
Abstract
Biomedical prediction is vital to the modern scientific view of life, but it is a challenging task due to high-dimensionality, limited-sample size (also known as HDLSS problem), non-linearity, and data types tend are complex. A large number of dimensionality reduction techniques developed, but, unfortunately, not efficient with small-sample (observation) size dataset. To overcome the pitfalls of the sample-size and dimensionality this study employed variational autoencoder (VAE), which is a powerful framework for unsupervised learning in recent years. The aim of this study is to investigate a reliable biomedical diagnosis method for HDLSS dataset with minimal error. Hence, to evaluate the strength of the proposed model six genomic microarray datasets from Kent Ridge Repository were applied. In the experiment, several choices of dimensions were selected for data preprocessing. Moreover, to find a stable and suitable classifier, different popular classifiers were applied. The experimental results found that the VAE can provide superior performance compared to the traditional methods such as PCA, fastICA, FA, NMF, and LDA.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Variational autoencoder
- High dimensional and small sample size dataset
- Biomedical diagnosis
- Computational biology
1 Introduction
Biomedical prediction is a vital research and application area. The purpose of prediction is to minimize the risk in decision-making. In the area of computational biology, genomic microarray data plays a crucial role to assess the pathological diagnosis and classification of diseases, but it is a challenging task due to the properties of gene expression data such as small sample, high dimensions (features), and data types tend are complex and may correspond to discrete sequence data [1]. There are many features of genomic microarray affecting the structure and function of the body. These might be difficult for doctors to diagnose quickly and accurately. Therefore, it is necessary to employ computational intelligence in diagnosis to assist doctors to diagnose faster with high accuracy. For this purpose, in the last few decades, computational intelligence techniques have been proposed and exploited for biomedical prediction.
Several applications, especially in the area of biomedical the measurements tend to be very expensive; consequently, the number of samples is very limited (can be below 100) whereas several thousand of features (dimensions). These datasets are called high dimension low sample size (HDLSS) datasets; are characterized with a large number of features P and relatively small number of samples n, often written \( P > > n \) [2]. These HDLSS problems create significant challenges for the development of computational science.
The accuracy of prediction tends to deteriorate in high-dimensions due to the curse of dimensionality [3]. Hence, dimension reduction is invaluable for the analysis of high-dimensional data and several methods have been proposed including principal components analysis (PCA), independent components analysis (ICA), feature analysis (FA), non-negative matrix factorization (NMF), and latent Dirichlet allocation (LDA).
However, existing dimensional reduction techniques are unable to cope with the nonlinear relationships of the data but also unable to perform well with HDLSS datasets [4, 5]. For that reason, there needs an investigation to find an effective method that can deal with HDLSS datasets and improvement of the accuracy. In recent years, variational autoencoder (VAE) has emerged as one of the most popular approaches to unsupervised learning of complicated distributions and successfully applied in the area of image processing, text mining, and computer vision which involve high-dimensional data. VAE reduces dimensions in a probabilistically way, theoretical foundations are solid.
The objective of this paper is to illustrate the value of dimensional reduction of HDLSS datasets. Moreover, the paper also demonstrates the potential difficulties and the over-fitting dangers of performing dimensional-reduction in small sample size situations. Several well-known dimension reduction techniques have been implemented and compared. Also, the effectiveness of the VAE is tested on HDLSS dataset and comparisons with various dimensions in the prediction shown.
2 Literature Review
Due to the curse of dimensionality, dimensionality reduction is often crucial. The complexity of many decision trees [8] and decision forest [9,10,11,12] algorithm is \( {\text{O}}\left( {{\text{nm}}^{2} } \right) \), where n is the number of records and m is the number of attributes. Over the past decades, many dimensionality reduction techniques have been proposed. An interesting approach [13] automatically computes weights for attributes, where a weight zero means complete deletion of the attribute, a weight 1 means full consideration of the attribute and anything in between 0 and 1 means a weighted inclusion of the attribute. Most commonly used dimensionality reduction methods are principal component analysis (PCA), independent component analysis (ICA), factor analysis (FA), linear discriminant analysis, and nonnegative matrix factorization (NMF). PCA has been applied in classification and clustering of relevant genes expression microarray or RNA-sequence data [4, 5]. Moreover, a number of prior techniques also investigated to classification and clustering of genes expression data for disease diagnosis [5, 6].
Traditional PCA is a frequently used method for reducing data for visualization and clustering [7]. In the case where sample sizes are larger than features \( (n > P) \), classical methods such as PCA, ICA, and FA are likely to perform well. PCA essentially depends on the empirical covariance matrix in which a large number of samples are necessary. This work considered the problem where P is much larger than n. Yeung and Ruzzo [4] explored PCA for clustering gene expression data, the experimental result showed that PCA not suitable for dimensionality reduction in \( P >> n \) datasets. Moreover, PCA and ICA have a major disadvantage in that they assume data is linearly separable, but the linear model is not always reliable in capturing nonlinear relationships of real-world problems, especially with limited samples [14].
NMF is another efficient way of high-dimensional data analysis [15]. In the bioinformatics area, NMF has been used for microarray data and protein sequence analysis [16, 17]. In PCA the principal components are defined by the number of samples using the eigenvalue decomposition, while for NMF the number of learned basis experiments is not limited. It appears that NMF can derive more features than samples for further analysis, and this may be why it got higher clustering accuracy as shown in the experimental results [18]. PCA, ICA, and FA are deterministic while NMF is stochastic; so NMF appears to be more suitable for HDLSS data analysis than PCA, ICA, and FA.
In the recent years, a particular class of probabilistic graphical model called topic models is found be a useful tool for mining microarray data. Latent Dirichlet allocation (LDA) is one of the most popular topic models, applied to mitigate overfitting of high-dimensionality in various fields including biomedical science [19,20,21,22]. Deep learning is a competent way for nonlinear dimensionality reduction which provides an appealing framework for handling high-dimensional datasets. Deep learning techniques have been successfully applied to extract information from high-dimensional data [23, 24].
Dimensionality reduction is effective if the loss of information due to mapping to a lower-dimensional space is less than the gain from simplifying the problem. A further challenge is that high-dimensionality and limited-sample size both increase the risk of overfitting and decrease the accuracy of classification [25, 29]. It is essential to building a classification model with good generalization ability, expected that perform equally well on the training set and independent testing set.
3 Methodology
The detailed design of the diagnosis system consists of three major states (see Fig. 1): preprocessing, variational autoencoder (VAE) based dimensionality reduction, and classification. Input dataset is first divided into two sub-datasets: a training set and testing set. Then variational autoencoder is applied to select desirable encoded dimensions of attributes which reduces computational burden and enhances the performance of classification. Finally, for disease classification, on the obtained reduced dimensional data the classifier is applied.

Diagnosis framework.
3.1 Data Preprocessing
Firstly, the dataset was divided into two parts: one for training and another for the testing, where 60% samples were used as training set, and remaining 40% were used as test set. More importantly, to overcome from overfitting and unbiased classification accuracy, separate datasets (training/testing) are used in the training and testing dimension reduction and classification. The training dataset used to train the dimension reduction technique and to build the classifier only same as the test dataset is used to test dimension reduction and classifier. Nonetheless, the experiment emphasis the importance of testing data unseen at any part of the dimensionality reduction and the classifier training.
3.2 Variational Autoencoder
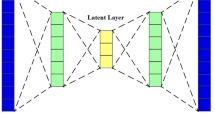
Variational autoencoder (VAE) introduced by Kingma and Welling [26] and Rezende et al. [27], an exciting development in machine learning for combined generative modeling and inference. The main ideas of the VAE are comprising of a probabilistic model over data and a variational model over latent variables. VAE is rooted in Bayesian inference, i.e., it wants to model the underlying probability distribution of data so that it could sample new data from that distribution. A VAE consists of an encoder, a decoder, and a loss function. Figure 2 shows the basic structure of the variational autoencoder.

Architecture of variation autoencoder based diagnosis.
Encoder.
The encoder compresses data x into a latent variable z (lower-dimensional space). The lower dimensional space is stochastic, the encoder output parameter is \( p_{\theta } \left( {z |x} \right) \), which is a Gaussian probability density. Data x can sample from this distribution to get noisy values of the representations z. \( \theta \) is the weight and bias parameter.
Decoder.
The decoder reconstructs the data is denoted by \( q_{\upphi} \left( {z |x} \right) \), gets input as the latent representation z and output the parameters of a probability distribution of the data. It goes from a smaller to a larger dimension. Information loss computed using the reconstruction log-likelihood, \( { \log }\, q_{\upphi} \left( {z |x} \right) \). This measure states how effectively the decoder has learned to reconstruct an input x given its latent representation z. ϕ is the weight and biases parameter.
Loss Function.
The loss function of the VAE is the negative log-likelihood with a regularizer. Because there are no global representations that are shared by all data points, loss function can decompose into only terms that depend on a single data point \( l_{i} \). The total loss is \( \sum\nolimits_{i = 1}^{n} {l_{i} } \) for n total data points. The loss function \( l_{i} \) for data point \( x_{i} \) is:
In Eq. (1), the first term is reconstruction loss or expected negative log-likelihood of the \( i{\text{th}} \) data point. The second term is a regularizer, the Kullback-Leibler divergence between the encoder’s distribution \( p_{\theta } \left( {z |x} \right) \) and p(z). This divergence measures how close q and p. If the encoder outputs representations z are different than a standard normal distribution, it gets a penalty.
3.3 Classification
The classification algorithm is applied to the obtained reduced dimensional data. An ideal classifier is only a fiction. Since the classifier model is never a perfect classifier, a substitute is usually chosen from the area of machine learning. Classification algorithms can be grouped into the Bayesian classifier, functions, lazy algorithm, meta-algorithm, rules, and trees algorithm [28]. Some of the widely used classification algorithms are ANN, decision tree, KNN, logistic regression, Naïve Bayes, fuzzy logic, and SVM. Each classifier has own strength, but the challenge is an appropriate selection in the application of a complex and growing dataset.
4 Simulation Results and Discussion
4.1 Dataset
In this research, six well-known high-dimensional genomic microarray datasets from the Kent Ridge Biomedical Dataset Repository used. More details of datasets are shown in Table 1.
4.2 Experiment Setup
The experiments are setup to diagnose the diseases using genomic microarray data; six datasets are tested for different reduced dimensions e.g., 60, 100, 200, 300, 400, 500, and 600. To demonstrate the effectiveness of the VAE two kinds of comparisons are investigated in this research (1) single layer VAE, and (2) multiple layers VAE from two to four layers. Firstly, the dimensionality reduction technique is applied. For reduction methods performance evaluation, principal components analysis (PCA) [29], independent components analysis (fastICA) [30], factor analysis (FA) [31], latent Dirichlet allocation (LDA) [32], mini-batch dictionary learning (MBDL) [33], and non-negative matrix factorization (NMF) [34] results are compared. Then, the classification algorithm is applied to the obtained reduced dimensional data. Classification performance is compared to nine widely used classifiers namely AdaBoost (AB) [35], decision tree (DT) [36], Gaussian Naive Bayes (GNB) [37], Gaussian process (GP) [38], kNeighbors (KN) [39], logistic regression (LR) [40], multilayer perceptron (MLP) [41], random forest (RF) [42], and support vector classification (SVC) [43].
The experiments carried out in Python Tensorflow Keras framework. The configuration of the machine that was used to run these experiments was: Intel(R) Core i3-23S0M, CPU speed 2.30 GHz, RAM 4.00 GB, OS Windows 10 Pro 64-bit, x64-based processor.
4.3 Performance Measures
To evaluate the classification performances accuracy used in this research. Accuracy is the most used standard for evaluation classification techniques as well as for the comparison of performance defined by Eq. (2).
where TP and FP are the number of true positive and false positive, respectively; TN and FN are the number of true and false negative, respectively.
4.4 Results Analysis and Discussions
Table 2 shows the classification accuracy of different classifiers by using the original features (all features) of the datasets. Average classification accuracies using different classifiers on a particular selected number of genes by several dimension reductions methods are shown in Figs. 3, 4, 5, 6, 7 and 8.

Average accuracy of different methods in different dimension of Breast cancer dataset. (PCA: principal components analysis; FastICA: independent components analysis; FA: factor analysis; LDA: latent Dirichlet allocation; MBDL: mini-batch dictionary learning; VAE: single layer VAE; L2VAE: 2-layer VAE; L3VAE: 3-layer VAE; L4VAE: 4-layer VAE)

Average accuracy of different methods in different dimension of CNS dataset. (PCA: principal components analysis; FastICA: independent components analysis; FA: factor analysis; LDA: latent Dirichlet allocation; MBDL: mini-batch dictionary learning; VAE: single layer VAE; L2VAE: 2-layer VAE; L3VAE: 3-layer VAE; L4VAE: 4-layer VAE)

Average accuracy of different methods in different dimension of Colon tumor dataset. (PCA: principal components analysis; FastICA: independent components analysis; FA: factor analysis; LDA: latent Dirichlet allocation; MBDL: mini-batch dictionary learning; VAE: single layer VAE; L2VAE: 2-layer VAE; L3VAE: 3-layer VAE; L4VAE: 4-layer VAE)

Average accuracy of different methods in different dimension of Lung cancer dataset. (PCA: principal components analysis; FastICA: independent components analysis; FA: factor analysis; LDA: latent Dirichlet allocation; MBDL: mini-batch dictionary learning; VAE: single layer VAE; L2VAE: 2-layer VAE; L3VAE: 3-layer VAE; L4VAE: 4-layer VAE)

Average accuracy of different methods in different dimension of Leukaemia dataset. (PCA: principal components analysis; FastICA: independent components analysis; FA: factor analysis; LDA: latent Dirichlet allocation; MBDL: mini-batch dictionary learning; VAE: single layer VAE; L2VAE: 2-layer VAE; L3VAE: 3-layer VAE; L4VAE: 4-layer VAE)

Average accuracy of different methods in different dimension of Ovarian cancer dataset. (PCA: principal components analysis; FastICA: independent components analysis; FA: factor analysis; LDA: latent Dirichlet allocation; MBDL: mini-batch dictionary learning; VAE: single layer VAE; L2VAE: 2-layer VAE; L3VAE: 3-layer VAE; L4VAE: 4-layer VAE)
In experiment 1, for the Breast cancer dataset train-test test, on the 60-dimension the average classification accuracy of PCA and VAE is 62% and 63% respectively, whereas in the case of using more dimension with VAE and multilayer VAE model provide better accuracy.
In experiment 2 for CNS dataset, the highest average accuracy of fastICA and 4-layer VAE is around 66% on the 60-dimension data. It is observed that as the dimension increased as the classification accuracy of LDA, MBDL, NMF, VAE and multi-layer VAEs has gained. Figure 4 shows that VAE and multi-layer VAEs performed better with high accuracy compare to other techniques. Moreover, NMF and LDA also obtained significantly better accuracy.
In experiment 3, for the Colon tumor dataset, this study’s method VAE and multi-layer VAEs provides significantly better accuracy than other traditional algorithms. Figure 5 shows that the classification accuracy for VAE and multi-layer VAEs has small growth with dimension relatively large.
In another experiment the Lung cancer dataset has 203 samples, class distribution is 139/17/6/21/20. Thus, it is an imbalanced dataset (139 and 6 sample in class one and three respectively). Figure 6 reveals a clear difference between the VAE and other methods. VAE and multi-layer VAEs perform significantly better accuracy compared to the other methods. The method of this study achieves a higher accuracy of 91% on the 300-dimension space, while multi-layer VAEs perform consistently better in all the different latent spaces (dimension).
The same is true for another two experiments for Leukemia and Ovarian dataset in Figs. 7 and 8 respectively. The results thus showcase the robustness of the proposed model compare to other methods. The experimental analysis found that the accuracy rate with application of 60 or 100 dimensions is comparatively smaller than 200 to 600 dimension and accuracy gained when dimension is relatively large.
Figure 9 shows the loss curve for 100 epochs of the training and validation data of the single layer VAE of the different datasets. It is observed that the loss function usually converged after 150 iterations, here used 200 iterations. More iterations can be used, but there is a risk of overfitting.

Loss curve of the different dimensions for the training and validation data of the single layer VAE of different datasets.
5 Conclusion
This study presents a variational autoencoder based dimensionality reduction for high-dimensional small-sample biomedical diagnosis. In contrast to PCA, ICA, and FA, while using VAE can reduce the dimension as suitable from the high-dimensional dataset that enhances the performance of the classification system. Here, the authors have demonstrated the effectiveness of the proposed model by testing it on six gene expression microarray datasets. Moreover, compared with the traditional methods of PCA, fastICA, FA, MBDL, LDA, and NMF. The performance comparison of different reduction techniques to several popular classifiers in term of accuracy presented. The experimental results show that the proposed model performs superior to traditional methods. This reliable prediction will aid for biomedical diagnosis.
It is difficult to design an efficient prediction system for the small-sample-size dataset because limited-sample can easily contaminate the performance. A large test sample is required to evaluate a model accurately. Notably, the classification accuracy of small-sample-size gene expression microarray datasets are still poor; there needs further investigation to find an effective model to improve the accuracy.
References
Clarke, R., et al.: The properties of high-dimensional data spaces: implications for exploring gene and protein expression data. Nat. Rev. Cancer 8(1), 37–49 (2008)
Friedman, J., Hastie, T., Tibshirani, R.: The Elements of Statistical Learning. Springer Series in Statistics, 2nd edn. Springer, New York (2008). https://doi.org/10.1007/978-0-387-84858-7
Köppen, M.: The curse of dimensionality. In: 5th Online World Conference on Soft Computing in Industrial Applications (WSC5) (2000)
Yeung, K.Y., Ruzzo, W.L.: Principal component analysis for clustering gene expression data. Bioinformatics 17(9), 763–774 (2001)
Dai, J.J., Lieu, L., Rocke, D.: Dimension reduction for classification with gene expression microarray data. Stat. Appl. Genet. Mol. Biol. 5(1), 1–21 (2006)
Mishra, D., Dash, R., Rath, A.K., Acharya, M.: Feature selection in gene expression data using principal component analysis and rough set theory. Adv. Exp. Med. Biol. 696, 91–100 (2011)
Jolliffe, I.: Principal Component Analysis, 2nd edn. Springer, New York (2002). https://doi.org/10.1007/b98835
Islam, M.Z.: EXPLORE: a novel decision tree classification algorithm. In: MacKinnon, L.M. (ed.) BNCOD 2010. LNCS, vol. 6121, pp. 55–71. Springer, Heidelberg (2012). https://doi.org/10.1007/978-3-642-25704-9_7
Islam, M.Z., Giggins, H.: Knowledge discovery through SysFor: a systematically developed forest of multiple decision trees. In: Proceedings of the Ninth Australasian Data Mining Conference (AusDM 2011), Ballarat, Australia. CRPIT, vol. 121 (2011)
Adnan, M.N., Islam, M.Z.: Forest PA: constructing a decision forest by penalizing attributes used in previous trees. Expert. Syst. Appl. (ESWA) 89, 389–403 (2017)
Siers, M.J., Islam, M.Z.: Novel algorithms for cost-sensitive classification and knowledge discovery in class imbalanced datasets with an application to NASA software defects. Inf. Sci. 459, 53–70 (2018)
Adnan, M.N., Islam, M.Z.: Optimizing the number of trees in a decision forest to discover a subforest with high ensemble accuracy using a genetic algorithm. Knowl. Based Syst. 110, 86–97 (2016). ISSN 0219-1377
Rahman, M.A., Islam, M.Z.: AWST: A novel attribute weight selection technique for data clustering. In: Proceedings of the 13th Australasian Data Mining Conference (AusDM 2015) (2015)
Gupta, A., Wang, H., Ganapathiraju, M.: Learning structure in gene expression data using deep architectures with an application to gene clustering. In: IEEE International Conference on Bioinformatics and Biomedicine (BIBM) (2015)
Berry, M.W., Brown, M., Langville, A.N., Paucac, P., Plemmons, R.J.: Algorithms and applications for the nonnegative matrix factorization. Comput. Stat. Data Anal. 52(1), 55–173 (2007)
Pascual-Montano, A., Carmona-Saez, P., Chagoyen, M., Tirado, F., Carazo, J.M., Pascual-Marqui, R.D.: bioNMF: a versatile tool for nonnegative matrix factorization in biology. BMC Bioinform. 7, 366 (2006)
Gao, Y., Church, G.: Improving molecular cancer class discovery through sparse non-negative matrix factorization. Bioinformatics 21(21), 3970–3975 (2005)
Liu, W., Kehong, Y., Datian, Y.: Reducing microarray data via nonnegative matrix factorization for visualization and clustering analysis. J. Biomed. Inform. 41, 602–606 (2008)
Zhao, W., Zou, W., Chen, J.J.: Topic modeling for cluster analysis of large biological and medical datasets. BMC Bioinform. 15, S11 (2014)
Lu, H.M., Wei, C.P., Hsiao, F.Y.: Modeling healthcare data using multiple-channel latent Dirichlet allocation. J. Biomed. Inform. 60, 210–223 (2016)
Kho, S.J., Yalamanchili, H.B., Raymer, M.L., Sheth, A.P.: A novel approach for classifying gene expression data using topic modeling. In: Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics (2017)
Tan, J., Ung, M., Cheng, C., Greene, C.S.: Unsupervised feature construction and knowledge extraction from genome-wide assays of breast cancer with denoising autoencoders. Pac. Symp. Biocomput. 20, 132–143 (2015)
Danaee, P., Ghaeini, R., Hendrix, D.A.: A deep learning approach for cancer detection and relevant gene identification. Pac. Symp. Biocomput. 22, 219–229 (2017)
Smialowski, P., Frishman, D., Kramer, S.: Pitfalls of supervised feature selection. Bioinformatics 26(3), 440–443 (2010)
Diciotti, S., Ciulli, S., Mascalchi, M., Giannelli, M., Toschi, N.: The ‘peeking’ effect in supervised feature selection on diffusion tensor imaging data. Am. J. Neuroradiol. 34(9), E107 (2013)
Kingma, D.P., Welling, M.: Auto-encoding variational Bayes. In: International Conference on Learning Representations (2014)
Rezende, D.J., Mohamed, S., Wierstra, D.: Stochastic backpropagation and approximate inference in deep generative models. In: Proceedings of the 31st International Conference on Machine Learning, vol. 32(2), pp. 1278–1286 (2014)
Witten, L.H., Frank, E.: Data Mining: Practical Machine Learning Tools and Techniques, 2nd edn. Morgan Kaufmann, San Francisco (2005)
Tipping, M.E., Bishop, C.M.: Mixtures of probabilistic principal component analysers. Neural Comput. 11(2), 443–482 (1999)
Hyvarinen, A., Oja, E.: Independent component analysis: algorithms and applications. Neural Netw. 13(4–5), 411–430 (2000)
Barber, D.: Bayesian Reasoning and Machine Learning, Algorithm 21.1. Cambridge University Press, Cambridge (2012)
Hoffman, M.D., Blei, D.M., Bach, F.: Online learning for latent Dirichlet allocation. In: Proceedings of the 23rd International Conference on Neural Information Processing Systems, vol. 1, pp. 856–864 (2010)
Mairal, J., Bach, F., Ponce, J., Sapiro, G.: Online dictionary learning for sparse coding. In: Proceedings of the 26th Annual International Conference on Machine Learning (2009)
Cichocki, A., Phan, A.H.: Fast local algorithms for large scale nonnegative matrix and tensor factorizations. IEICE Trans. Fundam. Electron. Commun. Comput. Sci. 92(3), 708–721 (2009)
Zhu, J., Zou, H., Rosset, S., Hastie, T.: Multi-class AdaBoost. Stat. Interface 2, 349–360 (2009)
Breiman, L., Friedman, J., Olshen, R., Stone, C.: Classification and Regression Trees. CRC Press, Boca Raton (1984)
Manning, C.D., Raghavan, P., Schuetze, H.: Introduction to Information Retrieval. Cambridge University Press, Cambridge (2008)
Rasmussen, C.E., Williams, C.: Gaussian Processes for Machine Learning. MIT Press, Cambridge (2006)
Altman, N.S.: An introduction to kernel and nearest-neighbor nonparametric regression. Am. Stat. 46(3), 175–185 (1992)
Yu, H.F., Huang, F.L., Lin, C.J.: Dual coordinate descent methods for logistic regression and maximum entropy models. Mach. Learn. 85(1–2), 41–75 (2011)
Hinton, G.E.: Connectionist learning procedures. Artif. Intell. 40(1), 185–234 (1989)
Breiman, L.: Random forests. Mach. Learn. 45(1), 5–32 (2001)
Wu, T.F., Lin, C.J., Weng, R.C.: Probability estimates for multi-class classification by pairwise coupling. J. Mach. Learn. Res. 5, 975–1005 (2004)
Acknowledgments
This research is funded in part by the National Natural Science Foundations of China (Grant No. 61472258 and 61473194) and the Shenzhen-Hong Kong Technology Cooperation Foundation (Grant No. SGLH20161209101100926).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Mahmud, M.S., Fu, X., Huang, J.Z., Masud, M.A. (2019). High-Dimensional Limited-Sample Biomedical Data Classification Using Variational Autoencoder. In: Islam, R., et al. Data Mining. AusDM 2018. Communications in Computer and Information Science, vol 996. Springer, Singapore. https://doi.org/10.1007/978-981-13-6661-1_3
Download citation
DOI: https://doi.org/10.1007/978-981-13-6661-1_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-6660-4
Online ISBN: 978-981-13-6661-1
eBook Packages: Computer ScienceComputer Science (R0)