancer Research
Gao D, Jiang L (2018) Exosomes in cancer therapy: a novel experimental strategy. Am J Cancer Res 8:2165–2175
Geis-Asteggiante L, Belew AT, Clements VK, Edwards NJ, Ostrand-Rosenberg S, El-Sayed NM, Fenselau C (2018) Differential content of proteins, mRNAs, and miRNAs suggests that MDSC and their exosomes may mediate distinct immune suppressive functions. J Proteome Res 17:486–498
Goulet CR, Champagne A, Bernard G, Vandal D, Chabaud S, Pouliot F, Bolduc S (2019) Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 19:137
Graner MW, Alzate O, Dechkovskaia AM, Keene JD, Sampson JH, Mitchell DA, Bigner DD (2009) Proteomic and immunologic analyses of brain tumor exosomes. FASEB J 23:1541–1557 Official Publication of the Federation of American Societies for Experimental Biology
Graner MW, Schnell S, Olin MR (2018) Tumor-derived exosomes, micrornas, and cancer immune suppression. Semin Immunopathol 40:505–515
Groh V, Wu J, Yee C, Spies T (2002) Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419:734–738
Gross C, Hansch D, Gastpar R, Multhoff G (2003) Interaction of heat shock protein 70 peptide with NK cells involves the NK receptor CD94. Biol Chem 384:267–279
Guo XL, Ma NN, Zhou FG, Zhang L, Bu XX, Sun K, Song JR, Li R, Zhang BH, Wu MC, Wei LX (2009) Up-regulation of hTERT expression by low-dose cisplatin contributes to chemotherapy resistance in human hepatocellular cancer cells. Oncol Rep 22:549–556
Haderk F, Schulz R, Iskar M, Cid LL, Worst T, Willmund KV, Schulz A, Warnken U, Seiler J, Benner A, Nessling M, Zenz T, Gobel M, Durig J, Diederichs S, Paggetti J, Moussay E, Stilgenbauer S, Zapatka M, Lichter P, Seiffert M (2017) Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci Immunol 2
Hagemann T, Biswas SK, Lawrence T, Sica A, Lewis CE (2009) Regulation of macrophage function in tumors: the multifaceted role of NF-κB. Blood 113:3139–3146
Han L, Lam EW, Sun Y (2019) Extracellular vesicles in the tumor microenvironment: old stories, but new tales. Mol Cancer 18:59
Harding C, Heuser J, Stahl P (1983) Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol 97:329–339
He L, Zhang G, He Y, Zhu H, Zhang H, Feng Z (2005) Blockade of B7-H1 with sPD-1 improves immunity against murine hepatocarcinoma. Anticancer Res 25:3309–3313
Hedlund M, Nagaeva O, Kargl D, Baranov V, Mincheva-Nilsson L (2011) Thermal- and oxidative stress causes enhanced release of nkg2d ligand-bearing immunosuppressive exosomes in leukemia/lymphoma t and b cells. PLoS ONE 6:e16899
Herrera M, Llorens C, Rodriguez M, Herrera A, Ramos R, Gil B, Candia A, Larriba MJ, Garre P, Earl J, Rodriguez-Garrote M, Caldes T, Bonilla F, Carrato A, Garcia-Barberan V, Pena C (2018) Differential distribution and enrichment of non-coding rnas in exosomes from normal and cancer-associated fibroblasts in colorectal cancer. Mol Cancer 17:114
Hsieh CH, Tai SK, Yang MH (2018) Snail-overexpressing cancer cells promote M2-like polarization of tumor-associated macrophages by delivering MiR-21-abundant exosomes. Neoplasia 20:775–788
Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, Vale RD (2017) T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355:1428–1433
Jang JY, Lee JK, Jeon YK, Kim CW (2013) Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor-associated macrophage infiltration and M2 polarization. BMC Cancer 13:421
Javidi-Sharifi N, Martinez J, English I, Joshi SK, Scopim-Ribeiro R, Viola SK, Edwards DKt, Agarwal A, Lopez C, Jorgens D, Tyner JW, Druker BJ, Traer E (2019) FGF2-FGFR1 signaling regulates release of Leukemia-Protective exosomes from bone marrow stromal cells. eLife 8
Jong AY, Wu CH, Li J, Sun J, Fabbri M, Wayne AS, Seeger RC (2017) Large-scale isolation and cytotoxicity of extracellular vesicles derived from activated human natural killer cells. J Extracell Vesicles 6:1294368
Kachapati K, Bednar KJ, Adams DE, Wu Y, Mittler RS, Jordan MB, Hinerman JM, Herr AB, Ridgway WM (2013) Recombinant soluble CD137 prevents type one diabetes in nonobese diabetic mice. J Autoimmun 47:94–103
Kim DK, Lee J, Kim SR, Choi DS, Yoon YJ, Kim JH, Go G, Nhung D, Hong K, Jang SC, Kim SH, Park KS, Kim OY, Park HT, Seo JH, Aikawa E, Baj-Krzyworzeka M, van Balkom BW, Belting M, Blanc L, Bond V, Bongiovanni A, Borras FE, Buee L, Buzas EI, Cheng L, Clayton A, Cocucci E, Dela Cruz CS, Desiderio DM, Di Vizio D, Ekstrom K, Falcon-Perez JM, Gardiner C, Giebel B, Greening DW, Gross JC, Gupta D, Hendrix A, Hill AF, Hill MM, Nolte-’t Hoen E, Hwang DW, Inal J, Jagannadham MV, Jayachandran M, Jee YK, Jorgensen M, Kim KP, Kim YK, Kislinger T, Lasser C, Lee DS, Lee H, van Leeuwen J, Lener T, Liu ML, Lotvall J, Marcilla A, Mathivanan S, Moller A, Morhayim J, Mullier F, Nazarenko I, Nieuwland R, Nunes DN, Pang K, Park J, Patel T, Pocsfalvi G, Del Portillo H, Putz U, Ramirez MI, Rodrigues ML, Roh TY, Royo F, Sahoo S, Schiffelers R, Sharma S, Siljander P, Simpson RJ, Soekmadji C, Stahl P, Stensballe A, Stepien E, Tahara H, Trummer A, Valadi H, Vella LJ, Wai SN, Witwer K, Yanez-Mo M, Youn H, Zeidler R, Gho YS (2015) Evpedia: a community web portal for extracellular vesicles research. Bioinformatics 31:933–939
Kitai Y, Kawasaki T, Sueyoshi T, Kobiyama K, Ishii KJ, Zou J, Akira S, Matsuda T, Kawai T (2017) DNA-containing exosomes derived from cancer cells treated with topotecan activate a sting-dependent pathway and reinforce antitumor immunity. J Immunol 198:1649–1659
Komatsu S, Ichikawa D, Takeshita H, Morimura R, Hirajima S, Tsujiura M, Kawaguchi T, Miyamae M, Nagata H, Konishi H, Shiozaki A, Otsuji E (2014) Circulating miR-18a: a sensitive cancer screening biomarker in human cancer. Vivo 28:293–297
Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T (2010) Secretory mechanisms and intercellular transfer of micrornas in living cells. J Biol Chem 285:17442–17452
Kruger S, Legenstein ML, Rosgen V, Haas M, Modest DP, Westphalen CB, Ormanns S, Kirchner T, Heinemann V, Holdenrieder S, Boeck S (2017) Serum levels of soluble programmed death protein 1 (sPD-1) and soluble programmed death ligand 1 (sPD-L1) in advanced pancreatic cancer. Oncoimmunology 6:e1310358
Kunigelis KE, Graner MW (2015) The dichotomy of tumor exosomes (TEX) in cancer immunity: is it all in the ConTeXt? Vaccines 3:1019–1051
Labiano S, Palazon A, Bolanos E, Azpilikueta A, Sanchez-Paulete AR, Morales-Kastresana A, Quetglas JI, Perez-Gracia JL, Gurpide A, Rodriguez-Ruiz M, Aznar MA, Jure-Kunkel M, Berraondo P, Melero I (2016) Hypoxia-induced soluble CD137 in malignant cells blocks CD137L-costimulation as an immune escape mechanism. Oncoimmunology 5:e1062967
Lan J, Sun L, Xu F, Liu L, Hu F, Song D, Hou Z, Wu W, Luo X, Wang J, Yuan X, Hu J, Wang G (2019) M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Can Res 79:146–158
Lawrence T (2009) The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol 1:a001651
Lehmann BD, Paine MS, Brooks AM, McCubrey JA, Renegar RH, Wang R, Terrian DM (2008) Senescence-associated exosome release from human prostate cancer cells. Can Res 68:7864–7871
Lespagnol A, Duflaut D, Beekman C, Blanc L, Fiucci G, Marine JC, Vidal M, Amson R, Telerman A (2008) Exosome secretion, including the DNA damage-induced p53-dependent secretory pathway, is severely compromised in tsap6/steap3-null mice. Cell Death Differ 15:1723–1733
Lin LY, Yang L, Zeng Q, Wang L, Chen ML, Zhao ZH, Ye GD, Luo QC, Lv PY, Guo QW, Li BA, Cai JC, Cai WY (2018) Tumor-originated exosomal lncUEGC1 as a circulating biomarker for early-stage gastric cancer. Mol Cancer 17:84
Lindenbergh MFS, Koerhuis DGJ, Borg EGF, van ‘t Veld EM, Driedonks TAP, Wubbolts R, Stoorvogel W, Boes M (2019) Bystander T-cells support clonal T-cell activation by controlling the release of dendritic cell-derived immune-stimulatory extracellular vesicles. Front Immunol 10:448
Liu H, Chen L, Liu J, Meng H, Zhang R, Ma L, Wu L, Yu S, Shi F, Li Y, Zhang L, Wang L, Feng S, Zhang Q, Peng Y, Wu Q, Liu C, Chang X, Yang L, Uemura Y, Yu X, Liu T (2017) Co-delivery of tumor-derived exosomes with alpha-galactosylceramide on dendritic cell-based immunotherapy for glioblastoma. Cancer Lett 411:182–190
Magistrelli G, Jeannin P, Herbault N, Benoit De Coignac A, Gauchat JF, Bonnefoy JY, Delneste Y (1999) A soluble form of CTLA-4 generated by alternative splicing is expressed by nonstimulated human T cells. Eur J Immunol 29:3596–3602
Marleau AM, Chen CS, Joyce JA, Tullis RH (2012) Exosome removal as a therapeutic adjuvant in cancer. J Transl Med 10:134
Mathivanan S, Simpson RJ (2009) Exocarta: a compendium of exosomal proteins and RNA. Proteomics 9:4997–5000
Matsumura T, Sugimachi K, Iinuma H, Takahashi Y, Kurashige J, Sawada G, Ueda M, Uchi R, Ueo H, Takano Y, Shinden Y, Eguchi H, Yamamoto H, Doki Y, Mori M, Ochiya T, Mimori K (2015) Exosomal microRNA in serum is a novel biomarker of recurrence in human colorectal cancer. Br J Cancer 113:275–281
Medrano RFV, Hunger A, Mendonca SA, Barbuto JAM, Strauss BE (2017) Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget 8:71249–71284
Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, Logtenberg MEW, Slagter M, Rozeman EA, Hofland I, Broeks A, Horlings HM, Wessels LFA, Blank CU, Xiao Y, Heck AJR, Borst J, Brummelkamp TR, Schumacher TNM (2017) Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549:106–110
Monleon I, Martinez-Lorenzo MJ, Monteagudo L, Lasierra P, Taules M, Iturralde M, Pineiro A, Larrad L, Alava MA, Naval J, Anel A (2001) Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J Immunol 167:6736–6744
Monypenny J, Milewicz H, Flores-Borja F, Weitsman G, Cheung A, Chowdhury R, Burgoyne T, Arulappu A, Lawler K, Barber PR, Vicencio JM, Keppler M, Wulaningsih W, Davidson SM, Fraternali F, Woodman N, Turmaine M, Gillett C, Franz D, Quezada SA, Futter CE, Von Kriegsheim A, Kolch W, Vojnovic B, Carlton JG, Ng T (2018) ALIX regulates tumor-mediated immunosuppression by controlling EGFR activity and PD-L1 presentation. Cell Rep 24:630–641
Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, Durbin JE, Biron CA (2002) Coordinated and distinct roles for IFN-αβ, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol 169:4279–4287
Ni F, Guo C, Sun R, Fu B, Yang Y, Wu L, Ren S, Tian Z, Wei H (2015) Microrna transcriptomes of distinct human nk cell populations identify miR-362-5p as an essential regulator of Nk cell function. Sci Rep 5:9993
Ni F, Gui Z, Guo Q, Hu Z, Wang X, Chen D, Wang S (2016) Downregulation of miR-362-5p inhibits proliferation, migration and invasion of human breast cancer MCF7 cells. Oncol Lett 11:1155–1160
Nishida-Aoki N, Tominaga N, Takeshita F, Sonoda H, Yoshioka Y, Ochiya T (2017) Disruption of circulating extracellular vesicles as a novel therapeutic strategy against cancer metastasis. Mol Ther J Am Soc Gene Ther 25:181–191
Noy R, Pollard JW (2014) Tumor-associated macrophages: from mechanisms to therapy. Immunity 41:49–61
Oaks MK, Hallett KM, Penwell RT, Stauber EC, Warren SJ, Tector AJ (2000) A native soluble form of CTLA-4. Cell Immunol 201:144–153
Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, Goud B, Benaroch P, Hacohen N, Fukuda M, Desnos C, Seabra MC, Darchen F, Amigorena S, Moita LF, Thery C (2010) Rab27a and rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 12:19–30, sup pp 11–13
Pan BT, Johnstone RM (1983) Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 33:967–978
Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, Yeoh KW, Kon OL, Tam JP, Sze SK (2019) Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microrna-mediated metabolic shift. Oncogene 38:5158–5173
Pathan M, Fonseka P, Chitti SV, Kang T, Sanwlani R, Van Deun J, Hendrix A, Mathivanan S (2019) Vesiclepedia 2019: a compendium of rna, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res 47:D516–D519
Peterson MF, Otoc N, Sethi JK, Gupta A, Antes TJ (2015) Integrated systems for exosome investigation. Methods 87:31–45
Piao YJ, Kim HS, Hwang EH, Woo J, Zhang M, Moon WK (2018) Breast cancer cell-derived exosomes and macrophage polarization are associated with lymph node metastasis. Oncotarget 9:7398–7410
Pitt JM, Charrier M, Viaud S, Andre F, Besse B, Chaput N, Zitvogel L (2014) Dendritic cell-derived exosomes as immunotherapies in the fight against cancer. J Immunol 193:1006–1011
Pitt JM, Andre F, Amigorena S, Soria JC, Eggermont A, Kroemer G, Zitvogel L (2016) Dendritic cell-derived exosomes for cancer therapy. J Clin Investig 126:1224–1232
Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A, Montabana E, Lang UE, Fu Q, Fong L, Blelloch R (2019) Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 177(414–427):e413
Qin X, Guo H, Wang X, Zhu X, Yan M, Wang X, Xu Q, Shi J, Lu E, Chen W, Zhang J (2019) Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1b and ING5. Genome Biol 20:12
Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y, Chen W, Liu F, Sun W, Li XF, Wang X, Wang Y, Xu ZY, Gao L, Yang Q, Xu B, Li YM, Fang ZY, Xu ZP, Bao Y, Wu DS, Miao X, Sun HY, Sun YH, Wang HY, Wang LH (2016) Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell 29:653–668
Raposo G, Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200:373–383
Rayamajhi S, Nguyen TDT, Marasini R, Aryal S (2019) Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater 94:482–494
Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS, Gangadhar TC, Hersey P, Dronca R, Joseph RW, Zarour H, Chmielowski B, Lawrence DP, Algazi A, Rizvi NA, Hoffner B, Mateus C, Gergich K, Lindia JA, Giannotti M, Li XN, Ebbinghaus S, Kang SP, Robert C (2016) Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 315:1600–1609
Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R (2017) Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 36:1770–1778
Ricklefs FL, Alayo Q, Krenzlin H, Mahmoud AB, Speranza MC, Nakashima H, Hayes JL, Lee K, Balaj L, Passaro C, Rooj AK, Krasemann S, Carter BS, Chen CC, Steed T, Treiber J, Rodig S, Yang K, Nakano I, Lee H, Weissleder R, Breakefield XO, Godlewski J, Westphal M, Lamszus K, Freeman GJ, Bronisz A, Lawler SE, Chiocca EA (2018) Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv 4:eaar2766
Rincon-Orozco B, Kunzmann V, Wrobel P, Kabelitz D, Steinle A, Herrmann T (2005) Activation of V gamma 9V delta 2 T cells by NKG2D. J Immunol 175:2144–2151
Ringuette Goulet C, Bernard G, Tremblay S, Chabaud S, Bolduc S, Pouliot F (2018) Exosomes induce fibroblast differentiation into cancer-associated fibroblasts through TGFβ signaling. Mol Cancer Res MCR 16:1196–1204
Rossille D, Gressier M, Damotte D, Maucort-Boulch D, Pangault C, Semana G, Le Gouill S, Haioun C, Tarte K, Lamy T, Milpied N, Fest T, Groupe Ouest-Est des Leucemies et Autres Maladies du S (2014) High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-cell lymphoma: results from a French multicenter clinical trial. Leukemia 28:2367–2375
Ruiz-Lopez L, Blancas I, Garrido JM, Mut-Salud N, Moya-Jodar M, Osuna A, Rodriguez-Serrano F (2018) The role of exosomes on colorectal cancer: a review. J Gastroenterol Hepatol 33:792–799
Saito Y, Ohnishi K, Miyashita A, Nakahara S, Fujiwara Y, Horlad H, Motoshima T, Fukushima S, Jinnin M, Ihn H, Takeya M, Komohara Y (2015) Prognostic significance of CD169 + lymph node sinus macrophages in patients with malignant melanoma. Cancer Immunol Res 3:1356–1363
Saunderson SC, McLellan AD (2017) Role of lymphocyte subsets in the immune response to primary B cell-derived exosomes. J Immunol 199:2225–2235
Schmittgen TD (2019) Exosomal miRNA cargo as mediator of immune escape mechanisms in neuroblastoma. Can Res 79:1293–1294
Seo N, Shirakura Y, Tahara Y, Momose F, Harada N, Ikeda H, Akiyoshi K, Shiku H (2018) Activated CD8(+) T cell extracellular vesicles prevent tumour progression by targeting of lesional mesenchymal cells. Nat Commun 9:435
Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, Mautino MR, Celis E, Sharpe AH, Francisco LM, Powell JD, Yagita H, Mellor AL, Blazar BR, Munn DH (2015) The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv 1:e1500845
Shenoy GN, Loyall J, Maguire O, Iyer V, Kelleher RJ Jr, Minderman H, Wallace PK, Odunsi K, Balu-Iyer SV, Bankert RB (2018) Exosomes associated with human ovarian tumors harbor a reversible checkpoint of T-cell responses. Cancer Immunol Res 6:236–247
Shi M, Jiang Y, Yang L, Yan S, Wang YG, Lu XJ (2018) Decreased levels of serum exosomal miR-638 predict poor prognosis in hepatocellular carcinoma. J Cell Biochem 119:4711–4716
Shifrin N, Raulet DH, Ardolino M (2014) NK cell self tolerance, responsiveness and missing self recognition. Semin Immunol 26:138–144
Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H (2015) T reg cells require the phosphatase PTEN to restrain TH1 and T FH cell responses. Nat Immunol 16:178–187
Skokos D, Botros HG, Demeure C, Morin J, Peronet R, Birkenmeier G, Boudaly S, Mecheri S (2003) Mast cell-derived exosomes induce phenotypic and functional maturation of dendritic cells and elicit specific immune responses in vivo. J Immunol 170:3037–3045
Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, Boyiadzis M (2011) Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-β1. Haematologica 96:1302–1309
Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, Takasugi M, Watanabe S, Kanemaki MT, Obuse C, Hara E (2017) Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun 8:15287
Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, Simpson RJ (2012) Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line lim1863-derived exosomes. Methods 56:293–304
Theodoraki MN, Yerneni SS, Hoffmann TK, Gooding WE, Whiteside TL (2018) Clinical significance of PD-L1(+) exosomes in plasma of head and neck cancer patients. Clin Cancer Res 24:896–905 An official Journal of the American Association for Cancer Research
Topalian SL, Taube JM, Anders RA, Pardoll DM (2016) Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 16:275–287
Torralba D, Baixauli F, Villarroya-Beltri C, Fernandez-Delgado I, Latorre-Pellicer A, Acin-Perez R, Martin-Cofreces NB, Jaso-Tamame AL, Iborra S, Jorge I, Gonzalez-Aseguinolaza G, Garaude J, Vicente-Manzanares M, Enriquez JA, Mittelbrunn M, Sanchez-Madrid F (2018) Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat Commun 9:2658
Van Deun J, Hendrix A, Consortium E-T (2017) Is your article ev-tracked? J Extracellular Vesicles 6:1379835
Viaud S, Terme M, Flament C, Taieb J, Andre F, Novault S, Escudier B, Robert C, Caillat-Zucman S, Tursz T, Zitvogel L, Chaput N (2009) Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Rα. PLoS ONE 4:e4942
Wahlgren J, Karlson Tde L, Glader P, Telemo E, Valadi H (2012) Activated human t cells secrete exosomes that participate in IL-2 mediated immune response signaling. PLoS ONE 7:e49723
Walsh PT, Buckler JL, Zhang J, Gelman AE, Dalton NM, Taylor DK, Bensinger SJ, Hancock WW, Turka LA (2006) PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J Clin Investig 116:2521–2531
Wang T, Gilkes DM, Takano N, Xiang L, Luo W, Bishop CJ, Chaturvedi P, Green JJ, Semenza GL (2014) Hypoxia-inducible factors and rab22a mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc Natl Acad Sci USA 111:3234–3242
Wang L, Wang H, Chen H, Wang WD, Chen XQ, Geng QR, Xia ZJ, Lu Y (2015) Serum levels of soluble programmed death ligand 1 predict treatment response and progression free survival in multiple myeloma. Oncotarget 6:41228–41236
Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, Chen L, Chen Y, Zhu G, Yin W, Zheng L, Zhou T, Badri T, Yao S, Zhu S, Boto A, Sznol M, Melero I, Vignali DAA, Schalper K, Chen L (2019a) Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176(334–347):e312
Wang X, Qin X, Yan M, Shi J, Xu Q, Li Z, Yang W, Zhang J, Chen W (2019b) Loss of exosomal miR-3188 in cancer-associated fibroblasts contributes to HNC progression. J Exper Clin Cancer Res CR 38:151
Ward FJ, Dahal LN, Wijesekera SK, Abdul-Jawad SK, Kaewarpai T, Xu H, Vickers MA, Barker RN (2013) The soluble isoform of CTLA-4 as a regulator of T-cell responses. Eur J Immunol 43:1274–1285
Whiteside TL (2013) Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem Soc Trans 41:245–251
Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL (2009) Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol 183:3720–3730
Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, Flament C, Pouzieux S, Faure F, Tursz T, Angevin E, Amigorena S, Zitvogel L (2001) Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med 7:297–303
Wu K, Yang L, Chen J, Zhao H, Wang J, Xu S, Huang Z (2015) miR-362-5p inhibits proliferation and migration of neuroblastoma cells by targeting phosphatidylinositol 3-kinase-C2β. FEBS Lett 589:1911–1919
Xu W, Yang Z, Lu N (2016) From pathogenesis to clinical application: insights into exosomes as transfer vectors in cancer. J Exper Clin Cancer Res CR. 35:156
Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, Monga SP, Geller DA, Lotze MT, Tsung A (2012) High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology 55:1863–1875
Yang M, Chen J, Su F, Yu B, Su F, Lin L, Liu Y, Huang JD, Song E (2011) Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol Cancer 10:117
Yang P, Ni F, Deng RQ, Qiang G, Zhao H, Yang MZ, Wang XY, Xu YZ, Chen L, Chen DL, Chen ZJ, Kan LX, Wang SY (2015) MiR-362-5p promotes the malignancy of chronic myelocytic leukaemia via down-regulation of GADD45α. Mol Cancer 14:190
Yang Y, Li CW, Chan LC, Wei Y, Hsu JM, Xia W, Cha JH, Hou J, Hsu JL, Sun L, Hung MC (2018) Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res 28:862–864
Yang J, Bi L, He X, Wang Z, Qian Y, Xiao L, Shi B (2019) Follicular helper T cell derived exosomes promote B cell proliferation and differentiation in antibody-mediated rejection after renal transplantation. Biomed Res Int 2019:6387924
Ye SB, Li ZL, Luo DH, Huang BJ, Chen YS, Zhang XS, Cui J, Zeng YX, Li J (2014) Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal microRNAs in human nasopharyngeal carcinoma. Oncotarget 5:5439–5452
Ye L, Zhang Q, Cheng Y, Chen X, Wang G, Shi M, Zhang T, Cao Y, Pan H, Zhang L, Wang G, Deng Y, Yang Y, Chen G (2018) Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1(+) regulatory B cell expansion. J Immunother Cancer 6:145
Yin Y, Cai X, Chen X, Liang H, Zhang Y, Li J, Wang Z, Chen X, Zhang W, Yokoyama S, Wang C, Li L, Li L, Hou D, Dong L, Xu T, Hiroi T, Yang F, Ji H, Zhang J, Zen K, Zhang CY (2014) Tumor-secreted miR-214 induces regulatory T cells: a major link between immune evasion and tumor growth. Cell Res 24:1164–1180
Ying X, Wu Q, Wu X, Zhu Q, Wang X, Jiang L, Chen X, Wang X (2016) Epithelial ovarian cancer-secreted exosomal miR-222-3p induces polarization of tumor-associated macrophages. Oncotarget 7:43076–43087
Yu X, Harris SL, Levine AJ (2006) The regulation of exosome secretion: a novel function of the p53 protein. Can Res 66:4795–4801
Yu J, Lin Y, Xiong X, Li K, Yao Z, Dong H, Jiang Z, Yu D, Yeung SJ, Zhang H (2019) Detection of exosomal PD-L1 RNA in saliva of patients with periodontitis. Front Genet 10:202
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TN, Lo RS, Ribas A (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375:819–829
Zhao R, Zhang Y, Zhang X, Yang Y, Zheng X, Li X, Liu Y, Zhang Y (2018) Exosomal long noncoding RNA HOTTIP as potential novel diagnostic and prognostic biomarker test for gastric cancer. Mol Cancer 17:68
Zhou M, Chen J, Zhou L, Chen W, Ding G, Cao L (2014) Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol 292:65–69
Zhou J, Mahoney KM, Giobbie-Hurder A, Zhao F, Lee S, Liao X, Rodig S, Li J, Wu X, Butterfield LH, Piesche M, Manos MP, Eastman LM, Dranoff G, Freeman GJ, Hodi FS (2017) Soluble PD-L1 as a biomarker in malignant melanoma treated with checkpoint blockade. Cancer Immunol Res 5:480–492
Zhu X, Shen H, Yin X, Yang M, Wei H, Chen Q, Feng F, Liu Y, Xu W, Li Y (2019) Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J Exper Clin Cancer Res CR 38:81
Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G (2015) Type I interferons in anticancer immunity. Nat Rev Immunol 15:405–414
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Hu, Y., Zhang, R., Chen, G. (2020). Exosome and Secretion: Action On?. In: Xu, J. (eds) Regulation of Cancer Immune Checkpoints. Advances in Experimental Medicine and Biology, vol 1248. Springer, Singapore. https://doi.org/10.1007/978-981-15-3266-5_19
Download citation
DOI: https://doi.org/10.1007/978-981-15-3266-5_19
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-3265-8
Online ISBN: 978-981-15-3266-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)