
Abstract
The NF-κB (Nuclear Factor kappa B) transcription factor plays crucial roles in the regulation of numerous biological processes including development of the immune system, inflammation, and innate and adaptive immune responses. Control over the immune cell functions of NF-κB results from signaling through one of two different routes: the canonical and noncanonical NF-κB signaling pathways. Present at the end of both pathways are the proteins NF-κB, IκB, and the IκB kinase (IKK). These proteins work together to deliver the myriad outcomes that influence context-dependent transcriptional control in immune cells. In the present chapter, we review the structural information available on NF-κB, IκB, and IKK, the critical terminal components of the NF-κB signaling, in relation to their physiological function.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- NF-κB (nuclear factor κB)
- IκB (inhibitor of NF-κB)
- IKK (IκB kinase)
- Signal transduction
- Transcription factor
10.1 General Overview: NF-κB and the Immune System
NF-κB was first identified in 1986 as a transcription factor with specific binding affinity for the decameric DNA sequence 5′-GGGACTTTCC-3′ located within the enhancer of the immunoglobulin kappa light chain gene in mature B and plasma cells [64]. It was subsequently shown that pathogen-derived products as well as cytokines and ultraviolet radiation induce NF-κB DNA-binding activity independent of new protein synthesis both in lymphocytes and nonlymphoid cells [63]. Today, more than 30 years and thousands of reported studies later, NF-κB is recognized as a master regulator of gene expression that profoundly influences many aspects of cellular biology. Chief among these are the roles NF-κB plays in immunology. NF-κB-dependent gene expression is required for proper development and formation of the immune system, lymphoid organogenesis, pathogen recognition and innate immune responses, immediate antimicrobial responses, inflammation, and initiation of the adaptive immune response [23, 24].
Despite its original identification as a nuclear factor in mature, antibody-producing B lymphocytes, NF-κB resides in the cytoplasm of most resting cells where its nuclear translocation and ability to activate target gene transcription can be induced as a consequence of signal transduction. Immediate control over NF-κB activity is carried out by two additional protein factors: a member of the IκB family of NF-κB inhibitor proteins and the IκB kinase (IKK) complex. After a brief introduction to the signal transduction pathways that trigger induction of NF-κB activity, the sections that follow describe how the structure and biochemistry of these three integral NF-κB pathway components inform their mechanism in regulating immune system functions.
10.2 Two Distinct NF-κB Signaling Pathways
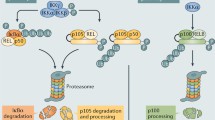
Activation of NF-κB results generally as a consequence of signal transduction through one of two different routes. These are referred to as the canonical and the noncanonical NF-κB signaling pathways (Fig. 10.1) [23, 46, 70]. Through the canonical NF-κB pathway, diverse stimuli engage immune receptors leading to rapid but transient activation of NF-κB (Fig. 10.1, left). Inflammatory cytokines such as TNFα or IL-1β, a wide variety of pathogen-associated molecular patterns (PAMPs), and antigen/immune-stimulatory signals induce NF-κB activity through the canonical NF-κB pathway. Upon recognition of the stimulatory ligand by its receptor, a signaling cascade is triggered that targets the IKK complex such that one of its catalytic subunits, known as IKK2/β, switches from its inactive state to a state of catalytic activity. This IKK2/β subunit activation requires phosphorylation of two serine (Ser) amino acid residues, Ser177 and Ser181, located within the activation loop of the IKK2/β protein kinase domain. Once activated, IKK targets IκB for phosphorylation. In resting cells, IκB proteins are tightly associated with dimeric NF-κB in a manner that prevents NF-κB from binding to DNA. In the specific case of the IκBα protein, the catalytically active IKK2/β subunit of IKK phosphorylates the NF-κB-associated inhibitor at Ser32 and Ser36. While it remains in complex with NF-κB, this newly phosphorylated IκBα then quickly undergoes ubiquitylation at residues Lys21 and Lys22 followed by its degradation via the 26 S Proteasome. Removal of IκBα activates NF-κB, typically a heterodimer of RelA (p65) and p50 subunits, which then rapidly translocates into the nucleus where it can bind to specific DNA sequences (known as κB sites or κB DNA) present within promoter or enhancer regions of distinct target genes and elevate their levels of transcription [33].

Canonical and noncanonical NF-κB pathways. Left, upon stimulation and cascade of different events, the IKK holocomplex becomes activated. The kinase activity of IKK phosphorylates IκBα, which resides in the cytoplasm bound to the NF-κB p50:RelA heterodimer. After IκBα phosphorylation and degradation by the proteasome, the NF-κB dimer becomes released and translocates into the nucleus. There, it binds to target promoters and activates gene transcription. Right, upon stimulation and cascade of different events, the NIK kinase is stabilized and phosphorylates IKK1. Upon IKK1 activation, p100 is phosphorylated and partially processed by the proteasome generating p52. Then, the heterodimer p52:RelB translocates into the nucleus and binds to target promoters to activate gene transcription
In comparison to the canonical NF-κB signaling pathway, activation of NF-κB through the noncanonical pathway generally exhibits delayed kinetics and results in a more persistent NF-κB transcriptional response (Fig. 10.1, right). Typically, the noncanonical NF-κB pathway is activated by developmental signals through specific receptors, including lymphotoxin β receptor (LTβR), B cell-activating factor receptor (BAFF-R), or CD40. The noncanonical pathway does not require the protein kinase catalytic activity of IKK2/β. Rather, it depends upon the sequential actions of NF-κB-inducing kinase (NIK) and the IKK1/α subunit to induce phosphorylation of the precursor IκB protein p100 at residues Ser866 and Ser870. In a manner similar to that described previously for IκBα, phosphorylation renders p100 a substrate for ubiquitylation via recruitment of the E3 ubiquitin-protein ligase β-TrCP1 [37]. Ubiquitylation of p100 leads to its partial processing, generating the mature NF-κB p52 subunit, which can either remain as a homodimer or associate with RelB to form the transcriptionally active NF-κB p52:RelB heterodimer. Interestingly, unlike the canonical pathway, signaling through the noncanonical NF-κB pathway does not require the NEMO/IKKγ subunit. It remains unclear, however, whether the IKK1/α targeted by inducers of the noncanonical pathway represents a distinct pool of free enzyme or if it is the same IKK1/α associated with the IKK holoenzyme complex implicated in the canonical NF-κB pathway.
10.3 The NF-κB Family Transcription Factors
In mammals, five different polypeptide subunits—RelA (p65), RelB, c-Rel, p50, and p52—associate with one another to generate functional NF-κB homo or heterodimers (Fig. 10.2). The p50 and p52 subunits represent the partially processed products of two larger precursor proteins known, respectively, as p105 and p100. In all, 15 different NF-κB homo or heterodimers are possible through combinatorial association of the five subunits. Some of these dimer combinations are abundant in diverse cell types while others are extremely rare. The NF-κB p50:RelA heterodimer, for example, is present at significant levels in most cell types. In contrast, c-Rel subunit-containing dimers are restricted primarily to lymphoid cells and a few other dimers, such as RelB homodimer and the p50:p52 heterodimer, have not been detected in cells.

Members of the NF-κB family. Left side, domains that characterize the NF-κB protein family are indicated. Abbreviations: RHR, Rel Homology Region; NTD, N-Terminal Domain; DD, Dimerization Domain; NLS, Nuclear Localization Signal; TA, Transactivation domain; LZ, Leucine Zipper; GRR, Glycine-Rich Region; ARD, Ankyrin Repeat Domain. Right side, structure of the ternary complex p50:RelA:DNA (indicated in purple, green, and blue, respectively). Important domains are highlighted. The article reference where this structure was originally published is also indicated
Each of the five NF-κB subunits shares a high degree of amino acid conservation within an N-terminal region that spans roughly 300 residues and which is referred to as the Rel homology region (RHR) (Fig. 10.2). X-ray crystallographic analyses have revealed that the RHR contains three independent structural elements: the N-terminal domain (NTD), the dimerization domain (DD), and the nuclear localization signal (NLS). Both NTD and DD are folded, globular domains, whereas the structure of the region including the NLS depends upon its interactions with other proteins. NF-κB RelA, RelB, and c-Rel subunits, but not p50 or p52, also contain unique amino acid sequences C-terminal to their respective RHR that are responsible for conveying transcriptional activation (TA). As a consequence, NF-κB dimers in possession of at least one of these three subunits exhibit inherent potential to activate target gene transcription. NF-κB p50 and p52 subunits lack this TA region entirely. Rather, the C-termini of these two subunits contain a glycine-rich region (GRR), apparently a remnant of their incomplete proteolytic processing from p105 and p100, respectively. Hence, homodimers of either p50 or p52 function as transcriptional repressors. However, these two NF-κB proteins can activate transcription of select target genes through their association with co-activating proteins in the nucleus [21, 29].
10.3.1 Rel Homology Region (RHR) Structure
The first glimpse into the structural details of the NF-κB pathway was afforded by two separate X-ray crystallographic analyses of the NF-κB p50:p50 homodimer RHR in complex with κB DNA [20, 50]. These two structures revealed a butterfly-like shape for the complexes with a double-stranded DNA “body” and p50 protein subunit RHR “wings” (Fig. 10.2). Both the NTD and DD of the RHR separately fold into variations on the common immunoglobulin fold. All DNA nitrogenous base and phosphate–ribose backbone contacts are mediated by loops that join individual beta-strands in both the NTD and DD. The NTD mediates all of the sequence-specific contacts with DNA bases, while the C-terminal DD, which is responsible for NF-κB subunit dimerization, contributes to nonspecific DNA backbone contacts. The NF-κB p50:p50 homodimer:DNA complex crystal structures demonstrated a novel mode of DNA binding whereby two copies of the p50 RHR cover one entire helical turn of the DNA [3, 49]. Since these first models were obtained, many additional NF-κB RHR:DNA X-ray crystal structures have been determined. Taken together, these models show striking similarities, but also subtle differences that have contributed significantly to deciphering the mechanism through which NF-κB selectively binds to different κB DNA sites [5,6,7,8, 11, 12, 14, 17, 18, 26, 47, 53].
10.3.2 NF-κB Subunit Dimerization
The DD of NF-κB proteins is capable of folding and assembling separately from the rest of the NF-κB RHR. This has enabled a detailed analysis of the biochemical processes that underlie NF-κB subunit dimerization selectivity. The DD is solely responsible for dimer formation, which occurs by juxtaposing two domains side-to-side with C2 point symmetry. A set of conserved amino acid residues from each subunit participate in mediating dimer formation. One of the most thermodynamically stable NF-κB dimer interfaces is the one that corresponds to the ubiquitous p50:RelA heterodimer (Fig. 10.2, top). Analysis of the respective crystallographic models yielded suggestions as to why the classical p50:RelA NF-κB heterodimer is more stable than either the p50:p50 or RelA:RelA homodimers [27]. The observed differences in dimer stability can be explained largely from variations in amino acid sequence at only two homologous positions. Asp254 and Tyr267 in murine p50 are changed to Asn200 and Phe213, respectively, in murine RelA. In the case of the p50:RelA heterodimer, the Asp and Asn of the two subunits form a stable hydrogen bond at center of the dimer interface. In contrast, within the context of either the p50:p50 or RelA:RelA homodimers, the juxtaposition of Asp-Asp or Asn-Asn at the interface is detrimental to dimer stability. Moreover, the hydroxyl group of Tyr267 in p50 forms hydrogen bonds to the peptide backbone of its dimerization partner, contributing to stabilization of both the p50:p50 homodimer as well as the p50:RelA heterodimer. Substitution of Phe at this position in the RelA subunit serves to further destabilize the RelA:RelA homodimer relative to p50:p50 or p50:RelA. Taken together, these structural observations predict a rank of dimerization stability in which p50:RelA > p50:p50 > RelA:RelA, which has subsequently been shown experimentally to be the case [54, 65].
Despite its success in explaining preferential dimerization between p50 and RelA, direct contact between complementary amino acids at the dimer interface fails to completely explain all the observed trends in NF-κB subunit dimerization. Mutational studies have revealed that some DD amino acid residues that do not mediate intersubunit contacts can influence the process. For example, changing murine RelA Cys216 to Ala affects homodimer formation [19]. However, the role of non-interfacial amino acid residues in dimerization is most strikingly illustrated in the case of the NF-κB RelB subunit. All the residues in RelB that participate directly in dimerization are either identical or homologous to those of other NF-κB subunits. And yet, RelB assembles into homodimers only through a completely unique domain-swapped arrangement [28]. The domain swapping observed in RelB:RelB homodimers occurs as a consequence of destabilization of the folded RelB dimerization domain, suggesting that domain stability is an important determinant for protein:protein interaction. In cells, decreased folding stability in both the N-terminal and dimerization domains contributes to its degradation by the proteasome, which explains why the RelB homodimer does not exist in vivo [40].
10.3.3 κB DNA binding by NF-κB
NF-κB recognizes 9–11 base pairs long specific double-stranded κB DNA elements located within the promoters or enhancers of numerous target genes. The κB site consensus sequence is 5′-GGGRNWYCC-3′, where R = A or G; N = any nucleotide; W = A or T, and Y = C or T. The critical feature of these κB sites is the presence of G:C bp at both 5′-ends, whereas the central region displays more sequence variation. The κB sites are pseudosymmetric with two nearly identical half sites. Each subunit in one NF-κB dimer may bind to a half site, with residues from the NTD and from a stretch of roughly 10 amino acids in length that flexibly link the NTD and DD mediating all base sequence-specific contacts. In general, the p50 and p52 subunits contact 5 bp half sites, whereas RelA, RelB, and c-Rel subunit proteins bind to 4 bp half sites. However, there are exceptions to this rule. For example, the NF-κB RelA:RelA homodimer can bind with near-perfect symmetry to some 9 bp κB sites with two 4 bp half sites. However, the same RelA homodimer binds asymmetrically to a different 10 bp κB site where one RelA subunit contacts the 5 bp half site in a manner reminiscent of the p50 subunit [8]. Perhaps the most striking biochemical observation of NF-κB is its ability to employ distinct modes and bind diverse κB sites. In one extreme case, the dimer binds κB DNA with only one consensus half site and a second half site that bears no resemblance to the consensus [7]. The modular architecture of the RHR, in which the dimerization function of the DD and the DNA base-contacting potential of the flexibly linked NTD can function independent of one another, permits a range of variability in DNA-binding mode. For example, in the case of the RelA:RelA homodimer binding to κB DNA in which only one half site is present, the NTD from one RelA RHR participates in normal base-specific contacts with its half site while the NTD from the other RelA subunit moves relative to the DD and participates in multiple nonspecific contacts with the DNA backbone. As a consequence, the DNA-binding affinity of RelA homodimer for the variant κB DNA is mostly preserved despite the specificity being significantly diminished.
Sequence-dependent preferred DNA conformations also play a vital role in the recognition of κB DNA by NF-κB. Targeting of DNA with A/T-rich sequences at their center allows for κB DNA with greater flexibility to accommodate NF-κB binding. Interestingly, A:T is by far the preferred central bp. Whereas the presence of a G:C or C:G bp at this central position is highly unusual, this position is never directly contacted by atoms from the NF-κB dimer, which further illustrates the role DNA structure plays in facilitating complex formation. Finally, it is worth noting that κB DNA sites can be located either very close to the transcription start site of target genes or as far as many kilobases away. Moreover, some genes contain a single κB site while others rely upon multiple copies.
10.4 The IκB Family
IκB proteins are vital players in the regulation of the NF-κB pathway. By biasing NF-κB nucleocytoplasmic dynamics and blocking its DNA binding, IκB proteins function generally as inhibitors of NF-κB through noncovalent binding. All proteins of the IκB family possess an ankyrin repeat domain (ARD). The IκB ARD is a structurally ordered domain consisting of six or seven copies of a repeating structural motif known as the ankyrin repeat (AR). Proteins of the IκB family can be classified functionally into three separate groups: classical IκB (IκBα, IκBβ, and IκBε), precursor IκB (p105/IκBγ and p100/IκBδ), and nuclear IκB (Bcl-3, IκBζ, IκBNS, and IκBη) (Fig. 10.3) [30]. Whereas all IκB proteins bind with high affinity to form stable complexes with dimeric NF-κB proteins, the classical and precursor IκB function primarily as inhibitors by retaining NF-κB in a transcriptionally inactive state in the cytoplasm. Interestingly, these inhibitors display differing binding preferences and specificities for various NF-κB subunits. Functional consequences of the third class of IκB proteins are more complex. As suggested by their name, these nuclear IκB proteins associate with particular NF-κB dimers primarily in the nucleus and have been shown to function as co-activators for expression of selected NF-κB target genes in a cell type-specific manner.

Members of the IκB family. Left side, domains that characterize the IκB protein family are indicated. Abbreviations: ARD, Ankyrin Repeat Domain; PEST, domain rich in Proline (P), Glutamate (E), Serine (S), and Threonine (T). Right side, structure of the ternary complex p50:RelA:IκBα (indicated in blue, red, and gray, respectively). Important domains are highlighted. The article reference where this structure was originally published is also indicated
10.4.1 Classical IκB Protein Structure
There are three classical IκB proteins: IκBα, IκBβ, and IκBε. These inhibitory proteins regulate the NF-κB pathway through at least two mechanisms. First, they control the nucleocytoplasmic distribution of RelA or c-Rel subunit-containing NF-κB dimers under non-stimulated conditions through interactions with their NLS. Second, most inducers of NF-κB activity ultimately lead to elevated expression of IκBα and IκBε and these newly synthesized IκB proteins can enter the nucleus on their own to competitively remove NF-κB from κB DNA and return it to the cytoplasm [4]. Importantly, the characteristic differences in kinetics of IκB protein expression coupled with their varied rates of constitutive and signal-induced degradation results in the observed periodic mobilization of NF-κB in waves of activity [25, 66].
The classical IκB proteins share a central ARD of six ankyrin repeats. Ankyrin repeats are a common tandem helical repeat motif of roughly 33 amino acids in length. The individual ankyrin repeats stack to form domains that are commonly present in proteins involved in protein:protein interactions. The ARD of classical IκB is flanked by sequences that are predicted to be unstructured. The N-terminal flexible region in IκBα, IκBβ, and IκBε contain two conserved Ser residues within the consensus sequence DSGXXS that are sites for phosphorylation by the IKK2/β subunit of the IKK complex. The classical IκB C-terminal region contains a structurally flexible region rich in the amino acids proline, glutamic acid, serine, and threonine that is called the PEST domain and which is a region commonly found in proteins that display rapid proteolytic turnover in cells. IκBα, by far the best-studied of all IκB proteins, is inherently thermodynamically unstable with respect to its folding and, consequently, is rapidly degraded in cells through a signal-independent mechanism dependent upon its own PEST domain and the 20 S Proteasome [43]. However, upon binding to NF-κB, the stability of IκBα increases significantly and its degradation becomes dependent upon specific phosphorylation- and ubiquitylation-dependent signaling events. The drastic change in folding stability for IκBα in complex with NF-κB is likely linked to the relatively high affinity of the two proteins, which bind one another with a dissociation constant in the high picomolar range.
The X-ray crystal structures of IκBα in complex with the NF-κB p50:RelA heterodimer and IκBβ:RelA homodimer complexes provide a molecular framework for understanding how IκB proteins regulate the function of NF-κB [31, 34, 39]. IκB uses its entire ARD as well as its proximal PEST domain to form a stable interaction with NF-κB. In the case of the IκBα:p50:RelA complex, the C-terminal 30 residues from the RelA RHR subunit, which were disordered in the NF-κB:DNA complex structures, adopt a helical structure upon binding to ankyrin repeats 1 and 2 of IκBα (Fig. 10.3). The RelA subunit NLS is contained within this C-terminal segment, which is directly masked through its ordering and association with IκBα. On the C-terminal end, the IκBα PEST domain residues and sixth ankyrin repeat combine to create a vast acidic surface that appears to electrostatically attract the RelA NTD into a drastically different position from that displayed in its DNA-bound conformations. For many years, it was speculated that this type of interaction might help explain how IκBα strips NF-κB off the DNA. More recently, NMR studies have revealed that the negatively charged PEST sequence of IκBα interacts with positively charged residues in the DNA-binding pocket of NF-κB electrostatically repelling DNA from NF-κB through a mechanism that has been termed “molecular stripping” [57, 69]. Such a mechanism suggests at least transient ternary complex formation of IκB upon the NF-κB:DNA complex prior to removal of NF-κB from the DNA. Data in support of this model include the recent observation that an IκBα PEST domain mutant in which the negatively charged residues were mutated to neutral polar residues yielded a detectable IκBα:NF-κB:DNA ternary complex [16]. It has further been shown that IκBα binding generates a twist in NF-κB heterodimer conformation such that it may not be able to remain bound to DNA facilitating disassociation of NF-κB from the DNA [59].
A similar mechanism is expected for IκBβ upon binding to NF-κB RelA homodimer due to the observation that interactions between the IκBβ ARD and the RelA NLS are nearly identical to those observed in the interaction of IκBα and RelA. However, some differences between the crystallographic models were observed such as the lack of involvement of IκBβ PEST domain in stabilizing the interaction [39]. Significant differences in domain architecture between IκBε and other classical IκB proteins include the relative absence of acidic amino acid residues within the C-terminal PEST region and an extended N-terminal region. More structural and in vitro biochemical studies are required in order to gain mechanistic insight into how IκBε regulates NF-κB activity [30].
10.4.2 The Precursor IκB Proteins
The two precursor IκB proteins are p105 and p100. These are referred to as precursors because, in addition to performing as bona fide cytoplasmic inhibitors of otherwise functional NF-κB dimers, p105 and p100 also operate as the immature precursors to NF-κB subunits p50 and p52, respectively. The degree to which the precursor IκB proteins contribute to NF-κB regulation is not insignificant, as p105 and p100 account for inhibition of nearly half the NF-κB dimers present in the cytoplasm in resting cells. Because of their NF-κB inhibitory activity, p105 and p100 are also sometimes referred to as IκBγ and IκBδ, respectively. The C-terminal regions of both proteins contain the ARD. Although it contains seven rather than six ankyrin repeats, the ARD of p105 and p100 is similar to all other IκB proteins in primary structure and is essential for inhibition NF-κB. Initial biochemical studies using Raw264.7 cells demonstrated that the NF-κB precursor protein p105 binds all NF-κB proteins barring RelB by forming multimeric high molecular weight complexes [61]. Similarly, p100 also forms multimeric high molecular weight complexes by binding to all NF-κB subunits except p50. These multi-protein inhibitory complexes have been referred to as “kappaBsomes” [71]. In support of these large multi-protein structures, in the native state, p100/IκBγ and p100/IκBδ are thought capable of binding to NF-κB dimers in two modes: by dimerization with NF-κB proteins through the p100/p105 RHR domains and by binding to NF-κB dimers through their respective C-terminal ARD.
The X-ray crystal structure of the C-terminal domain of p100/IκBδ partly explains how p100 forms the kappaBsome [71]. In the crystallographic model, two apparently stable IκBδ dimers contact each other through another protein–protein interface forming a tetramer. In this tetramer, one of the subunits in each dimer is not involved in tetrameric contacts, suggesting different functional consequences for the two types of ARDs in the tetramer. In general, residues at the dimer interface are highly conserved between p100 and p105. However, strikingly, these two molecules share no sequence similarity at the tetramer interface. This explains why p105 forms a 2:2 complex with NF-κB subunits whereas p100 can potentially form 4:4 complexes. Furthermore, cellular experiments using mutant versions defective in both dimer and tetramer formation reveal that the NF-κB inhibitory activity of IκBδ requires its ability to form the kappaBsome multimeric complex.
10.4.3 The Nuclear IκB Proteins
The nuclear IκB proteins are Bcl-3, IκBζ, IκBNS, and IκBη [62]. These proteins are classified as IκB proteins due to the fact that they contain ankyrin repeats and bind with specificity to NF-κB. Unlike classical IκB proteins, however, nuclear IκB proteins lack N-terminal signal-dependent phosphorylation sites or C-terminal PEST regions. Furthermore, when overexpressed in cells they accumulate in the nucleus. Another difference from classical IκB proteins is that they display binding preference toward NF-κB p50 and p52 homodimers [22, 74, 78]. The fact that the NF-κB proteins targeted by nuclear IκB do not contain their own inherent potential to activate expression of genes coupled with the observation that they are an absolute requirement for delayed expression of select pro-inflammatory factors that lag behind the initial NF-κB response genes suggest that nuclear IκB proteins are involved in coordinating a “second wave” of NF-κB-dependent gene expression [67, 77]. Whether this occurs via the regulation of NF-κB dimer exchange, the stabilization of NF-κB dimers on the DNA, or through recruitment of histone modifying enzymes remains an open question. However, recent observations implicate IκBζ and Akirin2 in linking NF-κB p50 homodimer to SWI/SNF chromatin remodeling complexes during innate immune responses leading to expression of the pro-inflammatory cytokines IL-6 and IL-12b [72].
The X-ray crystal structure of the free Bcl-3 ARD provides a template for understanding nuclear IκB function [45]. The Bcl-3 ARD is composed of seven individual ankyrin repeats. Several residues that are conserved between Bcl-3 and classical IκB proteins occupy similar positions on the surface and are likely involved in analogous interactions with the NLS and DD portions of its preferred p52:p52 homodimer-binding partner. Interestingly, when the IκBα:NF-κB complex crystal structure is employed to direct placement of the Bcl-3 ARD upon a p52:p52 homodimer:DNA complex crystal structure, the resulting composite model reveals severe clashes between ankyrin repeat 7 of Bcl-3 and the bound κB DNA. Therefore, there must either be some combination of movement of the Bcl-3 ARD relative to its NF-κB-binding partner and/or severe DNA bending in order to support ternary complex formation.
10.5 The IKK Complex
The IκB kinase (IKK) complex is composed of three subunits [33]. IKK1/α and IKK2/β are two very similar proteins at the levels of amino acid sequence homology through protein tertiary structural conservation (Fig. 10.4). Both contain functioning catalytic protein kinase domains at their N-terminal ends. The third IKK subunit is the necessary adapter protein known as NEMO/IKKγ. Other proteins have been suggested to participate in this complex. However, the identity of these additional factors and the nature of their association with the three core members of the IKK complex remain controversial topics. Initial biochemical characterization determined that endogenous IKK in activated HeLa cells exhibits a high molecular weight of around 700–900 kDa, suggesting that multiple copies of IKK1/α (85 kDa), IKK2/β (87 kDa) and NEMO/IKKγ (52 kDa) might be present [9]. It is possible, however, that the unique structural properties of NEMO might be responsible for this unusually high hydrodynamic radius. Therefore, IKK may not contain multiple copies of each subunit. In the absence of an experimentally determined three-dimensional structure of the native IKK complex, it remains speculative as to how the three IKK subunits assemble to form the holoenzyme complex.

Members of the IKK family. Left side, domains that characterize the IKK protein family are indicated. Abbreviations: KD, Kinase Domain; ULD, Ubiquitin-Like Domain, SDD, Scaffold Dimerization Domain; LZ, Leucine Zipper; HLH, Helix-Loop-Helix; NBD, NEMO-Binding Domain; UBD, ubiquitin-binding region embedded in the LZ domain; CC, Coiled-Coil; ZF, Zinc Finger domain. Right side, structures of IKK1 and IKK2 dimeric complexes that can form multimeric complexes and NEMO dimers (each monomer is indicated in a different color). Important domains are highlighted. The article references where these structures were originally published are also indicated
10.5.1 IKK Catalytic Subunit Structures
IKK1/α and IKK2/β share nearly 50% sequence identity and similar domain organization: an N-terminal kinase domain (KD) followed by a ubiquitin-like domain (ULD) and a scaffold/dimerization domain (SDD) (Fig. 10.4). The evolutionarily related IKKε and TANK-binding kinase 1 (TBK1) are the only other proteins known to share this domain structure. The C-terminal ~50 residues of both IKK1/α and IKK2/β interact with the N-terminus of NEMO. Moreover, IKK1/α but not IKK2/β contains a putative nuclear localization signal [68], possibly related to its reported activities in the nucleus. The first IKK X-ray crystal structure determined was IKK2/β from Xenopus laevis [76]. This was followed by two X-ray structures of human IKK2/β [38, 55]. These structures revealed that the KD and ULD are closely associated with one another. In addition, they revealed the SDD as an elongated helical domain dominated by three very long alpha-helices that contact the KD-ULD and mediate homodimerization. Previous to determination of the X-ray crystal structures, this portion of the protein had been predicted to contain a helix-loop-helix and leucine zipper-like motifs. Subsequent determination of high-resolution models for human IKK1 from combined X-ray crystallography and cryo-electron microscopy data has been reported recently [56].
As introduced in Sect. 10.2, signal-induced activation of IKK through either the canonical or noncanonical NF-κB signaling pathways requires the phosphorylation of two Ser residues (Ser176 and Ser180 for IKK1/α and Ser177 and Ser181 for IKK2/β) located within the activation loop of the respective protein kinase domains [15, 44]. However, the mechanism by which the kinase-containing subunits in the IKK complex become activated is not fully understood. Diverse kinases such as TAK1 or MEKK3 have been reported to function upstream to phosphorylate and consequently activate the IKK complex [51, 75]. However, other studies conclude that IKK activation occurs independent of an inducing kinase activity, invoking the possibility for a mechanism involving stimulus-dependent conformational change or oligomerization and autophosphorylation of the IKK proteins. Interestingly, the X-ray structures of IKK1 and IKK2 revealed unique multimeric organizations. Structural analysis of the catalytically active human IKK2/β showed that the enzyme adopts a unique open conformation that permits pairs of IKK2 enzymes to form higher order assemblies in which their catalytic domains are in close proximity to support trans autophosphorylation. Mutations at the surfaces on IKK2/β that mediate these homotypic interactions in the crystal resulted in IKK2/β enzymes that were greatly impaired in their ability to become activated in cells. Similar results were obtained when residues that mediate dimerization were mutated in the Xenopus IKK2/β.
In the case of the human IKK1/α, its structural analysis revealed that it also assembles in an oligomeric fashion [56]. Although dimerization mediated by the SDD occurs in a similar manner in both IKK1/α and IKK2/β, the two catalytic IKK complex subunits differ significantly in their surfaces that mediate the higher oligomerization states observed in the structural models. Interestingly, mutation of a unique set of surface exposed amino acid residues observed to participate in IKK1/α oligomerization into hexamers on the cryo-EM grid was shown to correlate with disruption of p100 processing in cells. This suggests a role for higher order assembly of IKK1/α during the noncanonical NF-κB signaling pathway, perhaps by interaction with NIK. Although more work is needed to fully understand the process of IKK activation, these observations strongly suggest that protein surfaces which are unique to either IKK1/α and IKK2/β contribute to their distinct propensities for oligomerization in ways that control specific signaling outcomes.
10.5.2 The Enigmatic NEMO/IKKγ
NEMO, also known as IKKγ, is the third component of the multisubunit IKK complex. Contrary to IKK1/α and IKK2/β, NEMO/IKKγ lacks kinase catalytic activity and instead serves an essential accessory role in activation of NF-κB via the canonical signaling pathway. The structure of NEMO/IKKγ is dominated by alpha-helical elements including two coiled-coil regions (CC1 and CC2), a helical ubiquitin-binding domain (UBD), and a leucine zipper (LZ). A zinc finger (ZF) domain caps off the NEMO/IKKγ structure at its extreme C-terminus. The N-terminal CC1 region binds through noncovalent interactions with the C-terminal portions of IKK1/α and IKK2/β, as mentioned previously [35]. Although the CC2 and LZ regions of NEMO have been suggested to mediate subunit trimerization [1] or tetramerization [73], the N-terminal domain encompassing CC1 has been reported to exist as a dimer [41]. Consequently, and in spite of the fact that no complete model for NEMO/IKKγ has yet to be determined experimentally, it structure is thought to consist of two long, alpha-helical coiled-coils each capped with a C-terminal zinc finger domain.
X-ray crystal structures of several portions of NEMO/IKKγ have been determined. The first consisted of NEMO/IKKγ residues 44–111, which constitute the minimal domain for stable interaction with the C-terminal peptides of IKK1/α and IKK2/β. Two X-ray crystal structures containing this domain in complex with IKK1/α and IKK2/β C-terminal peptides revealed that they form coiled-coil homodimers into which the IKK1/α and IKK2/β C-terminal peptides fit as helices to reinforce NEMO/IKKγ dimerization at the coiled-coil interface [60]. Determination of these models aided in understanding how phosphorylation of specific Ser residues in this region might affect IKK complex formation [52].
Several additional X-ray structures reveal how coiled-coil homodimerization is stabilized upon interaction of portions of NEMO/IKKγ with factors that are known to activate NF-κB. For example, crystallographic models of NEMO/IKKγ residues 150–272, containing portions of the CC1 and CC2 regions, in complex with the activator protein vFLIP from Kaposi’s Sarcoma-associated Herpesvirus (KSHV) show how the viral proteins hold the two coiled-coil protomers in place [2]. Of particular interest has been the manner by which coiled-coil NEMO/IKKγ homodimers are stabilized through their interaction with ubiquitin chains. The X-ray crystal structure of NEMO/IKKγ residues 250–339, which represents major portions of the CC2 and LZ regions, in complex with linear diubiquitin and K63-linked diubiquitin revealed how the ubiquitin proteins clamp down to hold the NEMO/IKKγ homodimer together [58, 79]. This discovery helped usher in the concept of linear ubiquitin as a constituent of immune cell signal transduction and served partially to explain the paradigm shifting observation that free ubiquitin is required for activation of IKK activity [10]. Additional NEMO/IKKγ-ubiquitin interaction events have been reported, including covalent attachment and K63-linked polyubiquitin chains, as well as demonstrations of how those ubiquitin chains facilitate the recruitment of other proteins to the IKK complex promote NF-κB activation [13].
10.6 Structural Immunology and NF-κB Signaling
Structural characterization of NF-κB, IκB, and IKK proteins has revealed their unique three-dimensional folds and arrangements in space. The important question, however, is how do these NF-κB pathway protein structures relate to immunology? As a rule, the high-resolution structure determination of complicated macromolecular assemblies serves not so much to explain biomolecule function as to better inform the generation of testable mechanistic hypotheses. By this yardstick, structural biology of the integral components of NF-κB signaling has proven an unqualified success.
Detailed structural analyses of NF-κB homo- and heterodimers in complex with different κB DNA sequences have generated a wealth of observations regarding DNA target site selection and the consequences of changes in DNA sequence on target gene expression levels. For instance, the observation that the DNA-binding RHR of NF-κB can adapt its structure to accommodate DNA sequences that differ significantly in one half site suggests that NF-κB is capable of interacting with a much larger cadre of targets than originally imagined. This suspicion is currently being confirmed as large scale, genomewide studies to identify NF-κB associated with genes for which there had not previously been a reason to suspect it [42]. Besides implicating NF-κB on more genes, the adaptability of NF-κB observed in X-ray crystal structures suggests that a single dimer can activate or repress hundreds of target genes by binding to diverse sequences. At the same time, the importance of particular base positions within the κB DNA consensus in determining whether or not transcription can start from that promoter suggests that even subtle structural changes can have profound effects on the consequence of NF-κB:DNA complex formation [36]. The NF-κB:DNA complex structures have allowed for visualization of the protein surfaces available to bind to other factors. As surface residues vary among NF-κB family subunits, the different dimers could interact with specific partners. Such interaction with additional nuclear factors on DNA represents another layer of complexity in regulation of target gene expression. Finally, many studies have consistently demonstrated that the activation domains of other transcription factors play roles in determining the stability and specificity of NF-κB RelA subunit interactions with DNA complexes via transient interactions with the DNA-binding RelA NTD. With the aid of high-resolution structural models, in vitro biochemistry, and computational modeling software, the portions of NF-κB and DNA involved in these interactions are being determined [48].
In addition to suggesting mechanisms for how the association with classical IκB proteins influences NF-κB subcellular localization and DNA binding, both of which continue to be supported by increasingly robust experimental approaches, the IκB:NF-κB complex crystal structures provide a template for directing binding toward specific NF-κB subunits. Interestingly, when researchers attempted to engineer the NF-κB p50 subunit by mutagenesis to gain the high-affinity binding to IκBα normally exhibited by RelA, they found that binding specificity results not only through direct contacts between IκBα and NF-κB, but also from amino acids at positions that are far from the binding interface [32]. However, it remains to be determined whether the other classes of IκB proteins rely upon similar determinants for NF-κB-binding partner specificity. Moreover, the question of how nuclear IκB proteins function through NF-κB on DNA promoters to regulate selected target gene expression remains to be worked out.
Three-dimensional structures of the catalytic IKK subunits have provided a wealth of information about their reliance upon oligomerization as a necessary step in converting from states of inactivity to their catalytically active forms. The observation that the IKK1/α and IKK2/β subunits rely upon chemically unique surfaces to mediate their own distinct, preferred oligomeric arrangements is striking—particularly in light of the fact that the two catalytic IKK subunits are so closely related, even at the level of primary amino acid sequence, and yet function distinctly in the canonical and noncanonical NF-κB signaling pathways. It remains to be seen whether the unique oligomerization surfaces of IKK1/α and IKK2/β are amenable to targeting by small molecules in order to selectively inhibit one pathway independently of the other. Finally, the incomplete NEMO/IKKγ structural work remains. Each of the portions of NEMO/IKKγ studied structurally thus far suggest that their coiled-coil structures are imperfect in their dimerization and must require additional stabilizing factors. This raises the question of whether at cellular concentrations, NEMO/IKKγ could exist as an unfolded monomer. Such a model could explain the necessary role of polyubiquitin or other factors that might serve to stabilize the NEMO dimer in a way that is essential for IKK activation.
References
Agou F, Ye F, Goffinont S, Courtois G, Yamaoka S, Isräel A, Veron M (2002) NEMO trimerizes through its coiled-coil C-terminal domain. J Biol Chem 277(20):17464–17475. https://doi.org/10.1074/jbc.M201964200
Bagneris C, Ageichik AV, Cronin N, Wallace B, Collins M, Boshoff C, Waksman G, Barrett T (2008) Crystal structure of a vFlip-IKKgamma complex: insights into viral activation of the IKK signalosome. Mol Cell 30(5):620–631. https://doi.org/10.1016/j.molcel.2008.04.029
Baltimore D, Beg AA (1995) DNA-binding proteins. A butterfly flutters by. Nature 373(6512):287–288. https://doi.org/10.1038/373287a0
Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA (2009) Kinetic enhancement of NF-κB:DNA dissociation by IκBα. Proc Natl Acad Sci USA 106(46):19328–19333. https://doi.org/10.1073/pnas.0908797106
Berkowitz B, Huang DB, Chen-Park FE, Sigler PB, Ghosh G (2002) The X-ray crystal structure of the NF-κB p50/p65 heterodimer bound to the interferonβ κB site. J Biol Chem 277(27):24694–24700. https://doi.org/10.1074/jbc.M200006200
Chen FE, Huang DB, Chen YQ, Ghosh G (1998) Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 391(6665):410–413. https://doi.org/10.1038/34956
Chen YQ, Ghosh S, Ghosh G (1998) A novel DNA recognition mode by the NF-κB p65 homodimer. Nat Struct Biol 5(1):67–73
Chen YQ, Sengchanthalangsy LL, Hackett A, Ghosh G (2000) NF-κB p65 (RelA) homodimer uses distinct mechanisms to recognize DNA targets. Structure 8(4):419–428
Chen ZJ, Parent L, Maniatis T (1996) Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell 84(6):853–862
Chen ZJ, Sun LJ (2009) Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 33(3):275–286. https://doi.org/10.1016/j.molcel.2009.01.014
Chen-Park FE, Huang DB, Noro B, Thanos D, Ghosh G (2002) The κB DNA sequence from the HIV long terminal repeat functions as an allosteric regulator of HIV transcription. J Biol Chem 277(27):24701–24708. https://doi.org/10.1074/jbc.M200007200
Cheng CS, Feldman KE, Lee J, Verma S, Huang DB, Huynh K, Chang M, Ponomarenko JV, Sun SC, Benedict CA, Ghosh G, Hoffmann A (2011) The specificity of innate immune responses is enforced by repression of interferon response elements by NF-κB p 50. Sci Signal 4(161):ra11. https://doi.org/10.1126/scisignal.2001501
Courtois G, Isräel A (2011) IKK regulation and human genetics. Curr Top Microbiol Immunol 349:73–95. https://doi.org/10.1007/82_2010_98
Cramer P, Larson CJ, Verdine GL, Müller CW (1997) Structure of the human NF-κB p 52 homodimer-DNA complex at 2.1 Å resolution. EMBO J 16(23):7078–7090. https://doi.org/10.1093/emboj/16.23.7078
Delhase M, Hayakawa M, Chen Y, Karin M (1999) Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science 284(5412):309–313
Dembinski HE, Wismer K, Vargas JD, Suryawanshi GW, Kern N, Kroon G, Dyson HJ, Hoffmann A, Komives EA (2017) Functional importance of stripping in NFκB signaling revealed by a stripping-impaired IκBα mutant. Proc Natl Acad Sci USA 114(8):1916–1921. https://doi.org/10.1073/pnas.1610192114
Escalante CR, Shen L, Thanos D, Aggarwal AK (2002) Structure of NF-κB p50/p65 heterodimer bound to the PRDII DNA element from the interferon-β promoter. Structure 10(3):383–391
Fusco AJ, Huang DB, Miller D, Wang VY, Vu D, Ghosh G (2009) NF-κB p52:RelB heterodimer recognizes two classes of κB sites with two distinct modes. EMBO Rep 10(2):152–159. https://doi.org/10.1038/embor.2008.227
Ganchi PA, Sun SC, Greene WC, Ballard DW (1993) A novel NF-κB complex containing p65 homodimers: implications for transcriptional control at the level of subunit dimerization. Mol Cell Biol 13(12):7826–7835
Ghosh G, van Duyne G, Ghosh S, Sigler PB (1995) Structure of NF-κB p50 homodimer bound to a κB site. Nature 373(6512):303–310. https://doi.org/10.1038/373303a0
Ghosh G, Wang VY, Huang DB, Fusco A (2012) NF-κB regulation: lessons from structures. Immunol Rev 246(1):36–58. https://doi.org/10.1111/j.1600-065X.2012.01097.x
Hatada EN, Nieters A, Wulczyn FG, Naumann M, Meyer R, Nucifora G, McKeithan TW, Scheidereit C (1992) The ankyrin repeat domains of the NF-κB precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-κB DNA binding. Proc Natl Acad Sci USA 89(6):2489–2493
Hayden MS, Ghosh S (2004) Signaling to NF-κB. Genes Dev 18(18):2195–2224. https://doi.org/10.1101/gad.1228704
Hayden MS, West AP, Ghosh S (2006) NF-κB and the immune response. Oncogene 25(51):6758–6780. https://doi.org/10.1038/sj.onc.1209943
Hoffmann A, Levchenko A, Scott ML, Baltimore D (2002) The IκB-NF-κB signaling module: temporal control and selective gene activation. Science 298(5596):1241–1245. https://doi.org/10.1126/science.1071914
Huang DB, Chen YQ, Ruetsche M, Phelps CB, Ghosh G (2001) X-ray crystal structure of proto-oncogene product c-Rel bound to the CD28 response element of IL-2. Structure 9(8):669–678
Huang DB, Huxford T, Chen YQ, Ghosh G (1997) The role of DNA in the mechanism of NFκB dimer formation: crystal structures of the dimerization domains of the p50 and p65 subunits. Structure 5(11):1427–1436
Huang DB, Vu D, Ghosh G (2005) NF-κB RelB forms an intertwined homodimer. Structure 13(9):1365–1373. https://doi.org/10.1016/j.str.2005.06.018
Huxford T, Ghosh G (2009) A structural guide to proteins of the NF-κB signaling module. Cold Spring Harb Perspect Biol 1(3):a000075. https://doi.org/10.1101/cshperspect.a000075
Huxford T, Hoffmann A, Ghosh G (2011) Understanding the logic of IκB:NF-κB regulation in structural terms. Curr Top Microbiol Immunol 349:1–24. https://doi.org/10.1007/82_2010_99
Huxford T, Huang DB, Malek S, Ghosh G (1998) The crystal structure of the IκBα/NF-κB complex reveals mechanisms of NF-κB inactivation. Cell 95(6):759–770
Huxford T, Mishler D, Phelps CB, Huang DB, Sengchanthalangsy LL, Reeves R, Hughes CA, Komives EA, Ghosh G (2002) Solvent exposed non-contacting amino acids play a critical role in NF-κB/IκBα complex formation. J Mol Biol 324(4):587–597
Isräel A (2010) The IKK complex, a central regulator of NF-κB activation. Cold Spring Harb Perspect Biol 2(3):a000158. https://doi.org/10.1101/cshperspect.a000158
Jacobs MD, Harrison SC (1998) Structure of an IκBα/NF-κB complex. Cell 95(6):749–758
Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U (2000) CIKS, a connection to IκB kinase and stress-activated protein kinase. Proc Natl Acad Sci USA 97(19):10494–10499. https://doi.org/10.1073/pnas.190245697
Leung TH, Hoffmann A, Baltimore D (2004) One nucleotide in a κB site can determine cofactor specificity for NF-κB dimers. Cell 118(4):453–464. https://doi.org/10.1016/j.cell.2004.08.007
Liang C, Zhang M, Sun SC (2006) β-TrCP binding and processing of NF-κB2/p100 involve its phosphorylation at serines 866 and 870. Cell Signal 18(8):1309–1317. https://doi.org/10.1016/j.cellsig.2005.10.011
Liu S, Misquitta YR, Olland A, Johnson MA, Kelleher KS, Kriz R, Lin LL, Stahl M, Mosyak L (2013) Crystal structure of a human IκB kinase β asymmetric dimer. J Biol Chem 288(31):22758–22767. https://doi.org/10.1074/jbc.M113.482596
Malek S, Huang DB, Huxford T, Ghosh S, Ghosh G (2003) X-ray crystal structure of an IκBβ:NF-κB p65 homodimer complex. J Biol Chem 278(25):23094–23100. https://doi.org/10.1074/jbc.M301022200
Marienfeld R, Berberich-Siebelt F, Berberich I, Denk A, Serfling E, Neumann M (2001) Signal-specific and phosphorylation-dependent RelB degradation: a potential mechanism of NF-κB control. Oncogene 20(56):8142–8147. https://doi.org/10.1038/sj.onc.1204884
Marienfeld RB, Palkowitsch L, Ghosh S (2006) Dimerization of the IκB kinase-binding domain of NEMO is required for tumor necrosis factor α-induced NF-κB activity. Mol Cell Biol 26(24):9209–9219. https://doi.org/10.1128/MCB.00478-06
Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, Weissman S, Snyder M (2003) Distribution of NF-κB-binding sites across human chromosome 22. Proc Natl Acad Sci USA 100(21):12247–12252. https://doi.org/10.1073/pnas.2135255100
Mathes E, Wang L, Komives E, Ghosh G (2010) Flexible regions within IκBα create the ubiquitin-independent degradation signal. J Biol Chem 285(43):32927–32936. https://doi.org/10.1074/jbc.M110.107326
Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A (1997) IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278(5339):860–866
Michel F, Soler-Lopez M, Petosa C, Cramer P, Siebenlist U, Müller CW (2001) Crystal structure of the ankyrin repeat domain of Bcl-3: a unique member of the IκB protein family. EMBO J 20(22):6180–6190. https://doi.org/10.1093/emboj/20.22.6180
Mitchell S, Vargas J, Hoffmann A (2016) Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med 8(3):227–241. https://doi.org/10.1002/wsbm.1331
Moorthy AK, Huang DB, Wang VY, Vu D, Ghosh G (2007) X-ray structure of a NF-κB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem κB sites. J Mol Biol 373(3):723–734. https://doi.org/10.1016/j.jmb.2007.08.039
Mulero MC, Shahabi S, Ko MS, Schiffer JM, Huang DB, Wang VY, Amaro RE, Huxford T, Ghosh G (2018) Protein cofactors are essential for high-affinity DNA binding by the nuclear factor κB RelA subunit. Biochemistry 57(20):2943–2957. https://doi.org/10.1021/acs.biochem.8b00158
Müller CW, Rey FA, Harrison SC (1996) Comparison of two different DNA-binding modes of the NF-κB p50 homodimer. Nat Struct Biol 3(3):224–227
Müller CW, Rey FA, Sodeoka M, Verdine GL, Harrison SC (1995) Structure of the NF-κB p50 homodimer bound to DNA. Nature 373(6512):311–317. https://doi.org/10.1038/373311a0
Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K (1999) The kinase TAK1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398(6724):252–256. https://doi.org/10.1038/18465
Palkowitsch L, Leidner J, Ghosh S, Marienfeld RB (2008) Phosphorylation of serine 68 in the IκB kinase (IKK)-binding domain of NEMO interferes with the structure of the IKK complex and tumor necrosis factor-α-induced NF-κB activity. J Biol Chem 283(1):76–86. https://doi.org/10.1074/jbc.M708856200
Panne D, Maniatis T, Harrison SC (2007) An atomic model of the interferon-β enhanceosome. Cell 129(6):1111–1123. https://doi.org/10.1016/j.cell.2007.05.019
Phelps CB, Sengchanthalangsy LL, Huxford T, Ghosh G (2000) Mechanism of IκBα binding to NF-κB dimers. J Biol Chem 275(38):29840–29846. https://doi.org/10.1074/jbc.M004899200
Polley S, Huang DB, Hauenstein AV, Fusco AJ, Zhong X, Vu D, Schröfelbauer B, Kim Y, Hoffmann A, Verma IM, Ghosh G, Huxford T (2013) A structural basis for IκB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS Biol 11(6):e1001581. https://doi.org/10.1371/journal.pbio.1001581
Polley S, Passos DO, Huang DB, Mulero MC, Mazumder A, Biswas T, Verma IM, Lyumkis D, Ghosh G (2016) Structural basis for the activation of IKK1/α. Cell Rep 17(8):1907–1914. https://doi.org/10.1016/j.celrep.2016.10.067
Potoyan DA, Zheng W, Ferreiro DU, Wolynes PG, Komives EA (2016) PEST control of molecular stripping of NFκB from DNA transcription sites. J Phys Chem B 120(33):8532–8538. https://doi.org/10.1021/acs.jpcb.6b02359
Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, Randow F, Wakatsuki S, Dikic I (2009) Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 136(6):1098–1109. https://doi.org/10.1016/j.cell.2009.03.007
Ramsey KM, Dembinski HE, Chen W, Ricci CG, Komives EA (2017) DNA and IκBα both induce long-range conformational changes in NFκB. J Mol Biol 429(7):999–1008. https://doi.org/10.1016/j.jmb.2017.02.017
Rushe M, Silvian L, Bixler S, Chen LL, Cheung A, Bowes S, Cuervo H, Berkowitz S, Zheng T, Guckian K, Pellegrini M, Lugovskoy A (2008) Structure of a NEMO/IKK-associating domain reveals architecture of the interaction site. Structure 16(5):798–808. https://doi.org/10.1016/j.str.2008.02.012
Savinova OV, Hoffmann A, Ghosh G (2009) The Nfkb1 and Nfkb2 proteins p105 and p100 function as the core of high-molecular-weight heterogeneous complexes. Mol Cell 34(5):591–602. https://doi.org/10.1016/j.molcel.2009.04.033
Schuster M, Annemann M, Plaza-Sirvent C, Schmitz I (2013) Atypical IkappaB proteins—nuclear modulators of NF-κB signaling. Cell Commun Signal 11(1):23. https://doi.org/10.1186/1478-811X-11-23
Sen R, Baltimore D (1986) Inducibility of kappa immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell 47(6):921–928
Sen R, Baltimore D (1986) Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46(5):705–716
Sengchanthalangsy LL, Datta S, Huang DB, Anderson E, Braswell EH, Ghosh G (1999) Characterization of the dimer interface of transcription factor NFκB p50 homodimer. J Mol Biol 289(4):1029–1040. https://doi.org/10.1006/jmbi.1999.2823
Shih VF, Kearns JD, Basak S, Savinova OV, Ghosh G, Hoffmann A (2009) Kinetic control of negative feedback regulators of NF-κB/RelA determines their pathogen- and cytokine-receptor signaling specificity. Proc Natl Acad Sci USA 106(24):9619–9624. https://doi.org/10.1073/pnas.0812367106
Shiina T, Konno A, Oonuma T, Kitamura H, Imaoka K, Takeda N, Todokoro K, Morimatsu M (2004) Targeted disruption of MAIL, a nuclear IκB protein, leads to severe atopic dermatitis-like disease. J Biol Chem 279(53):55493–55498. https://doi.org/10.1074/jbc.M409770200
Sil AK, Maeda S, Sano Y, Roop DR, Karin M (2004) IκB kinase-α acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature 428(6983):660–664. https://doi.org/10.1038/nature02421
Sue SC, Dyson HJ (2009) Interaction of the IκBα C-terminal PEST sequence with NF-κB: insights into the inhibition of NF-κB DNA binding by IκBα. J Mol Biol 388(4):824–838. https://doi.org/10.1016/j.jmb.2009.03.048
Sun SC (2017) The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol 17(9):545–558. https://doi.org/10.1038/nri.2017.52
Tao Z, Fusco A, Huang DB, Gupta K, Young Kim D, Ware CF, Van Duyne GD, Ghosh G (2014) p100/IκBdelta sequesters and inhibits NF-κB through kappaBsome formation. Proc Natl Acad Sci USA 111(45):15946–15951. https://doi.org/10.1073/pnas.1408552111
Tartey S, Matsushita K, Vandenbon A, Ori D, Imamura T, Mino T, Standley DM, Hoffmann JA, Reichhart JM, Akira S, Takeuchi O (2014) Akirin2 is critical for inducing inflammatory genes by bridging IκBζ and the SWI/SNF complex. EMBO J 33(20):2332–2348. https://doi.org/10.15252/embj.201488447
Tegethoff S, Behlke J, Scheidereit C (2003) Tetrameric oligomerization of IκB kinase gamma (IKKgamma) is obligatory for IKK complex activity and NF-κB activation. Mol Cell Biol 23(6):2029–2041
Trinh DV, Zhu N, Farhang G, Kim BJ, Huxford T (2008) The nuclear IκB protein IκBζ specifically binds NF-κB p50 homodimers and forms a ternary complex on κB DNA. J Mol Biol 379(1):122–135. https://doi.org/10.1016/j.jmb.2008.03.060
Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412(6844):346–351. https://doi.org/10.1038/35085597
Xu G, Lo YC, Li Q, Napolitano G, Wu X, Jiang X, Dreano M, Karin M, Wu H (2011) Crystal structure of inhibitor of κB kinase β. Nature 472(7343):325–330. https://doi.org/10.1038/nature09853
Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, Kaisho T, Kuwata H, Takeuchi O, Takeshige K, Saitoh T, Yamaoka S, Yamamoto N, Yamamoto S, Muta T, Takeda K, Akira S (2004) Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IκBζ. Nature 430(6996):218–222. https://doi.org/10.1038/nature02738
Yamazaki S, Muta T, Takeshige K (2001) A novel IκB protein, IκBζ, induced by proinflammatory stimuli, negatively regulates nuclear factor-κB in the nuclei. J Biol Chem 276(29):27657–27662. https://doi.org/10.1074/jbc.M103426200
Yoshikawa A, Sato Y, Yamashita M, Mimura H, Yamagata A, Fukai S (2009) Crystal structure of the NEMO ubiquitin-binding domain in complex with Lys 63-linked di-ubiquitin. FEBS Lett 583(20):3317–3322. https://doi.org/10.1016/j.febslet.2009.09.028
Acknowledgements
Research is funded by the National Institutes of Health Grant GM085490 to GG. Biochemistry research at SDSU is supported in part by the California Metabolic Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mulero, M.C., Huxford, T., Ghosh, G. (2019). NF-κB, IκB, and IKK: Integral Components of Immune System Signaling. In: Jin, T., Yin, Q. (eds) Structural Immunology. Advances in Experimental Medicine and Biology, vol 1172. Springer, Singapore. https://doi.org/10.1007/978-981-13-9367-9_10
Download citation
DOI: https://doi.org/10.1007/978-981-13-9367-9_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-9366-2
Online ISBN: 978-981-13-9367-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)