
Abstract
Renal fibrosis is the final common pathway of all chronic kidney diseases progressing to end-stage renal diseases. Autophagy, a highly conserved lysosomal degradation pathway, plays important roles in maintaining cellular homeostasis in all major types of kidney cells including renal tubular cells as well as podocytes, mesangial cells and endothelial cells in glomeruli. Autophagy dysfunction is implicated in the pathogenesis of various renal pathologies. Here, we analyze the pathological role and regulation of autophagy in renal fibrosis and related kidney diseases in both glomeruli and tubulointerstitial compartments. Further research is expected to gain significant mechanistic insights and discover pathway-specific and kidney-selective therapies targeting autophagy to prevent renal fibrosis and related kidney diseases.
Co-first authors with equal contributions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Autophagy
- Renal fibrosis
- Focal segmental glomerulosclerosis
- Diabetic kidney disease
- Acute kidney injury
- Podocytes
- Proximal tubular epithelial cells
1 Introduction: Basics of Autophagy
First proposed by Christian de Duve in 1963, autophagy is a term derived from Greek and refers to “self-eating” (Klionsky 2008). It is an evolutionarily conserved catabolic process from yeasts to mammals, by which portions of cytosolic components and organelles are delivered to the lysosomes for degradation and recycling (Mizushima et al. 2008; Mizushima and Komatsu 2011). There are three types of autophagy in mammalian cells: macroautophagy, microautophagy and chaperone-mediated autophagy, which differ in the type of cargo to be degraded and the way of delivering them to lysosomes. Macroautophagy (herein referred to as autophagy), the best characterized form and the focus of this chapter, begins with the enveloping of large cytosolic structures by double-membraned autophagosomes and then fuses with the lysosome. Microautophagy involves the direct engulfment of small cytoplasmic cargos within the invagination of the lysosomal membrane. Chaperone-mediated autophagy is a process of direct transport of selective unfolded protein via chaperonin across the lysosome membrane for degradation (Levine and Kroemer 2008; Mizushima et al. 2008; Ravikumar et al. 2010).
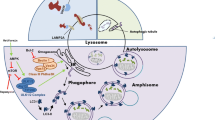
The process of autophagy consists of a cascade of cellular events (Fig. 28.1). It is initiated by the formation of a double-membrane, cup-shaped structure termed phagophore around sequestered cytoplasmic targets, followed by expansion and closure of the phagophore to form an autophagosome. The autophagosome then docks and fuses with a lysosome to form an autolysosome, in which the autophagosome inner membrane and the cytoplasmic substrates are degraded by acidic lysosomal hydrolases. Eventually, the resultant degradation products are released for recycling (Mizushima et al. 2008; Mizushima and Komatsu 2011). The complete dynamic flow of autophagy is thus called autophagic flux. Although the origin of the autophagosome membrane has been a matter of debate for decades, it is now generally agreed that the initiation of the phagophore is associated with the phosphatidylinositol 3-phosphate (PtdIns3P)-enriched membrane compartment that appears to be linked to the endoplasmic reticulum (ER). An omega-shaped protrusion (also called omegasome) forms on ER at the initiation site of phagophore and functions as a scaffold for autophagosome biogenesis (Yang and Klionsky 2010; Rubinsztein et al. 2012). Furthermore, the phagophore is in contact with many surrounding organelles either by vesicular transport to and from the phagophore or through transient membrane contacts and exchange of proteins and lipids to grow and expand (Yang and Klionsky 2010; Rubinsztein et al. 2012; Yu et al. 2018).

Basics of autophagy. The autophagy pathway and its molecular regulation are shown. Autophagy is initiated by the formation of omegasome, an omega-shaped protrusion from the ER, which is enriched in phosphatidylinositol 3-phosphate (PtdIns3P) and further forms the phagophore. Then, the autophagosome is formed by the expansion and closure of phagophore, which then docks and fuses with a lysosome to form an autolysosome. The autolysosome degrades the inner membrane of autophagosome and the cytoplasmic substrates by acidic lysosomal hydrolases to recycle the nutrients. The initiation of autophagy is regulated by the ULK1/2 complex and the class III phosphatidylinositol 3-kinase (PI3K) complex. The ULK1/2 complex senses upstream signals and relays them to the downstream central autophagy pathway via the coordination and interaction with other ATG proteins or complexes. The PI3K complex produces PtdIns3P at the site of phagophore nucleation to stabilize membrane curvature and to recruit more downstream factors for phagophore growth. ATG18/WIPIs (WD repeat phosphoinositide interacting proteins) and ATG9L contribute to delivery of membrane to the phagophore. The expansion and closure of phagophore to form autophagosome require two ubiquitin-like conjugation systems: the ATG12-ATG5-ATG16L complex and the microtubule-associated protein light chain 3-phosphatidyl ethanolamine (MAPLC3/LC3-PE). LC3 precursor (proLC3) is cleaved by a cysteine protease ATG4 to generate LC3-I. The PE then conjugated to LC3-I to form LC3-II by ATG7 and ATG3 (E2-like) enzymes. The ATG12-ATG5-ATG16L complex is formed by the ATG7 (E1-like) and ATG10 (E2-like) enzymes, which directs LC3-II into autophagosomal membrane. Specific soluble NSF attachment protein receptor (SNARE) complexes contribute to the fusion of autophagosomes with lysosomes. The mTORC1 and AMP-activated protein kinase (AMPK) are major autophagy regulators, sensing and integrating multiple signaling pathways. mTORC1 inhibits autophagy by phosphorylating ULK1 to inhibit its activity, while AMPK induces autophagy by either directly phosphorylating ULK1 or suppressing mTORC1. Nutrient starvation or low energy induces autophagy by suppressing mTORC1 and activating AMPK pathway
In mammals, autophagosome biogenesis is regulated coordinately at different steps by the core machinery that consists of more than 36 autophagy-related (Atg) genes (Yang and Klionsky 2010; Klionsky et al. 2011; Mizushima et al. 2011; Rubinsztein et al. 2012; Hurley and Young 2017). The initiation of phagophore formation is orchestrated by the ULK1/2 (Unc-51-like kinase 1/2) complex, which is composed of ULK1/2 serine–threonine kinase, ATG13, FIP200 (FAK family kinase-interacting protein of 200 kDa, also known as RB1CC1, RB1-inducible coiled-coil protein 1) and ATG101. The ULK1/2 complex senses upstream signals and relays them to the downstream central autophagy pathway via the coordination and interaction with other ATG proteins or complexes (Yang and Klionsky 2010; Rubinsztein et al. 2012). Phagophore nucleation is dependent on the class III phosphatidylinositol 3-kinase (PtdIns3K) complex that consists of PIK3C3/VPS34 (vacuolar protein sorting 34) lipid kinase, PIK3R4/VPS15, beclin 1/BECN1 and ATG14L. This complex produces PtdIns3P at the site of phagophore nucleation to stabilize membrane curvature and to recruit more downstream factors for phagophore growth (Yang and Klionsky 2010; Rubinsztein et al. 2012). The delivery of membrane from other sources to the forming autophagosome is regulated by ATG18/WIPIs (WD repeat phosphoinositide interacting proteins), a PtdIns3P scaffold and binding protein, and cycling of the transmembrane protein ATG9L (Yang and Klionsky 2010; Rubinsztein et al. 2012). Autophagosome elongation and completion require two ubiquitin-like conjugation systems: the ATG12-ATG5-ATG16L complex and the microtubule-associated protein light chain 3-phosphatidyl ethanolamine (MAPLC3/LC3-PE). ATG12, a ubiquitin-like protein, is conjugated to ATG5 following activation by ATG7 and ATG10, E1- and E2-like enzymes, respectively. ATG12-ATG5 then associates with ATG16L to form a large protein complex. LC3 precursor (proLC3) is cleaved by a cysteine protease ATG4 to generate LC3-I. Conjugation of PE to LC3-I to form LC3-II is mediated by ATG7 and ATG3 (E2-like) enzymes. The ATG12-ATG5-ATG16L complex directs LC3-II into autophagosomal membrane, and the conversion of cytosolic LC3-I to membrane-bound LC3-II is a biochemical and cell biologic hallmark of autophagy. A large set of molecules, including cytoskeleton components and related motor proteins, tethering factors, phospholipids, and specific soluble NSF attachment protein receptor (SNARE) complexes have been identified as important players in the maturation and fusion of autophagosome with lysosome (Yang and Klionsky 2010; Rubinsztein et al. 2012; Yu et al. 2018). Dynein, a microtubule motor, mediates the centripetal movement of autophagosomes to ensure a close spatial positioning between autophagosomes and lysosomes. UV radiation resistance-associated gene (UVRAG), via interaction with PtdIns3 K complex, activates the GTPase Rab7 (Ras-related protein 7) for tethering. As the core components of the fusion machinery, SNAREs can mediate membrane fusion on their own as well as via the regulation and interaction with other fusion factors. Upstream of the core machinery, autophagy is tightly regulated by a complex signaling network (He and Klionsky 2009; Mehrpour et al. 2010; Klionsky et al. 2016). The mechanistic target of rapamycin (mTOR) pathway, specifically mTOR complex 1 (mTORC1), serves as a sensor and a master negative regulator of autophagy. Multiple signaling pathways stimulated by nutrients, growth factors and energy may integrate and merge at mTORC1 to regulate autophagy. mTOR-independent mechanisms have also been implicated in autophagy regulation. A variety of cellular stress, including hypoxic stress, oxidative stress, ER stress and DNA damage, may also induce autophagy through various signaling pathways (Klionsky et al. 2016).
Multiple techniques have been developed to monitor autophagy in cells or tissues (Klionsky et al. 2016). Electron microscopy (EM) is a classic method for detecting and analyzing various autophagic structures including the phagophore, autophagosome and autolysosome. However, EM is less quantitative and could be problematic due to sampling artifacts. LC3-based assays have been widely used for the analysis of autophagy in cells and tissues. Especially, the conversion of LC3-I to LC3-II or expression of LC3-II measured by immunoblotting is a well-accepted biomarker for autophagy. In addition, LC3-I resides in cytosol whereas LC3-II is membrane-bound on autophagosomes. The change of LC3 localization can be revealed by microscopic examination of endogenous LC3 following immunostaining or exogenous green fluorescence protein (GFP)-LC3 following transfection. Considering the dynamic change of autophagic flux, several methods are utilized to detect the dynamic flow: (1) LC3 turnover assay that compares LC3-II abundance in the presence and absence of lysosome inhibitors; (2) examination of the turnover of autophagic substrates such as p62/sequestosome 1 (SQSTM1); and (3) the use of a tandem red fluorescent protein (mRFP)/mCherry-GFP-LC3 reporter for the monitoring of autophagosomes (red- and green-costained LC3 puncta) and autolysosomes (red-only LC3 puncta) (Klionsky et al. 2016).
Autophagy can non-selectively break down bulk cytosol and also selectively recognize and digest specific organelles such as mitochondria, ER and lysosomes, protein aggregates, lipid droplets and intracellular pathogens (Mizushima et al. 2008; Mizushima and Komatsu 2011; Zaffagnini and Martens 2016). Under physiological conditions, a basal level of autophagy in most cells is an important mechanism of removing potentially harmful or simply unneeded cytoplasmic materials, which is essential to the maintenance of cellular homeostasis. During starvation or nutrient deprivation, autophagy is activated to break down and recycle cytosolic contents to replenish pools of biosynthetic precursors (amino acids, lipids, nucleotides) and energy sources (Sionov et al. 2015; Dikic and Elazar 2018). Emerging studies have further shown the role of autophagy in a much broader range of cell stress and pathological conditions. Under these conditions, induction of autophagy primarily serves as an adaptive and defensive strategy for cell to deal with stress for survival (Sionov et al. 2015; Dikic and Elazar 2018). Conversely, dysregulation of autophagy may contribute to the pathogenesis of various diseases, such as cancer and cardiovascular disease (Huber et al. 2012; Choi et al. 2013; Sionov et al. 2015; Dikic and Elazar 2018).
2 Autophagy in Renal Resident Cells
Autophagy is an important mechanism for maintaining cellular homeostasis in all major types of renal resident cells including podocytes, mesangial cells, glomerular endothelial cells and renal tubular epithelial cells.
2.1 Autophagy in Podocytes
Podocytes, also called glomerular visceral epithelial cells, are highly specialized epithelial cells with a large cell body and primary processes that further branch into fine secondary foot processes. The foot processes from adjacent podocytes interdigitate and wrap around the outside of glomerular basement membrane (GBM) that surrounds the capillaries of glomeruli. Podocytes play a critical role in maintaining the selective permeability and structural integrity of the glomerular filtration barrier. Terminally differentiated, podocytes are incapable of proliferation, and the mechanism of podocyte replacement is limited (Pavenstädt et al. 2003). As a result, podocytes are the most vulnerable component of the glomerulus and can be irreversibly injured by various insults, leading to proteinuria and glomerulosclerosis that contribute to the pathogenesis of many glomerular diseases. As long-lived cells, podocytes rely on cellular quality control mechanisms to maintain their structural and functional homeostasis, and autophagy is one such mechanism under both normal and disease conditions (Zhang et al. 2014a).
Compared to other types of cells in the kidney, podocytes, especially differentiated podocytes, exhibit a high level of constitutive autophagy. In normal adult rats, abundant LC3 staining was detected in glomeruli but barely seen in proximal tubules. Punctate LC3 staining co-localized with podocalyxin, a podocyte marker. LC3-II accumulated in tissue lysates of isolated glomeruli. In conditionally immortalized mouse podocytes, a high level of basal autophagy was also primarily seen in differentiated podocytes (Asanuma et al. 2003). In GFP-LC3 transgenic mice, compared with tubular cells, podocytes displayed clearly detectable levels of GFP-LC3 puncta under basal conditions (Mizushima et al. 2004; Hartleben et al. 2010; Fang et al. 2013). However, in newborn GFP-LC3 mice, GFP-LC3 puncta were not seen until more mature or differentiated podocytes appeared in the late capillary loop stage (Hartleben et al. 2010; Fang et al. 2013). The formation of GFP-LC3 puncta was enhanced upon starvation (Mizushima et al. 2004). LC3 turnover assay further revealed remarkably increases in both the number of GFP-LC3 puncta and the accumulation of LC3-II by lysosomal inhibitor chloroquine, suggesting a complete process of autophagic flux in podocytes (Hartleben et al. 2010; Fang et al. 2013).
In doxycycline-inducible podocyte-specific Atg5 knockout mice, acute induction of Atg5 deletion in 12-week-old mice triggered a rapid onset of albuminuria, suggesting that autophagy is a fundamental mechanism for podocyte homeostasis (Hartleben et al. 2010; Fang et al. 2013). In contrast to this inducible knockout model, mice with constitutive podocyte-specific Atg5 knockout were indistinguishable from their wild-type littermates for up to 2–4 months after birth, suggesting that other adaptive pathways may compensate for the constitutive loss of Atg5 (Hartleben et al. 2010; Fang et al. 2013). Indeed, proteasome activity was significantly increased in glomerular lysates of 8-month-old podocyte-specific Atg5 knockout mice, preventing the accumulation of polyubiquitinated proteins. Inhibition of proteasome activity led to increased albuminuria in 6-month-old podocyte-specific Atg5 knockout mice. In differentiated podocytes in vitro, inhibition of proteasome activated autophagy. These results suggest that basal autophagy in podocytes acts in concert with the proteasome pathway to maintain podocyte homeostasis (Hartleben et al. 2010; Fang et al. 2013). Using paracellular permeability influx assay, Fang et al. found that inhibition of autophagy by 3-methyladenine in cultured mouse podocytes led to an increased leakage of albumin across the podocyte monolayer. The expression of podocyte slit diaphragm proteins such as nephrin and podocin was suppressed by 3-methyladenine and BECN1 siRNA, further suggesting an essential role of high basal autophagy in the maintenance of podocyte health (Hartleben et al. 2010; Fang et al. 2013). Since lysosome is crucial for autophagic degradation, lysosomal dysfunction in podocytes by gene ablation of mTOR, prorenin or VPS34 also resulted in severe glomerulosclerosis and proteinuria (Oshima et al. 2011; Cinà et al. 2012a; Chen et al. 2013). These results highlight the importance of an intact autophagic flux pathway in maintaining podocyte homeostasis.
In humans, Zeng et al. demonstrated a negative correlation between the podocyte autophagic activity and the progression of glomerular diseases (Zeng et al. 2014). In experimental models, autophagy was induced by adriamycin in cultured podocytes in vitro and in podocytes in mice. Importantly, inducible ablation of autophagy-related gene 7(ATG7) in podocytes exacerbated podocyte injury, glomerulopathy and proteinuria during adriamycin treatment, supporting a protective role of autophagy in podocytes (Yi et al. 2017). Recent studies have further suggested defective autophagy in podocytes of aged kidneys. Compared with young rats, the expression of LC3 and ATG7 was significantly decreased in old rats. p62/SQSTM1 and polyubiquitin aggregates accumulated in aged kidneys, accompanied with an accumulation of damaged mitochondria and induction of oxidative stress (Cui et al. 2012). Similar findings were also shown in aging mice (Wanner et al. 2014). In 20- to 24-month-old podocyte-specific Atg5 knockout mice, Atg5 deletion accelerated podocyte aging, leading to massive ER stress and oxidative stress in the knockout mice. The compensatory proteasome pathway that clears protein aggregates in young Atg5 knockout mice was also significantly reduced in the old mice. Ultimately, the Atg5-deficient old mice developed podocyte loss and progressive glomerulosclerosis (Hartleben et al. 2010; Fang et al. 2013). These results highlight the fundamental role of autophagy for the longtime maintenance of glomerular podocytes.
Notably, podocytes also display higher mTORC1 activity compared with other glomerular cells, which appears to be required for postnatal growth (Narita et al. 2011; Fukuda et al. 2012). Podocytes stop cell division in the capillary loop stage; thus, an increasing glomerular volume must be accompanied by mTORC1-dependent growth of every single podocyte to cover the glomerular capillaries (Hartleben et al. 2014; Inoki 2014). The high levels of both basal autophagy and mTORC1 in podocytes seem contradictory to the concept that mTORC1 negatively regulates autophagy; however, it may suggest the existence of a unique mechanism involving a mutual function and coordination of mTORC1 and autophagy in these cells. TOR-autophagy spatial coupling compartment (TASCC), a distinct cytoplasmic compartment, has been identified in podocytes (Narita et al. 2011). Located at the trans side of Golgi apparatus, TASCC is mostly occupied by mTORC1, lysosomes and autolysosomes but largely excludes autophagosomes. The formation of a spatial mTORC1 gradient within a podocyte by TASCC sequestration thus allows simultaneous activation of mTORC1 and autophagy in different areas of the same cell (Narita et al. 2011). Functionally, this system plays a beneficial role in generating sufficient secretory proteins with a constant energy and source supply derived from autophagy. More importantly, it also creates a self-regulating mechanism, in which the autolysosomal degradation products reinforce mTOR enrichment and activity to in turn suppress autophagy and recycle lysosomes. This feedback regulation, called autophagic lysosome reformation (ALR), is very important for a balance and fine-tuning between mTOR pathway and autophagy-lysosomal pathway (Yu et al. 2010, 2018). In line with this, mice with podocyte-specific mTOR knockout developed severe proteinuria and renal failure 3–5 weeks after birth. mTOR deficiency led to impaired autophagic flux in podocytes, and the failure of ALR appeared to account for this podocyte dysfunction (Cinà et al. 2012b).
2.2 Autophagy in Mesangial Cells
Glomerular mesangial cells are specialized contractile cells that are located within the mesangium. They provide structural support for the glomerular tufts and also form a functional unit together with adjacent podocytes and glomerular endothelial cells to regulate glomerular filtration. Mesangial cells produce ECM components in the mesangium and play an important role in maintaining mesangial matrix homeostasis.
The role of autophagy in mesangial cells is poorly understood. Transforming growth factor (TGF)-β1 induced autophagy in mouse mesangial cells and protected against serum deprivation-induced apoptosis. The induction of autophagy by TGF-β1 in mesangial cells was shown to be mediated by TGF-β-activated kinase 1 (TAK1) and PI3 K-protein kinase B(PKB)/Akt pathway. TGF-β1 failed to rescue autophagy-deficient mesangial cells from serum deprivation-induced apoptosis, further supporting a pro-survival role of autophagy in mesangial cells (Kim et al. 2012a). The protective effect of mesangial autophagy was also associated with its role in the maintenance of matrix protein homeostasis (Kim et al. 2012a). Primary mouse mesangial cells isolated from autophagy-deficient mice expressed a higher basal level of collagen I protein. In response to TGF-β1, both protein and mRNA levels of collagen I were induced, and notably, the increased collagen I protein was co-localized with LC3 and lysosomal marker lysosome-associated membrane protein 1 (LAMP1). Inhibition of autophagy by BECN1 knockdown or lysosomal inhibitors further increased collagen I protein accumulation without affecting mRNA expression. Upregulation of autophagy reduced collagen I protein in wild-type but not in autophagy-deficient mesangial cells. These results suggest a critical role of autophagy in negatively regulating to limit excessive ECM deposition in mesangial cells by promoting collagen I degradation (Kim et al. 2012a).
2.3 Autophagy in Glomerular Endothelial Cells
Glomerular endothelial cells localize in the inner side of GBM and are important components of the glomerular filtration barrier. The renal microvasculature also plays a key role in renal physiology by regulating vasomotor tone, vascular permeability, leukocyte recruitment and antithrombogenic responses. Glomerular endothelial dysfunction is associated with the progression of CKD and renal fibrosis; however, the underlying mechanisms remain largely unknown. Thus far, very few studies have examined the role of autophagy in glomerular endothelial cells. Xavier et al. showed that bone morphogenetic protein and activin membrane-bound inhibitor (BAMBI), a competitive receptor antagonist for the TGF-β receptor family, was increased in cultured mouse glomerular endothelial cells treated with TGF-β. By contrast, the decrease of BAMBI by serum starvation or rapamycin was completely impeded by lysosomal inhibitor bafilomycin A1 and partially inhibited by 3-methyladenine, but not by proteasome inhibitors. These results suggest a role of autophagy in regulating BAMBI turnover in endothelial cells, which may influence endothelial function via BAMBI-mediated regulation of TGF-β pathway (Xavier et al. 2010).
2.4 Autophagy in Proximal Tubular Epithelial Cells (PTECs)
PTECs are the key targets in both acute kidney injury (AKI) and chronic kidney diseases (CKDs). Under physiological conditions, PTECs exhibit a relatively low level of autophagy. Mice with proximal tubule-specific knockout of Atg5 or Atg7 showed progressive renal damage and also developed premature renal aging, as indicated by an accumulation of deformed mitochondria, p62/SQSTM1 and polyubiquitin-positive inclusion bodies as well as increased tubular cell apoptosis and renal interstitial fibrosis. These results suggest that a low but sufficient level of basal autophagy is needed to maintain cellular homeostasis in PTECs under normal conditions and a higher level of autophagy is required for the cells to deal with age-related stress (Kimura et al. 2011; Liu et al. 2012). Autophagy is remarkably activated in PTECs under various stress conditions and plays a renoprotective role against tubular injury and cell death (Huber et al. 2012; Jiang et al. 2012; Havasi and Dong 2016; Zhang et al. 2016; Tang et al. 2018).
3 Autophagy in Renal Fibrosis and Related Kidney Diseases
Renal fibrosis, characterized by the excessive deposition of extracellular matrix (ECM) in glomeruli and tubulointerstitium, is the common pathological hallmark of progressive CKD regardless of the initial causes. The pathogenesis of renal fibrosis involves an extremely complex interaction of multiple cellular events including excessive proliferation and activation of fibroblasts, increased deposition of ECM, infiltration of inflammatory cells, tubular atrophy, glomerulosclerosis and microvascular rarefaction (Liu 2011; Duffield 2014; Humphreys 2018). Recently, increasing evidence has demonstrated that dysregulated autophagy may also contribute to the pathogenesis of renal fibrosis and related kidney diseases.
3.1 Autophagy in Focal Segmental Glomerulosclerosis (FSGS)
FSGS is a heterogeneous fibrotic kidney disease with poor clinical outcome due to progressive loss of kidney function. It is either idiopathic or secondary to a number of other disorders. The cause of idiopathic or primary FSGS remains unclear. Although FSGS is characterized by lesions in the glomerulus, particularly podocytes, early functional defects in proximal tubular cells are also found in some patients with FSGS (Löwik et al. 2009; Kawakami et al. 2015). A pathological feature in the development of FSGS is podocyte injury and loss, leading to the adhesion of glomerular capillary tuft to Bowman’s capsule followed by occlusion of the capillaries and eventual total nephron degeneration (D’Agati 2012; Hartleben et al. 2014).
Using in vivo and in vitro models of puromycin aminonucleoside (PAN) nephrosis (an experimental model of human FSGS), Asanuma et al. showed that autophagy induction in podocytes was correlated with the recovery from PAN nephrosis, and provided the first evidence that podocyte autophagy may prevent the development of FSGS (Asanuma et al. 2003). Upregulation of autophagy in podocytes was further shown in human renal biopsy specimens from patients with FSGS, as indicated by increased Atg3 mRNA and LC3 puncta. Accordingly, podocyte-specific Atg5 knockout significantly sensitized mice to glomerulosclerosis induced by PAN, further suggesting that autophagy defends the integrity of podocytes against the development of FSGS (Hartleben et al. 2010). In an adriamycin-induced experimental model of FSGS, our recent work demonstrated an induction of autophagy in cultured podocytes and the podocytes in mice (Yi et al. 2017). In cultured podocytes, activation of autophagy by rapamycin suppressed adriamycin-induced apoptosis, whereas inhibition of autophagy by chloroquine sensitized the cells to apoptosis. In inducible podocyte-specific Atg7 knockout mice, adriamycin induced more severe podocyte injury, glomerulopathy and proteinuria as compared with wild-type mice, further supporting a protective role of podocyte autophagy in FSGS (Yi et al. 2017).
Using renal biopsies from patients with minimal change disease (MCD) or FSGS, a recent study examined the role of podocyte autophagy in controlling the progression of podocytopathies (Zeng et al. 2014). It showed that podocytes from MCD patients had higher levels of BECN1-mediated autophagy than podocytes from FSGS patients.
Tracking of podocyte autophagy in MCD patients by repeat renal biopsies showed that the patients with decreased autophagy activity in podocyte progressed to FSGS, whereas those with relatively high levels of podocyte autophagy maintained MCD status. In PAN-treated cultured podocytes, apoptosis was enhanced by autophagy inhibition through BECN1 knockdown or autophagy inhibitors (3-methyladenine or chloroquine), while alleviated by autophagy induction through rapamycin. In PAN-treated rats, inhibition of autophagy led to severe renal dysfunction and podocyte injuries with earlier onset and greater proteinuria, and more extensive foot process effacement and reduction in podocyte markers. Conversely, restoration of autophagy by rapamycin resulted in the attenuation of proteinuria and foot process effacement, and better preservation of podocyte markers. These results suggest that a gradually impaired autophagy in podocytes contributes to the progression of podocytopathies from MCD to FSGS (Zeng et al. 2014). In addition, compared with the podocytes from patients with MCD, the ubiquitin-proteasome activity was also compromised in podocytes from patients with FSGS, leading to abnormal protein accumulation and compensatory upregulation of autophagy (Beeken et al. 2014). These results further demonstrate that the two proteolytic systems may act in concert to protect podocytes and prevent the progression of FSGS.
To further determine the role of autophagy in the pathogenesis of FSGS, Kawakami et al. generated a mouse model in which Atg5 or Atg7 gene was mutated in the embryonic progenitor cells (Humphreys et al. 2008; Kawakami et al. 2015). The mutant mice showed mild albuminuria with normal kidney function and widespread podocyte foot process effacement without significant mesangial matrix deposition at 2 months old. At 4 months of age, these mice developed severe albuminuria, tubulointerstitial pathologies and glomerular changes. The mice also showed histologic features of FSGS in the kidneys, such as segmental lesions of scarring with capillary loop obliteration, tuft to capsule adhesion, lesions at the tubular pole and glomeruli with tuft collapse. The mutant mice died from kidney failure by 6 months. Ultrastructurally, podocytes and tubular cells from the mutant mice displayed vacuolization, abnormal mitochondria, evidence of ER stress and increased production of ROS, which recapitulated the features of human idiopathic FSGS kidney biopsy specimens. This over-production of ROS in podocyte and tubules, appearing early before any histological pathology, may be a result of autophagy deficiency-mediated impaired clearance of dysfunctional mitochondria and its proteins. These findings indicate that mitochondrial dysfunction and ER stress due to impairment of autophagic turnover may play a central role in FSGS development (Carney 2015; Kawakami et al. 2015).
3.2 Autophagy in Diabetic Kidney Disease (DKD)
DKD is a serious complication of diabetes mellitus and a leading cause of CKD and end-stage renal disease (ESRD) worldwide (Levin et al. 2017). The pathogenesis of DKD is extremely complex, involving multifactorial interactions between hyperglycemia-mediated metabolic alterations, hemodynamic abnormalities and intracellular stress (Brownlee 2005; Forbes and Cooper 2013). The clinical hallmark pathology of DKD is persistent albuminuria or proteinuria followed by decreased glomerular filtration rate (GFR), tubular cell damage and tubulointerstitial lesions, eventually leading to renal failure. Additional pathological features of DKD include an accumulation of ECM components, thickening of both GBM and tubular basement membrane, mesangial expansion, glomerulosclerosis, podocyte effacement, tubular atrophy, and afferent and efferent arteriolar hyalinosis (Abbate et al. 2006).
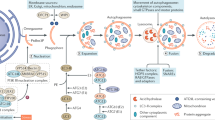
Emerging evidence has suggested that autophagy is impaired in diabetic kidneys (Fig. 28.2). The defective autophagy in DKD is associated with the abnormalities of multiple nutrient-sensing pathways including mTOR, AMP-activated protein kinase (AMPK) and sirtuins (SIRTs) (Kume et al. 2012; Levin et al. 2017; Yang et al. 2018). mTOR, especially mTORC1, is activated under excessive nutrient conditions by increased levels of glucose, amino acid and growth factors (Wellen and Thompson 2010; Zoncu et al. 2011). mTORC1 negatively regulates autophagy by phosphorylating ULK1 to inhibit its activity (Hosokawa et al. 2009; Jung et al. 2009). Upon nutrient/energy depletion, AMPK and SIRTs are activated, respectively, in response to increasing levels of intracellular AMP and nicotinamide adenine dinucleotide (NAD+) levels (Steinberg and Kemp 2009; Imai and Guarente 2010). In contrast to mTORC1, both AMPK and SIRTs are positive regulators of autophagy. AMPK either directly phosphorylates ULK1 to promote autophagy or inhibits mTORC1 for autophagy induction (Lee et al. 2010; Kim et al. 2011; Alers et al. 2012). SIRT1, the most studied member of the SIRTs family, promotes autophagy by deacetylating ATG5, ATG7 and LC3 (Lee et al. 2008). SIRT1 also deacetylates the transcriptional factor Forkhead box O3a (FoxO3a), leading to activation of BNIP3 (BCL2/adenovirus E1B 19-kDa interacting protein 3) (Kume et al. 2010). In addition, SIRT1 cross talks with AMPK and mTOR to regulate autophagy (Cantó et al. 2009; Ghosh et al. 2010). Dysregulation of these nutrient-sensing pathways under diabetic conditions contributes to the defective autophagy and the pathogenesis of DKD (Kume et al. 2012; Levin et al. 2017; Yang et al. 2018).

Autophagic insufficiency in diabetic kidneys. a TEM of kidney specimens from control subjects (normal renal tissues from patients with renal cell carcinoma) and patients with diabetic nephropathy (DN). The number of autophagic vacuoles (arrows) was reduced in podocytes from patients with DN (means ± SEM, n = 4 for control subjects, n = 5 for patients with DN; 51–57 images were selected from each group). Student’s t-test, *P < 0.05 versus control group. Scale bar = 500 nm. b Immunofluorescence staining for LC3 (red) and synaptopodin (green) in the renal cortex from control subjects and patients with DN. Quantification of the number of LC3—positive dots (yellow) indicated a reduction in the number of autophagosomes in podocytes from patients with DN (means ± SEM, n = 3, and 14–20 images from each group). Student’s t-test, *P < 0.05 versus control. Scale bar = 50 μm. c Western blot assay of beclin 1 expression in renal cortex from control subjects and patients with DN. Beclin 1 expression normalized against GAPDH was reduced in the renal cortex from patients with DN (means ± SEM, n = 3). Student’s t-test, *P < 0.05 versus control
Hyperactivation of mTORC1 was frequently seen in animal models and patients of both type 1 and 2 DKDs (Nagai et al. 2005; Mori et al. 2009; Zhang et al. 2014b). In non-diabetic mice, activation of mTORC1 specifically in podocytes induced renal damage that recapitulates features of DKD including GBM thickening, ECM expansion, podocyte loss and proteinuria (Inoki et al. 2011). The causative link between the hyperactivation of mTORC1 and the development of DKD was further verified in both mouse models and human DKD samples (Gödel et al. 2011). In diabetic PTECs, hyperactivation of mTORC1 also induced apoptosis and tubular hypertrophy (Sakaguchi et al. 2006; Velagapudi et al. 2011). On the contrary, inhibition of mTORC1 exerted a renoprotective role against DKD. Inhibiting mTORC1 pharmacologically by rapamycin alleviated kidney injury and attenuated expression of pro-inflammatory and profibrotic cytokines in STZ-induced diabetic rats (Yang et al. 2007; Wittmann et al. 2009). Rapamycin also reduced proteinuria, glomerulosclerosis, mesangial expansion and renal hypertrophy in both STZ-induced diabetic rats and db/db mice (Lloberas et al. 2006; Sakaguchi et al. 2006; Sataranatarajan et al. 2007; Mori et al. 2009; Stridh et al. 2015). Rapamycin rescued autophagy inhibition in cultured podocytes during prolonged high glucose treatment (Fang et al. 2013). The protective effects of rapamycin via inhibiting mTORC1 for autophagy activation were also shown in STZ-induced diabetic mice (Xiao et al. 2014). Pharmacological inhibition of mTORC1 by Torin1 also restored autophagy in db/db mice with high levels of advanced glycation end products (AGEs) and in AGEs-stimulated cultured podocytes (Zhao et al. 2018). In diabetic Wistar fatty rats, inhibition of mTORC1 by a very low protein diet restored autophagy in PTECs and protected against tubular cell injury, inflammation and interstitial fibrosis (Kitada et al. 2016). These findings suggest that hyperactivation of mTOR signaling pathway, via negatively regulating autophagy, plays a critical role in the pathogenesis of DKD.
The activity of AMPK was suppressed in both type 1 and 2 diabetic kidneys, which, importantly, could be reversed by several AMPK activators, leading to the restoration of autophagy and the attenuation of diabetic kidney injury. For example, berberine suppressed high glucose-induced podocyte apoptosis and protected diabetic mice by reactivating AMPK and autophagy (Zhao et al. 2014; Jin et al. 2017). Cinacalcet attenuated diabetic injury in db/db mice by activating autophagy via AMPK pathway (Lim et al. 2018). Consistently, other AMPK activators, such as resveratrol, metformin and AICAR (5-aminoimidazole-4-carboxamide-1β-riboside), also afforded renoprotective effects against kidney injury in both type 1 and 2 models of DKD (Kim et al. 2012b, 2013; Dugan et al. 2013). Similar to AMPK, SIRT1 was downregulated in renal cells in human and animal models of DKD, and activation of SIRT1 protected the kidney from diabetic injury. Mice with overexpressed SIRT1 specifically in proximal tubules were resistant to diabetes-related progression of podocyte damage and subsequent proteinuria (Hasegawa et al. 2013). Resveratrol, via restoring SIRT1 activity, provided beneficial effects in both podocytes and mesangial cells (Chuang et al. 2011; Wu et al. 2012). Recently, Ma et al. demonstrated a role of SIRT1 and consequent autophagy reactivation in the renoprotective effects of resveratrol in type 2 diabetic rats and in hypoxia-treated PTECs (Ma et al. 2016). Similarly, calorie restriction, via increasing SIRT1 activity, activated autophagy in a mouse model of type 2 diabetes and ameliorated glomerular and tubular injury of DKD (Kitada et al. 2011). These findings suggest that inactivation of AMPK and SIRT1 pathways under diabetic conditions suppresses autophagy and facilitates the development of DKD.
Although dysregulated nutrient-sensing pathways suppress autophagy in diabetic kidneys, as a compensatory response, autophagy is also induced by stress signaling to maintain cell integrity. The failure of this adaptive response may lead to abnormal accumulation of damaged organelles, such as mitochondria and ER, and progression of DKD. In this regard, both oxidative stress via reactive oxygen species (ROS) and ER stress have been suggested to regulate autophagy under diabetic conditions (Levin et al. 2017; Yang et al. 2018). Autophagy was induced in podocytes by high glucose within 24 h along with ROS generation, which was inhibited by antioxidant N-acetylcysteine (Ma et al. 2013). ROS-mediated autophagy was also seen in podocytes treated with palmitic acid, a saturated free fatty acid (FFA), for 24 h (Jiang et al. 2017). Oxidative stress stimulated autophagy to remove damaged mitochondria (mitophagy). This autophagy-mediated mechanism of mitochondrial quality control and subsequent reduction of ROS is indispensable for protecting against kidney injury under diabetic conditions (Higgins and Coughlan 2014). Autophagy also plays an important role in maintaining ER health during DKD. Defective autophagy under diabetic conditions may lead to prolonged ER stress, and activation of autophagy for ER degradation (ERphagy) is required to protect kidneys from cytotoxic ER stress. Tauroursodeoxycholic acid (TUDCA), a chemical chaperone, inhibited AGEs-induced podocyte apoptosis by reducing ER stress (Chen et al. 2008). Autophagy was restored in TUDCA-treated diabetic mice, accompanied by reduced podocyte injury and proteinuria (Fang et al. 2013; Cao et al. 2016a). In STZ-induced diabetic rats and db/db mice, 4-phenylbutyric acid (4-PBA), a chemical chaperone and ER stress inhibitor, suppressed ER stress-associated inflammation and kidney injury (Qi et al. 2011; Cao et al. 2016b). Both TUDCA and 4-PBA reactivated autophagy in db/db mice and in high glucose-treated podocytes, thus preventing ER stress-induced podocyte apoptosis under diabetic conditions (Cao et al. 2016b).
The changes of autophagy in podocyte seem to be time-dependent and correlated with the severity of podocyte injury and the progression of DKD. Autophagy was induced in immortalized murine podocytes within 24 h of high glucose treatment (Ma et al. 2013), but inhibited at 48 h (Fang et al. 2013). Similarly, in primary podocytes, autophagic flux was prompted by 24-h treatment of high glucose. Genetical inhibition of autophagy by Atg5 ablation sensitized the podocytes to high glucose-induced apoptosis. In a stable podocyte cell line (SVI), autophagy was activated at 48 h of high glucose treatment, while inhibited after 15 days of high glucose treatment (Lenoir et al. 2015). In diabetic mice, autophagy was induced in podocytes at 4 weeks after STZ injection, when mice were hyperglycemic but had not yet developed glomerular lesions. At 8-week post-STZ injection, along with the occurrence of glomerular lesions autophagy in podocytes was inhibited. Podocyte-specific Atg5 deletion accelerated glomerular injury in these mice. TEM further confirmed the presence of mesangial expansion and glomerulosclerosis in podocyte-specific autophagy-deficient mice, underscoring the communication between podocytes and mesangial cells under diabetic conditions (Lenoir et al. 2015). In renal biopsy samples from diabetic patients, autophagy was also decreased in podocytes (Fang et al. 2013). These results suggest that autophagy is induced for renoprotection by short term of high glucose (early stage diabetes), but it is suppressed by long term of high glucose exposure (late stage diabetes) contributing to aggravated glomerular injury and progression of DKD. Interestingly in cultured podocytes, along with autophagy impairment, ER stress was induced by prolonged high glucose treatment for up to 60 h (Fang et al. 2013). There was a switch from an adaptive unfolded protein response (UPR) to a cytotoxic ER stress response. ER stress inhibitors such as salubrinal or TUDCA restored autophagy and reduced podocyte injury, indicating that this adaptive-to-cytotoxic switch may be correlated to deficient autophagic turnover of damaged ER (Fang et al. 2013). Tagawa et al. further confirmed that autophagy deficiency in podocytes is pivotal for the progression of advanced DKD, particularly the development of massive proteinuria (Tagawa et al. 2016). Autophagy was impaired in the podocytes of type 2 diabetic patients and Otsuka Long-Evans Tokushima Fatty (OLETF) rats with massive proteinuria, but not in those with absent or minimal proteinuria. Compared with high-fat diet (HFD)-fed control mice that had minimal proteinuria, HFD-fed mice with podocyte-specific Atg5 deletion displayed more severe podocyte injury and tubulointerstitial fibrosis, damaged lysosomes accumulated in podocytes. These autophagy-deficient diabetic mice showed massive proteinuria, suggesting that autophagy is crucial for the clearance of damaged lysosomes in DKD and its impairment is involved in the progression of podocyte injury and proteinuria in DKD. The sera from diabetic patients and OLETF rats with massive proteinuria impeded autophagy–lysosomal pathway in cultured podocytes and prompted apoptosis, suggesting that serum-derived factors may negatively regulate podocyte autophagy in the progression of DKD (Tagawa et al. 2016).
The mechanism underlying diabetes-associated autophagy impairment in podocytes is under investigation. Nutrient-sensing pathways and intracellular stress signaling pathways are most explored. Several recent studies have provided novel insights. β-arrestins were upregulated in the kidney of diabetic mice and kidney biopsies from diabetic patients as well as in cultured podocytes exposed to high glucose. β-arrestins interacted with ATG7 to downregulate ATG12-ATG5 conjugation, thereby suppressing autophagy in podocytes (Liu et al. 2016). Sun et al. further demonstrated a negative regulation of podocyte autophagy by miR-217. In high glucose-induced podocytes, miR-217 was upregulated to induce podocyte injury and insulin resistance. Inhibition of miR-217 expression reactivated autophagy and reduced podocyte injury. Phosphatase and tensin homolog (PTEN) was a target of miR-217 in podocytes (Sun et al. 2017). A role of FoxO1 in regulating mitophagy in podocytes has also been suggested. Overexpression of FoxO1 promoted mitophagy via PTEN-induced putative kinase 1 (PINK1)/Parkin pathway, leading to the clearance of aberrant mitochondria and the amelioration of podocyte injury (Li et al. 2017). Furthermore, HDAC4 was induced in cultured podocytes by high glucose, AGEs and transforming growth factor (TGF)-β. By deacetylating signal transducers and activators of transcription factor 1 (STAT1), HDAC4 inhibited podocyte autophagy and induced podocyte injury. Inhibition of HDAC4 reactivated autophagy and alleviated podocyte injury. These results suggest that HDAC4 may suppress podocyte autophagy via STAT1 to accelerate the development of DKD (Wei and Dong 2014).
Hyperglycemia inhibits autophagy in proximal and distal tubules of diabetic animals. Sodium-glucose cotransporter 2 (SGLT2), mainly expressed in proximal tubules, regulates the high capacity reabsorption of glucose in PTECs via sodium gradient produced by sodium/potassium ATPase pumps. Inhibition of SGLT2 reduced the reabsorption of glucose in PTECs and decreased blood glucose concentrations (Nair and Wilding 2010). Deletion of Sglt2 alleviated STZ-induced abnormal accumulation of p62/SQSTM1 in kidney, indicating that SGLT2-mediated glucose uptake contributes to autophagy impairment in PTECs under diabetic conditions (Vallon et al. 2013). High glucose also induced p53 to upregulate the transcription and expression of miR-155 in PTECs. Overexpression of miR-155 targeted SIRT1 by binding to the SIRT1 3′UTR region to reduce its expression and activity on essential ATG proteins such as ATG5 and LC3, leading to autophagy impairment in PTECs (Wang et al. 2018). In addition, hyperactivated mTORC1 was involved in obesity-related autophagy inhibition in PTECs in type 2 diabetic mice and patients, as diet restriction or rapamycin restored autophagy under these conditions (Moruno-Manchon et al. 2018). Similarly in diabetic Wistar fatty rats, dietary restriction rescued autophagy deficiency in PTECs via suppression of mTORC1 and activation of SIRT1 pathways, which subsequently resulted in the protection against diabetes-induced tubular injury and renal dysfunction (Kitada et al. 2011, 2016). Recent studies further reveal an interconnection between autophagy impairment and AGEs accumulation in PTECs and its contribution to tubular injury in DKD. AGEs overload led to lysosomal dysfunction and disruption of autophagic flux in PTECs (Liu et al. 2015; Takahashi et al. 2017). AGEs overload upregulated LAMP1 in Atg5-competent primary PTECs, while it was suppressed in Atg5-deficient cells with AGEs overload, indicating that autophagy contributes to inducing lysosomal biogenesis and function under diabetic conditions. Similarly, the upregulation of lysosome was suppressed in PTEC-specific Atg5 deletion of diabetic mice compared to the diabetic control mice. Along with autophagy deficiency, the PTEC-specific Atg5 deletion of diabetic mice displayed enhanced accumulation of AGEs in PTECs, glomeruli and renal vasculature, and increased inflammation and renal fibrosis (Takahashi et al. 2017). These results suggest that autophagy may degrade accumulated AGEs by promoting lysosomal biogenesis and function in PTECs. As such, impaired autophagy under diabetic conditions may lead to failure in the turnover of lysosomes and consequent AGE accumulation, which further disrupts lysosomal function and blocks lysosomal degradation of AGEs. This vicious cycle subsequently results in irreversible injury in both glomeruli and tubulointerstitium for the progression of DKD (Takahashi et al. 2017).
The role of autophagy in mesangial cells and glomerular endothelial cells remains largely unclear. The expression of tissue inhibitors of metalloproteinase 3 (TIMP3) was reduced in STZ-induced diabetic mice and diabetic patients. Timp3-deficient mice had more severe diabetic kidney injury compared with control mice. Timp deletion in cultured mesangial cells suppressed autophagy via FoxO1/STAT pathway, and re-expression of TIMP in these mesangial cells reversed autophagy impairment (Fiorentino et al. 2013). In AGEs-treated mesangial cells, autophagy/mitophagy was activated via ROS and protected against AGEs-induced mitochondrial dysfunction and cell apoptosis (Xu et al. 2016a). In contrast, autophagy was suppressed in cultured rat mesangial cells in response to high glucose, accompanied with collagen I accumulation and cell hypertrophy. The inhibition of autophagy was mediated by miR-21/PTEN/Akt/mTOR pathway, and ursolic acid restored autophagy activity and attenuated mesangial injury and collagen I production (Lu et al. 2015). Using endothelial-specific Atg5 knockout mice, a recent study demonstrated direct evidence on the role of glomerular endothelial cell autophagy in DKD (Lenoir et al. 2015). Endothelial-specific Atg5 knockout non-diabetic mice exhibited mild lesions to the glomerular filtration barrier. These mice also had more severe glomerular endothelial injuries following STZ injection, showing GBM thickening and podocyte effacement. These results suggest that via a crosstalk between glomerular endothelial cells and surrounding podocytes autophagy in glomerular endothelial cells preserves both endothelial integrity and podocyte homeostasis (Lenoir et al. 2015).
3.3 Autophagy in Acute Kidney Injury (AKI) and Kidney Repair
AKI, mostly caused by nephrotoxic drugs, renal ischemia-reperfusion and sepsis, is a major renal disease associated with poor clinical outcomes in both short term (high morbidity and mortality) and long term (the development of CKD and ESRD) (Bellomo et al. 2012; Zuk and Bonventre 2016). The pathogenesis of AKI is multifactorial and involves a complex interplay among microvascular, tubular and inflammatory factors. Tubular cell injury and death are the key pathological features of this disorder (Bellomo et al. 2012; Linkermann et al. 2014; Zuk and Bonventre 2016). The activation of autophagy in AKI was initially demonstrated in experimental models of cisplatin-induced nephrotoxicity (Periyasamy-Thandavan et al. 2008; Yang et al. 2008). While Yang et al. showed autophagy activation in cultured renal tubular cells (Yang et al. 2008), we demonstrated it using both cell culture and mouse models (Periyasamy-Thandavan et al. 2008). Interestingly, both studies suggested a protective role of autophagy in renal tubular cells. Our follow-up study further verified autophagy activation and its protective role in renal ischemic/hypoxic AKI (Jiang et al. 2010). In 2012, we and other two groups independently demonstrated the protective role of tubular cell autophagy in AKI using renal tubule-specific autophagy gene knockout mouse models (Liu et al. 2012; Takahashi et al. 2012; Cheng et al. 2015). These findings have been summarized in recent reviews (Livingston and Dong 2014; Havasi and Dong 2016).
After injury, renal tubular cells have the capacity to regenerate for kidney repair. This process involves the activation of multiple signaling pathways in injured and regenerating tubular cells leading to the production and secretion of growth factors, cytokines and inflammatory mediators. Normal tubular repair begins with dedifferentiation, migration and proliferation of surviving cells to replace injured cells, followed by re-differentiation to restore normal epithelial structure and function. However, tubular repair following severe or multiple episodes of AKI is often incomplete and maladaptive, leading to renal interstitial fibrosis and CKD (Ferenbach and Bonventre 2015; Venkatachalam et al. 2015; Basile et al. 2016; He et al. 2017). Although the mechanisms underlying AKI to CKD transition remain to be elucidated, emerging evidence suggests a central role of proximal tubules in the disease progression. After severe or episodic AKI normal tubular epithelial cells undergo a phenotype change with persistent production and secretion of profibrotic proteins. These tubule-derived molecules may drive renal interstitial fibrosis and AKI to CKD transition via autocrine and paracrine functions (Ferenbach and Bonventre 2015; Venkatachalam et al. 2015; Basile et al. 2016).
Autophagy is induced in tubular cells during AKI and protects against kidney injury (Livingston and Dong 2014; Kaushal and Shah 2016). During recovery following AKI, resolution of autophagy in tubular cells may promote cell proliferation for tubular regeneration and repair (Li et al. 2014). Using autophagy reporter mice with a tandem RFP-EGFP-LC3 fusion protein expressing ubiquitously under the CAG promoter, Li et al. revealed dynamic changes of autophagy in renal tubules during ischemic AKI and the following recovery phase. At 1 day of reperfusion, there was increased formation of both autophagosomes and autolysosomes in proximal tubule cells. At 3 days of reperfusion, autolysosomes appeared, suggesting the resolution of autophagy during the recovery phase. Mechanistically, mTOR was activated after ischemic AKI. Some renal tubules showed defective mTOR activity and persistent RFP-LC3 puncta, and interestingly tubular proliferation was also inhibited, indicating that autophagic cells are less likely to divide for tubular repair (Li et al. 2014). These findings are consistent with an earlier study suggesting that rapamycin delays recovery from ischemic AKI (Lieberthal et al. 2006).
A recent study by Brooks et al. further revealed a novel mechanism of epithelial biology linking phagocytosis, autophagy and antigen presentation to regulation of the inflammatory response after injury (Brooks et al. 2015). Kidney injury molecule-1 (KIM-1) expressed on proximal tubular cells transformed the cells into phagocytes for the uptake of luminal apoptotic cell debris. This KIM-1-mediated phagocytosis was subsequently processed through autophagy for efficient clearance of apoptotic cells and autophagic degradation of phagosomes, leading to major histocompatibility complex (MHC) restricted antigen presentation that suppressed CD4 T cell proliferation but increased the percentage of regulatory T cells in an autophagy‐dependent manner. These results highlight the role of autophagy in downregulating the inflammatory response after injury and maintaining self-tolerance in proximal tubular cells, both of which would contribute to an improved tubular repair (Brooks et al. 2015).
The role of autophagy in maladaptive repair and AKI to CKD transition needs to be further elucidated. In this setting, autophagy may be less effective and thereby fail to mediate intracellular degradation of newly synthesized fibrotic proteins (Zuk and Bonventre 2016). Instead, autophagy, coordinated with several other proximal tubular responses such as dedifferentiation, cell cycle changes and metabolic changes, may be adaptive initially, but ultimately lead to maladaptive responses that promote interstitial fibrosis and AKI to CKD transition (He et al. 2014; Gewin 2018). Along this line, a profibrotic role of autophagy has been shown in a mouse model of post-ischemic kidney fibrosis (Baisantry et al. 2016). Sustained activation of autophagy (30 days) following ischemic AKI induced prosenescent changes in proximal tubules of the S3 segments during recovery phase. Selective deletion of Atg5 in these proximal tubules suppressed autophagy, which in turn inhibited the development of a senescent phenotype and AKI progression to CKD (Baisantry et al. 2016). Of interest, compared with wild-type mice, selective Atg5 knockout mice showed more tubular cell death at the S3 segment at 2 h after reperfusion but less tubular damage and inflammation at day 3, suggesting that autophagy inhibition may enhance cell death in severely damaged tubular cells during injury phase but is beneficial for adaptive tubular repair during recovery phase. When such compromised cells with intact autophagy escape from cell death pathway and persist, they may develop into a senescent phenotype and promote renal interstitial fibrosis (Baisantry et al. 2016).
3.4 Autophagy in Renal Interstitial Fibrosis Induced by Unilateral Ureteral Obstruction (UUO) or TGF-β1
So far, most of the studies on the role of autophagy in renal interstitial fibrosis were performed in models of UUO or TGF-β1 and the findings are controversial. In mice subjected to UUO, autophagy was activated in renal tubules along with tubular apoptosis (Li et al. 2010; Forbes et al. 2011; Xu et al. 2013). Under this condition, autophagy and apoptosis acted in concert to induce tubular atrophy and nephron loss (Li et al. 2010). Oxidative stress-mediated mitochondrial damage was likely to promote autophagy and apoptosis in renal tubules, which may play a role in facilitating tubular decomposition in UUO (Xu et al. 2013). Using a tetracycline-controlled mouse model with TGF-β1 overexpression specifically in renal tubules, Koesters et al. showed that persistent expression of TGF-β1 promoted autophagy in renal tubules, leading to tubular dedifferentiation with widespread peritubular fibrosis. Notably, such degenerating cells were not positive for TUNEL staining for apoptosis, indicating that autophagy could be a key driver of tubular atrophy in TGF-β1-induced renal fibrosis (Koesters et al. 2010). Using pharmacological and genetic inhibitory approaches, we further demonstrated a profibrotic role of autophagy in a mouse model of UUO and in TGF-β1-treated PTECs (Livingston et al. 2016). Autophagy was persistently activated in proximal tubules following UUO. Pharmacological and genetic blockade of autophagy attenuated interstitial fibrosis, accompanied with the alleviation of tubular cell apoptosis, interstitial macrophage infiltration and production of fibroblast growth factor 2. In primary culture of PTECs, TGF-β1 induced fibronectin accumulation and cell death in an autophagy-dependent method (Livingston et al. 2016). A recent study by Yan et al. further demonstrated a connection between sustained activation of autophagy and lipid accumulation in tubular epithelial cells during kidney fibrosis (Yan et al. 2018). UUO-induced lipid accumulation in tubular cells was significantly reduced by autophagy inhibitors, along with the attenuation of renal interstitial fibrosis, tubular cell apoptosis and tubular cell dedifferentiation. This fibrosis-related lipid accumulation was not associated with lipophagy–lysosome pathway but was dependent on BECN1. These results highlight a role of autophagy in regulation of the lipid metabolism in renal tubular cells (Yan et al. 2018). Of interest, during thioacetamide or CCl4-induced liver injury, autophagy was activated in hepatic stellate cells, which further broke down lipids to fuel the activation of these cells to promote liver fibrosis (Thoen et al. 2011; Hernández-Gea et al. 2012). Therefore, autophagy may involve in lipid metabolism through a bidirectional mechanism of inducing lipolysis to activate liver fibroblasts as well as promoting lipid accumulation to induce lipotoxicity in renal tubular cells, both of which contribute to the development of fibrosis in these organs. A role for autophagy in the activation of fibroblasts and its profibrotic effects were also shown in UUO kidneys and in cultured renal fibroblast cells exposed to TGF-β1. Protein kinase C (PKC)-α, via stimulating autophagic flux, drove renal fibroblast activation and kidney fibrosis (Xue et al. 2018).
On the contrary, several studies have demonstrated an antifibrotic role of autophagy in UUO-associated renal interstitial fibrosis. In a rat model of UUO, inhibition of autophagy by 3-methyladenine aggravated tubular cell apoptosis and interstitial fibrosis, indicating that autophagy may inhibit fibrosis by suppressing tubular apoptosis (Kim et al. 2012c). In primary culture of mouse kidney mesangial cells, both protein and mRNA levels of collagen I were induced by TGF-β1. Inhibition of autophagy by BECN1 knockdown or lysosomal inhibitors further increased collagen I protein accumulation without affecting its mRNA level, indicating a potential effect for autophagy in regulating ECM deposition in mesangial cells by facilitating the degradation of collagen I (Kim et al. 2012a). MAP1S, via interacting with LC3, activated autophagy to suppress fibrosis by mediating fibronectin turnover. Defective MAP1S impaired autophagic clearance of fibronectin and induced renal fibrosis in aged mice. Reduced expression of MAP1S in renal biopsies from patients with kidney fibrosis was also accompanied with elevated accumulation of fibronectin (Xu et al. 2016b). Ding Y et al. further showed the role for autophagy in promoting the degradation of mature TGF-β1 in UUO kidneys and in TGF-β1-treated PTECs, further suggesting that autophagy can inhibit renal interstitial fibrosis by negative regulation of TGF-β1 (Ding et al. 2014). Using a proximal tubule-specific Atg5 knockout mouse model, Li et al. suggested that autophagy deficiency promoted cell cycle G2/M arrest and accelerated renal interstitial fibrosis following UUO. Stimulation of Atg5-deficient primary proximal tubular cells with angiotensin II also led to G2/M arrest and increased production of collagen I, which was rescued by the restoration of autophagy-competent Atg5 in these cells (Li et al. 2016). Elucidation of the mechanisms of autophagy regulation in renal interstitial fibrosis is pivotal for identifying potential therapeutic targets to treat progressive CKD.
4 Conclusions and Perspectives
Basal level of autophagy is essential to the maintenance of cellular homeostasis in renal resident cells including podocytes, renal tubular cells, mesangial cells and glomerular endothelial cells. Autophagy defects in these cells have been implicated in the development of CKD such as FSGS and DKD. Autophagy is induced in response to AKI for renoprotection. After kidney injury, tightly regulated autophagy may participate in adaptive renal repair, whereas dysregulated autophagy may lead to maladaptive repair contributing to AKI to CKD transition. The role of autophagy in renal interstitial fibrosis is multifaceted and complex. Further research is needed to gain significant insights into the role of autophagy in the pathogenesis of renal fibrosis and related kidney diseases as well as the regulatory mechanisms of autophagy in these disease settings. A comprehensive understanding of the regulation and pathological roles of autophagy in renal fibrosis will facilitate the discovery of novel therapeutic strategy that can target autophagy for the prevention and treatment of fibrosis-related CKD.
References
Abbate M, Zoja C, Remuzzi G (2006) How does proteinuria cause progressive renal damage. J Am Soc Nephrol 17:2974–2984
Alers S, Löffler AS, Wesselborg S, Stork B (2012) Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 32:2–11
Asanuma K, Tanida I, Shirato I, Ueno T, Takahara H, Nishitani T et al (2003) MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J 17:1165–1167
Baisantry A, Bhayana S, Rong S, Ermeling E, Wrede C, Hegermann J et al (2016) Autophagy induces prosenescent changes in proximal tubular S3 segments. J Am Soc Nephrol 27:1609–1616
Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH et al (2016) Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol 27:687–697
Beeken M, Lindenmeyer MT, Blattner SM, Radón V, Oh J, Meyer TN et al (2014) Alterations in the ubiquitin proteasome system in persistent but not reversible proteinuric diseases. J Am Soc Nephrol 25:2511–2525
Bellomo R, Kellum JA, Ronco C (2012) Acute kidney injury. Lancet 380:756–766
Brooks CR, Yeung MY, Brooks YS, Chen H, Ichimura T, Henderson JM et al (2015) KIM-1-/TIM-1-mediated phagocytosis links ATG5-/ULK1-dependent clearance of apoptotic cells to antigen presentation. EMBO J 34:2441–2464
Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54:1615–1625
Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC et al (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458:1056–1060
Cao A, Wang L, Chen X, Guo H, Chu S, Zhang X et al (2016a) Ursodeoxycholic acid ameliorated diabetic nephropathy by attenuating hyperglycemia-mediated oxidative stress. Biol Pharm Bull 39:1300–1308
Cao AL, Wang L, Chen X, Wang YM, Guo HJ, Chu S et al (2016b) Ursodeoxycholic acid and 4-phenylbutyrate prevent endoplasmic reticulum stress-induced podocyte apoptosis in diabetic nephropathy. Lab Invest 96:610–622
Carney EF (2015) Glomerular disease: autophagy failure and mitochondrial dysfunction in FSGS. Nat Rev Nephrol 11:66
Chen Y, Liu CP, Xu KF, Mao XD, Lu YB, Fang L et al (2008) Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am J Nephrol 28:1014–1022
Chen J, Chen MX, Fogo AB, Harris RC, Chen JK (2013) mVps34 deletion in podocytes causes glomerulosclerosis by disrupting intracellular vesicle trafficking. J Am Soc Nephrol 24:198–207
Cheng H, Fan X, Lawson WE, Paueksakon P, Harris RC (2015) Telomerase deficiency delays renal recovery in mice after ischemia-reperfusion injury by impairing autophagy. Kidney Int 88:85–94
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368:651–662
Chuang PY, Dai Y, Liu R, He H, Kretzler M, Jim B et al (2011) Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS ONE 6:e23566
Cinà DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J et al (2012a) MTOR regulates autophagic flux in the glomerulus. Autophagy 8:696–698
Cinà DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J et al (2012b) Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol 23:412–420
Cui J, Bai XY, Shi S, Cui S, Hong Q, Cai G et al (2012) Age-related changes in the function of autophagy in rat kidneys. Age (Dordr) 34:329–339
D’Agati VD (2012) Pathobiology of focal segmental glomerulosclerosis: new developments. Curr Opin Nephrol Hypertens 21:243–250
Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19:349–364
Ding Y, Sl K, Lee SY, Koo JK, Wang Z, Choi ME (2014) Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 25:2835–2846
Duffield JS (2014) Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest 124:2299–2306
Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE et al (2013) AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest 123:4888–4899
Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W et al (2013) Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS ONE 8:e60546
Ferenbach DA, Bonventre JV (2015) Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11:264–276
Fiorentino L, Cavalera M, Menini S, Marchetti V, Mavilio M, Fabrizi M et al (2013) Loss of TIMP3 underlies diabetic nephropathy via FoxO1/STAT1 interplay. EMBO Mol Med 5:441–455
Forbes JM, Cooper ME (2013) Mechanisms of diabetic complications. Physiol Rev 93:137–188
Forbes MS, Thornhill BA, Chevalier RL (2011) Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol 301:F110–F117
Fukuda A, Chowdhury MA, Venkatareddy MP, Wang SQ, Nishizono R, Suzuki T et al (2012) Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol 23:1351–1363
Gewin LS (2018) Renal fibrosis: primacy of the proximal tubule. Matrix Biol 68–69:248–262
Ghosh HS, McBurney M, Robbins PD (2010) SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE 5:e9199
Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S et al (2011) Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121:2197–2209
Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S et al (2010) Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 120:1084–1096
Hartleben B, Wanner N, Huber TB (2014) Autophagy in glomerular health and disease. Semin Nephrol 34:42–52
Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K et al (2013) Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med 19:1496–1504
Havasi A, Dong Z (2016) Autophagy and tubular cell death in the kidney. Semin Nephrol 36:174–188
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93
He L, Livingston MJ, Dong Z (2014) Autophagy in acute kidney injury and repair. Nephron Clin Pract 127:56–60
He L, Wei Q, Liu J, Yi M, Liu Y, Liu H et al (2017) AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int 92:1071–1083
Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z et al (2012) Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142:938–946
Higgins GC, Coughlan MT (2014) Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy. Br J Pharmacol 171:1917–1942
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y et al (2009) Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 20:1981–1991
Huber TB, Edelstein CL, Hartleben B, Inoki K, Jiang M, Koya D et al (2012) Emerging role of autophagy in kidney function, diseases and aging. Autophagy 8:1009–1031
Humphreys BD (2018) Mechanisms of Renal Fibrosis. Annu Rev Physiol 80:309–326
Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS et al (2008) Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2:284–291
Hurley JH, Young LN (2017) Mechanisms of autophagy initiation. Annu Rev Biochem 86:225–244
Imai S, Guarente L (2010) Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci 31:212–220
Inoki K (2014) mTOR signaling in autophagy regulation in the kidney. Semin Nephrol 34:2–8
Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S et al (2011) mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest 121:2181–2196
Jiang M, Liu K, Luo J, Dong Z (2010) Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 176:1181–1192
Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z (2012) Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 82:1271–1283
Jiang XS, Chen XM, Wan JM, Gui HB, Ruan XZ, Du XG (2017) Autophagy protects against palmitic acid-induced apoptosis in podocytes in vitro. Sci Rep 7:42764
Jin Y, Liu S, Ma Q, Xiao D, Chen L (2017) Berberine enhances the AMPK activation and autophagy and mitigates high glucose-induced apoptosis of mouse podocytes. Eur J Pharmacol 794:106–114
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J et al (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20:1992–2003
Kaushal GP, Shah SV (2016) Autophagy in acute kidney injury. Kidney Int 89:779–791
Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE et al (2015) Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol 26:1040–1052
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141
Kim J, Shon E, Kim CS, Kim JS (2012a) Renal podocyte injury in a rat model of type 2 diabetes is prevented by metformin. Exp Diabetes Res 2012:210821
Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME (2012b) Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J Biol Chem 287:11677–11688
Kim WY, Nam SA, Song HC, Ko JS, Park SH, Kim HL et al (2012c) The role of autophagy in unilateral ureteral obstruction rat model. Nephrology (Carlton) 17:148–159
Kim MY, Lim JH, Youn HH, Hong YA, Yang KS, Park HS et al (2013) Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1α axis in db/db mice. Diabetologia 56:204–217
Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T et al (2011) Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 22:902–913
Kitada M, Takeda A, Nagai T, Ito H, Kanasaki K, Koya D (2011) Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabetes Res 2011:908185
Kitada M, Ogura Y, Suzuki T, Sen S, Lee SM, Kanasaki K et al (2016) A very-low-protein diet ameliorates advanced diabetic nephropathy through autophagy induction by suppression of the mTORC1 pathway in Wistar fatty rats, an animal model of type 2 diabetes and obesity. Diabetologia 59:1307–1317
Klionsky DJ (2008) Autophagy revisited: a conversation with Christian de Duve. Autophagy 4:740–743
Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM et al (2011) A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 7:1273–1294
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo AA et al (2016) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12:1–222
Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R et al (2010) Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol 177:632–643
Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S et al (2010) Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 120:1043–1055
Kume S, Thomas MC, Koya D (2012) Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes 61:23–29
Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE et al (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 105:3374–3379
Lee JW, Park S, Takahashi Y, Wang HG (2010) The association of AMPK with ULK1 regulates autophagy. PLoS ONE 5:e15394
Lenoir O, Jasiek M, Hénique C, Guyonnet L, Hartleben B, Bork T et al (2015) Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 11:1130–1145
Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB et al (2017) Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Li L, Zepeda-Orozco D, Black R, Lin F (2010) Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol 176:1767–1778
Li L, Wang ZV, Hill JA, Lin F (2014) New autophagy reporter mice reveal dynamics of proximal tubular autophagy. J Am Soc Nephrol 25:305–315
Li H, Peng X, Wang Y, Cao S, Xiong L, Fan J et al (2016) Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 12:1472–1486
Li W, Du M, Wang Q, Ma X, Wu L, Guo F et al (2017) FoxO1 promotes mitophagy in the podocytes of diabetic male mice via the PINK1/parkin pathway. Endocrinology 158:2155–2167
Lieberthal W, Fuhro R, Andry C, Patel V, Levine JS (2006) Rapamycin delays but does not prevent recovery from acute renal failure: role of acquired tubular resistance. Transplantation 82:17–22
Lim JH, Kim HW, Kim MY, Kim TW, Kim EN, Kim Y et al (2018) Cinacalcet-mediated activation of the CaMKKβ-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell Death Dis 9:270
Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z (2014) Regulated cell death in AKI. J Am Soc Nephrol 25:2689–2701
Liu Y (2011) Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7:684–696
Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N et al (2012) Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 8:826–837
Liu WJ, Shen TT, Chen RH, Wu HL, Wang YJ, Deng JK et al (2015) Autophagy-lysosome pathway in renal tubular epithelial cells is disrupted by advanced glycation end products in diabetic nephropathy. J Biol Chem 290:20499–20510
Liu J, Li QX, Wang XJ, Zhang C, Duan YQ, Wang ZY et al (2016) β-Arrestins promote podocyte injury by inhibition of autophagy in diabetic nephropathy. Cell Death Dis 7:e2183
Livingston MJ, Dong Z (2014) Autophagy in acute kidney injury. Semin Nephrol 34:17–26
Livingston MJ, Ding HF, Huang S, Hill JA, Yin XM, Dong Z (2016) Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 12:976–998
Lloberas N, Cruzado JM, Franquesa M, Herrero-Fresneda I, Torras J, Alperovich G et al (2006) Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol 17:1395–1404
Löwik MM, Groenen PJ, Levtchenko EN, Monnens LA, van den Heuvel LP (2009) Molecular genetic analysis of podocyte genes in focal segmental glomerulosclerosis—a review. Eur J Pediatr 168:1291–1304
Lu X, Fan Q, Xu L, Li L, Yue Y, Xu Y et al (2015) Ursolic acid attenuates diabetic mesangial cell injury through the up-regulation of autophagy via miRNA-21/PTEN/Akt/mTOR suppression. PLoS ONE 10:e0117400
Ma T, Zhu J, Chen X, Zha D, Singhal PC, Ding G (2013) High glucose induces autophagy in podocytes. Exp Cell Res 319:779–789
Ma L, Fu R, Duan Z, Lu J, Gao J, Tian L et al (2016) Sirt1 is essential for resveratrol enhancement of hypoxia-induced autophagy in the type 2 diabetic nephropathy rat. Pathol Res Pract 212:310–318
Mehrpour M, Esclatine A, Beau I, Codogno P (2010) Overview of macroautophagy regulation in mammalian cells. Cell Res 20:748–762
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15:1101–1111
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075
Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27:107–132
Mori H, Inoki K, Masutani K, Wakabayashi Y, Komai K, Nakagawa R et al (2009) The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem Biophys Res Commun 384:471–475
Moruno-Manchon JF, Uzor NE, Kesler SR, Wefel JS, Townley DM, Nagaraja AS et al (2018) Peroxisomes contribute to oxidative stress in neurons during doxorubicin-based chemotherapy. Mol Cell Neurosci 86:65–71
Nagai K, Matsubara T, Mima A, Sumi E, Kanamori H, Iehara N et al (2005) Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int 68:552–561
Nair S, Wilding JP (2010) Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus. J Clin Endocrinol Metab 95:34–42
Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S et al (2011) Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332:966–970
Oshima Y, Kinouchi K, Ichihara A, Sakoda M, Kurauchi-Mito A, Bokuda K et al (2011) Prorenin receptor is essential for normal podocyte structure and function. J Am Soc Nephrol 22:2203–2212
Pavenstädt H, Kriz W, Kretzler M (2003) Cell biology of the glomerular podocyte. Physiol Rev 83:253–307
Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z (2008) Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 74:631–640
Qi W, Mu J, Luo ZF, Zeng W, Guo YH, Pang Q et al (2011) Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metabolism 60:594–603
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW et al (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90:1383–1435
Rubinsztein DC, Shpilka T, Elazar Z (2012) Mechanisms of autophagosome biogenesis. Curr Biol 22:R29–R34
Sakaguchi M, Isono M, Isshiki K, Sugimoto T, Koya D, Kashiwagi A (2006) Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochem Biophys Res Commun 340:296–301
Sataranatarajan K, Mariappan MM, Lee MJ, Feliers D, Choudhury GG, Barnes JL et al (2007) Regulation of elongation phase of mRNA translation in diabetic nephropathy: amelioration by rapamycin. Am J Pathol 171:1733–1742
Sionov RV, Vlahopoulos SA, Granot Z (2015) Regulation of bim in health and disease. Oncotarget 6:23058–23134
Steinberg GR, Kemp BE (2009) AMPK in health and disease. Physiol Rev 89:1025–1078
Stridh S, Palm F, Takahashi T, Ikegami-Kawai M, Hansell P (2015) Inhibition of mTOR activity in diabetes mellitus reduces proteinuria but not renal accumulation of hyaluronan. Ups J Med Sci 120:233–240
Sun J, Li ZP, Zhang RQ, Zhang HM (2017) Repression of miR-217 protects against high glucose-induced podocyte injury and insulin resistance by restoring PTEN-mediated autophagy pathway. Biochem Biophys Res Commun 483:318–324
Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M et al (2016) Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes 65:755–767
Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H et al (2012) Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol 180:517–525
Takahashi A, Takabatake Y, Kimura T, Maejima I, Namba T, Yamamoto T et al (2017) Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes 66:1359–1372
Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z et al (2018) PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 1–18
Thoen LF, Guimarães EL, Dollé L, Mannaerts I, Najimi M, Sokal E et al (2011) A role for autophagy during hepatic stellate cell activation. J Hepatol 55:1353–1360
Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H et al (2013) Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304:F156–F167
Velagapudi C, Bhandari BS, Abboud-Werner S, Simone S, Abboud HE, Habib SL (2011) The tuberin/mTOR pathway promotes apoptosis of tubular epithelial cells in diabetes. J Am Soc Nephrol 22:262–273
Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK (2015) Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol 26:1765–1776
Wang Y, Zheng ZJ, Jia YJ, Yang YL, Xue YM (2018) Role of p53/miR-155-5p/sirt1 loop in renal tubular injury of diabetic kidney disease. J Transl Med 16:146
Wanner N, Hartleben B, Herbach N, Goedel M, Stickel N, Zeiser R et al (2014) Unraveling the role of podocyte turnover in glomerular aging and injury. J Am Soc Nephrol 25:707–716
Wei Q, Dong Z (2014) HDAC4 blocks autophagy to trigger podocyte injury: non-epigenetic action in diabetic nephropathy. Kidney Int 86:666–668
Wellen KE, Thompson CB (2010) Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 40:323–332
Wittmann S, Daniel C, Stief A, Vogelbacher R, Amann K, Hugo C (2009) Long-term treatment of sirolimus but not cyclosporine ameliorates diabetic nephropathy in the rat. Transplantation 87:1290–1299
Wu L, Zhang Y, Ma X, Zhang N, Qin G (2012) The effect of resveratrol on FoxO1 expression in kidneys of diabetic nephropathy rats. Mol Biol Rep 39:9085–9093
Xavier S, Gilbert V, Rastaldi MP, Krick S, Kollins D, Reddy A et al (2010) BAMBI is expressed in endothelial cells and is regulated by lysosomal/autolysosomal degradation. PLoS ONE 5:e12995
Xiao T, Guan X, Nie L, Wang S, Sun L, He T et al (2014) Rapamycin promotes podocyte autophagy and ameliorates renal injury in diabetic mice. Mol Cell Biochem 394:145–154
Xu Y, Ruan S, Wu X, Chen H, Zheng K, Fu B (2013) Autophagy and apoptosis in tubular cells following unilateral ureteral obstruction are associated with mitochondrial oxidative stress. Int J Mol Med 31:628–636
Xu G, Yue F, Huang H, He Y, Li X, Zhao H et al (2016a) Defects in MAP1S-mediated autophagy turnover of fibronectin cause renal fibrosis. Aging (Albany NY) 8:977–985
Xu L, Fan Q, Wang X, Zhao X, Wang L (2016b) Inhibition of autophagy increased AGE/ROS-mediated apoptosis in mesangial cells. Cell Death Dis 7:e2445
Xue X, Ren J, Sun X, Gui Y, Feng Y, Shu B et al (2018) Protein kinase Cα drives fibroblast activation and kidney fibrosis by stimulating autophagic flux. J Biol Chem 293:11119–11130
Yan Q, Song Y, Zhang L, Chen Z, Yang C, Liu S et al (2018) Autophagy activation contributes to lipid accumulation in tubular epithelial cells during kidney fibrosis. Cell Death Discov 5:2
Yang Z, Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22:124–131
Yang Y, Wang J, Qin L, Shou Z, Zhao J, Wang H et al (2007) Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am J Nephrol 27:495–502
Yang C, Kaushal V, Shah SV, Kaushal GP (2008) Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 294:F777–F787
Yang D, Livingston MJ, Liu Z, Dong G, Zhang M, Chen JK et al (2018) Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cell Mol Life Sci 75:669–688
Yi M, Zhang L, Liu Y, Livingston MJ, Chen JK, Nahman NS et al (2017) Autophagy is activated to protect against podocyte injury in adriamycin-induced nephropathy. Am J Physiol Renal Physiol 313:F74–F84
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J et al (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465:942–946
Yu L, Chen Y, Tooze SA (2018) Autophagy pathway: cellular and molecular mechanisms. Autophagy 14:207–215
Zaffagnini G, Martens S (2016) Mechanisms of selective autophagy. J Mol Biol 428:1714–1724
Zeng C, Fan Y, Wu J, Shi S, Chen Z, Zhong Y et al (2014) Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J Pathol 234:203–213
Zhang L, Livingston MJ, Chen JK, Dong Z (2014a) Autophagy in podocytes. Contrib Nephrol 183:83–100
Zhang MZ, Wang Y, Paueksakon P, Harris RC (2014b) Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes 63:2063–2072
Zhang D, Pan J, Xiang X, Liu Y, Dong G, Livingston MJ et al (2016) Protein kinase Cδ suppresses autophagy to induce kidney cell apoptosis in cisplatin nephrotoxicity. J Am Soc Nephrol
Zhao L, Sun LN, Nie HB, Wang XL, Guan GJ (2014) Berberine improves kidney function in diabetic mice via AMPK activation. PLoS ONE 9:e113398
Zhao X, Chen Y, Tan X, Zhang L, Zhang H, Li Z et al (2018) Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. J Pathol 245:235–248
Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12:21–35
Zuk A, Bonventre JV (2016) Acute kidney injury. Annu Rev Med 67:293–307
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Zhao, XC., Livingston, M.J., Liang, XL., Dong, Z. (2019). Cell Apoptosis and Autophagy in Renal Fibrosis. In: Liu, BC., Lan, HY., Lv, LL. (eds) Renal Fibrosis: Mechanisms and Therapies. Advances in Experimental Medicine and Biology, vol 1165. Springer, Singapore. https://doi.org/10.1007/978-981-13-8871-2_28
Download citation
DOI: https://doi.org/10.1007/978-981-13-8871-2_28
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-8870-5
Online ISBN: 978-981-13-8871-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)