
Abstract
Optic pathway gliomas are the most common cause of vision loss in patients with neurofibromatosis and are clinically seen in 1–5% patients and radiologically evident in 15%. Clinical surveillance with MRI is required for diagnosis. Treatment options include surgery, chemotherapy, and radiation with future prospects of targeted treatment involving the mTOR pathway.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Optic pathway glioma
- Optic nerve glioma
- Pilocytic astrocytoma
- Neurofibromatosis 1 (NF-1)
- Chiasm
- Pediatric oncology
Introduction
Optic pathway gliomas account for about 1% of all intracranial tumors [1]. These tumors are pilocytic astrocyte tumors that can occur sporadically or due to neurofibromatosis type I (NF-1). Ninety percent of optic pathway tumors are either benign childhood gliomas or optic nerve sheath meningiomas; adult malignant gliomas are especially rare [2]. Most glioma cases occur in children under the age of 20. A younger age of onset generally correlates with poorer outcomes whereas NF-1-related optic nerve gliomas have better outcomes.
Optic pathway gliomas can sometimes be asymptomatic. Patients with NF-1 are more commonly asymptomatic compared to those with spontaneous gliomas [3]. When they do occur, symptoms are generally progressive due to slow enlargement of the tumor leading to proptosis and eventually displacement of the globe as the tumor expands. Decreased visual acuity can be an accompanying symptom along with decreased visual fields, RAPD, and disc swelling. Rarely, nystagmus could be a presenting symptom in children with chiasma/hypothalamus involving tumors [4]. Posterior optic nerve gliomas can also present as obstructive hydrocephalus or endocrinopathies. Precocious puberty is the most common hormonal disturbance from an optic glioma endocrinopathy [5, 6]. Other endocrinopathies that have been associated with OPG include growth hormone deficiency, obesity with insulin resistance/impaired glucose tolerance, GH excess, ACTH deficiency, hypogonadotropic hypogonadism, and thyrotropin deficiency [6]. Acute vision loss is a very rare complication that occurs when there is a hemorrhage in a tumor.
Severity of the tumor generally depends on the location of the tumor and can be divided into anterior visual pathway tumors and posterior visual pathway tumors. Posterior visual pathway tumors can be more aggressive and are generally more severe because the tumor can compress the chiasm and the hypothalamus (Fig. 1). These are more commonly associated with a sporadic tumor leading to more symptomatic presentations compared to those with NF-1, which are more commonly anterior visual pathway tumors [3]. Studies have demonstrated that the risk of vision loss is higher in more posteriorly located tumors and in older individuals and hence it is recommended that children with posteriorly located tumors have a close follow-up and visual assessment till the age of 18 [1]. A vast majority of these tumors pathologically fall under the category of pilocytic astrocytomas with a benign outcome. However, a small proportion of these tumors could run a more aggressive course, known as diffuse non-pilocytic astrocytomas. Spontaneous regression of clinically symptomatic tumor, both in patients with and without NF 1, has been reported in literature and could be associated with variable degree of visual improvement. This is a factor that should be considered while planning management of these tumors [7].

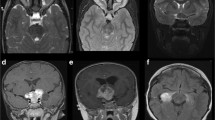
Optic chiasmal glioma imaged with MRI
Diagnosis of optic nerve gliomas is confirmed by MRI. MRI is considered the best imaging due to its ability to show the entire path of the optic nerve to the hypothalamus to ensure that there is no hypothalamic involvement.
Chemo- and radiation treatment options have been successful in preventing severe visual disturbances by halting the progression of tumor growth. This chapter discusses the advancements in screening and treatment of optic nerve gliomas.
Screening
Current recommendations for screening children suggest that all children with NF1 younger than 8 years should undergo an annual ophthalmological examination that should include measurement of visual acuity, confrontation visual field evaluation, color vision testing, and assessment of pupils, eyelids, ocular motility, irises, and fundi (Table 1) [8]. Formal computerized or kinetic testing of visual fields may be adjunctive if the patient is reliable, but is not necessary.
Guidelines for evaluation of children under 1 year of age are not very clear; however Liu et al. recommend neuroimaging in this age group if the diagnosis is confirmed and visual assessment is unreliable to guide management.
Once children grow past the age of 8, the risk of development of OPG significantly decreases and there are no firm guidelines for follow-up protocol in this subset of children. Until new evidence is discovered, it is recommended that children in this age group should receive complete eye examinations every 2 years until they become 18 years old. After the age of 18, adults may have routine eye assessments without the need for specialized testing or imaging. Assessing color vision during an examination is also important because the presence of a visual acuity defect in the absence of a color vision defect would suggest other causes of an abnormal exam such as refractive error or amblyopia.
Visual evoked potential visual tests (VEP), though a sensitive method to detect optic nerve gliomas in asymptomatic children, is not routinely recommended. This is because it would not alter the management in the case of an asymptomatic patient. Even in symptomatic children with either low visual acuity, an abnormal fundus examination, or abnormal visual fields, neuroimaging is necessary regardless of the VEP report. Follow-up VEPs are also not recommended as subtle changes in the value are of uncertain significance and in the setting of stable visual and radiologic testing would not be a basis to start treatment [8].
Listernick et al. also recommended that the children have annual height and weight measurements to screen for precocious puberty [9]. This was further confirmed by Segal et al. who reported a precocious puberty incidence of 14% in his retrospective chart review which followed 44 patients with OPG [10].
Risk for Progression
Using a multivariate analysis, Stokland et al. found factors that determine tumor progression in a childhood low-grade glioma including age, histology, and extent of resection. The risk of tumor progression decreases with age. A pathological diagnosis of fibrillary astrocytoma and pilomyxoid astrocytoma has poor outcomes in comparison to pilocytic astrocytoma. Finally, partial and incomplete surgical resection had poorer outcomes compared to complete tumor resection. They also found that the chiasmatic/hypothalamic group shows an early peak (4 years of age) and a declining incidence in subsequent age [10].
The possibility of sex playing a role in presentation was explored by Kelly et al. who found that females with NF1-associated optic gliomas were twice as likely to have undergone neuroimaging for visual symptoms and three times more likely than boys to require treatment due to visual decline, though the glioma location and size are not impacted by sex [11]. They further went on to explore the same in NF1 genetically engineered mice model and found that female mice were more likely than their male counterpart to have visual symptoms and were found to be associated with degeneration of retinal ganglion cells both in vivo and in vitro.
Imaging
Routine imaging for screening in NF1-positive patients is currently not recommended by AAP as the incidence and morbidity of these tumors are low. MRI is recommended only if symptoms such as visual changes, persistent headaches, or seizures occur as well as if there is a marked increase in head size or a plexiform neurofibroma of the head. A small percentage of the NF1 population that has a deletion of the entire NF1 gene with flanking DNA will also get a MRI [12]. CT scans do not have any advantage over MRIs and are not recommended for any imaging because they have additional risk of radiation exposure in children which can lead to secondary tumors.
Segal et al. discovered no additional benefit from routine baseline MRI imaging. The research group found that an earlier diagnosis did not end up having an effect on clinical outcome. Additionally, frequent imaging leads to repeated sedation and psychological effects on both the child and parents for a generally benign tumor that would require no further intervention. These adverse effects in addition to the financial burden of repeated imaging with no significant change in care support the idea of not requiring routine MRI [13].
The current protocol for imaging includes an MRI of the orbits and brain with thin, coronal, sagittal, and axial images both with and without contrast. It also includes both T1- and T2-weighted images with sections through the optic nerve. OPGs have a typical appearance on MRI with fusiform enlargement and a downward kink in the mid orbit. Chiasmal tumors are better seen on coronal sections. Enhancement with gadolinium is a possible measure of activity in the tumor and could be used as a possible guide for treatment and follow-up (quote reference).
The tumor is typically hypointense on T1W images (Fig. 2c), and slightly hyperintense on T2W (Fig. 2d). Optic nerve gliomas (ONG) show increased diffusion on DWI; they exhibit high apparent diffusion coefficient (ADC) and low fractional anisotropy (FA) values. This is attributed to their low cellularity and low proliferative indices. ONGs show ADC values in the range of 1.2–2.09 × 10–3 mm2/s, which cannot however distinguish between clinically stable and aggressive tumors. In his cohort study, Yeom et al. found that a higher ADC predicted earlier tumor progression. However, there is insufficient data to include this in a routine protocol for imaging in OPG patients. They suggest that there is likely no benefit to treating a patient prior to onset of symptoms regardless of the ADC, but may prove to be useful in setting an appropriate clinical surveillance schedule and evaluating treatment responses [14].

6-Year-old girl with right optic nerve glioma presented with mild proptosis (a), decreased visual acuity, afferent pupillary defect, and disc edema (b). MRI shows fusiform right optic nerve lesion, which is showing contrast enhancement on T1 (c) and isointense on T2 sequence (d)
DTI tractography can be used in the presurgical evaluation of ONG by demonstrating integrity of the optic nerve in patients with resectable lesions and can help reduce postsurgical morbidity and visual field loss [15].
De Blanc et al. suggest that in a MRI with diffusion tensor imaging (DTI), a decrease in FA (fractional anisotropy) of the optic radiations is associated with abnormal visual acuity in NF1-associated OPGs and may be predictive of visual acuity loss during the following year. DTI allows identification and quantitation of white matter pathways, including optic nerves, tracts, and radiations [16].
As research continues to be conducted on the best imaging techniques, MRI continues to be the standard of care for imaging of OPGs when it is necessitated.
Recommendations for Management
Management options for OPGs include observation, chemotherapy, radiation, or surgery (Table 2). Choosing a treatment is often a difficult decision due to the unpredictable course of the tumor. Parsa et al. reported spontaneous regression in 12 out of the 13 eyes with improvement in visual function [17]. Smaller, asymptomatic tumors can be observed with periodic surveillance as per protocol. Considering that only one-third of OPGs progress, intervention is considered in patients when symptomatic. Indications for treatment include visual deterioration, tumor growth, increase in enhancement of MRI, visual field loss, endocrine dysfunction, hydrocephalus, or mass effect [18]. Liu et al. state that optic nerve gliomas without chiasmal involvement at presentation are more likely to remain localized without extension to the chiasm. Treatment for these patients for possible extension into chiasm is unwarranted.
Initiation of nonsurgical treatment is recommended in the case of clear evidence of bilateral visual deterioration. This can take the form of chemotherapy or radiation. A child with low vision unilaterally at presentation can still be observed if assessment in the child is reliable and they can be followed up closely every 1–2 months until further progressive deterioration is documented.
Chemotherapy is considered the primary treatment option in progressive OPG (documented as clinical or radiologic deterioration from baseline) of the chiasma or posterior optic pathway, when associated with NF1. In patients without NF1, a biopsy should be considered to ascertain the diagnosis. If a diagnosis of pilocytic astrocytoma is made, then chemotherapy is the option in symptomatic patients. A tumor with any other pathologic diagnosis in the optic pathway should be managed in a similar fashion to tumors of the same etiology arising elsewhere in CNS with chemotherapy or radiation [19].
The non-NF1 tumors involving the chiasm or hypothalamus are known to be chronically relapsing tumors which can eventually enter a static phase. Hence the general consensus in treatment of these tumors is to wait till tumor progression [20].
Studies have demonstrated that the risk of vision loss is higher in more posteriorly located tumors and in older individuals and hence it is recommended that children with posteriorly located tumors have a close follow-up and visual assessment till the age of 18.
Surgery is considered in patients of glioma involving optic nerve when they present with proptosis, mass effect, or significant deterioration of vision [21]. In chiasm involving tumors, surgery is used as the primary mode of management only for exophytic chiasmatic tumors, large tumors for de-bulking, or hydrocephalus [22]. These patients require close follow-up with two monthly scans for at least 6 months.
A multicenter retrospective cohort study aimed at determining the natural history of optic nerve gliomas found that during a mean follow-up of 5.6 years, 59% of the tumors progressed, 23% remained stable, and 18% (all with neurofibromatosis type 1) displayed some degree of spontaneous regression [23]. The authors concluded that radiological progression and visual deterioration occur in greater percentages in NF1 than in the general population of patients with OPGs. Response to chemotherapy may be better in this group, and its use should be considered early in the course of the disease.
Based on the Warburg hypothesis, mouse studies have shown regression of gliomas in response to ketogenic diet [24]. This is based on the theory that tumor cells thrive mainly on glycolysis for nutrition.
Radiation
Radiation therapy has not been recommended for treatment of OPG for quite some time now due to the risk of developing secondary tumors especially in children younger than the age of 5. The optic pathway glioma taskforce consensus report states that various studies suggested that radiotherapy appeared to improve the 5- and 10-year disease-free rates in patients with progressive gliomas. At 20 years, however, the overall disease-free survival rates are essentially equivalent between the patients who were treated with radiotherapy and those who were not [25].
Radiation therapy continues to have a role in the treatment for OPGs when it is refractive to other treatments such as chemotherapy. Combs et al. described fractionated stereotactic radiotherapy (FSRT) which was well tolerated in children and could be considered in unresectable tumors involving the chiasm. The 5-year survival rate after FSRT was 90%, and there were no secondary malignancies during this period [26]. It is important to note the short follow-up period in this study. FSRT as opposed to conventional radiotherapy has the ability to deliver a high-dose radiation to the tumor while sparing the normal brain tissue.
Muller et al. published a subgroup analysis from the low-grade glioma (LGG) trial in 1996 on patients who underwent either stereotactic brachytherapy (SBT) or external fractionated radiotherapy (EBRT). He reported a 10-year overall survival rate and progression-free survival (PFS) rate of 97% and 70%, respectively. There was no significant difference in either the SBT or the EBRT groups. The overall dose required was effectively reduced with these techniques without jeopardizing tumor control [27].
Coombs et al. also re-irradiated gliomas with signs of malignant transformation when the tumors were unifocal and contrast enhancing with a maximal diameter of 4 cm. He developed a prognostic score to predict the outcome following re-irradiation which consisted of scoring based on the histology of tumor, the age of patient, and the time since previous radiation. The additive score was then generated using the three factors on the scale (range 0–4 points). Patients that scored 3–4 represented the worst outcome, and patients that scored 0 had the best outcome after re-irradiation. For treatment decisions, patients scoring 0–2 present a clear benefit from re-irradiation over those who had a higher score [28]. It should be noted that the histology of grade IV tumors, i.e., glioblastomas, has a higher score and this itself has a poor prognosis as an independent factor. This scoring system was later validated by Kessel et al. in an independent large cohort of 199 patients who underwent re-irradiation (Table 3) [29].
Chemotherapy
Results from the multicenter treatment trial of LGG in 1996 showed that chemotherapy with carboplatin and vincristine in gliomas involving the chiasma or hypothalamus is an effective therapy to delay any need for radiotherapy with an overall survival of 93% and radiotherapy-free survival of 83%. However, the conventional use of these agents has been reported to have a poorer response in children less than 1 year of age. Patients less than 1 year old have to adapt their treatment with the addition of another drug or a change in the chemotherapy regimen to improve outcomes in that patient population [30,31,32].
Some centers have reported up to 30% of children with OPGs having tumor progression despite first-line management with the standard chemo regimen. Azizi et al. evaluated the next line of management for these patients using Web-based questionnaire to members of SIOPE (Societé Internationale d’Oncologie Pédiatrique) brain tumor group. Components suggested for second line were vinblastine (62%), cisplatin (34%), and cyclophosphamide (26%). Bevacizumab (BVZ) was considered a suitable drug as a third line of chemotherapy, often with irinotecan and vinblastine, and has had favorable outcome [33, 34]. In addition to systemic therapy 38% of respondents would consider a neurosurgical option (if safely feasible) in combination with further chemotherapy [33]. A majority of the respondents in this study also stated that they would not use radiotherapy as a second- or third-line option upon failure of chemotherapy. Radiotherapy was considered as an option only after failure of the second- or third-line chemo drugs, and only once the children were older than age 7 [33].
Reports from a RCT involving 18 institutions and 11 countries to investigate the addition of etoposide by the International Society of Pediatric Oncology-Low Grade Glioma (SIOP LGG) Committee concluded that addition of etoposide to the standard regimen of carboplatin and vincristine did not significantly alter the 5-year overall survival or disease-free progression [35].
Bavle et al. described the use of BRAF inhibitor (dabrafenib) in the treatment of a patient with disseminated OPG with a BRAFv600e mutation refractory to MEK inhibition (selumetinib) [36].
A multicenter retrospective analysis of visual outcomes in children with NF1-associated OPG following carboplatin-based chemotherapy regimens reported that one-third of children had visual acuity improvement and another 40% had stable acuity. They concluded that tumor location was the most important prognostic factor to predict visual outcomes [18]. Prior studies had focused on radiologic progression for response of chemotherapy, which is now considered a poor predictor for outcomes. Therapy and follow-up should focus on visual changes as an outcome measure.
Other agents that are being used for chemotherapy include temozolomide [37], cisplatin-etoposide-ifosfamide (PEI), and 6-thioguanine-procarbazine-vincristine-CCNU (TPVC) [30]. Chemotherapy, when used as second-line management, is known to be equally effective as the first line and is known to have a 5-year overall survival and progression-free survival of 86 ± 6% and 37 ± 8%, respectively [38]. These values are comparable to first-line chemotherapy. A randomized control trial comparing the chemotherapy regimens of vincristine with carboplatin (CV) to TPCV (thioguanine, procarbazine, lomustine, and vincristine) reported that the 5-year event-free survival (EFS) was 39% ± 4% for CV and 52% ± 5% for TPCV. EFS with the TPCV regimen was similar to that of CV in the first 2 years, but the EFS was higher in the long term for TPCV.
The main side effects of using chemotherapy include an allergic response, a drug hypersensitivity, myelosuppression, bleeding, a higher rate of infection, and other drug-specific side effects such as hemorrhagic cystitis with the use of cyclophosphamide and neuropathy with Vinca alkaloids (vincristine).
Role and Evolution of Surgery in OPG
Biopsy in an OPG is considered only in radiologically atypical tumors, usually only in NF-1-negative patients, and only if it can be done safely and avoid damage to any neighboring structures. Suitable methods of collecting the biopsy include endoscopic biopsy, stereotactic biopsy, and open biopsy via craniotomy [39].
Surgery as a treatment is used in OPG when they involve optic chiasma or hypothalamus for de-bulking the tumor when they are symptomatic. A VP shunt can also be planned in the case of hydrocephalus and is a relatively safe procedure [40]. Though a total tumor resection of low-grade gliomas is associated with better 5-year survival rate there is always a potential risk of damage to vital structures. Gooden et al. reported an overall survival of 92% in patients who underwent primary surgery for tumors involving the chiasma or hypothalamus, with or without adjuvant therapy [41]. A more recent paper based on a retrospective analysis of children who underwent surgery for symptomatic OPG by Liu et al. reported the 5-year overall survival rate (OS) and progression-free survival rate (PFS) of 84.1% and 70.6%, respectively. The majority of the patients had a partial resection of tumor with the primary intent of de-bulking and all the patients received chemotherapy after surgery. Also, children >5 years old received 3D conformal radiotherapy and this subset of patients were reported to have a better OS and PFS when compared to those who did not.
Recently the use of intraoperative imaging in the form of ultrasound, CT, or MRI has emerged as a very useful tool to guide surgeons to enhance the possibility of maximal tumor bulk resection while preserving the vital surrounding structures. Ulrich et al. reported a resection rate of 82% while using 3D real-time USG intraoperatively. This technique could be supplemented with the use of neurosurgical navigation systems. Ultrasound has limitations in the fact that it is highly operator dependent and intraoperative spatial orientation could be cumbersome. However, it could be significantly cost effective and is easily installed. Use of intraoperative CT is not recommended in pediatric population due to exposure to radiation [41].
Another useful tool that has emerged is the use of intraoperative MRI [42]. It reduces the rate of re-surgery by around 7–10% by allowing maximal resection at a single sitting. It helps to localize residual tumors in planned complete tumor resections and their proximity to surrounding vital tissue. This allows the surgeon to plan accordingly to achieve maximal benefit with minimal neurological compromise [43, 44]. Although great in theory, this leads to greater acquisition time as well as higher costs.
Genetics and Possible Future Pathways for Treatment
Genetic research is continuingly being done in search for treatments that can possibly target one specific factor or pathway that would allow patients to theoretically have a smaller array of side effects compared to using chemo- or radiation therapy. Rodriguez et al. investigated BRAF mutations and the MAPK pathway in optic gliomas as possible targets for new therapy of optic nerve gliomas [45]. OPGs have also been found to be associated with activation of the mTOR/Akt pathway and inhibition of this pathway as a possible therapeutic agent was investigated in phase 2 trials with the use of everolimus in chemotherapy-resistant radiographically progressive pediatric low-grade gliomas. This has shown promise with significant tumor stability during treatment [46]. Decreased tumor perfusion is also seen, which is a positive effect of these drugs that could be used as marker to document tumor response with angiography. Cabezas et al. found heterogeneity among the tumors with activation of either mTORC1 or mTORC2 pediatric low-grade gliomas, which could probably explain variability of response of these tumors when a single pathway is targeted [47]. Essentially, the mTOR is a protein kinase which can control cell growth and proliferation in response to nutrients and growth factors and is frequently dysregulated in cancers [48]. These studies are encouraging and suggest that mTOR inhibition may become an important component of pediatric low-grade glioma treatment.
Key Points
-
Optic pathways are clinically seen in 1–5% of neurofibromatosis 1 patients and is radiologically evident in 15%.
-
Ophthalmic surveillance is essential to rule out a progressive optic pathway glioma.
-
MRI is the primary mode of diagnosis and serial MRI could be necessary till age 8 years to rule out progression.
-
Treatment options include surgery, chemotherapy, and radiation with lot of future treatment options available involving the mTOR pathway.
References
Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol. 1994;38(5):427–52.
Wilhelm H. Primary optic nerve tumours. Curr Opin Neurol. 2009;22:11–8.
Robert-Boire V, Rosca L, Samson Y, Ospina LH, Perreault S. Clinical presentation and outcome of patients with optic pathway glioma. Pediatr Neurol. 2017;75:55–60.
Toledano H, Muhsinoglu O, Luckman J, Goldenberg-Cohen N, Michowiz S. Acquired nystagmus as the initial presenting sign of chiasmal glioma in young children. Eur J Paediatr Neurol. 2015;19(6):694–700. https://doi.org/10.1016/j.ejpn.2015.06.007. Epub 2015 Jul 9.
Silverman B, Listernick R, Charrow J. Precocious puberty in children with neurofibromatosis type 1. J Pediatr. 1995;126:364–7.
Sani I, Albanese A. Endocrine long-term follow-up of children with neurofibromatosis type 1 and optic pathway glioma. Horm Res Paediatr. 2017;87(3):179–88. https://doi.org/10.1159/000458525. Epub 2017 Mar 27.
Parsa CF, Hoyt CS. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol. 2001;119(4):516–2.
Liu GT, Malloy P, Needle M, Phillips P. Optic gliomas in neurofibromatosis type 1: role of visual evoked potentials. Pediatr Neurol. 1995;12:89–90.
Listernick R, Ferner RE, Liu GT, Gutmann DH. Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann Neurol. 2007;61:189–98.
Stokland T, et al. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro-Oncology. 2010;12(12):1257–68. PMC. Web. 8 Jan. 2018.
Diggs KA. Sex is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol. 2014;75(2):309–16. Wiley 2014-2 0364-5134.
Hersh JH, American Academy of Pediatrics Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics. 2008;121:633–42. https://doi.org/10.1542/peds.2007-3364.
Segal L, Darvish-Zargar M, Dilenge ME, Ortenberg J, Polomeno RC. Optic pathway gliomas in patients with neurofibromatosis type 1: follow-up of 44 patients. J AAPOS. 2010;14:155–8.
Yeom KW, Lober RM, Andre JB. Prognostic role for diffusion-weighted imaging of pediatric optic pathway glioma. J Neuro-Oncol. 2013;113:479.
Purohit BS, et al. Orbital tumours and tumour-like lesions: exploring the armamentarium of multiparametric imaging. Insights Imaging. 2016;7(1):43–68.
Kennedy de Blank PM, Jeffrey Berman I, Liu G, Leslie Roberts TP, Fisher M. Fractional anisotropy of the optic radiations is associated with visual acuity loss in optic pathway gliomas of neurofibromatosis type 1. Neuro-Oncology. 2013;15(8):1088–95.
Parsa CF, Hoyt CS, Lesser RL, et al. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol. 2001;119:516–29.
Fisher M, Loguidice M, Gutmann D, Listernickr R, Liu G. Visual outcomes in children with neurofibromatosis type 1–associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro-Oncology. 2012;14(6):790–7.
Thomas RP, Gibbs IC, Xu LW. Treatment options for optic pathway gliomas. Curr Treat Options Neurol. 2015;17:2.
Stokland T, Liu JF, Ironside JW, Ellison DW, Taylor R, Robinson KJ, Picton SV, Walker DA. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro-Oncology. 2010;12(12):1257–68.
Balcer LJ, Liu GT, Heller G, et al. Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: relation to tumor location by magnetic resonance imaging. Am J Ophthalmol. 2001;131:442–5.
Astrup J. Natural history and clinical management of optic pathway glioma. Br J Neurosurg. 2003;17:327–35.
Shofty B, Ben-Sira K, Jallo G, Isolated Optic Nerve Abnormalities (IONA) Collaboration. Isolated optic nerve gliomas: a multicenter historical cohort study. J Neurosurg Pediatr. 2017;20(6):549–55.
Stafford P, Abdelwahab MG, do Kim Y, Preul MC, Rho JM, Scheck AC. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr Metab (Lond). 2010;10(7):74.
Listernick R. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143–9. Wiley 1997-2;0364-5134.
Combs SE, Schulz-Ertner D, Moschos D, Thilmann C, Huber PE, Debus J. Fractionated stereotactic radiotherapy of optic pathway gliomas: tolerance and long-term outcome. Int J Radiat Oncol Biol Phys. 2005;62:814–9.
Müller K. Radiotherapy in pediatric pilocytic astrocytomas. A subgroup analysis within the prospective multicenter study HIT-LGG 1996 by the German Society of Pediatric Oncology and Hematology (GPOH). Strahlenther Onkol. 2013;189(8):647–55. Springer 2013-8;0179-7158.
Combs SE, Edler L, Rausch R, Welzel T, Wick W, Debus J. Generation and validation of a prognostic score to predict outcome after re-irradiation of recurrent glioma. Acta Oncol. 2013;52(1):147–52.
Kessel KA, Hesse J, Straube C. Validation of an established prognostic score after re-irradiation of recurrent glioma. Acta Oncol. 2017;56(3):422–6.
Mirow C, Pietsch T, Berkefeld S, Kwiecien R, Warmuth-Metz M, Falkenstein. Children <1 year show an inferior outcome when treated according to the traditional LGG treatment strategy: a report from the German multicenter trial HIT-LGG 1996 for children with low grade glioma (LGG). Pediatr Blood Cancer. 2014;61:457–63.
Ater J, Zhou T, Homes E, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30:2641–7.
Ater JL, Holmes E, Zhoy T, et al. Randomized study of two chemotherapy regimens for low grade glioma in young children: results of COG protocol A9952. Pediatr Blood Cancer. 2008;53.
Azizi AA, Schouten-van Meeteren AYN. Current and emerging treatment strategies for children with progressive chiasmatic-hypothalamic glioma diagnosed as infants: a web-based survey. J Neuro-Oncol. 2018;136:127.
Packer RJ, Jakacki R, Horn M, Rood B, Vezina G, MacDonald T, Fisher MJ, Cohen B. Objective response of multiply recurrent low-grade gliomas to bevacizumab and irinotecan. Pediatr Blood Cancer. 2009;52(7):791–5.
Gnekow AK, Walker DA, Kandels D, Picton S. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma—a final report. Eur J Cancer. 2017;81:206–25.
Bavle A, Jones J, Lin FY, Malphrus A, Adesina A, Su J. Dramatic clinical and radiographic response to BRAF inhibition in a patient with progressive disseminated optic pathway glioma refractory to MEK inhibition. Pediatr Hematol Oncol. 2017;34(4):254–9.
Gururangan S, Fisher MJ, Allen JC, Herndon JE II, Quinn JA, Reardon DA, et al. Temozolomide in children with progressive low-grade glioma. Neuro-Oncology. 2007;9(2):161–8.
Scheinemann K, Bartels U, Tsangaris E, Hawkins C, Huang A, Dirks P, Fried I, Bouffet E, Tabori U. Feasibility and efficacy of repeated chemotherapy for progressive pediatric low-grade gliomas. Pediatr Blood Cancer. 2011;57:84–8.
Walker DA, Liu J, Kieran M, Jabado N, Picton S, Packer R, et al. A multi-disciplinary consensus statement concerning surgical approaches to low-grade, high-grade astrocytomas and diffuse intrinsic pontine gliomas in childhood (CPN Paris 2011) using the Delphi method. Neuro-Oncology. 2013;15:462–8.
Liu Y, et al. Analysis of survival prognosis for children with symptomatic optic pathway gliomas who received surgery. World Neurosurg. 2018;109:e1–e15. https://doi.org/10.1016/j.wneu.2017.09.144.
Ulrich NH, Burkhardt JK, Serra C, et al. Resection of pediatric intracerebral tumors with the aid of intraoperative real-time 3-D ultrasound. Childs Nerv Syst. 2012;28:101–9.
Goodden J, Pizer B, Pettorini B, Williams D, Blair J, Didi M, Thorp N, Mallucci C. The role of surgery in optic pathway/hypothalamic gliomas in children. J Neurosurg Pediatr. 2014;13(1):1–12.
Shah MN, Leonard JR, Inder G. Intraoperative magnetic resonance imaging to reduce the rate of early reoperation for lesion resection in pediatric neurosurgery. J Neurosurg Pediatr. 2012;9:259–64.
Giordano M, Arraez C, Samii A, et al. Childs Nerv Syst. 2016;32:1915.
Rodriguez FJ, Ligon AH, Horkayne-Szakaly I, et al. BRAF duplications and MAPK pathway activation are frequent in gliomas of the optic nerve proper. J Neuropathol Exp Neurol. 2012;71(9):789–94.
Segal D, Gardner S, Allen J, Karajannis M. EPT-21 Efficacy of everolimus in pediatric brain tumors: a single-institution patient series. Neuro-Oncology. 2016;18(Suppl 3):iii28.
Hütt-Cabezas M, et al. Activation of mTORC1/mTORC2 signaling in pediatric low-grade glioma and pilocytic astrocytoma reveals mTOR as a therapeutic target. Neuro-Oncology. 2013;15(12):1604–14.
Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314–41.
Acknowledgment
This work was supported in part by an unrestricted institutional grant from Research to Prevent Blindness, NY, NY.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Narain, S., Kini, A., Ramasubramanian, A. (2019). Advancements in the Management of Optic Pathway Gliomas. In: Ramasubramanian, A. (eds) Ocular Oncology . Current Practices in Ophthalmology. Springer, Singapore. https://doi.org/10.1007/978-981-13-7538-5_4
Download citation
DOI: https://doi.org/10.1007/978-981-13-7538-5_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-7537-8
Online ISBN: 978-981-13-7538-5
eBook Packages: MedicineMedicine (R0)