
Abstract
Proteases (EC 3.4), enzymes originally used to cleave the amide bonds of proteins by hydrolysis, have been utilized for the enzymatic synthesis of peptide compounds. This enzymatic synthesis of polypeptides is a biomass-based, environmentally benign, atom-economical, and stereo-/regioselective method that can replace petroleum-derived chemical polypeptide syntheses. Enzymatic polymerization of amino acid derivatives using proteases proceeds via the reverse reaction of hydrolysis, which is aminolysis, in an equilibrium. Thermodynamic and kinetic controls in the aminolysis reaction rationally optimize enzymatic polymerization efficiency. Polymerization is regulated by the substrate specificity of proteases, namely, a combination of amino acid monomers and proteases. A great number of polypeptides, including homopolymers, random/block copolymers, and specific polymer architectures, such as star-shaped polymers, are synthesized by a protease-catalyzed polymerization technique. In this chapter, versatile designs and syntheses of polypeptide materials using various types of proteases are entirely reviewed in detail.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
8.1 Introduction
Polypeptides are physiologically essential components as the main proteins in living systems. Proteins are synthesized via the translation of mRNA sequential information by ribosomes in the central dogma. The diversity of their physiological functions and properties is associated with their primary, secondary, and higher-order structures. In addition to proteins possessing well-defined amino acid sequences, artificial polypeptide materials, including homopolymers, random or block copolymers, and special polymeric architectures, such as star-shaped polymers, have attracted intensive attention as bio-based polymeric materials. The material properties of these polypeptides can be tuned by amino acid monomer units and their sequences, promising useful applications in various fields.
There are three approaches to synthesizing artificial polypeptides: chemical, biological, and biochemical synthetic methods. The chemical method includes solid phase peptide synthesis (SPPS), which was developed by Merrifield,[1] and ring opening polymerization of amino acid N-carboxyanhydrides (NCA).[2] In the SPPS method, polypeptides are synthesized on insoluble solid supports to propagate the peptide chain in a stepwise manner, enabling precise control of the amino acid sequence of polypeptides. On the other hand, the polymerization of amino acid NCA derivatives can provide polypeptides of higher molecular weight; however, this method is only available for the synthesis of homopolymers and random/block copolymers. These chemical synthetic methods employ the polymerization of amino acid derivatives, which are chemically synthesized in advance with organic solvents. In spite of their beneficial advantages, a recent trend to avoid petroleum-derived chemicals for a future sustainable society pushes us to pursue an environmentally benign alternative method of polypeptide synthesis.
The biological method includes the biosynthesis of proteins in vivo, which is generally processed by combination of biomacromolecules, such as enzymes. A key enzyme involved in biosynthesis is ligase. Free amino acids are activated by aminoacyl-tRNA synthetases to generate amino-acid-ligated tRNAs, and then, they iteratively react with the C-terminal of a propagating peptide according to the order that the mRNA regulates by its codon sequence. Since gene engineering technology was established, many natural and engineered proteins have been biosynthesized in living cells, particularly in genetically modified microbes. Biosynthesis using a sophisticated protein expression system in nature is a powerful way to strictly control amino acid sequences based on the codon sequences preserved in mRNA. Desired polypeptide sequences can be obtained by designing proper DNA constructs to transform host genes. Not only natural proteins but also artificial polypeptides can be designed for biosynthesis. Tirrell et al. pioneered the biosynthesis of desired polypeptides using living organisms and demonstrated that artificial polypeptides were successfully designed and expressed by in vivo biosynthesis using microbes transformed with plasmids encoding target sequences. The synthetic polypeptides consisting of periodic sequences, such as GluAsp(Glu17Asp)4GluGlu or [(AlaGly)3ProGluGly]4, were designed and encoded in plasmid DNA to transform Escherichia coli [3, 4]. This biosynthetic technique enables the precise, sophisticated design of functional polypeptides; however, practical use of this biosynthesis system suffers from some disadvantages. Expression of the target proteins is occasionally suppressed to a low level by various factors, mainly because of the cytotoxicity of the expressed proteins. A tedious purification process to isolate target polypeptides from cell lysates also hampers cost-effective production.
The biochemical method uses enzymes as catalysts in vitro to synthesize peptides by enzyme-mediated polymerizations. Enzymatic polymerization is another promising candidate for an alternative green synthetic method for polypeptide synthesis. In contrast to biosynthesis with ligase, the enzymes used for the enzymatic polymerization of amino acid monomers are proteases, which originally cleave the peptide bonds in polypeptides. This protease-catalyzed hydrolysis of polypeptides is a reversible reaction; hence, the reverse reaction can propagate peptide chains under optimized reaction conditions as described in the next section. Protease-catalyzed polymerization has several advantages over conventional chemical synthetic methods. Amino acid monomers, generally in a form of ester, are polymerized in the presence of a protease in an aqueous buffer solution, resulting in the formation of polypeptides with only small alcoholic by-products. Therefore, this enzymatic polymerization is considered as an eco-friendly synthetic method with high atom economy compared to conventional condensation techniques. In addition, the substrate specificity of proteases rationally recognizes an appropriate chiral isomer (generally the l-isomer) and reacting groups attached to an α-carbon. This feature allows enzymatic polymerization to proceed in a stereo- and regioselective manner. Some amino acids, therefore, can be polymerized without protection of the reactive side groups. In addition, the enzymatic reaction is readily scalable because the synthetic protocol simply mixes amino acid monomers and proteases in moderate conditions.
Enzymatic amide formation has been continually developed to synthesize small peptide compounds since the 1950s [5]. To date, many comprehensive studies on enzymatic peptide synthesis have not only elucidated the mechanisms of the involved reactions but also synthesized novel functional polypeptide materials. Because protease-catalyzed aminolysis generates amide bonds, an enzymatic polymerization technique using proteases can be applied to not only polypeptides but also other synthetic polyamides. Indeed, some reports have employed unnatural amino acids for enzymatic synthesis in copolymerization with natural amino acids as described below (Sect. 8.5.6 Unnatural Amino Acids). In this chapter, the target building blocks are mainly focused on polypeptides [6,7,8], and not only fundamental aspects, including polymerization mechanisms and characteristics of proteases, but also structural variations of polypeptides synthesized by enzymatic polymerization are thoroughly reviewed.
8.2 Mechanisms of Enzymatic Reactions
Proteases catalyze not only the hydrolysis of peptide bonds but also their aminolysis, which results in the formation of a peptide bond between two amino acids. There are two strategies to synthesize peptides by proteases [9,10,11,12,13,14,15,16], that is, thermodynamically and kinetically controlled syntheses.
8.2.1 Thermodynamic Control
Thermodynamically controlled synthesis of peptides can be applied to all proteases. This synthetic method focuses on an equilibrium between the hydrolysis and aminolysis reactions. As shown in Fig. 8.1, protease-catalyzed peptide bond formation can be divided into two equilibrium reactions. One reaction is the equilibrium between the ionization and deionization of carboxylic acid/amine, and the other is the equilibrium between hydrolysis and aminolysis. Thus, these two steps must be considered to obtain peptides from amino acids with free carboxylic acid and amine groups. In the first step, non-charged substrates must be formed by the deionization of carboxylic acid and amine groups. Subsequently, the aminolysis reaction occurs via the formation of an acyl intermediate within proteases. Therefore, proteases are involved in the second step to accelerate the hydrolysis/aminolysis reaction. When neutral amino acids are used as the starting substrates, the first equilibrium tends to drive to the left side. This effect can be explained by the equilibrium constant and the ΔG of the reaction [17]. In cases using two unprotected amino acids, the equilibrium constant of the first step is 10–7.5, and the ΔG is approximately 10 kcal·mol−1, which means that the equilibrium favors not the nonionic but rather the ionic reactants. The equilibrium constant of the second step is 103.7, and its ΔG is about −5 kcal mol−1. Therefore, the equilibrium constant of the whole reaction is 10–3.8, and its ΔG is approximately 5 kcal·mol−1. This value indicates that the equilibrium reaction proceeds toward an endothermic reverse reaction. Great disincentives to forming peptide bonds in this step are a slow reaction speed and a low yield because of the low equilibrium constant and positive ΔG. When the aminolysis reaction in homopolymerization proceeds, the equilibrium constant of the reaction becomes lower, and the ΔG of the reaction becomes higher. Hence, it is difficult to obtain the polypeptide by iterative aminolysis reactions. Thermodynamically controlled synthesis is a method to control the thermodynamic equilibrium constants of these two elementary reactions. There are two strategies to increase the equilibrium constant of aminolysis. One strategy is the control of the first reaction, and the other is the control of the second reaction.

Equilibrium in thermodynamically controlled peptide synthesis
In the first step, deionization of an ionized substrate occurs by proton transfer from ammonium to carboxylate groups at a pH above its isoelectric point. However, amino acids exist as zwitterions at pH 5–9, a pH at which most proteases show high catalytic activity. In this pH range, the neutral form of amino acids is a minor substrate because the equilibrium constant of first step is much lower than 1. In contrast, the equilibrium constant of the reaction between N-protected and C-protected amino acids is 10–4.0. This value is still lower than 1, but it is much higher than that of protection-free amino acids. Therefore, the combination of N- and C-protected amino acids is favorable for driving the equilibrium toward neutral forms. This method is limited to the synthesis of oligopeptides via a coupling between two reactants and is not favorable for the synthesis of polypeptides, which generally result from multistep reactions. There are also other factors that can control the first equilibrium reaction, such as pH and ionic strength. For example, the addition of water-miscible organic solvents induces a reduction in ionic strength and thereby a decrease in the frequency of the proton transfer from ammonium to carboxylate groups. By using the N- or C-protected amino acids as substituents and organic/water-miscible solution as solvents, the equilibrium constant can increase, and the equilibrium constant can move to the right side, namely, peptide synthesis. Homandberg et al. examined the equilibrium constant of the reaction between benzyloxycarbonyl-protected tryptophan (Z-Trp) and glycinamide (Gly-NH2) in water, 60 v/v% triethylene glycol solution, or 85 v/v% 1,4-butanediol solution [18]. The equilibrium constant in 85 v/v% 1,4-butanediol solution showed the largest value. The reaction rate and yields can be controlled by the content of organic solvent and water.
Precipitation of peptide products is useful to effectively drive the second equilibrium toward the right (Fig. 8.1). When products precipitate from the solution, the concentration of products dissolved in the solution becomes lower, and the equilibrium shifts toward the right side. In this way, peptide formation proceeds by removing products from a system. In other words, by using a biphasic condition, products can be removed from water media. The reaction occurs around the surface between water and organic layers. A nonpolar organic solvent, such as ethyl acetate, chloroform, and toluene, is used as the organic layer. Cassells and Halling reported protease-catalyzed peptide synthesis in water/organic two-phase systems [19]. The reaction rate of Z-Phe-Phe-OMe synthesis with a silica-supported thermolysin in a reaction mixture composed predominantly of ethyl acetate was as fast as that with thermolysin in the corresponding high-water emulsion system. In this case, the reaction proceeds as follows: substrates dissolved in the organic layer diffused to water media in which proteases exist and reacted to afford products. Substrates are continuously supplied from the organic layer to water media, whereas products are removed from water to the organic layer. This removal of the peptide product drives the equilibrium toward aminolysis and prevents hydrolysis of the product. However, the reaction rate is very slow because of the slow diffusion of the substrate, and the reaction suffers from denaturing of proteases at the surface.
Hydrolysis of peptides is induced by nucleophilic attack of water molecules (H2O). Reducing the amount of H2O or the activity of H2O is a useful method to prevent hydrolysis. An organic solvent suppresses the activity of H2O. However, proteases do not maintain their structures and functions in organic solvent. Therefore, water-miscible organic solvents (such as ethanol, dimethylsulfoxide, N,N-dimethylformamide, dioxane, acetone, and acetonitrile) are used with 2–5% water as the equilibrium condition [18, 20, 21].
8.2.2 Kinetic Control
The synthetic scheme of kinetically controlled peptide synthesis is shown in Fig. 8.2 [22]. An ester group is generally introduced at a carboxyl end and used as a mildly activated acyl donor. The ester donor rapidly reacts at the catalytic center in proteases and an acyl-protease intermediate forms. Subsequently, the acyl-protease intermediate is nucleophilically attacked by nucleophiles, i.e., H2O or an amino group of another amino acid substrate. Thus, hydrolysis/aminolysis occurs by deacylation with H2O and free amino groups. Hydrolysis and aminolysis are not reversible reactions but rather cooperative reactions in kinetically controlled peptide synthesis. Therefore, the degree of the nucleophilic attack affects the rate of hydrolysis/aminolysis, which is why this effect is called “kinetic control.” At the same time, thermodynamic hydrolysis proceeds. Nucleophilic hydrolysis and aminolysis are much faster than the equilibrium reaction. In the initial stage of the reaction, the concentration of peptide product rapidly increases. In contrast, in the latter half of the reaction, the concentration of peptide product reaches a plateau. Qin et al. reported the polymerization of l-Glu diethyl ester catalyzed by papain [23]. They examined the relationship between consumption of the starting material and reaction times. The l-Glu diethyl ester was consumed immediately after the reaction started. The yield reached 80% by 20 min and did not significantly increase afterward. This result indicates that the aminolysis reaction rapidly occurs under kinetically controlled peptide synthesis.

Mechanism of kinetically controlled peptide synthesis
Serine or cysteine proteases can be used for kinetically controlled synthesis to create peptide bonds. The hydroxyl group of serine and the thiol group of cysteine play an important role in forming the acyl-protease intermediate. Figure 8.3 shows molecular behaviors around the active site of serine proteases. When carboxylic ester comes close to the catalytic center, a tetrahedral intermediate forms with a C–O covalent bond between the ester and the catalytic serine residue. Next, the acyl-protease intermediate is generated by releasing an alcohol corresponding to the ester groups of the starting substrates. Subsequently, the tetrahedral intermediate is formed again by nucleophilic attack of nucleophiles. Finally, an amide bond is formed through the equilibrium between the tetrahedral intermediate and a product.

The behavior of acyl donors and nucleophiles at the active site of serine proteases
There are many factors that enhance the rate of aminolysis. The concentration of amine nucleophilic groups, pH, and temperature affect the yield of peptide products. The rate of nucleophilic attack by amines to form acyl-protease intermediates is increased by raising the concentration of amine substrates. The amine nucleophilic group exists in its reactive neutral form at high pH. A suitable pH is in a range of 5–9 for the enzymatic reaction in the thermodynamic control method, whereas the kinetic control method needs a higher pH condition [24]. In addition, higher temperature induces a lower reaction rate of aminolysis. Based on these characteristics of aminolysis by proteases, frozen aqueous synthesis has been developed by many research groups [25,26,27]. In frozen aqueous conditions, namely, −20 to 5 °C, the rate of hydrolysis is reduced, and the yield of peptide formation is increased.
In contrast to the thermodynamic control method, the use of biphasic solution is inappropriate for kinetically controlled synthesis [28,29,30,31]. Proteases exist in an aqueous phase, whereas amino acid ester substrates exist in both phases. Therefore, the concentration of acyl-protease intermediates is low and hardly controlled in this system. On the other hand, the activity of H2O toward the acyl-protease is reduced in a miscible water/organic solvent system, which suppresses the competing hydrolysis reaction. Kisee et al. [32] and Viswanathan et al. [33] reported a miscible water/organic (methanol, ethanol, tetrahydrofuran, and so on) solvent system. The yield of resulting peptides was dependent on the content of water and organic solvent. Thus, the water/organic solvent system is an interesting reaction system. However, it is noted that organic solvent has the potential to cause protease denaturation and dysfunction.
8.2.3 Specificity of Proteases
The substrate specificity of proteases affects the rate of aminolysis/hydrolysis. An acyl ester donor is recognized as a substrate at the active sites of proteases, and acyl-proteases are formed. Therefore, the affinity of acyl esters to proteases is the most important in forming acyl-protease intermediates. Furthermore, the affinity of amine nucleophilic groups to the catalytic site depends on the species of amino acids. Fastrez and Fersht reported a comparison of the deacylation rate of Acyl-Phe-chymotrypsin.[34] The rate of aminolysis with l-glycine amide (Gly-NH2) was 11 times faster than that of hydrolysis with H2O, whereas the rate of aminolysis with l-alanine amide (Ala-NH2) was 44 times faster than that of hydrolysis with H2O. This result means that the affinity of Ala-NH2 to chymotrypsin is higher than that of Gly-NH2 to chymotrypsin. The substrate specificity of proteases can be evaluated by the partition constant p. The partition constant p is calculated from the reaction rates of hydrolysis and aminolysis. Schellenberger reported on the partition constant of nucleophilic amino acids using maleyl-(3-carboxyacryloyl)-Phe-chymotrypsin as an acyl-protease [35]. The order of nucleophilicity toward the protease is Arg-NH2 > Leu-NH2 > Val-NH2 > Ala-NH2 > Gly-NH2 > H2O. Since the affinity of amino acids to proteases affected the reaction rate of aminolysis, the yields and degree of polymerization (DP) of the polypeptides depend on the selection of proteases and amino acids.
Proteases have some subsites that align on both sides of the catalytic site, inducing substrate specificity [36, 37]. These subsites determine the cleavage point of hydrolysis and the reaction rate of aminolysis. Schechter and Berger reported the subsite of papain [36]. The size of the binding site of papain was estimated using the oligo(l-Ala) with or without d-Ala; the existence of d-Ala reduces the rate of hydrolysis. Based on this experimental result, the size of the papain subsite was estimated at over 25 Å. Furthermore, they proposed that there are four subsites (S1–S4) located on one side and three subsites (S1′–S3′) located on the other (Fig. 8.4), and each site is able to accommodate one amino acid residue of a substrate (P1–P4 and P1′–P3′). Peptide bonds far from the d-Ala residue were labile to papain-catalyzed hydrolysis, whereas peptide bonds next to the d-Ala residue (P1 and P1′ position) hardly reacted. This experimental result indicates that the chirality of amino acid substrates is most importantly recognized in S1 and S′1. On the other hand, d-residues are acceptable to some extent in occupying the subsites away from the catalytic center. Furthermore, they concluded that active site S2 tends to bind the hydrophobic amino acids, such as Phe, Leu, and Val. In the case of enzymatic polymerization, this specificity affects the efficiency of the reaction. Detailed insights into the specificity of subsites in each protease are summarized in Sect. 8.4.

Schematic illustration of the catalytic site (black triangle) and subsites (denoted as Sn and Sn′) in the papain active site. Each subsite can accommodate an amino acid residue in polypeptide substrates (denoted as Pn and Pn′), and subsites S1 and S1′ strictly regulate the stereospecificity of oligoalanine substrates. Peptide bonds next to the l-alanine residue are preferred for hydrolytic cleavage compared to the bonds next to the d-alanine residue [36]
8.3 General Synthetic Procedure for Enzymatic Polymerization
The homopolymerization and copolymerization of α-amino acids by kinetically controlled enzymatic polymerization have been reported by many research groups. The general procedure of protease-catalyzed polypeptide synthesis is as follows:
-
1. C-terminal-activated amino acid is dissolved in a buffer solution.
-
2. A solution of protease in buffer solution is added.
-
3. The mixture is stirred at a certain temperature for several hours.
-
4. After the reaction, the protease is removed, and the polypeptide is isolated.
Amino acid esters or oligopeptide esters [38], such as methyl, ethyl, and benzyl ester derivatives, have been used as a monomer for enzymatic homo- [39,40,41,42,43] and copolymerization [44, 45]. To maintain pH conditions, aqueous buffer solutions are used as a reaction media. A suitable range of pH for protease function is 5–9. For example, phosphate, carbonate, and citrate buffers have been used as a reaction solvent. Generally, 25–60 °C is suitable for enzymatic polymerizations because of proteases’ thermal stability. The final concentration of amino acid substrate needs to be high enough to overcome the hydrolysis reaction competing with aminolysis.
After the reaction, there are several methods to isolate the resultant polypeptides from the polymerization solution. If the polypeptides are insoluble in the polymerization solution, we can obtain the peptides as a precipitate. In this case, the precipitate is washed with water to remove the remaining proteases and unreacted monomers. On the other hand, when the polypeptides are soluble in the polymerization solution, the polypeptides, proteases, unreacted monomers, and by-products are dissolved in the reaction solution. Thus, an additional isolation process is required to separate the polypeptides from other components. If the molecular weight of the polypeptides, which is generally less than 5000 in enzymatic polymerization, is much smaller than that of the protease, the protease can be removed from the mixture by centrifuge ultrafiltration with a suitable molecular weight cutoff (MWCO) membrane filter. Qin et al. isolated polyLys and N ε-protected polyLys with different solubilities in a buffer solution with the following procedures [23]. In the case of water-soluble polyLys, the mixture was centrifuged using a commercially available centrifugal filter with a 3000 MWCO membrane, and then the filtrate was dialyzed to isolate the polyLys product. In contrast, in the case of water-insoluble N ε-protected polyLys, the precipitate was easily collected by simple centrifugation of the reaction mixture, and purification was done by washing with a pH = 5 diluted HCl solution three times.
8.4 Enzymes Available for Enzymatic Polypeptide Synthesis
The enzymes used in enzymatic polypeptide synthesis include cysteine proteases (papain and bromelain), serine proteases (α-chymotrypsin, proteinase K, and trypsin), and metalloproteases (thermolysin). In addition to these proteases, enzymes with distinct properties (carboxypeptidase Y, d-aminopeptidase, alkaline d-peptidase, and lipase) are also available. The choice of the enzyme is one of the key factors in successful polypeptide synthesis because each enzyme has its own substrate specificity and optimal condition for catalytic activity.
8.4.1 Papain
Papain is found in papaya fruit (Carica papaya) and is one of the most investigated plant cysteine proteases in terms of structure-function relationships. It consists of a single polypeptide chain with 212 amino acid residues (23,429 Da). The three-dimensional structure exhibits two domains with a V-shaped cleft at which substrates can bind (Fig. 8.5). On the top of the V-shaped cleft, Cys25 and His159 form the catalytic diad. Although Cys25 and His159 are essential for catalysis, additional residues, including Gln19 [46], Asn175 [47], and Trp177 [48], have been suggested to play an important role in catalytic activity. Schechter and Berger proposed that the papain active site contains seven subsites as described above (Fig. 8.4) [36]. Two decades later, Turk and co-workers redefined five subsites (S1–S3 and S1′–S2′) [49]. The subsites are located on both sides of the catalytic site, three on the N-terminal side (S1–S3) and two on the C-terminal side (S1′–S2′). Substrate specificity is mainly determined by the S2 subsite, which prefers hydrophobic amino acid residues, such as Val, Phe, Tyr, and Leu [50]. Although the S1 subsite exhibits some preference for Arg and Lys [51], the other sites lack the selectivity for amino acid residues of the substrate. In addition, the subsites (except for S1 and S1′) have been shown to accept d-amino acid residues [36]. Taken together, papain favors substrates possessing hydrophobic residues at the P2 position, but it exhibits broad substrate specificity.

(a) Crystal structure of papain (PDB: 1PPN), (b) a 90 °C-rotated view of (a), (c) crystal structure of papain with a peptide-like substrate (inhibitor, succinyl-QVVAA-p-nitroanilide) (PDB: 1PIP), (d) an enlarged view of the boxed area in (b)
In the field of enzymatic polypeptide synthesis, papain has been widely used owing to its availability at low cost and broad substrate specificity. To date, papain has successfully catalyzed the polymerization of various α-amino acid derivatives. Papain-catalyzed polymerization has often been performed at 40 °C in alkaline buffer solution to obtain a high yield and DP of peptide products. Geng and co-workers examined the papain-catalyzed polymerization of a side chain-protected l-Glu ethyl ester (γ-ethyl l-Glu-OEt) at various pH to elucidate the relationship between reaction pH and product yield [40]. This study indicated that the suitable pH range was between 5.5 and 8.0, giving an over 60% product yield and an average DP (DPavg) of 8.7 ± 0.3 (determined by 1HNMR). Since papain maintains its catalytic activity in the presence of organic solvents, water-miscible cosolvents can be applied to papain-catalyzed polymerization of poorly water-soluble amino acid substrates. The polymerization of l-Phe methyl ester (l-Phe-OMe) was achieved by papain in 0.25 M sodium phosphate buffer containing 10% dimethylsulfoxide, 20% methanol, and 18% acetonitrile, resulting in 38%, 50%, and 50% of the product yield, respectively [33].
The efficiency of papain-catalyzed polymerization (i.e., yield and DP of peptide products) can be influenced not only by reaction conditions (temperature, pH, and concentrations of the enzyme and substrate) but also by substrate properties. Schwab and co-workers investigated papain-catalyzed homopolymerizations using four hydrophobic amino acid methyl esters (l-Leu-OMe, l-Tyr-OMe, l-Phe-OMe, and l-Trp-OMe) as the substrate [43]. Based on the resulting yield and DP of each homopolymer, the reactivity of the substrates was classified as l-Tyr-OMe > l-Leu-OMe > l-Phe-OMe > l-Trp-OMe. Another comparative study using different l-Ala and Gly esters (methyl, ethyl, benzyl, and tert-butyl esters) revealed that the yield of poly(l-Ala) and polyGly increased in the order of tert-butyl (no polymer was obtained) < methyl < ethyl < benzyl esters [52]. The highest yield obtained from the benzyl esters was likely due to the higher affinity of papain toward aromatic substrates. Proper choice of the ester group can enhance substrate affinity toward papain, leading to efficient polymerization. In addition to the ester group, the side chain-protecting group can affect polymerization efficiency. Qin and co-workers reported the polymerization of side chain-protected l-Lys derivatives (N ε-Z-l-Lys-OMe and N ε-Boc-l-Lys-OMe) catalyzed by papain [23]. The resulting poly(N ε-Z-l-Lys) and poly(N ε-Boc-l-Lys) exhibited narrower molecular weight distributions and higher DPavg in comparison to the products obtained from the polymerization of unprotected l-Lys-OMe. The benefit of side chain protection of l-Lys is improved binding affinity for the S2 subsite, which prefers bulky hydrophobic side chains. Another benefit is the precipitation of products, which shifts the equilibrium toward product formation and decreases the propensity toward unfavorable reactions, such as transamidation and hydrolysis. Moreover, the precipitated products are easily isolated from the reaction media by centrifugation.
Reports on papain-catalyzed copolymerization of different amino acid esters have been increasing. Uyama and co-workers first demonstrated that papain successfully catalyzed the copolymerization of γ-ethyl-l-Glu-OEt with various amino acid esters, including l-Met-OMe, l-Ala-OEt, l-Leu-OEt, l-Phe-OEt, l-Tyr-OEt, l-Phe-OEt, and β-ethyl l-Asp-OEt [44]. Papain was also used to synthesize several random copolymers, such as poly(l-Glu-co-l-Cys) [53], poly(l-Tyr-co-l-Lys) [54], poly(l-Lys-co-l-Ala) [54], and poly(l-Ala-co-Gly) [52]. Another report showed papain-catalyzed binary and ternary copolymerization using four hydrophobic amino acid methyl esters (l-Leu-OMe, l-Tyr-OMe, l-Phe-OMe, and l-Trp-OMe) [43]. It was also reported that papain enabled the conversion of a dipeptide (l-Ala-Gly-OEt) monomer into a polypeptide with an alternating sequence [38]. From the examples introduced above, one can conclude that papain is the most versatile enzyme in enzymatic peptide synthesis and broad substrate specificity is its most important property. This broad specificity can contribute to product variety with respect to amino acid sequences.
8.4.2 Bromelain
Specific enzymes derived from the pineapple plant are known as bromelains. Among these enzymes, stem bromelain, which is present in plant stem extracts, is generally used for enzymatic peptide synthesis. Stem bromelain is referred to as bromelain in this chapter. Bromelain is a cysteine protease of the papain family. It is a glycosylated, monomeric protein of 24.5 kDa that contains seven Cys residues and most likely three disulfide bonds. The oligosaccharide component has been shown to provide enzymatic stability and tolerance to denaturants, including guanidine hydrochloride [55], urea [56], and sodium dodecyl sulfate (SDS) [57]. The enzyme’s secondary structure is maintained between pH 7–10, but the enzyme is irreversibly denatured above pH 10 [58]. Although bromelain exhibits broad substrate specificity similar to papain, bromelain has been reported to prefer polar amino acid residues at the P1 and P1′ positions of substrates [59]. Additionally, it shows high hydrolysis efficiency toward substrates containing Arg and His at P2 and Pro at P3 [51].
Qin and co-workers used four proteases (papain, bromelain, α-chymotrypsin, and trypsin) for poly(l-Lys) synthesis and compared their activity [60]. Among them, bromelain showed the highest value of both DPavg (4.1) and monomer conversion (84 ± 2%) by maintaining the reaction pH between 7.6 and 7.8. This study also indicated that bromelain was sensitive to moderate pH although it catalyzed polymerizations over a wide range of temperatures (10–40 °C). Monomer conversion varied from 23 ± 1% to 76 ± 2% and 46 ± 5% for pH values of 6, 7, and 8, respectively. DPavg also varied between 2.8 and 4.1 from pH 6 to 9. Interestingly, bromelain was shown to be more efficient than papain for poly(l-Lys) synthesis, even though both enzymes are known to exhibit high catalytic activity against substrates containing Lys at the Pl position. The different efficiency between these enzymes is explained by their different preferences for the Lys residue at the P2 position: bromelain shows a higher preference for Lys than papain does. This example suggests that bromelain is an efficient enzyme to catalyze the homopolymerization of polar amino acids, although further comparative studies using various amino acid substrates and proteases are needed.
Bromelain-catalyzed polymerization of hydrophobic l-Phe-OEt was shown to proceed in 20% MeOH-containing phosphate buffer and provided maximum product yields (40–45%) at pH 7–8 and 40 °C [33]. In another study, bromelain displayed relatively high catalytic ability for copolymerizations of l-Leu-OEt with γ-ethyl l-Glu-OEt compared to α-chymotrypsin and protease SG [45]. Moreover, the catalytic ability of bromelain was comparable to that of papain, which favors hydrophobic amino acid residues, such as Leu. These studies indicate that bromelain can efficiently catalyze the polymerization of hydrophobic amino acid substrates as well as polar ones.
8.4.3 α-Chymotrypsin
Chymotrypsin, a serine protease, is produced in an inactive form (chymotrypsinogen) by the acinar cells of the pancreas and then converted into its active form of α- or γ-type chymotrypsin. α-Chymotrypsin contains three separate polypeptide chains linked by five disulfide bridges. His57, Asp102, and Ser195, located at the entrance of a substrate-binding pocket (Fig. 8.6), form the catalytic triad that is essential for hydrolysis/aminolysis reactions. In addition to these three residues, Ser214 contributes to catalytic activity by forming hydrogen bonds with Asp102 and stabilizing the charge of the buried Asp102 [61]. Gly193 and Ser195 are also considered to be key residues for catalysis activity because they serve as the oxyanion hole that stabilizes a tetrahedral intermediate [62]. The substrate specificity of α-chymotrypsin originates from the P1–S1 interaction. S1 is a hydrophobic pocket with high specificity for aromatic (Phe, Trp, and Tyr) and bulky nonpolar (Met) side chains of the substrate [62].

(a) Crystal structure of α-chymotrypsin (PDB: 1YPH), (b) an enlarged view of the boxed area in (a)
In addition to cysteine proteases (papain and bromelain), serine proteases, such as α-chymotrypsin, can be applied to kinetically controlled peptide synthesis. In the 1950s, Brenner and co-workers found that α-chymotrypsin polymerizes the isopropyl esters of l-Met, l-Thr, l-Phe, and l-Tyr [63, 64]. Recently, α-chymotrypsin was used for the homopolymerization of l-Cys-OEt [27]. Since disulfide bond formation and ester hydrolysis simultaneously occurred for l-Cys-OEt under an unfrozen condition, polymerization was performed under a frozen condition at −20 °C. α-Chymotrypsin-catalyzed polymerization successfully provided poly(l-Cys) with DP ranging from 6 to 11, although papain did not catalyze the polymerization of l-Cys-OEt at −20 °C. In another report, α-chymotrypsin was reported to catalyze the rapid conversion of l-Lys-l-Leu-OEt to poly(l-Lys-alt-l-Leu) at pH 8.5, whereas l-Lys-l-Leu-OEt polymerization by papain resulted in random sequence oligopeptides [65]. This finding indicated that sequence-controlled polymerization was achieved by α-chymotrypsin but not by papain. α-Chymotrypsin can be an alternative catalyst for enzymatic peptide synthesis, particularly when substrates contain a thiol group that inhibits the catalytic activity of cysteine proteases.
8.4.4 Proteinase K
Proteinase K is a serine protease and the main proteolytic enzyme produced by the fungus Tritirachium album Limber [66]. The designation “K” was chosen to indicate that it can even hydrolyze native keratin [67]. Proteinase K is composed of a single polypeptide chain with 278 amino acids (MW 28,930). It has a catalytic triad (Asp39, His69, and Ser224) and requires calcium ions for its full activity [68]. One characteristic of proteinase K is its broad substrate specificity, although its S1 subsite prefers aromatic and hydrophobic amino acids [69]. Another feature is that proteinase K retains its activity at relatively high temperatures and in the presence of denaturants, including urea, 0.5% (w/v) SDS, and 1% (w/v) Triton X (surfactant based on 4-octylphenol ethoxylates) [70]. These easy-to-use features make proteinase K one of the most widely used proteases in molecular biology.
Ageitos et al. first investigated proteinase K-catalyzed synthesis of poly(l-Phe) [71]. The highest DPavg of peptide products (12 ± 0.5) was obtained from the polymerization performed with 0.6 M l-Phe-OEt and 1.0 mg/mL proteinase K in sodium phosphate buffer (pH 8.0) at 40 °C for 3 hours. Furthermore, a star-shaped polypeptide was successfully synthesized by the proteinase K-catalyzed polymerization of l-Phe-OEt with a trifunctional terminal modifier, tris(2-aminoethyl)amine (TREN) [71]. Other star-shaped polypeptides were also constructed by proteinase K [72]. The star-shaped polypeptides consist of the three-armed TREN center and cationic polypeptide branches of poly(l-Lys), poly(l-Arg), or poly(l-Lys-co-l-Arg). Proteinase K was shown to catalyze copolymerization of l-Cys-OEt with l-His-OEt or γ-ethyl l-Glu-OEt. The resultant poly(l-Cys-co-l-His) and poly(l-Cys-co-γ-ethyl-l-Glu) exhibited DPavg values of 7.3 ± 1.1 and 9.5 ± 0.6, respectively [73]. So far, reports on proteinase K-catalyzed polymerization are limited compared to those on papain. This limitation might be due to the relatively higher cost of proteinase K compared to papain.
8.4.5 Trypsin
Trypsin is a serine protease that preferentially cleaves peptide bonds after Arg or Lys residues of substrates. It has a catalytic triad consisting of His57, Asp102, and Ser195. Trypsin strongly prefers substrates that possess an Arg or Lys residue at the P1 position [74], whereas it exhibits decreased catalytic activity for substrates with an Arg, Ile, Leu, Lys, or Phe residue at P2 and Pro at P3 [50]. The residue at P4 does not affect activity [75]. There are a few reports on enzymatic polypeptide synthesis catalyzed by trypsin. Poly(l-Arg) was synthesized by trypsin-catalyzed polymerization [76]. More than 40% of the substrate was converted into l-Arg-l-Arg within 1 h at 25 °C in 0.2 M carbonate buffer (pH 10). Trypsin was also shown to catalyze the polymerization of l-Lys-OEt [77]. The resultant poly(l-Lys) ranged from a dimer to an octamer. The highest monomer conversion was 70.7%, which was afforded by an optimal reaction condition (1 mM trypsin, 200 μM l-Lys-OEt, pH 10, 25 °C, 2 h). Interestingly, an addition of NaCl into the reaction mixture enhanced monomer conversion by 30% without affecting the polymerization degree, although the reasons for this enhancement were unclear. Trypsin is useful for the polymerization of polar substrates, including Arg and Lys, since it shows relatively high specificity against such residues.
8.4.6 Thermolysin
Thermolysin is a thermostable zinc metalloproteinase isolated from Bacillus thermoproteolyticus Rokko [78]. In addition to the Zn2+ ion, this enzyme binds four Ca2+ ions for its thermal stability [79]. The overall structure is divided into an N-terminal beta-sheet-rich domain and a C-terminal alpha-helix-rich domain. The two domains are spanned by a central alpha-helix located at a cleft running across the middle of the molecule. The central helix contains a zinc-binding HEXXH motif that is essential for hydrolysis activity. In the HEXXH motif, the two histidine residues coordinate with a Zn2+ ion, and the glutamate residue plays an important role in the hydrolysis reaction [80].
Although it is very rare to exploit thermolysin as a catalyst for amino acid polymerization, this enzyme has been applied to the industrial-scale synthesis of Z-l-Asp-l-Phe-OMe, the precursor of an artificial sweetener, aspartame [81]. Owing to this successful example, thermolysin has been extensively studied in terms of its catalytic ability for condensation reactions affording oligopeptides [82,83,84,85]. Wayne and Fruton investigated the substrate specificity of thermolysin in the condensation reaction [86]. Substrates with Phe or Leu at the P1′ position were efficiently condensed into oligopeptides by thermolysin. Additionally, the presence of aromatic residues (Phe and Trp) at P1 and hydrophobic residues (Ala, Met, and Phe) at P2 enhanced the condensation efficiency. Since thermolysin-catalyzed condensation proceeds through a thermodynamically controlled mechanism, the reaction conditions must be optimized to shift the equilibrium toward peptide bond formation. Kitaguchi and Klibanov have proposed an improved cosolvent system consisting of 90% organic solvent (tert-amyl alcohol), 9% water mimic (formamide or ethylene glycol), and 1% water that provides a high yield of peptide product [87]. Another approach is non-covalent immobilization of thermolysin by absorption on a porous support material (Celite) in a hydrophobic organic solvent, such as toluene [88]. The Celite-immobilized enzyme maintained its catalytic activity even in hydrophobic organic solvents owing to an aqueous phase around the Celite surface, thereby enabling highly efficient peptide condensation. Ulijn and co-workers have employed thermolysin for SPPS in which an amine substrate was linked to a solid support [89]. The immobilized amine substrate contributes to an equilibrium shift toward condensation, and its hydrophobicity is critical for efficiency [90]. One advantage of this strategy is that the reactions can achieve high conversion efficiency in bulk aqueous solution without needing an organic cosolvent and activated carboxylic acid. Although thermolysin has often been used for condensation reactions in previous studies, polymerization catalyzed by this enzyme is also interesting.
8.4.7 Others
In addition to the proteases described above, several enzymes with distinct properties are of interest as a catalyst for enzymatic polypeptide synthesis. For example, exopeptidases cleaving only the C-terminal amide bond are distinguishable from endo-type proteases, such as papain, which digest amide bonds within the polypeptide backbone. Exopeptidases, including carboxypeptidase Y (CPDY) [91], are expected to prevent unexpected hydrolysis of polypeptide backbones during polymerization, leading to a narrow molecular weight distribution of products. Indeed, CPDY-catalyzed polymerization successfully provided poly(l-Leu) with a relatively narrow molecular weight distribution [92]. Another example is d-stereospecific peptidase, which recognizes d-amino acid residues of substrates. Owing to its specificity for d-amino acids, d-stereospecific peptidase can be applied to the synthesis of peptides consisting of d-amino acids. Komeda and Asano demonstrated poly(d-Phe) synthesis catalyzed by alkaline d-peptidase from Bacillus cereus [93]. Synthesis of poly(d-Ala) was also achieved by immobilized d-aminopeptidase from Ochrobactrum anthropi in organic solvents [94]. Polymerization of amino acid esters can also be catalyzed by hydrolases, such as lipase. Lipase-catalyzed polymerization of l-Asp diethyl ester afforded high molecular weight polypeptides with a high yield, although peptide bond formation proceeded less regioselectively compared to protease-catalyzed polymerization [95, 96]. These distinct enzymes can be utilized as a special tool to provide novel peptides that are difficult to synthesize by commonly used proteases.
8.5 Amino Acid Esters for Enzymatic Polymerization
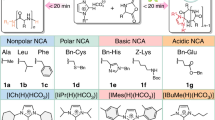
To date, there have been a great number of reports on the synthesis of polypeptides via enzymatic polymerization of various amino acid derivatives, especially amino acid esters. The amino acids used in enzymatic polymerization can be roughly classified into six different types in terms of their side groups, namely, hydrophobic, aromatic, acidic, basic, neutral, and unnatural amino acids (Fig. 8.7). These amino acids differ in their chemical and physical properties, and hence, enzymatic polymerization conditions differ for each amino acid.

Classification of natural amino acids based on their side group characteristics
8.5.1 Hydrophobic Amino Acids
Hydrophobic amino acids are appropriate substrates for enzymatic polymerization using proteases with a relatively high affinity for hydrophobic amino acids. One great advantage to using hydrophobic amino acids for enzymatic polymerization is that the resulting polypeptides precipitate from water due to their hydrophobicity. This precipitation is useful for easy isolation of the polypeptides obtained by simple centrifugation or filtration. The feature also restricts further chain elongation and hydrolysis of the obtained polypeptides after precipitation. The molecular weight of the obtained polypeptides is up to 3000 Da. However, fine-tuning of the reaction parameters, such as temperature and pH, can improve further polypeptide elongation.
There have been many reports on the enzymatic polymerization of hydrophobic amino acid esters, such as Ala and Leu. As a pioneering work of enzymatic polymerization, enzymatic synthesis of poly(l-Leu) was reported by Dannenberg and Smith using proteinase I from the bovine lung [97]. They found that the yield of polypeptide products remarkably relied on the pH of the reaction medium, which was monitored by tracking amine consumption using a ninhydrin assay. Poly(l-Leu) was also synthesized by the polymerization of l-Leu methyl ester using papain, and the average DP of poly(l-Leu) was 8–9 [98]. They revealed that the reaction proceeded with a pronounced induction period at a low substrate concentration before the precipitation of poly(l-Leu). The induction period can be shortened by the addition of a corresponding dimer or trimer, which implies that dimerization rate is very slow and that chain growth is rapid from the trimer onward. This finding means that chain elongation proceeds stepwise.
Ala derivatives are another hydrophobic amino acid monomer used in enzymatic polymerization. Poly(l-Ala) tends to adopt water-insoluble β-sheet structures, which facilitate rapid precipitation during enzymatic polymerization. Poly(l-Ala) can be readily synthesized by papain-catalyzed polymerization of l-Ala ethyl ester [42]. The reaction parameters influence the resulting poly(l-Ala) chain length to a certain extent. This influence was investigated by the papain-catalyzed polymerization of l-Ala ethyl ester in two different pH buffer solutions (i.e., 1 M sodium phosphate buffer at pH 7.0 and 1 M sodium carbonate buffer at pH 11.0). A higher yield (67.1%) was obtained at pH 7.0 than at pH 11.0 (35.7%). Interestingly, maximal DP increased at pH 11.0 (DPmax = 16) compared to that obtained at pH 7.0 (DPmax = 11) as determined by MALDI-TOF mass spectrometry. The resultant poly(l-Ala) formed characteristic microfibrils with the predominantly β-sheet structure, which was confirmed by atomic force microscopy and infrared (IR) spectroscopy.
As a highly promising structural material, silk fibers from animal species, such as silkworms and spiders, have attracted a wide range of attention from the scientific community due to their extraordinary strength and toughness. Silk proteins have a relatively longer repetitive domain between their conserved N- and C-terminal domains. In spider dragline silk proteins, an oligo(l-Ala) sequence in the repetitive domain forms β-sheet structures, whereas a glycine-rich domain forms random coil and helical structures. The higher-order structures constructed by these domains in silk fibers realize the excellent mechanical property of silk fibers in nature. Polypeptides mimicking spider silk proteins were synthesized utilizing enzymatic polymerization as shown in Fig. 8.8 [99]. The β-sheet-forming poly(l-Ala) motif was prepared by papain-catalyzed polymerization. The DP of poly(l-Ala) was controlled by adding poor organic solvents, such as methanol, in aqueous media to match the oligo(l-Ala) sequence length in spider silk proteins. On the other hand, a more flexible glycine-rich motif was prepared by random copolymerization of Gly and l-Leu esters using papain. This poly(Gly-r-l-Leu) acts as a “soft” motif expressed between the “hard” polyAla motifs in spider silk proteins. The post-polycondensation of these hard and soft polypeptide motifs was accomplished using polyphosphoric acid (PPA) as a condensing agent at an elevated temperature. The resulting multiblock polypeptide, poly(l-Ala)-b-poly(Gly-r-l-Leu), adopted characteristic secondary structures and crystallinity based on the polyAla and Gly-rich sequences similar to natural spider silks. This chemically ligated tandem sequence is a biomimetic polypeptide of spider dragline silks, proving that enzymatic polymerization is greatly applicable to the synthesis of polypeptide motifs in structural proteins.

(a) Synthesis of multiblock polypeptides inspired by spider dragline silk proteins via enzymatic polymerization followed by polycondensation, (b)enzymatic polymerization of l-AlaGly dipeptide ethyl ester affording alternating AlaGly sequence, (c) random copolymerization of l-Ala and Gly benzyl esters catalyzed by papain
Another β-sheet-forming peptide motif is GAGAGX, which is seen in Bombyx mori silkworm silk proteins. Two strategies were used to synthesize polypeptides similar to the GAGAGX motif (Fig. 8.8). Qin et al. used a dipeptide ester l-alanylglycine ethyl ester (l-AlaGly–OEt) to obtain a polypeptide with a strictly alternating sequence poly(l-Ala-alt-Gly) via enzymatic polymerization [38]. Stoichiometrically equivalent quantities of alternating l-Ala and Gly units were obtained, which was confirmed by MALDI-TOF mass spectrometry. To obtain alternating sequences of polypeptides, dipeptide esters can be used as monomers in enzymatic polymerization. On the other hand, Ageitos and co-workers used the strategy of copolymerization of l-Ala and Gly esters for the synthesis of polypeptides containing motifs similar to GAGAG [52]. This technique cannot control the exact sequence but can fine-tune the Gly/Ala content of the resulting polypeptides. Papain shows less affinity for Gly than Ala, resulting in a significant mismatch between Gly and Ala reactivities. To overcome this mismatch, l-Ala and Gly benzyl esters with similar reactivities were used for papain-catalyzed copolymerization. By changing the feed ratio of l-Ala/Gly benzyl esters, the compositions of l-Ala/Gly in the copolypeptides could be controlled. The resulting polypeptides differed in secondary structures and thermal stabilities depending on their l-Ala content.
8.5.2 Aromatic Amino Acids
Aromatic amino acids also exhibit a high affinity for proteases because of their hydrophobic aromatic side group. These aromatic amino acid esters are good substrates for enzymatic polymerization, but rapid precipitation usually causes a low DP for the resulting polypeptides. Enzymatic polymerization of poly(l-Phe) was reported by Dannenberg and Smith using proteinase I from the bovine lung [97]. The DPavg of the obtained poly(l-Phe) was 6. Gross and co-workers reported enzymatic synthesis of poly(l-Phe) using bromelain in various reaction conditions [33]. Ageitos et al. reported enzymatic polymerization of l-Phe esters using proteinase K [71]. In aqueous solutions, the obtained linear poly(l-Phe) showed a unique self-assembly property. In addition to linear poly(l-Phe), three-armed, star-shaped oligoPhe was synthesized by polymerization of l-Phe ethyl ester with tris(2-aminoethyl)amine. This star-shaped poly(l-Phe) self-assembled to form fibrous networks.
Tyrosine is an aromatic amino acid with a relatively polar side group. The phenol moiety of Tyr is polar and reactive; therefore, polypeptides containing Tyr as a monomer unit can be easily functionalized by further chemical modification. Adhesive polypeptides from the copolymerization of l-Tyr and l-Lys ethyl esters were developed using enzymatic polymerization, which was inspired by a natural adhesive protein, foot protein 5 (Mefp-5), produced by a blue mussel (Mytilus edulis) [54]. This polymerization was achieved by two-step synthesis (Fig. 8.9). First, copolypeptides of l-Tyr and l-Lys units were synthesized by papain-catalyzed polymerization, and then, enzymatic conversion of l-Tyr to l-3,4-dihydroxyphenylalanine (DOPA) was carried out using tyrosinase. The poly(l-Tyr-r-l-Lys-r-l-DOPA) with a Tyr/Lys/DOPA ratio of 50/25/25 at pH 12 showed excellent adhesiveness (adhesion strength, 0.95 MPa; Young’s modulus, 6 MPa), and this adhesiveness was higher than that of a commercially available superglue as determined by an adhesive lap joint shear strength test using mica sheets. Lampel et al. demonstrated that another functional polypeptide material, tyrosine-containing tripeptides prepared by enzymatic synthesis, acted as a polymeric pigment with tunable coloration ability [100].

Two-step enzymatic synthesis of adhesive polypeptides consisting of l-Tyr, l-Lys, and l-DOPA residues
8.5.3 Acidic Amino Acids
Glutamic acid and aspartic acid are acidic and hydrophilic amino acids since they contain a carboxylic acid residue in their side chain. There are no reports of successful enzymatic polymerization of Glu and Asp monomers with a free carboxylic acid side group. This lack of successful polymerization is probably because the carboxylic acids interact with the catalytic center of proteases by undesired proton transfer. However, the conversion of carboxylic acid to its corresponding ester causes a polarity change, causing the amino acid to become hydrophobic, which allows Glu to be a good substrate. Indeed, l-Glu diethyl ester is frequently used for enzymatic polymerization using various proteases. The polymerization of l-Glu diethyl ester to give poly(γ-ethyl l-glutamate) [poly(l-Glu-OEt)] using proteases, including papain, bromelain, and α-chymotrypsin, was reported by Uyama et al. (Fig. 8.10) [44]. Poly(l-Glu-OEt) was generated as white precipitate, and the average DP was estimated at approximately 9 by 1H NMR spectroscopy. The polymerization of γ-methyl l-glutamate, which possesses an ethyl ester group only at the side chain, did not proceed under similar reaction conditions using papain. This fact revealed that protease-catalyzed polymerization regioselectively proceeds to generate an α-peptide linkage in the resulting polymer backbone. Copolymerization of l-Glu diethyl ester with l-Met methyl ester, l-Ala ethyl ester, l-Leu ethyl ester, l-Phe ethyl ester, l-Tyr ethyl ester, and l-Asp diethyl ester proceeded using papain, providing various random copolypeptides.

(a) Papain-catalyzed polymerization of l-Glu diethyl ester with remarkable regioselectivity, (b) enzymatic polymerization of l-Asp diethyl ester using various enzymes
The other acidic amino acid, aspartic acid, is an analog of glutamic acid with only one less methylene group. However, the reactivities of Glu and Asp in enzymatic polymerization are quite different. Geng et al. reported that papain-catalyzed polymerization of l-Glu diethyl ester in 0.9 M phosphate buffer for 10 min resulted in poly(l-Glu-OEt) at 80% yield [40]. In contrast, l-Asp diethyl ester was not polymerized in the presence of papain, probably because of the poor affinity of papain for Asp substrates. Matsumura et al. reported that polymerization of l-Asp diethyl ester by alkalophilic proteinase from Streptomyces sp. at a temperature between 4 and 50 °C yielded poly[(β-ethyl α-l-aspartate)-co-(α-ethyl β-l-aspartate)] [101]. Soeda et al. later polymerized l-Asp diethyl ester by a bacterial protease from Bacillus subtilis (BS) using an organic solvent at a temperature ranging from 30 to 50 °C (Fig. 8.10). The resulting α-linked poly(β-ethyl α-l-aspartate) [poly(l-Asp-OEt)] with a weight average molecular weight (M w) up to 3700 was obtained at 85% yield [102]. Lipase, a subclass of esterases that hydrolyzes lipids, can also catalyze the polymerization of Asp ester derivatives. Zhang et al. reported that lipase (Candida antarctica lipase B) catalyzed the polymerization of diethyl d- or l-Asp diethyl ester, providing poly(α-ethyl β-d-aspartate) or poly(α-ethyl β-l-aspartate) in a solvent-free condition, respectively. The DPavg was found to be 60 with up to 96% β-linkages, indicating that lipase provided dominantly amide bonds with β-linkages (Fig. 8.10) [95]. However, recently, Gross and co-workers reported the polymerization of l-Asp diethyl ester with improved regioselectivity using an immobilized lipase at 80 °C for 24 h [96]. The reaction successfully yielded approximately 95% α-linked poly(l-Asp-OEt) with a 70% yield, and the DPavg was approximately 50. They concluded that polymerization proceeds in a controlled manner by a chain-growth mechanism with up to 90% conversion, and then, a step-growth mechanism competes with the chain-growth mechanism.
8.5.4 Basic Amino Acids
Amino acids possessing a basic side group, such as l-Lys, l-Arg, and l-His, fall in this category. No protection of the amine group in the side chain is required for the enzymatic polymerization of these amino acids because of sufficient regioselectivity in protease-catalyzed polymerization. The resulting cationic polypeptides have been employed as cell-penetrating peptides and peptide carriers for gene delivery systems [103, 104]. Enzymatic synthesis of poly(l-Lys) from l-Lys ethyl ester in an aqueous medium using four proteases (papain, bromelain, α-chymotrypsin, and trypsin) was studied to determine their activity in Lys polymerization at pH values ranging from 6 to 11 [60]. Bromelain was found to be the most effective protease since it gave the highest yield and DPavg of poly(l-Lys). Qin et al. further explored the polymerization of N ε-protected l-Lys esters (Fig. 8.11). This protection of the ε-amino group improved the yield of the resulting N ε-protected poly(l-Lys) by enhancing the hydrophobicity of the polymer, which facilitated a water-insoluble precipitate [23]. The protecting groups of the ε-amino group included Boc and Z groups. Poly(N ε-Boc-l-Lys) and poly(N ε-Z-l-Lys) resulted from enzymatic polymerization of the corresponding Lys monomers. These poly(l-Lys) with hydrophobic moieties were easily collected by centrifuging the precipitates.

(a) Enzymatic polymerization of l-Lys ethyl ester derivatives, (b) proteinase K-catalyzed copolymerization of l-His and l-Cys
Diblock and random polypeptides of l-Lys and l-Ala were synthesized by one-pot enzymatic polymerization using papain [105]. Characterization by 1H NMR spectroscopy and MALDI-TOF mass spectrometry revealed that the sequential addition of an l-Lys ethyl ester followed by an l-Ala ethyl ester resulted in the formation of diblock polypeptides. Observation by optical microscope clearly revealed that the crystal morphology of the copolymers was dependent on the monomer distribution in the copolymers and the pH of the solution. Diblock copolypeptides formed cubic or hexagonal crystals with a hollow structure at pH 3.0. This type of unique crystal with low cytotoxicity can be used as a biomedical material, e.g., as a carrier for drug delivery systems. Utilizing dipeptide esters as a monomer is another method to synthesize the copolypeptide, as reported by Gross and co-workers [38, 65]. An alternating copolypeptide of l-Lys and l-Leu units was enzymatically synthesized using α-chymotrypsin at pH 8.5 [65]. The resulting poly(l-Lys-alt-l-Leu) in the aqueous reaction solution underwent a sol-gel transition in which β-sheet structures assembled into a nanofibril network. This polypeptide hydrogel system can be a promising stimulus-responsive material that has potential in biomedical applications.
There have been a few reports on the enzymatic polymerization of Arg and His. The homo- and copolymerization of l-Arg ethyl ester was conducted using proteinase K in a condition similar to the polymerization of Lys esters [72]. Ma et al. reported the copolymerization of l-Cys and l-His ethyl esters with changing ratios of His/Cys using proteinase K (Fig. 8.11) [73]. The resulting copolypeptides with various His/Cys ratios were successfully obtained in 40–50% yields. Poly(l-His-co-l-Cys), inspired by a catalytic diad of cysteine proteases, exhibited protease-like activity to cleave an amide bond using fluorescein isothiocyanate-labeled casein as a substrate. This feature was utilized to peel off living cells from a culture plate to obtain a monolayer cell sheet.
8.5.5 Neutral Polar Amino Acids
Neutral amino acids include various side groups. This category focuses on amino acids with polar side groups rather than those with hydrophobic side groups, which were already described in the section on hydrophobic amino acids. Amino acids containing a sulfur in their side groups, namely, cysteine and methionine, play an important role in both natural proteins and synthetic polypeptides because of their specific functionality and availability as reactive residues for chemical modification. Cysteine has a reactive thiol group in the side chain that can be easily oxidized to form a disulfide bond. This feature hampers polypeptide formation during the polymerization process by undesired cross-linking of the polypeptide via disulfide bonds. Therefore, the thiol group of Cys must be protected prior to polymerization in conventional synthetic methods. Narai-Kanayama et al. reported that the poly(l-Cys) was synthesized by enzymatic polymerization of l-Cys ethyl ester using α-chymotrypsin in a frozen aqueous medium [27]. The thiol groups of the obtained polymer were intact during polymerization and purification, indicating that the polyCys was successfully synthesized without a tedious protection/deprotection process. The DP of the obtained polyCys ranges from 6 to 11 as confirmed by MALDI-TOF mass spectrometry. Ma et al. synthesized poly(l-Cys) by enzymatic polymerization using proteinase K, and the DP of the polyCys was as high as 17 [106].
Methionine is another sulfur-containing amino acid with a sulfide moiety in the side group. The sulfide group can be utilized for modification of the polypeptide by chemical reactions, such as oxidation and alkylation by electrophiles. One early example was the enzymatic synthesis of poly(l-Met) and its derivatization (Fig. 8.12) [107, 108]. Poly(l-Met) was synthesized from l-Met methyl ester by papain-catalyzed polymerization in a citrate buffer at pH 5.5. However, characterization of the product was limited due to the insolubility of the obtained poly(l-Met) in common solvents. The chemical conversion of sulfide groups in l-Met residues to sulfoxide or sulfone by treatment with acids increased the water solubility of the resulting polypeptide, poly(l-methionine sulfoxide). From 1H NMR spectroscopy, the DPavg was found to be up to 8.

Papain-catalyzed synthesis of poly(l-Met) and its oxidation to sulfoxide and sulfone derivatives
Serine and threonine, which possess a hydroxy group in their side chain, were used in enzymatic polymerization without protecting the hydroxy group, as demonstrated in early reports [63, 64]. To date, there are no reports on the enzymatic polymerization of glutamine and asparagine derivatives, although some short peptides containing these amino acids were synthesized by protease-catalyzed coupling reactions [109, 110].
8.5.6 Unnatural Amino Acids
Incorporation of unnatural motifs into the polypeptide backbone is an innovative method to fabricate various polypeptide materials with novel functionalities. The polymerization of unnatural amino acid esters using proteases was demonstrated in order to synthesize polypeptides containing unnatural structures, although proteases show poor substrate specificity for these substrates. Yazawa et al. demonstrated that ester derivatives of ω-aminoalkanoic acids, monomer units of nylon, were not polymerized alone using papain but were successfully introduced into polypeptides via papain-catalyzed copolymerization with natural amino acids, such as l-Leu and l-Glu esters, in a phosphate buffer [111, 112]. In general, polypeptides exhibit neither melting behavior nor glass transition because of intermolecular multiple hydrogen bonds, resulting in a lack of thermoplasticity. The insertion of nylon units into the polypeptide is assumed to drastically reduce intermolecular interactions via hydrogen bonds and induce melting behavior in the polypeptides. Papain-catalyzed copolymerization of l-Glu diethyl ester with various nylon monomers, including ethyl 3-aminopropionate, ethyl 4-aminobutyrate, and methyl 6-aminohexanoate, afforded polypeptides containing nylon units [111]. Only papain can polymerize copolymerization among many proteases due to its relatively broad substrate specificity. In the resulting polypeptides, one nylon unit was incorporated at the C-terminal of poly(l-Glu-OEt)s as evidenced by 1H NMR spectroscopy and MALDI-TOF mass spectrometry. On the other hand, in the case of the copolymerization of l-Leu and nylon esters using papain, approximately 15 mol% of the nylon unit was introduced into the poly(l-Leu) backbone [112]. The obtained copolymers showed a broad melting behavior at approximately 200 °C below the decomposition temperature. This result indicates that the introduction of artificial units, such as nylon, into the polypeptide backbone imparted thermoplasticity (Figs. 8.13 and 8.14).

Introduction of unnatural amino acid units in polypeptide backbones via enzymatic polymerization using (a) 4-aminobutyric acid (nylon) and (b) 2-aminoisobutyric acid (Aib)

Enzymatic synthesis of special polypeptide architectures; (a) three-armed, star-shaped poly(l-Phe), (b) telechelic poly(l-Ala)
Another example of an artificial amino acid is 2-aminoisobutyric acid (Aib), an α,α-disubstituted amino acid that has a tendency to form helical structures [113, 114]. Enzymatic polymerization of Aib ethyl esters resulted in no polymer formation since Aib is hardly recognized by the catalytic sites of proteases, even in copolymerization with natural amino acid esters. To overcome the incompatibility, the Aib unit was sandwiched in between l-Ala residues to form a tripeptide ester, which resulted in effective recognition by papain (Fig. 8.13b). Enzymatic polymerization of the Aib-containing tripeptide ester, l-Ala-Aib-l-Ala ethyl ester, proceeded smoothly in the presence of papain [115]. IR and circular dichroism (CD) analyses revealed that the obtained poly(l-Ala-Aib-l-Ala) adopted a helical conformation in both solid and solution states. The tripeptide strategy is an effective method to incorporate unnatural amino acids, which are poorly recognized by proteases, into polypeptides. Because the introduction of artificial synthetic amino acids into polypeptide backbones can tune their physical properties, it is useful to produce novel polypeptide materials with a distinctive property compared to natural polypeptides (proteins).
Using multifunctional monomers can afford polymers with a variety of special architectures. These include special structures, such as star-shaped polymers, hyperbranched polymers, polymer brushes, and cross-linked gels, that cannot be synthesized by biological methods. Two types of special polypeptides have been reported using enzymatic polymerization: three-armed, star-shaped polypeptides and telechelic polypeptides. The three-armed, star-shaped polypeptide was formed by polymerization of l-Phe ethyl ester in the presence of TREN catalyzed by proteinase K in an aqueous buffer [71]. The triamine TREN acts as a trifunctional terminal modifier at the C-terminus of the polypeptide. 1H NMR proved that the resulting polyPhe was a star-shaped polypeptide with a TREN core. Another three-armed star-shaped polypeptide consisting of l-Lys, l-Arg, or a copolymer of the two was prepared by proteinase K-catalyzed polymerization in the presence of TREN [72]. Cationic star-shaped polypeptides are frequently used as peptide/plasmid DNA complexes for gene delivery. The high efficiency of gene transfection into human embryonic kidney 293 cells was confirmed using these cationic star-shaped polypeptides.
Telechelic polypeptide architecture, where two polypeptide chains propagate from two initiation points in N- to C-terminal direction, was synthesized by enzymatic polymerization [116]. In this study, a novel telechelic bifunctional compound with an amino acid ester at both terminals was prepared, and then, papain-catalyzed enzymatic polymerization of Gly or l-Ala ethyl ester in the presence of this bifunctional compound was performed. The formation of telechelic poly(l-Ala) and polyGly was confirmed by 1H NMR spectroscopy and MALDI-TOF mass spectrometry. From atomic force microscopy observations, the crystals of telechelic poly(l-Ala) showed long nanofibrils with a high aspect ratio in contrast to the authentic linear poly(l-Ala), which assembled into granule-like crystals. Due to its self-assembling feature into a specific structure, telechelic poly(l-Ala) was utilized as a reinforcing agent for structural materials, such as regenerated silk fibroin films [117, 118].
8.6 Perspective
Enzymatic polymerization of amino acid derivatives using proteases is a powerful way to synthesize various types of polypeptides with a green synthetic protocol. The broad structural versatility of enzymatically synthesized polypeptides in recent studies is beneficial for realizing polypeptide materials in our forthcoming sustainable society. Further improvement of this technique is inevitable for practical applications of polypeptide materials. It should be noted that the molecular weight of the obtained polypeptides remains in an oligomeric range except for some examples. On the other hand, enzymatic polymerization of some amino acids, such as proline and glutamine, has not yet been achieved, which hampers the design of specific sequences of polypeptides. Developing more efficient polymerization systems will allow us to control the synthesis of well-defined polypeptides with high molecular weight and fine-tune amino acid sequences.
References
Merrifield B (1986) Solid phase synthesis. Science 232(4748):341–347
Deming TJ (2007) Synthetic polypeptides for biomedical applications. Prog Polym Sci 32(8–9):858–875
McGrath KP, Fournier MJ, Mason TL et al (1992) Genetically directed syntheses of new polymeric materials. Expression of artificial genes encoding proteins with repeating -(AlaGly)3ProGluGly- elements. J Am Chem Soc 114(2):727–733
Zhang G, Fournier MJ, Mason TL et al (1992) Biological synthesis of monodisperse derivatives of poly(α,l-glutamic acid): model rodlike polymers. Macromolecules 25(13):3601–3603
Bordusa F (2002) Proteases in organic synthesis. Chem Rev 102(12):4817–4867
Yazawa K, Numata K (2014) Recent advances in chemoenzymatic peptide syntheses. Molecules 19(9):13755
Numata K (2015) Poly(amino acid)s/polypeptides as potential functional and structural materials. Polym J 47(8):537–545
Tsuchiya K, Numata K (2017) Chemoenzymatic synthesis of polypeptides for use as functional and structural materials. Macromol Biosci 17(11):1700177
Jakubke HD, Kuhl P, Könnecke A (1985) Basic principles of protease-catalyzed peptide bond formation. Angew Chem Int Ed Engl 24(2):85–93
Morihara K (1987) Using proteases in peptide synthesis. Trends Biotechnol 5(6):164–170
Bongers J, Heimer EP (1994) Recent applications of enzymatic peptide synthesis. Peptides 15(1):183–193
Kumar D, Bhalla TC (2005) Microbial proteases in peptide synthesis: approaches and applications. Appl Microbiol Biotechnol 68(6):726–736
Guzmán F, Barberis S, Illanes A (2007) Peptide synthesis: chemical or enzymatic. Electron J Biotechnol 10(2):279–314
Yagasaki M, Hashimoto S (2008) Synthesis and application of dipeptides; current status and perspectives. Appl Microbiol Biotechnol 81(1):13–22
Chen F, Zhang F, Wang A et al (2010) Recent progress in the chemo-enzymatic peptide synthesis. Afr J Pharm Pharmacol 4(10):721–730
Białkowska AM, Morawski K, Florczak T (2017) Extremophilic proteases as novel and efficient tools in short peptide synthesis. J Ind Microbiol Biotechnol 44(9):1325–1342
Carpenter FH (1960) The free energy change in hydrolytic reactions: the non-ionized compound convention. J Am Chem Soc 82(5):1111–1122
Homandberg GA, berg MJA et al (1978) Synthesis of peptide bonds by proteinases. Addition of organic cosolvents shifts peptide bond equilibria toward synthesis. Biochemistry 17(24):5220–5227
Cassells JM, Halling PJ (1989) Low- water organic two-phase systems and problems affecting it. Biotechnol Bioeng 33:1489–1494
Halling PJ (1994) Thermodynamic predictions for biocatalysis in nonconventional media: theory, tests, and recommendations for experimental design and analysis. Enzym Microb Technol 16(3):178–206
Deschrevel B, Vincent JC, Ripoll C et al (2003) Thermodynamic parameters monitoring the equilibrium shift of enzyme-catalyzed hydrolysis/synthesis reactions in favor of synthesis in mixtures of water and organic solvent. Biotechnol Bioeng 81(2):167–177
Schellenberger V, Jakubke HDD (1991) Protease-catalyzed kinetically controlled peptide synthesis. Angew Chem Int Ed Engl 30(11):1437–1449
Qin X, Xie W, Tian S et al (2014) Influence of N ε-protecting groups on the protease-catalyzed oligomerization of L-lysine methyl ester. ACS Catal 4(6):1783–1792
Christensen U (1994) Effects of pH on carboxypeptidase-Y-catalyzed hydrolysis and aminolysis reactions. Eur J Biochem 220(1):149–153
Hansler M, Jakubke HD (1996) Reverse action of hydrolases in frozen aqueous solutions. Amino Acids 11:379–395
Jönsson Å, Wehtje E, Adlercreutz P (1997) Low reaction temperature increases the selectivity in an enzymatic reaction due to substrate solvation effects. Biotechnol Lett 19(1):85–88
Narai-Kanayama A, Hanaishi T, Aso K (2012) Α-chymotrypsin-catalyzed synthesis of poly-l-cysteine in a frozen aqueous solution. J Biotechnol 157(3):428–436
Kuhl P, Konnecke A, Doring G et al (1980) Enzyme-catalyzed peptide synthesis in biphasic aqueous-organic systems. Tetrahedron Lett 21:895–896
Eggers DK, Blanch HW, Prausnitz JM (1989) Extractive catalysis: solvent effects on equilibria of enzymatic reactions in two-phase systems. Enzym Microb Technol 11(2):84–89
Gaertner H, Puigserver A (1989) Kinetics and specificity of serine proteases in peptide synthesis catalyzed in organic solvents. Eur J Biochem 181(1):207–213
Nadim A, Stoineva IB, Galunsky B et al (1992) Mass transfer induced interchange of the kinetic and thermodynamic control of enzymic peptide synthesis in biphasic water-organic systems. Biotechnol Tech 6(6):539–544
Kisee H, Fujimoto K, Noritomi H (1988) Enzymatic reactions in aqueous-organic media. VI. Peptide synthesis by α-chymotrypsin in hydrophilic organic solvents. J Biotechnol 8(4):279–290
Viswanathan K, Omorebokhae R, Li G et al (2010) Protease-catalyzed oligomerization of hydrophobic amino acid ethyl esters in homogeneous reaction media using l-phenylalanine as a model system. Biomacromolecules 11(8):2152–2160
Fastrez J, Fersht AR (1973) Demonstration of the acyl-enzyme mechanism for the hydrolysis of peptides and anilides by chymotrypsin. Biochemistry 12(11):2025–2034
Schellenberger V, Jakubke H-D (1986) A spectrophotometric assay for the characterization of the S’ subsite specificity of α-chymotrypsin. Biochim Biophys Acta Protein Struct Mol Enzymol 869(1):54–60
Schechter I, Berger A (1967) On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27(2):157–162
Berger A, Schechter I (1970) Mapping the active site of papain with the aid of peptide substrates and inhibitors. Philos Trans R Soc B 257(813):249–264
Qin X, Khuong AC, Yu Z et al (2013) Simplifying alternating peptide synthesis by protease-catalyzed dipeptide oligomerization. Chem Commun 49(4):385–387
Fukuoka T, Tachibana Y, Tonami H et al (2002) Enzymatic polymerization of tyrosine derivatives. Peroxidase- and protease-catalyzed synthesis of poly(tyrosine)s with different structures. Biomacromolecules 3(4):768–774
Geng L, Vaidya A, Viswanathan K et al (2006) Rapid regioselective oligomerization of l-glutamic acid diethyl ester catalyzed by papain. Macromolecules 39(23):7915–7921
Viswanathan K, Li G, Gross RA (2010) Protease catalyzed in situ C-terminal modification of oligoglutamate. Macromolecules 43(12):5245–5255
Baker PJ, Numata K (2012) Chemoenzymatic synthesis of poly(l-alanine) in aqueous environment. Biomacromolecules 13(4):947–951
Schwab LW, Kloosterman WMJ, Konieczny J et al (2012) Papain catalyzed (co)oligomerization of α-amino acids. Polymers 4(1):710–740
Uyama H, Fukuoka T, Komatsu I et al (2002) Protease-catalyzed regioselective polymerization and copolymerization of glutamic acid diethyl ester. Biomacromolecules 3(2):318–323
Li G, Raman VK, Xie W et al (2008) Protease-catalyzed co-oligomerizations of l-leucine ethyl ester with l-glutamic acid diethyl ester: sequence and chain length distributions. Macromolecules 41:7003–7012
Ménard R, Carrière J, Laflamme P et al (1991) Contribution of the glutamine 19 side chain to transition-state stabilization in the oxyanion hole of papain. Biochemistry 30(37):8924–8928
Vernet T, Tessier DC, Chatellier J et al (1995) Structural and functional roles of asparagine 175 in the cysteine protease papain. J Biol Chem 270:16645–16652
Gul S, Hussain S, Thomas MP et al (2008) Generation of nucleophilic character in the Cys25/His159 ion pair of papain involves Trp177 but not Asp158. Biochemistry 47(7):2025–2035
Turk D, Guncar G, Podobnik M et al (1998) Revised definition of substrate binding sites of papain-like cysteine proteases. Biol Chem 379(2):137–147
Harris JL, Backes BJ, Leonetti F et al (2000) Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci 97(14):7754–7759
Choe Y, Leonetti F, Greenbaum DC et al (2006) Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem 281(18):12824–12832
Ageitos JM, Yazawa K, Tateishi A et al (2016) The benzyl ester group of amino acid monomers enhances substrate affinity and broadens the substrate specificity of the enzyme catalyst in chemoenzymatic copolymerization. Biomacromolecules 17(1):314–323
Viswanathan K, Schofield MH, Teraoka I et al (2012) Surprising metal binding properties of phytochelatin-like peptides prepared by protease-catalysis. Green Chem 14(4):1020–1029
Numata K, Baker PJ (2014) Synthesis of adhesive peptides similar to those found in blue mussel (mytilus edulis) using papain and tyrosinase. Biomacromolecules 15(8):3206–3212
Rasheedi S, Haq SK, Khan RH (2003) Guanidine hydrochloride denaturation of glycosylated and deglycosylated stem bromelain. Biochem Mosc 68(10):1097–1100
Ahmad B, Rathar GM, Varshney A et al (2009) pH-dependent urea-induced unfolding of stem bromelain: unusual stability against urea at neutral pH. Biochemistry (Mosc) 74(12):1337–1343
Bhattacharya R, Bhattacharyya D (2009) Resistance of bromelain to SDS binding. Biochim Biophys Acta, Proteins Proteomics 1794(4):698–708
Dave S, Mahajan S, Chandra V et al (2010) Specific molten globule conformation of stem bromelain at alkaline pH. Arch Biochem Biophys 499(1–2):26–31
Napper AD, Bennett SP, Borowski M (1994) Purification and characterization of multiple forms of the pineapple-stem-derived cysteine proteinases ananain and comosain. Biochem J 301(Pt3):727–735
Qin X, Xie W, Su Q et al (2011) Protease-catalyzed oligomerization of l-lysine ethyl ester in aqueous solution. ACS Catal 1(9):1022–1034
McGrath ME, Craik CS, Fletterick RJ et al (1992) Perturbing the polar environment of Asp102 in trypsin: consequences of replacing conserved Ser214. Biochemistry 31(12):3059–3064
Blow DM (1976) Structure and mechanism of chymotrypsin. Acc Chem Res 9(4):145–152
Brenner M, Müller HR, Pfister RW (1950) Eine neue enzymatische peptidsynthese. 1. Mitteilung. Helv Chim Acta 33(3):568–591
Brenner M, Pfister RW (1951) Enzymatische peptidsynthese. 2. Mitteilung. Isolierung von enzymatisch gebildeteml-methionyl-l-methionin und l-methionyl-l-methionyl-l-methionin; vergleich mit synthetischen produkten. Helv Chim Acta 34(6):2085–2096
Qin X, Xie W, Tian S et al (2013) Enzyme-triggered hydrogelation via self-assembly of alternating peptides. Chem Commun 49(42):4839–4841
Ebeling W, Hennrich N, Klockow M et al (1974) Proteinase K from Tritirachium album limber. Eur J Biochem 47(1):91–97
Saenger W (2013) Proteinase K, vol 3. Elsevier Ltd., Amsterdam
Bajorath J, Saenger W, Pal GP (1988) Autolysis and inhibition of proteinase K, a subtilisin-related serine proteinase isolated from the fungus tritirachium album limber. Biochim Biophys Acta, Proteins Proteomics 954((C)):176–182
Kraus E, Kiltz HH, Femfert UF (1976) The specificity of proteinase K against oxidized insulin b chain. Hoppe-Seyler’s Z. Physiol Chem 357(2):233–237
Sweeney PJ, Walker JM (1993) Proteinase K (EC 3.4.21.14). Methods Mol Biol 16(5):305–311
Ageitos JM, Baker PJ, Sugahara M et al (2013) Proteinase K-catalyzed synthesis of linear and star oligo(l-phenylalanine) conjugates. Biomacromolecules 14(10):3635–3642
Ageitos JM, Chuah JA, Numata K (2015) Chemo-enzymatic synthesis of linear and branched cationic peptides: evaluation as gene carriers. Macromol Biosci 15(7):990–1003
Ma Y, Li Z, Numata K (2016) Synthetic short peptides for rapid fabrication of monolayer cell sheets. ACS Biomater Sci Eng 2(4):697–706
Craik C, Largman C, Fletcher T et al (1985) Redesigning trypsin: alteration of substrate specificity. Science 228(4697):291–297
Baird T, Wang B, Lodder M et al (2000) Generation of active trypsin by chemical cleavage. Tetrahedron 56(48):9477–9485
May R (1979) Trypsin-catalyzed synthesis of the arginyl-arginine dipeptide from l-arginine ethyl ester. Biotechnol Lett 1(2):102–102
Aso K, Kodaka H (1992) Trypsin-catalyzed oligomerization of l-lysine esters. Biosci Biotechnol Biochem 56(5):755–758
Endo S (1962) Studies on protease produced by thermophilic bacteria. J Ferment Technol 40:346–353
Feder J, Garrett LR, Wildi BS (1971) Studies on the role of calcium in thermolysin. Biochemistry 10(24):4552–4556
Lipscomb WN, Sträter N (1996) Recent advances in zinc enzymology. Chem Rev 96(7):2375–2434
Isowa Y, Ohmori M, Ichikawa T et al (1979) The thermolysin-catalyzed condensation reactions of N-substituted aspartic and glutamic acids with phenylalanine alkyl esters. Tetrahedron Lett 20(28):2611–2612
Isowa Y, Ohmori M, Sato M et al (1977) The enzymatic synthesis of protected valine-5 angiotensin II amide-I. Bull Chem Soc Jpn 50(10):2766–2772
Isowa Y, Ohmori M, Sato M et al (1977) The synthesis of peptides by means of proteolytic enzymes. Bull Chem Soc Jpn 50(10):2762–2765
Isowa Y, Ichikawa T, Ohmori M (1978) Peptide syntheses with proteinases. Fragment condensation of ZLeuGlnGlyOH or ZGlnGlyOH with HLeuValNH2 using metalloproteinases. Bull Chem Soc Jpn 51(1):271–276
Isowa Y, Ichikawa T (1979) Syntheses of N-acyl dipeptide derivatives by metalloproteinases. Bull Chem Soc Jpn 52(3):796–800
Wayne SI, Fruton JS (1983) Thermolysin-catalyzed peptide bond synthesis. Proc Natl Acad Sci U S A 80(11):3241–3244
Kitaguchi H, Klibanov AM (1989) Enzymatic peptide synthesis via segment condensation in the presence of water mimics. J Am Chem Soc 111(26):9272–9273
Basso A, De Martin L, Ebert C et al (2000) High isolated yields in thermodynamically controlled peptide synthesis in toluene catalysed by thermolysin adsorbed on celite R-640. Chem Commun 6:467–468
Ulijn RV, Baragaña B, Halling PJ et al (2002) Protease-catalyzed peptide synthesis on solid support. J Am Chem Soc 124(37):10988–10989
Ulijn RV, Bisek N, Halling PJ et al (2003) Understanding protease catalysed solid phase peptide synthesis. Org Biomol Chem 1(8):1277–1281
Thust S, Koksch B (2004) Discovery of carboxypeptidase Y as a catalyst for the incorporation of sterically demanding α-fluoroalkyl amino acids into peptides. Tetrahedron Lett 45(6):1163–1165
Nitta S, Komatsu A, Ishii T et al (2016) Synthesis of peptides with narrow molecular weight distributions via exopeptidase-catalyzed aminolysis of hydrophobic amino-acid alkyl esters. Polym J 48(9):955–961
Komeda H, Asano Y (1999) Synthesis of d-phenylalanine oligopeptides catalyzed by alkaline d-peptidase from bacillus cereus DF4-B. J Mol Catal B Enzym 6(3):379–386
Kato Y, Asano Y, Nakazawa A et al (1990) Synthesis of d-alanine oligopeptides catalyzed by d-aminopeptidase in non-aqueous media. Biocatal Biotransformation 3(3):207–215
Zhang Y, Xia B, Li Y et al (2016) Solvent-free lipase-catalyzed synthesis: unique properties of enantiopure d- and l-polyaspartates and their complexation. Biomacromolecules 17(1):362–370
Totsingan F, Centore R, Gross RA (2017) CAL-B catalyzed regioselective bulk polymerization of l-aspartic acid diethyl ester to alpha-linked polypeptides. Chem Commun 53(28):4030–4033
Dannenberg AM, Smith EL (1955) Action of proteinase I of bovine lung. Hydrolysis of the oxidized b chain of insulin; polymer formation from amino acid esters. J Biol Chem 215(1):55–66
Sluyterman LAÆ, Wijdenes J (1972) Sigmoidal progress curves in the polymerization of leucine methyl ester catalyzed by papain. Biochim Biophys Acta, Enzymol 289(1):194–202
Tsuchiya K, Numata K (2017) Chemical synthesis of multiblock copolypeptides inspired by spider dragline silk proteins. ACS Macro Lett 6(2):103–106
Lampel A, McPhee SA, Park H-A et al (2017) Polymeric peptide pigments with sequence-encoded properties. Science 356(6342):1064
Matsumura S, Tsushima Y, Otozawa N et al (1999) Enzyme-catalyzed polymerization of l-aspartate. Macromol Rapid Commun 20(1):7–11
Soeda Y, Toshima K, Matsumura S (2003) Sustainable enzymatic preparation of polyaspartate using a bacterial protease. Biomacromolecules 4(2):196–203
Martin ME, Rice KG (2007) Peptide-guided gene delivery. AAPS J 9(1):E18–E29
Heitz F, Morris MC, Divita G (2009) Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol 157(2):195–206
Fagerland J, Finne-Wistrand A, Numata K (2014) Short one-pot chemo-enzymatic synthesis of l-lysine and l-alanine diblock co-oligopeptides. Biomacromolecules 15(3):735–743
Ma Y, Sato R, Li Z et al (2016) Chemoenzymatic synthesis of oligo(l-cysteine) for use as a thermostable bio-based material. Macromol Biosci 16(1):151–159
Anderson G, Luisi PL (1979) Papain-induced oligomerization of α-amino acid esters. Helv Chim Acta 62(2):488–494
Jost R, Brambilla E, Monti JC et al (1980) Papain catalyzed oligomerization of α-amino acids. Synthesis and characterization of water-insoluble oligomers of l-methionine. Helv Chim Acta 63(2):375–384
Nuijens T, Piva E, Kruijtzer JAW et al (2011) Fully enzymatic n→c-directed peptide synthesis using C-terminal peptide α-carboxamide to ester interconversion. Adv Synth Catal 353(7):1039–1044
Wang M, Qi W, Yu Q et al (2011) Kinetically controlled enzymatic synthesis of dipeptide precursor of l-alanyl-l-glutamine. Biotechnol Appl Biochem 58(6):449–455
Yazawa K, Numata K (2016) Papain-catalyzed synthesis of polyglutamate containing a nylon monomer unit. Polymers 8(5):194
Yazawa K, Gimenez-Dejoz J, Masunaga H et al (2017) Chemoenzymatic synthesis of a peptide containing nylon monomer units for thermally processable peptide material application. Polym Chem 8(29):4172–4176
Galoppini E, Fox MA (1996) Effect of the electric field generated by the helix dipole on photoinduced intramolecular electron transfer in dichromophoric α-helical peptides. J Am Chem Soc 118(9):2299–2300
Solà J, Helliwell M, Clayden J (2010) N- versus C-terminal control over the screw-sense preference of the configurationally achiral, conformationally helical peptide motif Aib8GlyAib8. J Am Chem Soc 132(13):4548–4549
Tsuchiya K, Numata K (2017) Chemoenzymatic synthesis of polypeptides containing the unnatural amino acid 2-aminoisobutyric acid. Chem Commun 53(53):7318–7321
Tsuchiya K, Numata K (2016) Papain-catalyzed chemoenzymatic synthesis of telechelic polypeptides using bis(leucine ethyl ester) initiator. Macromol Biosci 16(7):1001–1008
Tsuchiya K, Masunaga H, Numata K (2017) Tensile reinforcement of silk films by the addition of telechelic-type polyalanine. Biomacromolecules 18(3):1002–1009
Tsuchiya K, Takaoki I, Masunaga H et al (2018) Spider dragline silk composite films doped with linear and telechelic polyalanine: effect of polyalanine on the structure and mechanical properties. Sci Rep 8:3654
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Tsuchiya, K., Miyagi, Y., Miyamoto, T., Gudeangadi, P.G., Numata, K. (2019). Synthesis of Polypeptides. In: Kobayashi, S., Uyama, H., Kadokawa, Ji. (eds) Enzymatic Polymerization towards Green Polymer Chemistry. Green Chemistry and Sustainable Technology. Springer, Singapore. https://doi.org/10.1007/978-981-13-3813-7_8
Download citation
DOI: https://doi.org/10.1007/978-981-13-3813-7_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3812-0
Online ISBN: 978-981-13-3813-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)