
Abstract
The synapse between the inner hair cells (IHCs) and the spiral ganglion neurons (SGNs) in mammalian cochleae is characterized as having presynaptic ribbons and therefore is called ribbon synapse. The special molecular organization is reviewed in this chapter in association with the functional feature of this synapse in signal processing. This is followed by the review on noise-induced damage to this synapse with a focus on recent reports in animal models in which the effect of brief noise exposures is observed without causing significant permanent threshold shift (PTS). In this regard, the potential mechanism of the synaptic damage by noise and the impact of this damage on hearing are summarized to clarify the concept of noise-induced hidden hearing loss, which is defined as the functional deficits in hearing without threshold elevation. A controversial issue is addressed in this review as whether the disrupted synapses can be regenerated. Moreover, the review summarizes the work of therapeutic research to protect the synapses or to promote the regeneration of the synapse after initial disruption. Lastly, several unresolved issues are raised for investigation in the future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
3.1 Introduction
According to the current standard (ISO1999: 2013(E)), noise-induced hearing loss (NIHL) is defined by sustaining a permanent threshold shift (PTS). However, this definition has been challenged by the fact that noise exposure can cause massive damage to the synapses between inner hair cells (IHCs) and spiral ganglion neurons (SGNs) in the cochleae of laboratory animals without a significant PTS [24, 43, 48, 50, 88, 90, 94]. The synaptic damage and the associated functional deficits in signal coding by auditory nerve fibers (ANFs) have been labelled as noise-induced cochlear synaptopathy. Since coding deficits in the absence of a PTS cannot be detected by routine audiological evaluations that are currently focused on seeking thresholds, they are umbrellaed under the concept of noise-induced hidden hearing loss (NIHHL) [42, 47, 51, 57, 64, 94]. This chapter will review the current knowledge of the potential mechanisms of noise-induced synaptic damage and the following repair processes after a brief review on the special anatomy of the synapse between IHCs and SGNs. The chapter will then shift its focus to the therapeutic methods promoting the regeneration of the synapses.
3.2 Anatomic and Functional Features of Cochlear Ribbon Synapse
The synapse between IHCs and SGNs is characterized by the presence of an electron-dense, ribbon-like structure and therefore called a ribbon synapse. It is mainly found in the retina, the inner ear, and in the pinealocytes. The structural features of the ribbon synapses between IHCs and SGNs are summarized in Fig. 3.1. The synaptic ribbons found in mature hair cells are anchored to the plasma membrane, one ribbon per active zone (AZ). A small number of “floating” ribbons (<5%) were observed and probably reflected the turnover of these subcellular organelles [40, 114]. The synaptic ribbons in the IHC are shaped like an American football, with a tee structure underneath that is formed by a protein named Bassoon. This bar structure anchors the ribbon to the AZ [59].

Schematic view of IHC ribbon synapses
The main protein forming the framework of the ribbon is called the Ribeye, which consists of two domains: the A-domain is located inside and appears to have a structural role, whereas the B-domain points to the cytoplasmic face of the ribbon, where it interacts with other proteins and tethers synaptic vesicles [2]. The aminoterminal A-domain is not homologous with any other protein in the public databases and therefore specific to the ribbon synapse, whereas the carboxyterminal B-domain is largely identical to the nuclear corepressor protein, C-terminal binding protein 2 (CtBP2). The gene encoding the Ribeye is called the CtBP2 gene, which encodes two proteins: the unique Ribeye(A + B) in the ribbon synapses and the CtBP2, which is also expressed in the cellular nucleus [52, 100, 106, 114]. The Ribeye in photoreceptor cells contains CtBP1 [102], which has not been verified in IHC ribbons.
The scaffold of synaptic ribbons is built up from multiple Ribeyes [52]. The Ribeye A-domain has three interaction docking sites that mediate homotypic interactions with other RIBEYE(A)-domains. In addition, homotypic B-domain interactions can be formed as well as heterotypic interactions between the RIBEYE A- and B-domains, which are regulated (inhibited) by nicotinamide adenine dinucleotide hydride (NADH). In the photoreceptors, ribbon size dynamically changes in response to light: ribbons are disassembled in bright light and reassembled in dark [1, 75, 84, 95, 103]. It is not clear if the ribbons in the IHCs are dynamically disassembled/reassembled.
The most striking functional characteristic of the IHC ribbon synapse is its ability to make fast response to transient signals in the meantime to keep its long-lasting response to persistent stimuli. These features require special mechanisms to enable fast neurotransmitter release (exocytosis) and replenishment, as well as fast recycling of neurotransmitters via endocytosis. It is not entirely clear how these processes are realized. However, they must be related to the special protein compositions and the structure of the cytomatrix of the active zone (CAZ). Several proteins that are important for transmission across conventional synapses are not seen in IHCs. Those include synaptotagmins 1 and 2, synapsins, synaptophysins, synaptogyrin complexins, neuronal SNAREs as well as priming factors of the Munc13 and CAPS families (see reviews [56, 81]). Instead, the function of those proteins seems to be replaced by a single protein, otoferlin, which is located between the ribbon and the presynaptic membrane and strongly interacts with adaptor protein 2 (AP-2) [17, 38, 61].
Bassoon and Piccolo are two big proteins (>400 kDa) that are seen in conventional synapses. Their function in synaptic transmission is not clear. In ribbon synapses, Bassoon is responsible for anchoring the ribbons to the CAZ. The knockout of this protein in the cochlea of mice results in the loss of ribbons and the deterioration of temporal resolution of the auditory nerve fibers (ANFs), without significant change in hearing sensitivity [7, 37]. Piccolo is present in ribbon synapses as a shortened variant, called Piccolino, which is distributed over the entirety of the ribbon. Knockdown of this protein resulted in a lack of dynamic ribbon assembly in the retina of mice [23, 76]. However, it is not clear what role the Piccolino plays in IHC ribbon synapses.
The postsynaptic terminal of the ribbon synapses exhibits similarities with the conventional excitatory synapses. Glutamate has been confirmed as the neurotransmitter in the IHC ribbon synapse [27, 28, 58]. Once the neurotransmitter is released into the synaptic cleft, it activates an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) residing within a receptor cluster at the postsynaptic density of afferent ANFs [27]. Glutamate receptor subtypes (GluR) 2, 3, and 4 are abundant in IHC ribbon synapses [53]. GluR2 is not expressed until the onset of hearing, while GluR3 and GluR4 are present earlier during development. N-methyl-d-aspartate (NMDA) receptors (NR1, NR2A/B) are also present at the afferent synapses in the cochlea [58, 69, 80]. They are not activated for fast transmission as they are blocked by magnesium at resting membrane potential [31]. However, they modulate the reaction of AMPAR to glutamate at the type I afferent terminals [9, 28].
3.3 Synaptic Damage by Noise
3.3.1 Potential Mechanisms
The finding that noise induces cochlear synaptopathy reveals a novel locus of cochlear damage for NIHL. The damage to the postsynaptic terminal occurs through a similar mechanism as seen in conventional excitatory synapses: the glutamate-induced excitotoxicity. This mechanism is supported by the fact that cochlear infusion with glutamate or agonists mimics noise-induced damage [68, 71, 78]. Calcium influx and accumulation in the postsynaptic terminal is the initial step toward excitotoxicity [98]. Among the ionotropic glutamate receptors (iGluRs), AMPARs, NMDARs, and kainite-Rs, NMDARs have been considered the major contributors to the calcium influx and accumulation in the postsynaptic terminals. Therefore they are mainly responsible for the excitotoxicity in general [65, 98] and in the mammalian cochlea [55, 98]. While the neurotransmission between IHCs and SGNs is mainly mediated by the AMPAR, it is not considered responsible for the calcium influx. However, this opinion has been challenged by a recent report [85]. In this study, three subunits of AMPARs (GluA2, GluA3, and GluA4) were identified, and only GluA2 lacks calcium permeability. Moreover, calcium influx to the postsynaptic neurons was found to occur mainly via the Ca-permeable AMPARs (CP-AMPARs), but not NMDARs as previously recognized. This conclusion is supported by the fact that the CP-AMPAR blocker, IEM1460 (N, N, N-trimethyl-5-(tricyclo [3.3.1.13,7] dec-1-ylmethyl) amino-1-pentanaminium bromide hydrobromide), significantly reduces calcium accumulation in the postsynaptic auditory neurons, whereas the NMDAR blocker, APV (DL-2-amino-5-phosphonovaleric acid), shows no effect.
It is not clear if there is/are presynaptic mechanism(s) for the synaptic damage by noise or other toxic factors other than the Ca2+-mediated glutamate release. The presynaptic ribbons within photoreceptors are dynamic: they are dissembled in bright light and reassembled in dark [1, 75, 84, 95, 103]. This dynamic change serves as a mechanism of adaptation to stimulation and results in the change of neurotransmitters released. However, there is no evidence to date that supports the ribbons in the IHCs being dynamic in their response to acoustic overstimulation. Based upon the immunohistochemistry observation, the noise-induced reduction of the presynaptic ribbons is parallel with the breakdown of the postsynaptic terminals [89, 94]. In the photoreceptors, ribbons are dissembled when the cells are hyperpolarized by light that causes a reduction of [Ca2+]i. In the IHCs, the response to acoustic stimulation is a depolarization of membrane potential (not hyperpolarization) and an increase of [Ca2+]i. Therefore, if there is a disassembly/reassembly process in the IHCs, it must undergo a different mechanism. It is possible that the presynaptic ribbons in the IHCs are broken down after the postsynaptic terminals are destroyed. More research is needed to identify the fate of the ribbon protein after they are broken down by noise.
3.3.2 Selective Damage to Synapses with Low Spontaneous Rate Units
One IHC synapses with more than ten SGNs, and the synapses are distributed around the bottom of the IHC. The susceptibility of the synapses to noise damage appears to be location dependent: the synapses at the modiolar side of the IHC are more easily damaged. Although, the underlying mechanism is not entirely clear, this bias has been linked to the morphological variation around IHCs when identified in immunohistochemistry against CtBP2 and AMPAR (Fig. 3.2). The synapses close to the modiolar side of an IHC have larger ribbons and smaller postsynaptic terminals, whereas the synapses distributed toward the pillar side are the opposite [46]. This difference is functionally important because the synapses located at the modiolar side of IHCs innervate auditory nerve fibers (ANFs) that have lower spontaneous spike rates (SR), higher thresholds, and larger dynamic ranges. These ANFs are considered critical for hearing in noisy backgrounds, where only the high spontaneous SR units are saturated [10, 18, 33, 63, 112].

Spatial variations of ribbon synapses around the IHC. The synapses on the modiolar side appear to have a larger ribbon but a smaller postsynaptic terminal. The synapses on this side are also more sensitive to noise-induced damage
Several potential mechanisms have been proposed to explain the difference in the noise susceptibility between the synapses around IHCs. Firstly, there is a heterogeneity of Ca2+ channels around IHCs: synapses at modiolar side appear to have more Ca2+ channels per CAZ and display a higher Ca2+ influx and potentially a larger neurotransmitter release [20, 54]. The activation of the Ca2+channels at the modiolar side requires a larger degree of depolarization [54, 109, 110]. This heterogeneity has been linked to the spatial variations in the threshold and dynamic range of ANFs and to synaptic damage by noise around the IHCs [56]. Secondly, the larger amount of neurotransmitter release may be related to the larger ribbon size. The larger ribbons at the modiolar side can harbor more neurotransmitter vesicles close to the CAZ [27, 45, 93]. However, it is not clear if the vesicular priming and replenishment occurs faster for the ribbon synapses at the modiolar side of IHCs. Thirdly, the clearance of the released glutamate likely occurs slower at the modiolar side due to the lower amount of glutamate-aspartate transporters (GLAST) [25, 26, 74]. Fourthly, iGluRs (including AMPARs and NMDARs) are responsible for the glutamate-induced excitotoxic cell death in many neurologic diseases [29, 104]. Previously, NMDARs were thought to play a major role in noise-induced postsynaptic cochlear damage [4, 41], but more recently, this role has been attributed to Ca2+-permeable AMPAR as discussed above [85]. Nevertheless, it is not clear if the NMDARs are selectively distributed to the synapses at the modiolar side of the IHCs [83]. Interestingly, heterogeneity in the relative distribution of both Ca2+-permeable and Ca2+-impermeable subunits of AMPARs has been demonstrated across the IHC-SGN synapses. However, it is not clear how the heterogeneity is related to the synaptic distribution around the IHCs.
The functional significance of the selective damage to ANF synapses with the low-SR units remains to be confirmed. Theoretically, the selective loss of synapses with the low-SR ANFs will impair signal coding in strong background noise, one of the major problems seen in aging subjects [42, 56, 57, 88]. However, this coding deficit remains a speculation and has not been confirmed in single-unit data.
3.3.3 Can the Disrupted Ribbon Synapse Be Rebuilt?
It is currently debated whether the synaptic disruption by noise is reversible. In a pioneering study with CBA mice, no significant recovery of synapse counts was found after the threshold recovery that occurred in 1 week after the noise exposure [43]. Therefore, a single brief noise exposure caused up to 50% loss of synapses, permanently. However, studies from our labs in both Canada and China found that the decrease in synapse count was largely reversible in guinea pigs [50, 89, 94]. This reversibility was also reported in mice other than the CBA strain [90, 91]. It is worthy to notice that the concept of synaptic repair may involve two different phenotypes: (1) the rebuild of the synapses that are destroyed (by synaptogenesis) and (2) the repair of survived synapses that are damaged but not disconnected. The synaptic repair of the second type was reported after the initial noise-induced damage by the group of scientists who first reported noise-induced synaptic damage in the cochlea [67, 70, 71, 77]. In those early reports, the synaptic damage was observed using transmission electron microscopy (TEM). This technique limited the observation on the synapses that were partially damaged, but not destroyed. More importantly, the observation was not quantitative for the counting of total synapses. More recently, the dynamic changes of the number of ribbon synapses were reported in a study using AMPA infusion [79]. In our labs, the rebuild of the disrupted synapses was demonstrated by recovery of synapse counts. Functional data supported the synaptogenesis in that the recovery of the synapse count was matched by the recovery of compound action potential (CAP) measures (Fig. 3.3 a and b) [50, 89, 94]. In addition, the repair is also supported by the morphological changes of the synapses in the noise-damaged cochlea. Shortly after noise-induced damage, some synapses were found to be located up to the level of IHC nuclei and with extremely large ribbons, seen in immunohistochemical observation [89]. The synapse distribution returned to normal several weeks after the noise exposure, suggesting the re-established synapses were formed at a location close to the protein synthesis organelle. Furthermore, the repaired presynaptic ribbons appeared to have uneven sizes, with bigger hollow cores. In addition, many synapses observed weeks after the noise exposure had multiple ribbons to one AZ in TEM observation (Fig. 3.3c) [94]. This feature is seen in naïve ribbon synapses during early development [81] and is consistent with the regeneration of the synapses after they are destroyed by AMPA [79].

Evidence for synaptic repair after noise-induced damage. (a) Immunostaining images of pre- and postsynaptic components in control (ctrl) IHC and those 1 day, 1 week, and 1 month post noise (1DPN, 1WPN, and 1MPN). (b) Percentage changes of maximal CAP amplitude and synapse counts after noise. (c) TEM images of IHC ribbon synapses taken at 1MPN, showing hollow cores in some ribbons and double ribbons in some synapses
3.4 Synaptic Protection and Regeneration in Noise-Induced Synaptopathy
3.4.1 Synapse Protection
Noise-induced ribbon synapse damage involves the structural breakdown of both presynaptic ribbons and postsynaptic terminals [43, 89]. The mechanisms for the noise-induced damage on the postsynaptic terminal are clear and likely due to the glutamate-mediated excitotoxicity [70,71,72, 79]. Ca2+ overload via GluRs and voltage-gated Ca2+ channels (VGCCs) has been recognized as playing a critical role in noise-induced cochlear damage, both on HCs and postsynaptic terminals [5, 62, 65, 85]. Application of VGCC blockers (both L- and N-types) has shown the ability to protect the cochlear HCs from noise damage, consistent with the distribution of those calcium channels on HCs [34, 39, 49, 87, 101, 113]. However, it is not clear if the application of the blocker can prevent noise-induced synaptic damage.
Since noise causes synaptic damage via GluRs, blockage of these receptors may protect the synapses against noise. HC damage has been seen as part of excitotoxicity in zebrafish larvae, in which iGluRs are found to be expressed in the HCs [86]. Several studies have shown that NMDAR blockers can prevent tinnitus induced by salicylate [12, 66] and noise [4, 32]. Further research is needed to verify potential mechanisms [82, 83]. It is also important to note that blocking of iGluRs may have unforeseen effects. For example, long-term blockage of NMDA has been found to hinder the regeneration of the IHC-SGN synapses after excitotoxic damage [11, 79].
Previously, NMDAR antagonists have been tested for this potential protection [14, 15, 60]. However, the most significant effect of protection was seen on HCs, not on SGNs. Application of the NMDAR antagonists has been reported to reduce the swelling of the afferent dendrites synapsed with the IHCs in guinea pigs [14, 15]. However, the method for synapse quantification in those studies is questionable, since the number of the damaged synapses by noise in those studies was much fewer than that reported more recently using immunohistochemistry staining [43, 50, 89, 94]. Clearer evidence of synaptic protection against noise by the NMDAR antagonist was reported more recently [4]. However, in this study, the antagonist was administered at least 2 days after the noise exposure. Therefore, it is not clear what mechanism is underneath the reduction in synaptic damage. Presumably, the effect of the NMDARs in noise-induced damage to the afferent dendrites is based upon their role as a ligand-gated calcium channel. However, a recent study indicated that the sound-induced calcium entry was not mediated by NMDARs but by Ca2+-permeable AMPARs at the site [85]. This finding has shaken the theoretical basis of using NMDAR antagonists to reduce noise-induced synaptic damage in the cochlea. Previously, one study showed the protective effect of a blocker (caroverine) against both AMPA and NMDA receptors. It reduced the HC loss caused by impulse noise [16]. However, the protective effect on the synapses was not investigated. Therefore, further studies are needed to verify if NMDAR and/or AMPAR antagonists can protect the synapses from noise.
3.4.2 Synapse Regeneration
Since the synaptic damage induced by noise is partially reversible, there exists an endogenous mechanism to maintain the stability of the synaptic connections between SGNs and IHCs. Various studies have indicated the role of neurotrophic factors (NTFs) in synapse formation during development, plasticity, and the maintenance of synaptic stability in the cochlea (see review [21, 22, 73, 111]). Using NTFs appears to be a practical approach to rescue the damaged auditory nerves and their synapses to HCs [3]. Neurotrophins are a subclass of NTFs that are ubiquitously expressed and are very extensively studied. There are four types of neurotrophins in mammals: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4/5 (NT-4/5). BDNF and NT-3 are the major types of neurotrophins seen in the mammalian cochleae [73]. Within these two, BDNF is highly expressed during early development and declines to undetectable levels in adulthood. NT-3 is the only neurotrophin that exists in the adult cochleae, in addition to glial cell line-derived neurotrophic factor (GDNF) [13, 21, 30]. While the p75 neurotrophin receptor (p75NTR) is the receptor shared by BDNF, NT-3, and GDNF, each of the three factors has its own specific binding site(s): NT-3 binds mainly to the receptor C of the tropomyosin-related kinase (TrkC), BDNF to TrkB, and GDNF to RET-GFRα1 complex [73]. However, TrkA, TrkB, and TrkC are all expressed in the adult cochlea, even though their corresponding ligands (NGF to TrkA and BDNF to TrkB) are not detectable [30]. Therefore, BDNF and NGF can also be used for therapy in addition to NT-3, for the regeneration of the synapses after disruption.
NT-3 has been examined in several studies for its ability to promote the regeneration of the synapse. In the mammalian cochlea, NT-3 is expressed in both HCs and supporting cells [19, 22]. NT-3 overexpression by gene knock-in has been reported to increase the synapse density between IHCs and SGNs and decreased ABR threshold in mice [105]. The study suggested that the supporting cells are a more important source of NT-3 because the selective knock-in of NT-3 in the supporting cells promoted the regeneration of the synapses after disruption by noise exposure. However, the effect of selective knock-in in the HCs on synapses regeneration was not examined in this study.
Two studies showed a rescue effect of exogenous NT-3 to IHC-SGN synapses, when applied through the round window after noise trauma [92, 96]. In the first study done in mice, NT-3 was administered via the round window 24 hours after a 2 h noise exposure at 100 dB SPL, with the synapse count performed days later [96]. The NT-3 was delivered via slow-release gel placed in the round window niche. The protective effect of NT-3 was evaluated in both functional tests of auditory brainstem response (ABR) amplitude and synapse count. However, a large individual variation in the protective outcome made the authors divide the subjects into “effective” versus “ineffective” subgroups. Presumably, the “ineffective” was likely due to the failure in NT-3 application. A weak significance was seen when all of the NT-3 treated subjects were grouped together. In the second study listed above, guinea pigs were used. An equal mixture of NT-3 and BDNF was applied to the round window immediately after the noise exposure, which was given either at 95 or 105 dB SPL for 2 hours [92]. The synapse count observed 2 weeks after the noise exposure showed a significantly greater number in the ear treated with neurotrophins for the subjects receiving the 95 dB noise exposure. Since no data was reported from the subjects receiving the 105 dB noise exposure, and no control subjects were assessed, the interpretation of this data is difficult. Furthermore, the mixture of the two neurotrophins makes it difficult to measure the contribution from each neurotrophin.
Instead of using exogenous neurotrophins, the gene therapy type of approach appears to be more attractive in that it can provide long-term protection against repeated noise exposures. Using the gene knock-in technique, it has been found that overexpression of the NT-3 (but not BDNF) gene in supporting cells could significantly promote the ribbon synapse regeneration after noise-induced damage. However, no such protection was seen if the overexpression was only done in the IHCs [105]. In this study of normal-hearing guinea pigs, the overexpression of NT-3 in supporting cells and IHCs surprisingly increased the synaptic density of IHCs. Furthermore, the increase in synaptic density was accompanied by an increased ABR wave I amplitude and a decreased ABR threshold [105].
Due the ethical considerations, gene knock-in is unlikely to be used on human subjects. Instead, local gene transfection is an approach that can be translated to human clinics. To date, viral vectors appear to be much more effective in gene transfection. Among the viral vectors available, adeno-associated virus (AAV) is the most attractive due to its safety and the ability to cause long-lasting expression of the transfected gene. Several human trials of gene therapy are ongoing using AAV vectors. In a recent report, AAV-mediated NT-3 overexpression was found to cause considerable regeneration of synapses between IHCs and ANFs in guinea pigs that were deafened by aminoglycosides [6]. However, the benefit of NT-3 overexpression in the cochlea has been challenged by a study in which the overexpression was mediated by using (AAV) or adenovirus (Adv) [44]. In this study, subjects receiving the transfection either by AAV or Adv experienced ABR threshold elevations, more with Adv transfection. A decreased synapse count was seen in the subjects receiving Adv, but not AAV. The authors concluded that the elevation of NT-3 levels in the cochlea can disrupt synapses and impair hearing. A comparison between the two studies is impossible because one was done with normal-hearing subjects, while the other was done in subjects deafened by aminoglycosides. Furthermore, neither of the studies provided data for the transfection rate; and the study was done on deafened guinea pigs; no data was reported from a control group [6].
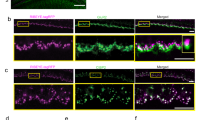
The safety of AAV vectors in cochlear gene transfection has been widely supported in the literature [35, 97, 99] and by our own published work [36, 107]. Recently conducted in our labs, the AAV of serotype 8 that had a surface tyrosine mutation at the residue of 733th amino acid on the capsid (rAAV8-mut773, at the titer of 6.92 × 1013, provided by the Retinal Gene Therapy Group, University of Florida, USA) was applied to transfect NT-3 into the cochlear cells of guinea pigs [8]. Figure 3.4 shows that the transfections of IHCs reached ~100% at the base and spread up to the second turn of the cochlea (10 mm from the apex or 4 kHz region). Therefore, it is a good model to test if the overexpression of NT-3 by AAV could promote synapse regeneration after noise-induced damage. After baseline ABR tests, transfection with AAV-NT-3 was done in one ear of each of the seven guinea pigs, whereas the other ear was given a sham surgery with the injection of the equivalent amount of saline. The ABR was retested 1 week after the transfection surgery. No threshold differences were seen between the ears (Fig. 3.5a). Then the animals were exposed to a high-pass noise with the cutoff at 4 kHz at 105 dB SPL for 2 h to create synaptopathy. A third ABR was administered 2 weeks after the noise exposure, followed by a near-field test of CAP with a round electrode. After the functional tests, the animals were sacrificed, and their cochleae were harvested for morphological evaluation for ribbon synapse counts. Another six animals were recruited as no-noise blank control.

AAV-mediated NT-3 transfection. (a) Representative IHCs image from 16 kHz region showing the transfected cells (green) across the cochlea. (b) Transfection cochleogram showing the mean (solid line) and +/-SEM (dashed lines) transfection of IHCs from three cochleae. (This figure is adapted from Chen et al. [8] Gene Therapy 25(4): 251–259)

Physiological function. (a) ABR threshold of two noise groups tested before surgery and 2 weeks after noise exposure. (b) The click-evoked CAP input-output function. (This figure is adapted from Chen et al. [8] Gene Therapy 25(4): 251–259)
Consistent with our previous reports, there was no significant difference in ABR thresholds between the baseline and 2 weeks after the noise exposure (Fig. 3.5a). To evaluate the impact of the synaptic damage on cochlear output, CAP was measured with clicks of different levels (Fig. 3.5b). The ears injected with AAV and saline are labelled as the two noise groups. In both groups, the noise exposure reduced CAP amplitude by more than half, and the input-output (I/O) functions from the noise groups were overlapped. Significant differences among the noise groups were seen in a one-way ANOVA, performed for the maximal CAP amplitude at 90 dB SPL (F = 57.6, P < 0.001). Post hoc tests (Bonferroni method) showed that the differences between the no-noise control group and the two noise groups were significant (control vs noise-NT-3: t = 9.3, p < 0.001; control vs noise-saline t = 9.3, p < 0.001), but not significant between the two noise groups (t = 0.1, p = 1).
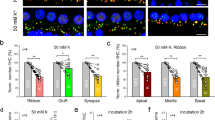
To evaluate the synaptic loss induced by the noise, the presynaptic ribbons (CtBP2) and postsynaptic densities (PSDs) were examined in immunohistochemistry (Fig. 3.6). The number of synapses was counted with the puncta of CtBP2s and PSDs that were paired. At each frequency point in each ear, the synapses were counted over eight IHCs to calculate the average synapse density (# of synapses per IHC). The noise-induced synaptic loss was mainly seen in the high-frequency region (>8 kHz, Fig. 3.7a). The effect of the NT-3 overexpression was demonstrated by less synaptic loss in the frequency region between 11.3 and 22.6 kHz (Fig. 3.7a). Over the high-frequency region (>8 kHz), the average synapse densities were 16.4 ± 0.2, 15.2 ± 0.2, and 18.4 ± 0.1 per IHC, for the noise-NT-3 group, noise-saline group, and no-noise control group, respectively. When compared to the no-noise control, this finding resulted in a 17.4% synaptic density reduction in the noise-saline group and a 10.9% reduction in the noise-NT-3 group. Compared between the noise-exposed groups, NT-3 overexpression appeared to reduce the synapse loss by ~38.5% in the high-frequency region. A significant effect of grouping was seen in a one-way ANOVA (F2,477 = 81.3, p < 0.001). The Bonferroni’s post hoc tests revealed the differences between the no-noise control group and the two noise groups (control vs noise-NT-3: t = 7.8, p < 0.001; control vs noise-saline t = 12.6, p < 0.001) and between the two noise groups (t = 4.9, p < 0.001) (Fig. 3.7b).

Images of CtBP2 and PSD staining from the three groups at 16 kHz region. (a) (b), and (c) were noise-NT-3, noise-saline, and no-noise control groups, respectively. The dashed lines indicate the outlines of IHCs and their nuclei. Only paired CtBP2 (red) and PSD (green) puncta were counted as synapses

NT-3 overexpression reduced the noise-induced synaptic loss. (a) The density-frequency curves of paired CtBP2 and PSD puncta. (b) The averaged counts of synapse density in the high-frequency region (between 8 and 32 kHz). One-way ANOVA followed by Bonferroni’s post hoc test revealed the significant differences between the no-noise control group and the two noise groups (not shown) and between the two noise groups. (a) error bar represent mean±SEM, (b) error bar represent mean±SEM. Asterisks indicate a significant level for the comparison between the two noise-exposed ears. ***p < 0.001. (This figure is adapted from Chen et al. [8] Gene Therapy 25(4): 251–259)
The nonsignificant difference result for NT-3, seen in the CAP I/O function, is likely due to the frequency range of the click-evoked CAP being biased to low-frequency regions, where no protection in the synapse count was seen. The power spectrum of clicks of 0.1 ms pulses was below 5 kHz. Even with the up-spread of cochlear vibration at a high intensity (90 dB SPL), the auditory nerves with characteristic frequencies higher than 8 kHz are unlikely to be excited. It is interesting to note that a synapse reduction of less than 5% was seen in the low-frequency region, while the CAP amplitude was reduced more than 50%. This suggests that the surviving/repaired synapses are functionally abnormal at this frequency.
In this study, we did not dynamically track the change in synapse counts at different time points after the noise exposure, nor did we compare the change across groups. Therefore, we do not know if the small reduction of synapse counts in the NT-3 overexpressed group resulted from the reduction of the initial loss of the synapses or the promoting effect of NT-3 on the regeneration of the synapses. However, based upon the working mechanism of NT-3 on synapse formation, and the rescue effect of NT-3 observed after noise exposure [92, 96, 108], we hypothesize that the major effect of NT-3 overexpression by AAV transfection in the present study is due to its effect on promoting synapse regeneration.
Based upon the study with the knock-in mouse model, NT-3 from supporting cells appear to be more effective than NT-3 from IHCs for promoting synapse regeneration [105]. In this study, the overexpression of the NT-3 was reportedly not effective at all. However in the present study (accepted), a significant protective effect is seen even though the NT-3 overexpression is limited to only IHCs (Fig. 3.4). While the quantitative comparison is impossible between the two studies due to the use of different species and different techniques for the overexpression, the protective effect in our study may have been limited by the confined transfection mediated by rAAV8-mut773 in the IHCs. We are exploring the use of new AAV that will transfect both the HCs and supporting cells for better protection against noise.
3.5 Conclusion and Future Direction
Gene therapy via cochlear gene transfection is an attractive approach to reduce noise-induced synaptopathy. The significance of this therapy is emphasized by the high probability of exposure to noise that can potentially produce such damage. Since NT-3 in both IHCs and supporting cells contributes to the synaptic regeneration, the AAV vector should be improved to transfect both the IHCs and supporting cells.
More research is needed to understand why synapses to the low-SR ANFs are more sensitive to noise damage. Research is also needed to investigate if there is a dis/reassembly mechanism of ribbons that act adaptively to reduce the traumatic glutamate release in response to intense noise. If this occurs, investigation into how this mechanism is regulated should be pursued. Research on gene therapy should be associated with the mechanisms for the neural transmission across this special synapse. Understanding the mechanisms of noise-induced synaptic damage in association with the working mechanism of ribbon synapses will provide insight toward reducing noise-induced damage and then increasing the amount of repair.
References
Adly MA, Spiwoks-Becker I, Vollrath L (1999) Ultrastructural changes of photoreceptor synaptic ribbons in relation to time of day and illumination. Invest Ophthalmol Vis Sci 40:2165–2172
Alpadi K, Magupalli VG, Kappel S, Koblitz L, Schwarz K, Seigel GM, Sung CH, Schmitz F (2008) RIBEYE recruits Munc119, a mammalian ortholog of the Caenorhabditis elegans protein unc119, to synaptic ribbons of photoreceptor synapses. J Biol Chem 283:26461–26467
Bezdjian A, Kraaijenga VJ, Ramekers D, Versnel H, Thomeer HG, Klis SF, Grolman W (2016) Towards clinical application of neurotrophic factors to the auditory nerve; assessment of safety and efficacy by a systematic review of neurotrophic treatments in humans. Int J Mol Sci 17
Bing D, Lee SC, Campanelli D, Xiong H, Matsumoto M, Panford-Walsh R, Wolpert S, Praetorius M, Zimmermann U, Chu H, Knipper M, Ruttiger L, Singer W (2015) Cochlear NMDA receptors as a therapeutic target of noise-induced tinnitus. Cell Physiol Biochem 35:1905–1923
Brassai A, Suvanjeiev RG, Ban EG, Lakatos M (2015) Role of synaptic and nonsynaptic glutamate receptors in ischaemia induced neurotoxicity. Brain Res Bull 112:1–6
Budenz CL, Wong HT, Swiderski DL, Shibata SB, Pfingst BE, Raphael Y (2015) Differential effects of AAV.BDNF and AAV.Ntf3 in the deafened adult guinea pig ear. Sci Rep 5:8619
Buran BN, Strenzke N, Neef A, Gundelfinger ED, Moser T, Liberman MC (2010) Onset coding is degraded in auditory nerve fibers from mutant mice lacking synaptic ribbons. J Neurosci 30:7587–7597
Chen H, Xing Y, Xia L, Chen Z, Yin S, Wang J (2018) AAV-mediated NT-3 overexpression protects cochleae against noise-induced synaptopathy. Gene Ther 25:251–259
Chen Z, Peppi M, Kujawa SG, Sewell WF (2009) Regulated expression of surface AMPA receptors reduces excitotoxicity in auditory neurons. J Neurophysiol 102:1152–1159
Costalupes JA (1985) Representation of tones in noise in the responses of auditory nerve fibers in cats. I. Comparison with detection thresholds. J Neurosci 5:3261–3269
d’Aldin CG, Ruel J, Assie R, Pujol R, Puel JL (1997) Implication of NMDA type glutamate receptors in neural regeneration and neoformation of synapses after excitotoxic injury in the guinea pig cochlea. Int J Dev Neurosci 15:619–629
Deng L, Ding D, Su J, Manohar S, Salvi R (2013) Salicylate selectively kills cochlear spiral ganglion neurons by paradoxically up-regulating superoxide. Neurotox Res 24:307–319
Despres G, Romand R (1994) Neurotrophins and the development of cochlear innervation. Life Sci 54:1291–1297
Diao M, Zhang Y, Liu H, Han H, Gao W (2005) Observation on the protective effect of MK-801 against hearing loss in acoustic trauma. Lin Chuang Er Bi Yan Hou Ke Za Zhi 19:27–30
Duan M, Agerman K, Ernfors P, Canlon B (2000) Complementary roles of neurotrophin 3 and a N-methyl-D-aspartate antagonist in the protection of noise and aminoglycoside-induced ototoxicity. Proc Natl Acad Sci U S A 97:7597–7602
Duan M, Chen Z, Qiu J, Ulfendahl M, Laurell G, Borg E, Ruan R (2006) Low-dose, long-term caroverine administration attenuates impulse noise-induced hearing loss in the rat. Acta Otolaryngol 126:1140–1147
Duncker SV, Franz C, Kuhn S, Schulte U, Campanelli D, Brandt N, Hirt B, Fakler B, Blin N, Ruth P, Engel J, Marcotti W, Zimmermann U, Knipper M (2013) Otoferlin couples to clathrin-mediated endocytosis in mature cochlear inner hair cells. J Neurosci 33:9508–9519
Eggermont JJ (2015) Animal models of auditory temporal processing. Int J Psychophysiol 95:202–215
Farinas I, Jones KR, Backus C, Wang XY, Reichardt LF (1994) Severe sensory and sympathetic deficits in mice lacking neurotrophin-3. Nature 369:658–661
Frank T, Khimich D, Neef A, Moser T (2009) Mechanisms contributing to synaptic Ca2+ signals and their heterogeneity in hair cells. Proc Natl Acad Sci U S A 106:4483–4488
Fritzsch B, Silos-Santiago I, Bianchi LM, Farinas I (1997) The role of neurotrophic factors in regulating the development of inner ear innervation. Trends Neurosci 20:159–164
Fritzsch B, Tessarollo L, Coppola E, Reichardt LF (2004) Neurotrophins in the ear: their roles in sensory neuron survival and fiber guidance. Prog Brain Res 146:265–278
Fuchs M, Brandstatter JH, Regus-Leidig H (2014) Evidence for a clathrin-independent mode of endocytosis at a continuously active sensory synapse. Front Cell Neurosci 8:60
Furman AC, Kujawa SG, Liberman MC (2013) Noise-induced cochlear neuropathy is selective for fibers with low spontaneous rates. J Neurophysiol 110:577–586
Furness DN, Lehre KP (1997) Immunocytochemical localization of a high-affinity glutamate-aspartate transporter, GLAST, in the rat and guinea-pig cochlea. Eur J Neurosci 9:1961–1969
Furness DN, Lawton DM (2003) Comparative distribution of glutamate transporters and receptors in relation to afferent innervation density in the mammalian cochlea. J Neurosci 23:11296–11304
Glowatzki E, Fuchs PA (2002) Transmitter release at the hair cell ribbon synapse. Nat Neurosci 5:147–154
Glowatzki E, Grant L, Fuchs P (2008) Hair cell afferent synapses. Curr Opin Neurobiol 18:389–395
Gonzalez J, Jurado-Coronel JC, Avila MF, Sabogal A, Capani F, Barreto GE (2015) NMDARs in neurological diseases: a potential therapeutic target. Int J Neurosci 125:315–327
Green SH, Bailey E, Wang Q, Davis RL (2012) The Trk A, B, C’s of neurotrophins in the cochlea. Anat Rec (Hoboken) 295:1877–1895
Guilarte TR, Chen MK (2007) Manganese inhibits NMDA receptor channel function: implications to psychiatric and cognitive effects. Neurotoxicology 28:1147–1152
Guitton MJ, Dudai Y (2007) Blockade of cochlear NMDA receptors prevents long-term tinnitus during a brief consolidation window after acoustic trauma. Neural Plast 2007:80904
Heil P, Peterson AJ (2015) Basic response properties of auditory nerve fibers: a review. Cell Tissue Res 361(1):129–158
Heinrich UR, Maurer J, Mann W (1999) Ultrastructural evidence for protection of the outer hair cells of the inner ear during intense noise exposure by application of the organic calcium channel blocker diltiazem. ORL J Otorhinolaryngol Relat Spec 61:321–327
Iizuka T, Kanzaki S, Mochizuki H, Inoshita A, Narui Y, Furukawa M, Kusunoki T, Saji M, Ogawa K, Ikeda K (2008) Noninvasive in vivo delivery of transgene via adeno-associated virus into supporting cells of the neonatal mouse cochlea. Hum Gene Ther 19:384–390
Jie H, Tao S, Liu L, Xia L, Charko A, Yu Z, Bance M, Yin S, Robertson GS, Wang J (2015) Cochlear protection against cisplatin by viral transfection of X-linked inhibitor of apoptosis protein across round window membrane. Gene Ther 22:546–552
Jing Z, Rutherford MA, Takago H, Frank T, Fejtova A, Khimich D, Moser T, Strenzke N (2013) Disruption of the presynaptic cytomatrix protein bassoon degrades ribbon anchorage, multiquantal release, and sound encoding at the hair cell afferent synapse. J Neurosci 33:4456–4467
Jung S, Maritzen T, Wichmann C, Jing Z, Neef A, Revelo NH, Al-Moyed H, Meese S, Wojcik SM, Panou I, Bulut H, Schu P, Ficner R, Reisinger E, Rizzoli SO, Neef J, Strenzke N, Haucke V, Moser T (2015) Disruption of adaptor protein 2mu (AP-2mu) in cochlear hair cells impairs vesicle reloading of synaptic release sites and hearing. EMBO J 34:2686–2702
Kansu L, Ozkarakas H, Efendi H, Okar I (2011) Protective effects of pentoxifylline and nimodipine on acoustic trauma in guinea pig cochlea. Otol Neurotol 32:919–925
Khimich D, Nouvian R, Pujol R, Tom Dieck S, Egner A, Gundelfinger ED, Moser T (2005) Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature 434:889–894
Knipper M, Van Dijk P, Nunes I, Ruttiger L, Zimmermann U (2013) Advances in the neurobiology of hearing disorders: recent developments regarding the basis of tinnitus and hyperacusis. Prog Neurobiol 111:17–33
Kobel M, Le Prell CG, Liu J, Hawks JW, Bao J (2017) Noise-induced cochlear synaptopathy: past findings and future studies. Hear Res 349:148–154
Kujawa SG, Liberman MC (2009) Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci 29:14077–14085
Lee MY, Kurioka T, Nelson MM, Prieskorn DM, Swiderski DL, Takada Y, Beyer LA, Raphael Y (2016) Viral-mediated Ntf3 overexpression disrupts innervation and hearing in nondeafened guinea pig cochleae. Mol Ther Methods Clin Dev 3:16052
Lenzi D, Crum J, Ellisman MH, Roberts WM (2002) Depolarization redistributes synaptic membrane and creates a gradient of vesicles on the synaptic body at a ribbon synapse. Neuron 36:649–659
Liberman LD, Wang H, Liberman MC (2011) Opposing gradients of ribbon size and AMPA receptor expression underlie sensitivity differences among cochlear-nerve/hair-cell synapses. J Neurosci 31:801–808
Liberman MC, Kujawa SG (2017) Cochlear synaptopathy in acquired sensorineural hearing loss: manifestations and mechanisms. Hear Res 349:138–147
Lin HW, Furman AC, Kujawa SG, Liberman MC (2011) Primary neural degeneration in the guinea pig cochlea after reversible noise-induced threshold shift. J Assoc Res Otolaryngol 12:605–616
Liu J, Niu YG, Li WX, Yuan YY, Han WJ, Yu N, Yang SM, Li XQ (2012) Interaction of a calcium channel blocker with noise in cochlear function in guinea pig. Acta Otolaryngol 132:1140–1144
Liu L, Wang H, Shi L, Almuklass A, He T, Aiken S, Bance M, Yin S, Wang J (2012) Silent damage of noise on cochlear afferent innervation in guinea pigs and the impact on temporal processing. PLoS One 7:e49550
Lobarinas E, Spankovich C, Le Prell CG (2017) Evidence of “hidden hearing loss” following noise exposures that produce robust TTS and ABR wave-I amplitude reductions. Hear Res 349:155–163
Magupalli VG, Schwarz K, Alpadi K, Natarajan S, Seigel GM, Schmitz F (2008) Multiple RIBEYE-RIBEYE interactions create a dynamic scaffold for the formation of synaptic ribbons. J Neurosci 28:7954–7967
Matsubara A, Laake JH, Davanger S, Usami S, Ottersen OP (1996) Organization of AMPA receptor subunits at a glutamate synapse: a quantitative immunogold analysis of hair cell synapses in the rat organ of Corti. J Neurosci 16:4457–4467
Meyer AC, Frank T, Khimich D, Hoch G, Riedel D, Chapochnikov NM, Yarin YM, Harke B, Hell SW, Egner A, Moser T (2009) Tuning of synapse number, structure and function in the cochlea. Nat Neurosci 12:444–453
Monaghan DT, Jane DE (2009) Pharmacology of NMDA receptors. In: Van Dongen AM (ed) Biology of the NMDA receptor. CRC Press, Boca Raton
Moser T, Vogl C (2016) New insights into cochlear sound encoding. F1000Res 5
Moser T, Starr A (2016) Auditory neuropathy—neural and synaptic mechanisms. Nat Rev Neurol 12:135–149
Nordang L, Cestreicher E, Arnold W, Anniko M (2000) Glutamate is the afferent neurotransmitter in the human cochlea. Acta Otolaryngol 120:359–362
Nouvian R, Beutner D, Parsons TD, Moser T (2006) Structure and function of the hair cell ribbon synapse. J Membr Biol 209:153–165
Ohinata Y, Miller JM, Schacht J (2003) Protection from noise-induced lipid peroxidation and hair cell loss in the cochlea. Brain Res 966:265–273
Pangrsic T, Reisinger E, Moser T (2012) Otoferlin: a multi-C2 domain protein essential for hearing. Trends Neurosci 35:671–680
Paoletti P (2011) Molecular basis of NMDA receptor functional diversity. Eur J Neurosci 33:1351–1365
Plack CJ, Barker D, Prendergast G (2014) Perceptual consequences of “hidden” hearing loss. Trends Hear 18
Plack CJ, Leger A, Prendergast G, Kluk K, Guest H, Munro KJ (2016) Toward a diagnostic test for hidden hearing loss. Trends Hear 20
Prentice H, Modi JP, Wu JY (2015) Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxidative Med Cell Longev 2015:964518
Puel JL (2007) Cochlear NMDA receptor blockade prevents salicylate-induced tinnitus. B-ENT 3(Suppl 7):19–22
Puel JL, Ruel J, Gervais d’Aldin C, Pujol R (1998) Excitotoxicity and repair of cochlear synapses after noise-trauma induced hearing loss. Neuroreport 9:2109–2114
Puel JL, Ruel J, Guitton M, Pujol R (2002) The inner hair cell afferent/efferent synapses revisited: a basis for new therapeutic strategies. Adv Otorhinolaryngol 59:124–130
Puel JL, Ladrech S, Chabert R, Pujol R, Eybalin M (1991) Electrophysiological evidence for the presence of NMDA receptors in the guinea pig cochlea. Hear Res 51:255–264
Puel JL, d’Aldin C, Ruel J, Ladrech S, Pujol R (1997) Synaptic repair mechanisms responsible for functional recovery in various cochlear pathologies. Acta Otolaryngol 117:214–218
Pujol R, Puel JL (1999) Excitotoxicity, synaptic repair, and functional recovery in the mammalian cochlea: a review of recent findings. Ann N Y Acad Sci 884:249–254
Pujol R, Puel JL, Gervais d’Aldin C, Eybalin M (1993) Pathophysiology of the glutamatergic synapses in the cochlea. Acta Otolaryngol 113:330–334
Ramekers D, Versnel H, Grolman W, Klis SF (2012) Neurotrophins and their role in the cochlea. Hear Res 288:19–33
Rebillard G, Ruel J, Nouvian R, Saleh H, Pujol R, Dehnes Y, Raymond J, Puel JL, Devau G (2003) Glutamate transporters in the guinea-pig cochlea: partial mRNA sequences, cellular expression and functional implications. Eur J Neurosci 17:83–92
Regus-Leidig H, Specht D, Tom Dieck S, Brandstatter JH (2010) Stability of active zone components at the photoreceptor ribbon complex. Mol Vis 16:2690–2700
Regus-Leidig H, Fuchs M, Lohner M, Leist SR, Leal-Ortiz S, Chiodo VA, Hauswirth WW, Garner CC, Brandstatter JH (2014) In vivo knockdown of Piccolino disrupts presynaptic ribbon morphology in mouse photoreceptor synapses. Front Cell Neurosci 8:259
Robertson D (1982) Effects of acoustic trauma on stereocilia structure and spiral ganglion cell tuning properties in the guinea pig. Hear Res 7:55–74
Ruel J, Chen C, Pujol R, Bobbin RP, Puel JL (1999) AMPA-preferring glutamate receptors in cochlear physiology of adult guinea-pig. J Physiol 518(Pt 3):667–680
Ruel J, Wang J, Rebillard G, Eybalin M, Lloyd R, Pujol R, Puel JL (2007) Physiology, pharmacology and plasticity at the inner hair cell synaptic complex. Hear Res 227:19–27
Ruel J, Chabbert C, Nouvian R, Bendris R, Eybalin M, Leger CL, Bourien J, Mersel M, Puel JL (2008) Salicylate enables cochlear arachidonic-acid-sensitive NMDA receptor responses. J Neurosci 28:7313–7323
Safieddine S, El-Amraoui A, Petit C (2012) The auditory hair cell ribbon synapse: from assembly to function. Annu Rev Neurosci 35:509–528
Sahley TL, Hammonds MD, Musiek FE (2013) Endogenous dynorphins, glutamate and N-methyl-d-aspartate (NMDA) receptors may participate in a stress-mediated Type-I auditory neural exacerbation of tinnitus. Brain Res 1499:80–108
Sanchez JT, Ghelani S, Otto-Meyer S (2015) From development to disease: diverse functions of NMDA-type glutamate receptors in the lower auditory pathway. Neuroscience 285:248–259
Schmitz F (2009) The making of synaptic ribbons: how they are built and what they do. Neuroscientist 15:611–624
Sebe JY, Cho S, Sheets L, Rutherford MA, von Gersdorff H, Raible DW (2017) Ca(2+)-permeable AMPARs mediate glutamatergic transmission and excitotoxic damage at the hair cell ribbon synapse. J Neurosci 37:6162–6175
Sheets L (2017) Excessive activation of ionotropic glutamate receptors induces apoptotic hair-cell death independent of afferent and efferent innervation. Sci Rep 7:41102
Shen H, Zhang B, Shin JH, Lei D, Du Y, Gao X, Wang Q, Ohlemiller KK, Piccirillo J, Bao J (2007) Prophylactic and therapeutic functions of T-type calcium blockers against noise-induced hearing loss. Hear Res 226:52–60
Shi L, Chang Y, Li X, Aiken SJ, Liu L, Wang J (2016) Coding deficits in noise-induced hidden hearing loss may stem from incomplete repair of ribbon synapses in the cochlea. Front Neurosci 10:231
Shi L, Liu L, He T, Guo X, Yu Z, Yin S, Wang J (2013) Ribbon synapse plasticity in the cochleae of guinea pigs after noise-induced silent damage. PLoS One 8:e81566
Shi L, Liu K, Wang H, Zhang Y, Hong Z, Wang M, Wang X, Jiang X, Yang S (2015) Noise induced reversible changes of cochlear ribbon synapses contribute to temporary hearing loss in mice. Acta Otolaryngol 135(11):1–10
Shi L, Guo X, Shen P, Liu L, Tao S, Li X, Song Q, Yu Z, Yin S, Wang J (2015) Noise-induced damage to ribbon synapses without permanent threshold shifts in neonatal mice. Neuroscience 304:368–377
Sly DJ, Campbell L, Uschakov A, Saief ST, Lam M, O’Leary SJ (2016) Applying neurotrophins to the round window rescues auditory function and reduces inner hair cell synaptopathy after noise-induced hearing loss. Otol Neurotol 37:1223–1230
Sobkowicz HM, Slapnick SM, August BK (2002) Differentiation of spinous synapses in the mouse organ of Corti. Synapse 45:10–24
Song Q, Shen P, Li X, Shi L, Liu L, Wang J, Yu Z, Stephen K, Aiken S, Yin S, Wang J (2016) Coding deficits in hidden hearing loss induced by noise: the nature and impacts. Sci Rep 6:25200
Spiwoks-Becker I, Glas M, Lasarzik I, Vollrath L (2004) Mouse photoreceptor synaptic ribbons lose and regain material in response to illumination changes. Eur J Neurosci 19:1559–1571
Suzuki J, Corfas G, Liberman MC (2016) Round-window delivery of neurotrophin 3 regenerates cochlear synapses after acoustic overexposure. Sci Rep 6:24907
Suzuki J, Hashimoto K, Xiao R, Vandenberghe LH, Liberman MC (2017) Cochlear gene therapy with ancestral AAV in adult mice: complete transduction of inner hair cells without cochlear dysfunction. Sci Rep 7:45524
Szydlowska K, Tymianski M (2010) Calcium, ischemia and excitotoxicity. Cell Calcium 47:122–129
Tao Y, Huang M, Shu Y, Ruprecht A, Wang H, Tang Y, Vandenberghe LH, Wang Q, Gao G, Kong WJ, Chen ZY (2017) Delivery of adeno-associated viral vectors in adult mammalian inner ear cell subtypes without auditory dysfunction. Hum Gene Ther 29(4)
tom Dieck S, Altrock WD, Kessels MM, Qualmann B, Regus H, Brauner D, Fejtova A, Bracko O, Gundelfinger ED, Brandstatter JH (2005) Molecular dissection of the photoreceptor ribbon synapse: physical interaction of bassoon and RIBEYE is essential for the assembly of the ribbon complex. J Cell Biol 168:825–836
Uemaetomari I, Tabuchi K, Nakamagoe M, Tanaka S, Murashita H, Hara A (2009) L-type voltage-gated calcium channel is involved in the pathogenesis of acoustic injury in the cochlea. Tohoku J Exp Med 218:41–47
Valente C, Spano S, Luini A, Corda D (2005) Purification and functional properties of the membrane fissioning protein CtBP3/BARS. Methods Enzymol 404:296–316
Vollrath L, Spiwoks-Becker I (1996) Plasticity of retinal ribbon synapses. Microsc Res Tech 35:472–487
Vyklicky V, Korinek M, Smejkalova T, Balik A, Krausova B, Kaniakova M, Lichnerova K, Cerny J, Krusek J, Dittert I, Horak M, Vyklicky L (2014) Structure, function, and pharmacology of NMDA receptor channels. Physiol Res 63(Suppl 1):S191–S203
Wan G, Gomez-Casati ME, Gigliello AR, Liberman MC, Corfas G (2014) Neurotrophin-3 regulates ribbon synapse density in the cochlea and induces synapse regeneration after acoustic trauma. eLife 3
Wan L, Almers W, Chen W (2005) Two ribeye genes in teleosts: the role of ribeye in ribbon formation and bipolar cell development. J Neurosci 25:941–949
Wang H, Murphy R, Taaffe D, Yin S, Xia L, Hauswirth WW, Bance M, Robertson GS, Wang J (2012) Efficient cochlear gene transfection in guinea-pigs with adeno-associated viral vectors by partial digestion of round window membrane. Gene Ther 19:255–263
Wang Q, Green SH (2011) Functional role of neurotrophin-3 in synapse regeneration by spiral ganglion neurons on inner hair cells after excitotoxic trauma in vitro. J Neurosci 31:7938–7949
Wong AB, Jing Z, Rutherford MA, Frank T, Strenzke N, Moser T (2013) Concurrent maturation of inner hair cell synaptic Ca2+ influx and auditory nerve spontaneous activity around hearing onset in mice. J Neurosci 33:10661–10666
Wong AB, Rutherford MA, Gabrielaitis M, Pangrsic T, Gottfert F, Frank T, Michanski S, Hell S, Wolf F, Wichmann C, Moser T (2014) Developmental refinement of hair cell synapses tightens the coupling of Ca2+ influx to exocytosis. EMBO J 33:247–264
Yang T, Kersigo J, Jahan I, Pan N, Fritzsch B (2011) The molecular basis of making spiral ganglion neurons and connecting them to hair cells of the organ of Corti. Hear Res 278:21–33
Young ED, Barta PE (1986) Rate responses of auditory nerve fibers to tones in noise near masked threshold. J Acoust Soc Am 79:426–442
Yu YF, Wu WY, Xiao GS, Ling HY, Pan C (2016) Protection of the cochlear hair cells in adult C57BL/6J mice by T-type calcium channel blockers. Exp Ther Med 11:1039–1044
Zenisek D, Horst NK, Merrifield C, Sterling P, Matthews G (2004) Visualizing synaptic ribbons in the living cell. J Neurosci 24:9752–9759
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Wang, J., Yin, S., Chen, H., Shi, L. (2019). Noise-Induced Cochlear Synaptopathy and Ribbon Synapse Regeneration: Repair Process and Therapeutic Target. In: Li, H., Chai, R. (eds) Hearing Loss: Mechanisms, Prevention and Cure. Advances in Experimental Medicine and Biology, vol 1130. Springer, Singapore. https://doi.org/10.1007/978-981-13-6123-4_3
Download citation
DOI: https://doi.org/10.1007/978-981-13-6123-4_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-6122-7
Online ISBN: 978-981-13-6123-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)