
Abstract
Oligo- and polysaccharides are important macromolecules in living systems, showing their multifunctional characteristics in the construction of cell walls, energy storage, cell recognition, and their immune response. The chemical synthesis of oligo- and polysaccharides is feasible though it can be laborious since multiple protection, deprotection, and purification steps are required. In contrast to this, phosphorylases are useful synthetic tools for the preparation of natural oligo- and polysaccharides, glycoconjugates, and their analogs. Since phosphorylases are rather tolerant with respect to utilizing modified donors and acceptor substrates, they can be used to prepare oligo- and polysaccharide analogs and for diversification of natural products. Their strict primer-dependence allows synthesis of interesting hybrid materials. Furthermore, enzymatic reaction, such as that using phosphorylase, is one of the most promising environmentally benign technologies with a simple operation under mild conditions, eliminating undesirable side reactions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
3.1 Introduction: Overview on Phosphorylase-Catalyzed Enzymatic Polymerization
Polysaccharides are composed of monosaccharide residues linked together through glycosidic bonds, a type of covalent linkage that joins a monosaccharide residue at its anomeric position to another group, typically another saccharide moiety, such that it is principally a result of dehydrative condensation between the components’ two hydroxy groups. Polysaccharides in nature are constructed by a wide variety of monosaccharide units linked through the different glycosidic bonds in stereo- and regioarrangements, resulting in quite complicated chemical structures [1, 2]. The structural diversity of polysaccharides leads to providing a whole range of biological functions, in which it is well accepted that a subtle change in their structure has a profound effect on the properties and functions of these molecules.
Polysaccharides are an abundant source of raw materials that are interesting due to the biodegradable, biocompatible, and renewable character. Saccharides are expected to play an increasingly bigger role as raw material in the future and to replace petrol-based materials. Already, polysaccharides find their way in many disparate fields of industry. A short overview is given in Table 3.1.
Conventional chemical synthetic approaches are, in many cases, inadequate to provide substantial quantities of saccharides [3]. The difficulties arise from realizing complete regio- and stereocontrol of the glycosylating process. At present, no such methods are available because, in chemical synthesis, most of the difficulties arise from the laborious regio- and stereochemical control [4,5,6]. Most synthetic approaches are therefore based on the modification or degradation of naturally occurring polysaccharides resulting in less than perfect products.
In contrast to this, enzymatic approaches have been identified as a powerful tool to synthesize polysaccharides with well-defined structure because enzymatic reaction generally proceeds with highly stereo- and regiocontrolled manners [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21]. Two approaches have dominated enzyme-catalyzed saccharide synthesis: glycosyl transferase and glycosidase-catalyzed glycosidic bond formation. The first uses the normal biosynthetic machinery of living organisms. In the second, enzymes that normally catalyze transfer of an enzyme-bound glycosyl residue to water are induced to transfer it instead to a different acceptor.
Phosphorylases belong to the class of glycosyltransferases (GTs) which are part of the class of transferases (Enzyme Classification (EC), Class No. 2). These enzymes catalyze reactions in which a group is transferred from one compound to another. Groups that are transferred are Cl, aldehydic or ketonic residues, acyl, glycosyl, alkyl, nitrogenous, phosphorus, and sulfur-containing groups [22]. GTs are important biological catalysts in cellular systems generating complex cell surface glycans involved in adhesion and signaling processes. Recent advances in glycoscience have increased the demands to access significant amount of glycans representing the glycome. GTs catalyze the transfer of a sugar moiety from an activated donor sugar onto saccharide and nonsaccharide acceptors. GTs can be divided into the Leloir and non-Leloir types according to the type of glycosyl donors they use. Non-Leloir GTs typically use glycosyl phosphates as donors, while Leloir GTs utilize sugar nucleotides as donors and transfer the monosaccharide with either retention (retaining enzymes) or inversion (inverting enzymes) of the configuration of the anomeric center. Most of GTs responsible for the biosynthesis of mammalian glycoproteins and glycolipids are Leloir glycosyltransferases. GTs are now playing a key role for in vitro synthesis of oligosaccharides, and the bacterial genome is increasingly utilized for cloning and overexpression of active transferases in glycosylation reactions [23,24,25,26,27,28,29,30,31,32,33].
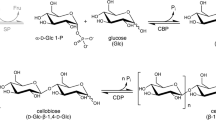
All of the phosphorylases, which have been found in nature, show strict stereo- and regiospecificities, which thus catalyze the phosphorolysis of a specific glycosidic linkage at the nonreducing end of the saccharide in the presence of inorganic phosphate (Pi) to form a monosaccharide 1-phosphates (Fig. 3.1) [34,35,36]. Therefore, phosphorylases are classified by the anomeric forms of the resulting monosaccharide 1-phosphates or by the anomeric forms of glycosidic linkages in substrates, which are phosphorolyzed. Alternatively, phosphorylases are classified in terms of retention or inversion at the anomeric position in the reaction. The reversible nature in phosphorylase-catalyzed reactions is conceived because the bond energy of phosphate esters in the product is comparable to that of the glycosidic linkage in the saccharide chain. With respect to reversibility, therefore, phosphorylases catalyze enzymatic glycosylation according to reaction conditions. In the reactions, monosaccharide 1-phosphates act as glycosyl donors, and the monosaccharide residue is transferred from the donor to the nonreducing end of a specific glycosyl acceptor to form glycosidic linkages with strictly controlled stereo- and regioarrangements, with liberating Pi. Some phosphorylases catalyze the reactions for a synthetic way to polysaccharides or oligosaccharides with relatively high DPs via consecutive glycosylations, whereas other phosphorylases only catalyze the reversible phosphorolysis of disaccharide substrates to produce the corresponding monosaccharide 1-phosphates and other monosaccharides. As mentioned mainly in this chapter, only α-glucan phosphorylase (GP) and cellodextrin phosphorylase (CDP)-catalyzed reactions (EC 2.4.1.1 and 2.4.1.49, respectively) have been used for the practical synthesis of poly- or oligosaccharides with relatively higher DPs, which have also provided polysaccharide- and oligosaccharide-based functional materials [19]. Several works for practical synthesis of oligosaccharides catalyzed by kojibiose phosphorylase (EC 2.4.1.230) have also been reported (vide infra) [37]. Besides them, β-1,3-oligoglucan phosphorylase (EC 2.4.1.30) [38] and β-1,2-oligoglucan phosphorylase [39] also catalyze the phosphorolysis of glucans with the higher DPs than 2. However, there have not been many studies on the synthesis of poly- or oligosaccharides catalyzed by these enzymes.

Reversible phosphorolysis reaction catalyzed by phosphorylase
As abovementioned, GP (systematic name: (1 → 4)-α-d-glucan:phosphate α-d-glucosyltransferase; EC 2.4.1.1) is one of the enzymes which have been used as catalysts for practical synthesis of polysaccharides [40,41,42]. While this enzyme is responsible for the depolymerization of linear α(1 → 4)-glucosidic chains in vivo, it can also be used to synthesize linear α(1 → 4)-glucosidic chains (amylose) in vitro. The existence of a phosphorylating enzyme in a higher plant was first reported by Iwanoff who observed that an enzyme he found in the germinating vetches, Vicia sativa, liberates inorganic phosphate from organic phosphorous compounds [43]. Shortly after, the same enzyme was found in other vetches and wheat [44, 45], rice and coleseed [46], barley and malt, etc. Bodńar was the first to report a progressive disappearance of inorganic phosphate (thus the reverse reaction) while incubating suspended flour from ground peas in a phosphate buffer [47]. Cori and Cori demonstrated that animal tissues contain an enzyme which acts upon glycogen as well [48,49,50,51]. Cori, Colowick, and Cori suggested that the product of this reaction is α-glucopyranose-1-phosphoric acid (also called Cori-ester), which was confirmed later by Kiessling [52] and Wolfrom and Pletcher [53].
In GPs, glycogen GPs belong to the group of vitamin B6 enzymes bearing a catalytic mechanism that involves the participation of the phosphate group of pyridoxal-5′-phosphate (PLP). The proposed mechanism is a concerted one with front-side attack as can be seen in Fig. 3.2 [54]. In the forward direction, e.g., phosphorolysis of α(1 → 4)-glycosidic bonds in oligo- or polysaccharides, the reaction is started by protonation of the glycosidic oxygen by orthophosphate, followed by stabilization of the incipient oxocarbonium ion by the phosphate anion and subsequent covalent binding of the phosphate to form α-D-glucose 1-phospate (Glc-1-P). The product, Glc-1-P, dissociates and is replaced by a new incoming phosphate. In the reverse direction, protonation of the phosphate of Glc-1-P destabilizes the glycosidic bond and promotes formation of a glucosyl oxocarbonium ion–phosphate anion pair. In the subsequent step, the phosphate anion becomes essential for promotion of the nucleophilic attack of a terminal glucosyl residue on the carbonium ion. This sequence of reactions brings about α-1,4-glycosidic bond formation and primer elongation. This mechanism accounts for retention of configuration in both directions without requiring sequential double inversion of configuration. It also provides for a plausible explanation of the essential role of pyridoxal-5′-phosphate in glycogen GP catalysis, as the phosphate of the cofactor, pyridoxal-5′-phosphate, and the substrate phosphates approach each other within a hydrogen-bond distance allowing proton transfer and making the phosphate of pyridoxal-5′-phosphate into a proton shuttle which recharges the substrate phosphate anion.

Catalytic mechanism of glycogen phosphorylases. The reaction scheme accounts for the reversibility of phosphorolysis of oligosaccharides (R) in the presence of orthophosphate (upper half) and primer-dependent synthesis in the presence of glucose-1-phosphate (lower half). PL = enzyme-bound pyridoxal; BH+ = a general base contributed by the enzyme protein. (Reprinted with permission from ref. [54]. Copyright 1990 American Chemical Society)
The fact that glycogen GP can be used to polymerize amylose was first demonstrated by Schäffner and Specht [55] in 1938 using yeast phosphorylase. Shortly after, the same behavior was also observed for other phosphorylases from yeast by Kiessling [56]; muscles by Cori, Schmidt, and Cori [57]; pea seeds [58]; potatoes by Hanes [58]; and preparations from liver by Ostern and Holmes [59]; Cori, Cori, and Schmidt [60]; and Ostern, Herbert, and Holmes [61]. These results opened up the field of enzymatic polymerizations of amylose using Glc-1-P as monomer and can be considered the first experiments ever to synthesize biological macromolecules in vitro.
One of the remarkable properties of GP is that it is unable to synthesize amylose unless a primer is added (poly- or oligomaltosaccharide); n (Glc-1-P) + primer ⇆ amylose + n (orthophosphate (Pi)). As the GP-catalyzed polymerization is conceived analogously to a living polymerization, accordingly, the molecular weights of the produced amylose can relatively be controlled by the monomer (Glc-1-P)/primer feed ratios. As the complete isolation of amylose from natural starch resources is not facilely achieved, the GP-catalyzed enzymatic polymerization is well accepted as a powerful tool to obtain a pure amylose sample with desired molecular size. Furthermore, a single amylose chain exhibits water solubility but spontaneously forms an antiparallel double helix to each other owing to its left-handed helical conformation to produce a water-insoluble assembly [62, 63]. By means of this behavior, amylosic materials with hierarchically controlled structure have been fabricated [19, 64,65,66,67,68,69].
The kinetic behavior of the polymerization of amylose with potato GP with various saccharides as primers was first studied by Hanes [58]. Green and Stumpf [70] failed to detect priming action with maltose but were able to confirm all other results by Hanes. Weibull and Tiselius [71] found that the maltooligosaccharide of lowest molecular weight to exhibit priming activity was maltotriose which was confirmed by Whelan and Bailey [72], who also showed that maltotriose (Glc3) is the lowest member of the series of oligosaccharides to exhibit priming activity. Whelan and Bailey were also able to clarify the polymerization mechanism of the enzymatic polymerization with GP [72]. Their results showed that the polymerization follows a “multichain” scheme in contrast to a “single-chain” scheme that was also proposed by some authors. In the “multichain” polymerization scheme, the enzyme–substrate complex dissociates after every addition step, whereas in the “single-chain” scheme, each enzyme continuously increases the length of a single primer chain without dissociation.
By studying the polydispersities of amyloses obtained by enzymatic polymerization with potato GP from maltooligosaccharides of various lengths, Pfannemüller and Burchard were able to show that the reaction mechanism of the polymerization with Glc3 as primer varies from its higher homologues [73]. While the amyloses built by polymerization from maltotetraose (Glc4) or higher showed a Poisson distribution [74] that can be expected from a polymerization following a “multichain” scheme (random synthesis occurs, and all the primer chains grow at approximately equal rates), a bimodal broad distribution was observed when Glc3 was used as primer. The authors found that in the case of Glc3 as a primer, the reaction can be divided into a start reaction and the following propagation, the rate of the first reaction being 400 times slower than the rate of the propagation. Due to this start reaction, not all chains start to grow at the same time which results in a broader distribution. The propagation follows again a “multichain” reaction scheme. Suganuma et al. [75] were able to determine the exact kinetic parameters of the synthetic as well as the phosphorolytic reaction using Glc3 and higher maltooligosaccharides as primer and were able to confirm the results of Whelan and Bailey and Pfannemüller and Burchard.
Although GP catalysis implies strict stereo- and regiospecificities, i.e., the formation of α(1→4)-glycosidic arrangement, the enzyme exhibits weak specificity for the recognition of monosaccharide structures in the 1-phosphate substrates. Accordingly, GP has also catalyzed glycosylations using analog substrates of Glc-1-P as glycosyl donors to obtain nonnatural oligosaccharides having different monosaccharide residues at the nonreducing end [76,77,78]. For example, potato GP has been found to recognize α-d-mannose, 2-deoxy-α-d-glucose, α-d-xylose, α-d-glucosamine, and N-formyl-α-d-glucosamine 1-phosphates (Man-1-P, dGlc-1-P, Xyl-1-P, GlcN-1-P, and GlcNF-1-P, respectively) as glycosyl donors in glycosylations using Glc4 as a glycosyl acceptor, to produce nonnatural α-mannosylated, 2-deoxy-α-glucosylated, α-xylosylated, α-glucosaminylated, and N-formyl-α-glucosaminylated pentasaccharides, respectively (Fig. 3.3) [79,80,81,82,83,84]. In these reactions, only a monosaccharide residue is transferred from the donor to the acceptor, but consecutive glycosylations, i.e., enzymatic polymerization, are not progressed. The produced α-glucosaminylated Glc4 by glucosaminylation using GlcN-1-P is a basic oligosaccharide owing to the presence of an amino group at the C-2 position of GlcN unit, while the other products are neutral oligosaccharides. Thermostable GP has shown more tolerance for recognition specificity of the glycosyl donor than that obtained from potato. For example, potato GP does not recognize α-d-glucuronic acid 1-phosphate (GlcA-1-P), while thermostable GP from Aquifex aeolicus VF5 [85] recognizes GlcA-1-P and catalyzes glucuronylation of Glc3 as a glycosyl acceptor to produce an acidic tetrasaccharide having a GlcA residue at the nonreducing end [86].

GP-catalyzed enzymatic glycosylations using analog substrates as glycosyl donors to produce nonnatural oligosaccharides
By means of the enzymatic glucuronylation, dendritic acidic α-glucans have been synthesized [87]. A highly branched cyclic dextrin was employed as a polymeric glycosyl acceptor with plural reactive sites. This material is a water-soluble dextrin, which is produced from amylopectin by cyclization catalyzed by the branching enzyme (EC 2.4.1.18, Bacillus stearothermophilus) [88,89,90]. As the branched dextrin has a number of the nonreducing α(1→4)-glucan ends, this acts as multi-glycosyl acceptors for the GP-catalyzed glucuronylation. The thermostable GP-catalyzed glucuronylation of the branched dextrin (M n = 1.25 × 105, the number of nonreducing ends = ca. 59) with GlcA-1-P was performed to produce acidic α-glucans. The glucuronylation ratios of the GlcA residues to the nonreducing ends were controlled by the donor/acceptor feed ratios. The subsequent thermostable GP-catalyzed glucosaminylation of the acidic products using GlcN-1-P was examined to obtain dendritic amphoteric α-glucans having both GlcA and GlcN residues at the nonreducing ends [91]. The amphoteric polysaccharides having the different GlcA/GlcN ratios were produced by the successive glucuronylation/glucosaminylation in several glycosyl donor feed ratios of GlcA-1-P/GlcN-1-P. Their inherent isoelectric points were calculated by ζ-potential measurement, which were reasonably changed in accordance with the GlcA/GlcN ratios in the products.
A series of studies on the potato GP-catalyzed glycosylations using the above analog substrates as glycosyl donors has suggested that if a single monosaccharide residue is transferred to the nonreducing end of the maltooligosaccharide acceptor, further glycosylations do not take place because the new structure different from the Glc residue at the nonreducing end is no longer recognized by potato GP. On the other hand, it has been found that when the thermostable GP (from Aquifex aeolicus VF5) is alternatively employed as a catalyst in the glycosylations of Glc3 using excess molar ratios of Man-1-P and GlcN-1-P, consecutive mannosylations/glucosaminylations took place to obtain nonnatural heterooligosaccharides composed of α(1→4)-linked mannose/glucosamine chains at the nonreducing end of Glc3 [92]. The matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF MS) of the products of the thermostable GP-catalyzed mannosylation/glucosaminylation with a donor to acceptor feed ratio of 10:1 showed several peaks corresponding to the molecular masses of tetra-octa-saccharides having one–five Man or GlcN residues with Glc3. Further chain elongation for higher DPs, however, was inhibited by Pi produced from the glycosyl donors, which is a naturally occurring substrate for phosphorolysis by GP catalysis as aforementioned.
An attempt was made to remove Pi as a precipitate in the thermostable GP-catalyzed glucosaminylation by conducting the reaction in an ammonium buffer (0.5 M, pH 8.6) containing MgCl2 [93] (Fig. 3.4), based on the fact that Pi forms an insoluble salt with ammonium and magnesium ions [94]. Consequently, the thermostable GP-catalyzed polymerization in the buffer system at 40 °C for 7 days of GlcN-1-P with the Glc3 primer (30:1) efficiently occurred to produce the α(1→4)-linked glucosamine polymer with a DP of the GlcN units of ~20 (M n = 3760), that is, an amylose analog aminopolysaccharide, called “amylosamine.” The produced polysaccharide is water-soluble and has not formed a controlled higher-order assembly in water as observed for the amylose double helix. It has been found that the cationic amylosamine forms a double helix with anionic amylouronic acid, another amylose analog polysaccharide, by electrostatic interaction [95]. In addition, the thermostable GP-catalyzed enzymatic copolymerizations of Glc-1-P with GlcN-1-P and with Man-1-P have been successfully progressed under the same conditions to obtain nonnatural glucosaminoglucan composed of Glc/GlcN units and mannoglucan composed of Glc/Man units [96, 97]. The thermostable GP-catalyzed enzymatic polymerization of GlcN-1-P using maltooligosaccharide-functionalized amylouronic acid (α(1→4)-linked GlcA polysaccharide with a short α(1→4)-linked Glc chain at the nonreducing end) under the same operation yielded an amylose analog amphoteric block polysaccharide composed of GlcN and GlcA chains [98].

GP-catalyzed enzymatic polymerization of GlcN-1-P in ammonia buffer containing Mg2+
CDP is an enzyme that catalyzes the reversible phosphorolysis of cello-oligosaccharides larger than cellotriose to produce Glc-1-P [99]. Cello-oligosaccharides have been synthesized by the CDP-catalyzed oligomerization using various cellobiose acceptors and Glc-1-P as the glycosyl donor [100, 101]. When cellobiose was used as a glycosyl acceptor, various cello-oligosaccharides ranging from water-soluble products to crystalline assembles were obtained, depending on the concentration of the acceptor. The precision determination of molecular-weight distributions of cello-oligosaccharides synthesized by CDP from Clostridium stercorarium or Clostridium thermocellum as catalysts was investigated (Fig. 3.5) [102]. The oligocellulose molar mass distribution was analyzed using different methods, such as MALDI-ToF MS. The molar mass distribution of the synthesized oligocellulose was only dependent on the concentration of cellobiose used in the reaction. Although glucose has been identified as a non-glycosyl acceptor for CDP-catalyzed reaction, a significant amount of insoluble cellulose was precipitated without accumulation of soluble cello-oligosaccharides in this enzymatic reaction system [103]. This result was explained in terms of the large difference in the reactivity of acceptors between glucose and cello-oligosaccharides.

Images and MALDI-ToF MS for molecular-weight distributions of cello-oligosaccharides synthesized by CDP from Clostridium stercorarium or Clostridium thermocellum (Reprinted with permission from ref [102]. Copyright 2015 American Chemical Society)
The CDP-catalyzed synthesis of cello-oligosaccharides substituted at their reducing ends has been investigated using various cellobiose derivatives and analogs as the glycosyl acceptors. Cellulose analog oligosaccharides, that is, β(1→3,1→4)-oligosaccharides and thiooligosaccharides, have been synthesized by the CDP-catalyzed oligomerization of Glc-1-P using β(1→3)-linked oligosaccharide acceptors [104, 105]. Moreover, cellobiosylated dimer and trimer and cellobiose-coated polyamidoamine dendrimers have been used as glycosyl acceptors for CDP-catalyzed oligomerization of Glc-1-P to give the corresponding materials composed of cello-oligosaccharide chains at the nonreducing end [106]. Based on the fact that CDP from Clostridium stercorarium was found to display a broad donor and acceptor specificity, the enzymatic glycosylations of sophorolipid and glucolipid acceptors with either Glc-1-P or α-d-galactose 1-phosphate as glycosyl donors were achieved to produce novel glycolipids [107]. The transfer of a glucose residue afforded a mixture of products that precipitated from the solution, resulting in near quantitative yields. The transfer of a galactose residue, on the other hand, a single product was generated, which remained in solution at thermodynamic equilibrium.
Kojibiose phosphorylase is an enzyme that catalyzes the phosphorolysis of kojibiose into 𝛽-d-glucose 1-phosphate and glucose [37]. This enzyme also catalyzes the reverse reaction for chain elongation greater than the disaccharide to synthesize koji-oligosaccharides composed of α(1→2)-linked glucose units. The kojibiose phosphorylase-catalyzed oligomerization has been extensively examined using various glycosyl acceptors to obtain novel oligosaccharides composed of koji-oligosaccharide chains [108,109,110,111].
3.2 Enzymatic Preparation of Glycomaterials Using Functional Primers
The GP-catalyzed enzymatic polymerization can be conducted using modified maltooligosaccharide primers, where their reducing ends, which do not take part in the polymerization, are covalently immobilized on various substances, such as low molecular-weight compounds, polymers, and surfaces [19, 64,65,66,67,68,69].
Pfannemüller et al. showed that it is possible to obtain carbohydrate-containing amphiphiles with various alkyl chains via amide bond formation. For this, maltooligosaccharides were oxidized to the according aldonic acid lactones which could subsequently be coupled to alkylamines [112,113,114,115,116,117,118,119]. Such sugar-based surfactants are important industrial products finding their applications in cosmetics, medical applications, etc. [120,121,122]. The authors were also able to extend the attached maltooligosaccharides with enzymatic polymerization with potato GP which resulted in products with very interesting solution properties [123, 124].
Amylosic diblock copolymers have been synthesized by the GP-catalyzed enzymatic polymerization using such polymeric primers having a maltooligosaccharide moiety at the chain end. For example, GP-catalyzed enzymatic polymerization using maltoheptaose (Glc7)-functionalized polystyrene was performed to produce (amylose-block-polystyrene)s [125,126,127]. Products with various compositions formed micellar aggregates in water and tetrahydrofuran (THF). The analytical results observed the presence of a single-chain, small aggregates, and large micellar species of the block copolymers in THF. Crew-cut micelles, in contrast, were formed in water from the same block copolymers by a single-solvent approach, elevated temperature, and pressure. These crew-cut aggregates were much more uniform than the respective star aggregates in THF. Similarly, amylose-block-polyTHF, amylose-block-poly(d-lactide), and amylose-block-poly(2-vinylpyridine) have been obtained by the GP-catalyzed enzymatic polymerization using the corresponding polymeric primers [128,129,130].
Amphiphilic amylose-block-methoxy-terminated poly(ethylene glycol) (MPEG) has been synthesized by GP-catalyzed enzymatic polymerization using Glc5-functionalized MPEO primer [131, 132]. A pure amylose is insoluble in chloroform, whereas the product forms reverse micelles, with hydrophobic methyl orange being entrapped in the amylose helix in the micelles in chloroform. Previously various linear block copolymers with PEG – of the AB, ABA and ABC type – with enzymatically polymerized amylose blocks were reported. Ziegast and Pfannemüller converted the hydroxy end groups of PEG into amino groups via tosylation and further reaction with 2-aminoalkylthiolate [133,134,135]. To the resulting mono- and di-amino-functionalized PEG, maltooligosaccharide lactones were attached and subsequently elongated to amylose via enzymatic polymerization [133]. Pfannemüller et al. performed a very detailed study on the solution properties of the synthesized A-B-A triblock copolymers as they can be considered as model substances for “once broken rod” chains [117]. With static and dynamic light scattering, they found that the flexible joint between the two rigid amylose blocks has no detectable effect on the common static and dynamic properties of the chain. With dielectric measurements it however became obvious that the directional properties of the electric dipoles of the broken rigid chains showed a different behavior to the non-broken rods (pure amylose).
Glycogen is known to be a water-soluble and high molecular-weight natural polysaccharide, composed of linear α(1→4)-glucan chains containing an average of 10 to 14 glucose residues, which are further interlinked by α(1→6)-glycosidic linkages, leading to a highly branched structure and spherical morphology in water [136, 137]. Accordingly, glycogen has the similar chemical structure as the abovementioned cyclic dextrin, but the molecular weight is much higher. Besides glycogen’s role in in vivo phosphorolysis with GP as an energy resource, therefore, it can be also used as polymeric primer for the GP-catalyzed enzymatic polymerizations because of the presence of a number of nonreducing α(1→4)-glucan chain ends. When the GP-catalyzed enzymatic polymerization of Glc-1-P on glycogen was carried out in aqueous acetate buffer solution, followed by the standing of the reaction mixture under ambient atmosphere for 24 h, the solution fully turned into a hydrogel [138]. Hydrogelation is induced by the formation of double-helix cross-linking points by the elongated amylose chains among glycogen molecules because amyloses are well-known to spontaneously form double helixes in water (Fig. 3.6). The hydrogel was then converted into a cryogel by lyophilization. The scanning electron microscope (SEM) image of the cryogel observed porous morphology. The X-ray diffraction (XRD) profile of the cryogel showed diffraction peaks ascribed to the crystalline structure of amylose double helix. This result indicated that the networks in the cryogels were constructed based on the double helical entanglement of the elongated amylose chains, resulting in the formation of the porous morphologies.

GP-catalyzed enzymatic polymerization using glycogen to produce hydrogel and consecutive enzymatic reactions to produce amphoteric glycogen hydrogel
By means of the thermostable GP-catalyzed consecutive glucosaminylation/glucuronylation of glycogen using GlcN-1-P and GlcA-1-P, amphoteric glycogens having both basic GlcN and acidic GlcA residues could also be synthesized (Fig. 3.6) [139]. The functionalities of the GlcA/GlcN residues depended on the feed ratios of glycosyl donors. The GP-catalyzed enzymatic polymerization of Glc-1-P with the non-functionalized, nonreducing ends of the amphoteric glycogens was then examined to produce amphoteric glycogen hydrogels through the formation of double helix cross-link by the elongated amylose chains (Fig. 3.6). The resulting hydrogels exhibited pH-responsive behavior, in which they were solubilized under alkaline conditions and returned to hydrogels upon acidification of the system. Furthermore, the hydrogels exhibited pH-dependent shrinking/swelling behavior.
The GP-catalyzed enzymatic polymerization of Glc-1-P using maltooligosaccharide-grafted polymeric primers, which are prepared by the covalent bond formation at the reducing end on the polymeric substrates, has been performed to produce amylose-grafted polymeric materials (chemoenzymatic approach) [19, 64,65,66,67,68,69]. For example, a styrene macromonomer having an amylose chain was prepared by the GP-catalyzed enzymatic polymerization using a primer having a polymerizable group at the reducing end. Radical polymerization of the macromonomer produced an amylose-grafted polystyrene [140]. This type of the graft material was also synthesized by radical polymerization of a styrene macromonomer having a maltooligosaccharide, followed by the GP-catalyzed enzymatic polymerization from the nonreducing end of the maltooligosaccharide primer on the product [140, 141]. By the similar approach, amylose-grafted dimethylsiloxane, polyacetylene, and poly(vinyl alcohol) have been synthesized [142,143,144]. A series of amylose-based star polymers (1, 2, 4, and 8 arms) have been synthesized by GP-catalyzed enzymatic polymerization using Glc5-functionalized PEG (Fig. 3.7) [145]. The obtained eight-arm primer serves as a gelator when triggered enzymatically.

Chemical structures and illustrations of Glc5-functionalized PEG primers. (Reprinted with permission from ref. [145]. Copyright 2015 American Chemical Society)
The chemoenzymatic approach is also appropriately conducted for the amylosic modification of spherical and planner surface, such as Au and Si. The Au and Si surfaces were amino-functionalized with self-assembled monolayers of cystamine and 3-aminopropyldimethylethoxysilane (APDMES), respectively. Glc7 was covalently attached to the amino-functionalized Au and Si surfaces via reductive amination. Amylose brushes were grown from Glc7-modified surfaces with GP-catalyzed enzymatic polymerization (Fig. 3.8) [146, 147].

Synthesis of amylose brushes via GP-catalyzed enzymatic polymerization from silica (left) and gold surfaces (right). (Adapted from ref. [146])
The chemoenzymatic approach has extensively been applied to the synthesis of amylosic conjugates with biopolymeric main-chains. Interestingly, the gelling system from amylose with suitable biopolymeric components has been reported and is constructed by the formation of amylose double helix as non-covalent cross-linking points through the GP-catalyzed enzymatic polymerization. For example, amylose-grafted heteropolysaccharides composed of abundant polysaccharide main-chains have been synthesized by this approach. The maltooligosaccharides have been introduced onto the main-chain polysaccharides by appropriate chemical reactions, such as reductive amination and condensation. Using the reductive amination, chitosan, a basic polysaccharide with amino groups at the C-2 position in its glucosamine repeating units, was functionalized to obtain a maltooligosaccharide-grafted chitosan, which could be further converted into maltooligosaccharide-grafted chitin by N-acetylation. From the maltooligosaccharide primer ends of the chitin/chitosan derivatives, amylose chains were elongated by the GP-catalyzed enzymatic polymerization of Glc-1-P to yield amylose-grafted chitin/chitosan (Fig. 3.9) [148, 149]. A hydrogel of the amylose-grafted chitosan was produced by slowly drying the polymerization mixture at 40–50 °C. An amylose-grafted cellulose was also synthesized using a similar procedure [150]. A partially aminated cellulose derivative at the C-6 position was initially prepared by the successive partial tosylation of its C-6 hydroxy groups, displacement of the tosylates by azido groups, and their subsequent reduction to amine groups. The resulting cellulose derivative was then reacted with a maltooligosaccharide by reductive amination to give a maltooligosaccharide primer-grafted cellulose, which was used for the GP-catalyzed enzymatic polymerization to produce the amylose-grafted cellulose (Fig. 3.9). The produced heteropolysaccharide is made up of two representative glucose polymers, cellulose and amylose, which are composed of the same repeating units but linked through opposite stereoarranged glycosidic bonds, β(1→4)– and α(1→4)–, respectively. The polymerization mixture totally turned into gel form when it was left standing at room temperature for several days. Drying the hydrogel under ambient atmosphere gave a solid material. The addition of water to the solid resulted in hydrogelation again. Such conversion cycle was repeated by the wetting and drying process. Moreover, the reaction mixture of the enzymatic polymerization was spread thinly on a glass plate and subsequently left standing at room temperature, resulting in a film.

Chemical structures of amylose-grafted chitosan, cellulose, and carboxymethyl cellulose
Amylose-grafted chitin nanofibers were also fabricated by the chemoenzymatic approach including reductive amination [151]. Nanofibrillated materials from native chitin, so-called chitin nanofibers, have increasingly attracted much attention and are expected to find applications as new functional materials. As native chitin sources are made up of nanofibrous assemblies, several methods for disentanglement of the assemblies have efficiently been developed to fabricate nanofiber dispersions in water. Re-dispersible amidinium chitin nanofibers are also obtained from an amidinated chitin by CO2 gas bubbling with ultrasonic treatment in water [152]. For the chemoenzymatic approach, maltooligosaccharide primers were first introduced by reductive amination with amino groups present on the amidinium chitin nanofibers partially having amino groups. Elongating of amylose chains on the nanofibers was then carried out by the GP-catalyzed enzymatic polymerization from the maltooligosaccharide graft chains to produce amylose-grafted chitin nanofiber materials. The reaction mixtures turned into hydrogels upon increasing the Glc-1-P/primer feed ratios (Fig. 3.10). The formation of double helixes from a part of amylose graft chains closely preset among the nanofibers contributed to forming the smaller networks, resulting in hydrogelation. Lyophilization of the hydrogels gave rise to the formation of the controlled microstructures, which were changed from fibrous to porous morphologies in accordance with the molecular weights of the amylose graft chains. Most of the amylose graft chains with the higher molecular weights, which did not take part in double helixes, formed amorphous membranes inside the nanofiber networks by lyophilization, to construct the porous structure.

Chemoenzymatic synthesis of amylose-grafted chitin nanofibers
The condensation reaction using an amine-functionalized maltooligosaccharide at the reducing end was also employed to prepare modified primers from acidic polysaccharides, such as alginate, xanthan gum, and carboxymethyl cellulose (CMC) having carboxylate groups. The GP-catalyzed enzymatic polymerization from the maltooligosaccharide chain ends of the condensation products was then carried out to obtain amylose-grafted alginate, xanthan gum, and CMC (Fig. 3.9) [153,154,155,156]. A film of the amylose-grafted CMC was formed by drying the thinly spread alkaline solution (0.040 g polysaccharide in 1.5 mL 0.50 M aqueous NaOH solution). The SEM image of the film showed highly entangled nanofibers. After washing out the residual NaOH from the film by immersion in water, the SEM image showed that the nanofibers were merged at the interface, while the arrangement of fiber was retained.
The chemoenzymatic approach has been successfully conducted to produce amylose-grafted polypeptides. For example, a maltopentaosyl amine was treated with carboxylate groups of poly(l-glutamic acid) using the condensation agent to produce a maltopentaose-grafted poly(l-glutamic acid). The product was employed as a polymeric primer for the GP-catalyzed enzymatic polymerization to obtain amylose-grafted poly(l-glutamic acid) [157]. Another polypeptide, poly(l-lysin) (PLL) having amino groups, was also used for the chemoenzymatic approach. Maltooligosaccharide was introduced into cholesterol-bearing poly(l-lysine) material by reductive amination [158]. The product, ChMaPLL31 (31 maltopentaoses per 100 lysine residues), forms positively charged nanogel with an average diameter of 50 nm via self-assembly in water. The nanogel was suited as a primer for the GP-catalyzed enzymatic polymerization. The average diameter after the enzymatic polymerization decreased to 30 nm as observed by AFM measurement (Fig. 3.11). The produced nanogel exhibited a weak negative charge, suggesting the shielding of the cationic charged surfaces by the elongated amylose graft chains.

AFM images and schematic illustrations before and after the enzymatic polymerization of ChMaPLL31 nanogels. (Reprinted with permission from ref. [158]. Copyright 2013 American Chemical Society)
A natural polypeptide, poly(γ-glutamic acid) (PGA), has been employed for the chemoenzymatic approach [159] because it has carboxylate groups and is a well-known material for multifarious potential applications in foods, pharmaceuticals, healthcare, water treatment, and other fields. Amine-functionalized maltooligosaccharide primers were first introduced on PGA main-chain by condensation using the condensing agent in aqueous NaOH. The GP-catalyzed enzymatic polymerization was then conducted from the primer chain ends of the product to obtain amylose-grafted PGAs (Fig. 3.12). The products formed hydrogels in reaction media depending on Glc-1-P/primer feed ratios. The amylose graft chains formed double helixes, which contributed to constructing network structure as cross-linking points for hydrogelation. The SEM images of the cryogels, which were obtained by lyophilization of the hydrogels, observed regularly controlled porous morphologies. Furthermore, pore sizes increased upon increasing Glc-1-P/primer feed ratios, while the degrees of substitution of primer on the PGA main-chain did not obviously affect pore sizes.

Chemoenzymatic synthesis of amylose-grafted PGA
3.3 Fabrication of Supramolecules and Controlled Assemblies in Phosphorylase-Catalyzed Enzymatic Polymerization Field
Amylose acts as a host molecule owing to its left-handed helical conformation to form inclusion complexes with various guest molecules, typically monomeric and oligomeric low molecular-weight compounds [160, 161]. The driving force for the binding of guest molecules is hydrophobic interactions, as the amylose cavity is hydrophobic, and therefore, in aqueous solvents, hydrophobic guest molecules are spontaneously included in the amylose cavity (Fig. 3.13). However, a limited number of investigations have been reported on the complexation of amylose with polymeric guest molecules (Fig. 3.13). As the difficulty in the inclusion of polymeric guest molecules into the amylose cavity arises from the necessity for a weak hydrophobic interaction as driving force of the complexation, amylose does not have a sufficient ability to direct the inclusion of long polymeric chains into its cavity. For the direct incorporation of polymeric guests, hydrophilic groups have been introduced at the polymer chain ends, which enhance the complexation ability by amylose in aqueous media [162, 163]. Additional methods for directly forming amylose–polymer inclusion complexes have been achieved by inclusion polymerization and guest-exchange approaches [164,165,166].

Amylose forms inclusion complex with relatively low molecular-weight (small) hydrophobic molecule but largely does not form it with polymeric molecule
An efficient method for the direct construction of amylose–polymer inclusion complexes has been developed by means of the GP-catalyzed enzymatic polymerization. In this enzymatic polymerization system, which involves dispersion with appropriate hydrophobic guest polymers in an aqueous polymerization solvent, the propagation is progressed with the formation of inclusion complexes between the produced amylose and guest polymers [65, 167,168,169,170,171,172]. The elongation of the short α(1→4)-glucan (maltooligosaccharide primer) to the longer α(1→4)-glucan (amylose) is considered to provide sufficient field for more facile complexation of polymeric guests compared to the direct complexation between the polymeric amylose host and guest. The process of this host-guest interaction method for the generation of such inclusion complexes is similar to the way that vines of plants grow and twine around a rod, which is the reason why this polymerization approach has been named “vine-twining polymerization” (Fig. 3.14). Hydrophobic polyethers such as poly(tetrahydrofuran) (PTHF) and poly(oxetane) (POXT) [173, 174]; polyesters such as poly(δ-valerolactone) (PVL), poly(ε-caprolactone) (PCL), and poly(glycolic acid-co-ε-caprolactone) [175,176,177]; polycarbonates such as poly(tetramethylene carbonate) [178]; and poly(ester-ether)s (–CH2CH2C(C=O)OCH2CH2CH2CH2–) [176] have been found to act as guest polymers and included into amylose using this polymerization technique. To investigate the effect of the structures of polyethers on inclusion complexation in vine-twining polymerization, the GP-catalyzed enzymatic polymerization of G-1-P was conducted using polyethers with different alkyl chain lengths, that is, PTHF (4 methylenes), poly(oxetane) (POXT, 3 methylenes), and PEG (2 methylenes). Consequently, the hydrophobic POXT formed an inclusion complex with amylose, whereas vine-twining polymerization with PEG did not induce inclusion complexation. A hydrophilic poly(ester-ether) (–CH2CH2C(=O)OCH2CH2O–) with a shorter methylene length also did not form an inclusion complex. In addition, complexation via vine-twining polymerization has not been achieved with strongly hydrophobic polymers like a polyether, poly(oxepane) with 6 methylenes, due to their aggregation in aqueous buffer solvents as a result of their longer alkyl chains when compared to PTHF and POXT. It, therefore, is concluded that the moderate hydrophobicity of guest polymers is the important factor in determining whether or not amylose will form inclusion complexes during a vine-twining polymerization.

Image for vine-twining polymerization and typical guest polymers
An attempt of a parallel enzymatic polymerization system was made to achieve the formation of an inclusion complex with a strongly hydrophobic polyester [179]. In this approach, two enzymatic polymerizations, the GP-catalyzed enzymatic polymerization of Glc-1-P from the Glc7 primer to produce amylose and the lipase-catalyzed polycondensation of a diol (1,8-octanediol) and a dicarboxylic acid (sebacic acid) to produce a strongly hydrophobic aliphatic polyester as the guest polymer [180, 181], were simultaneously conducted in an aqueous buffer solvent (Fig. 3.15). The analysis of the product supported the formation of the inclusion complex of amylose with the polyester.

Parallel enzymatic polymerization system to produce amylose-polyester inclusion complex
Amylose showed the selective inclusion behavior in certain structures, molecular-weight distributions, and chiralities of the guest polymers in the vine-twining polymerization system. By means of the vine-twining polymerization technique, the selective inclusion complexation toward two resemblant polymers by amylose has been achieved. For example, amylose selectively included one polyether, that is, PTHF from a mixture of PTHF/POXT in the vine-twining polymerization [182]. The selective inclusion by amylose was also found when the vine-twining polymerization was investigated in the presence of a mixture of two resemblant polyesters, PVL/PCL, in which PVL was selectively included by amylose [183].
Amylose also showed selective inclusion behavior toward a specific range of molecular weights of guest polymers in vine-twining polymerization [169]. Synthetic polymers are considered to be mixtures of different molecular-weight analogs, which possess different properties. For example, the molecular weight of PTHF polymers affects its hydrophobicity and water solubility, where low molecular-weight PTHF exhibits water solubility, whereas those with larger molecular weight are hydrophobic and show insolubility in water. When several vine-twining polymerization systems were examined using PTHFs with different average molecular weights, the specific range of molecular weights of all PTHFs was suitably recognized by amylose to form inclusion complexes.
Aside from the chemical structure and molecular weight, amylose showed selectivity toward the chirality in guest polymers in vine-twining polymerization. The selective inclusion of chiral molecules by amylose was achieved using chiral polyesters, poly(lactide)s (PLAs) as guest polymers with three stereoisomers, i.e., poly(l-lactide) (PLLA), poly(d-lactide) (PDLA), and racemic poly(dl-lactide) (PLDLA) [184]. When vine-twining polymerization was conducted using PLLA, an inclusion complex was formed, while from the PDLA and PDLLA, inclusion complexation was not observed. The similar selective inclusion was also observed in vine-twining polymerization using chiral polyalanine (PAlas) stereoisomers as guest polymers [185]. An inclusion complex was formed with poly(d-alanine) (PDAla), whereas inclusion complexes were not obtained with poly(l-alanine) (PLAla) or poly(dl-alanine) (PDLAla). The stereoselective inclusion behavior of amylose based on chirality in vine-twining polymerization is explained by the helical direction of the host and guest polymers. The left-handed helical conformation of PLLA and PDAla is the same direction as that of the host amylose, resulting in their efficient inclusion. In contrast, the opposite and irregular helical conformations of the other stereoisomers are not suitable for binding by the amylose helix.
The vine-twining polymerization approach by the GP-catalyzed enzymatic polymerization has been applied to the preparation of amylosic supramolecular networks based on amylose–polymer inclusion complexes. Such supramolecular network materials, e.g., hydrogels, which are hierarchically composed of inclusion complexes as cross-linking points, were designed as the vine-twining polymerization products by using graft copolymers with hydrophobic graft chains. As shown in Fig. 3.16, the enzymatically produced amylose chains by GP catalysis potentially include the hydrophobic graft chains as guest polymers to form inclusion complexes, which act as cross-linking points to hierarchically construct the supramolecular network structure in aqueous media, giving rise to hydrogels. The hydrophobicity of the graft chains as guest polymers is necessary, while the graft copolymer should generally be water-soluble to efficiently act as a component of hydrogels.

Preparation of amylosic supramolecular networks by vine-twining polymerization using graft copolymers having hydrophilic main-chains and hydrophobic guest graft chains
For example, the hierarchical construction of hydrogels was achieved by the GP-catalyzed enzymatic polymerization of Glc-1-P from Glc7, according to the vine-twining polymerization manner in the presence of a water-soluble graft copolymers composed of hydrophobic PVL or PCL graft chains, that is, poly(acrylic acid sodium salt-graft-δ-valerolactone) (P(AA-Na-g-VL)) [186], CMC (sodium salt)-graft-PCL (NaCMC-g-PCL) [187], and poly(γ-glutamic acid-graft-ε-caprolactone) (PGA-g-PCL) [188]. The enzymatic reaction mixtures completely turned into the hydrogel form, where the hydrophilic main-chains, PAA, NaCMC, and PGA, acted as the main components in the hydrogels. The enzymatically produced amylose included the PVL graft chains between the intermolecular (P(AA-Na-g-VL)s to form inclusion complexes as the polymerization progressed, which acted as cross-linking points for hydrogelation. The ability of the resulting hydrogel to reversibly facilitate enzymatic disruption and reproduction was successfully demonstrated by the combined use of β-amylase-catalyzed hydrolysis of the amylose component in the hydrogel, followed by its reformation by the GP-catalyzed enzymatic polymerization. A film was further formed by adding water, followed by drying, to a powdered sample, which was prepared by lyophilization of the hydrogel from NaCMC-g-PCL.
The mechanical properties of the hydrogels obtained by the above systems using PAA-Na-g-PVL and NaCMC-g-PCL, however, were not sufficient for further applications. To improve the mechanical properties of the hydrogels, PGA has been used as the main-chain of a graft copolymer, based on the viewpoint of its better water retention and moisturizing properties. Indeed, vine-twining polymerization using PGA-g-PCL resulted in the formation of a hydrogel with self-standing properties, indicating much better mechanical properties compared to the abovementioned hydrogels. It was found, moreover, that the resulting hydrogel showed the macroscopic interfacial healing through the GP-catalyzed enzymatic polymerization. The hydrogel initially formed, as the vine-twining polymerization product, was cut into two pieces, and a sodium acetate buffer solution dissolving Glc-1-P and GP was dropped on the surfaces of the hydrogel pieces. After the surfaces were placed in contact with one another, the materials were left standing for the progress of the GP-catalyzed enzymatic polymerization. Consequently, the two pieces were fused at the contacted area. Such healing behavior of the hydrogels on a macroscopic level was induced by the complexation of the enzymatically produced amyloses with the PCL graft chains at the interface. In addition, a porous cryogel and an ion gel were produced by lyophilization and soaking of the hydrogel in an ionic liquid of 1-butyl-3-methylimidazolium chloride (BMIMCl).
The abovementioned maltooligosaccharide-grafted PGA (Glc7-grafted PGA) has also been used for vine-twining polymerization to construct amylosic network structure. The GP-catalyzed enzymatic polymerization of Glc-1-P initiated from the primer chain ends of the graft copolymer was carried out in the presence of different feed ratios of a guest polymer, PCL according to vine-twining polymerization manner [189]. The predominant formation of double helixes from the amylose graft chains in the products in the absence of PCL (as aforementioned) or in the presence of less amount of PCL gave rise to the hierarchical construction of the lager macroscopic network, leading to hydrogelation of the reaction mixtures. In the presence of larger amount of PCL, on the other hand, the smaller network mostly constructed from inclusion complexes was formed according to the vine-twining polymerization manner, resulting in the production of aggregates with the smaller macroscopic network in the reaction mixtures. Furthermore, it was revealed that in the presence of moderate amount of PCL, inclusion complex was initially formed by the shorter amylose chains with the progress of the GP-catalyzed enzymatic polymerization, and subsequently, double helix was produced by the longer amylose chains (Fig. 3.17).

Formation of smaller network structure by inclusion complex and larger network structure by double helix in GP-catalyzed enzymatic polymerization
Supramolecular polymers composed of the continuums of amylose-PTHF and amylose-PLLA inclusion complexes were successfully synthesized by vine-twining polymerizations using Glc7-block-PTHF and Glc7-block-PLLA as primer–guest conjugates (Fig. 3.18a) [190, 191]. In these systems, an enzymatically propagating amylose chain included a PTHF or PLLA segment of another conjugate, and such inclusion complexation among the conjugates consecutively takes place, giving rise to the formation of the linear inclusion supramolecular polymers.

Formation of (a) linear and (b) hyperbranched supramolecular polymers by vine-twining polymerization using primer–guest conjugates
The relative chain orientations of amylose and the two stereoisomers of PLA in inclusion complexes formed in the GP-catalyzed enzymatic polymerization were precisely investigated by using four different primer–guest conjugates, which consisted of a Glc7 segment functionalized at the carboxylate or hydroxy termini of both PLLA and PDLA [129]. The amylose-PLLA supramolecular polymers were formed in the enzymatic polymerization in the presence of both of the two PLLA conjugates, suggesting that the amylose cavity included the guest polymer, regardless of the chain orientation of PLLA. In contrast, the products from two PDLA conjugates were the amylose–PDLA diblock copolymers due to the noninclusion owing to the recognition behavior of amylose for chirality, regardless of the chain orientation of PDLA. The left-handed helixes from both the amylose and PLLA induce inclusion complexation, whereas complexation was not significantly affected by the orientation of the methyl substituents in PLA, which oppositely change according to the relative chain orientation.
Vine-twining polymerization using a branched maltoheptaose-(poly(l-lactide))2 (G7-PLLA2) conjugate resulted in the production of a hyperbranched supramolecular polymer (Fig. 3.18b) [192]. The hyperbranched product formed an ion gel with BMIMCl, which was further converted into a hydrogel upon exchange of the dispersion media by soaking in water. Lyophilization of the resulting hydrogel produced a porous cryogel.
The CDP-catalyzed enzymatic oligomerization of Glc-1-P has been employed to prepare post-functionalizable two-dimensional crystalline cellulose nanosheets. An azido-functionalized cellulose oligomer was obtained by the enzymatic oligomerization of Glc-1-P using β-glucosyl azide as a primer [193]. The products aligned and formed nanosheets of an average thickness of 5.5 nm with antiparallel chain arrangement (cellulose II allomorph). As the azido groups of the cellulose oligomers at the reducing end were located on the sheet surface, the post-functionalization with 1-ethynylpyrene was achieved by copper(I)-catalyzed Huisgen cycloaddition to produce a pyrene-conjugated nanosheet (Fig. 3.19). The nanosheet has been found to act as an artificial hydrolytic enzyme [194]. The as-prepared nanosheets exhibited relatively low hydrolytic activities. However, distorted and smaller nanosheets with larger surface areas, which were prepared by sonication-based mechanical treatment, exhibited significantly greater hydrolytic activities. Through the CDP-catalyzed enzymatic oligomerization from the primer 2-(glucosyloxy)ethyl methacrylate (GEMA), a novel type of two-dimensional methacrylate-containing cellulose nanosheets with a thickness of about 6 nm was directly synthesized by a bottom-up method [195]. The obtained nanosheet was covalently incorporated into PEG matrix through thiol−ene Michael addition, fabricating a series of GEMA-cellulose nanosheet-based nanocomposite hydrogels.

Schematic illustration of the post-functionalization of surface-azidized 2D crystalline cellulose oligomers with 1-ethynyl pyrene through copper(I)-catalyzed Huisgen cycloaddition reactions; cellulose oligomers with azide groups at the reducing end were synthesized by CDP-catalyzed enzymatic oligomerization using Glc-1-P monomers and β-glucosyl azide primers. Reprinted from ref. [193] (Royal Society of Chemistry) under Creative Commons Attribution 3.0 Unported License
The enzymatically synthesized cellulose oligomers by the CDP-catalyzed oligomerization using a cellobiose primer appeared to align perpendicularly to the base plane of the nanoribbons in an antiparallel manner [196]. The analysis of reaction time dependence suggested that the production of nanoribbon network structures was kinetically controlled by the amount of water-insoluble cellulose oligomers produced. A solution state with high macromolecular concentrations, so-called macromolecular crowding, was used to promote the crystallization-driven self-assembly of the enzymatically synthesized cellulose oligomer by CDP catalysis. The enzymatic oligomerization was conducted in the concentrated solutions of water-soluble polymers, such as dextran, PEG, and poly(N-vinylpyrrolidone). The reaction mixtures turned into cellulose oligomer hydrogels composed of well-grown crystalline nanoribbon networks irrespective polymer species [197]. This method was further applied to the one-pot preparation of double network hydrogels composed of the nanoribbons and physically cross-linked gelatin molecules. The effect of solution viscosity on hydrogelation in the enzymatic reaction system was investigated using a highly branched polymer, Ficoll, which created macromolecular crowding conditions with relatively low solution viscosity [198]. The results suggested that a certain level of solution viscosity for the enzymatic synthesis is an essential requirement of hydrogelation. CDP-catalyzed bottom-up synthesis of mechanically and physicochemically stable nanoribbon network hydrogels composed of crystalline cellulose oligomers in which cellulose nanocrystals (CNCs) as model colloidal particles are immobilized spatially [199]. The stiffness of the hydrogels increased with the amount of CNCs incorporated. Protein adsorption property of the cellulose oligomer nanoribbons with primary amino groups was also investigated [200]. The enzymatic synthesis was carried out by the CDP-catalyzed oligomerization using 2-aminoethyl-β-glucose as a primer. The primary amino groups on the nanoribbon surfaces effectively attracted negatively charged proteins but not positively charged ones.
3.4 Amylose Engineering Applications by Phosphorylase-Catalyzed Enzymatic Polymerization
The GP-catalyzed enzymatic polymerization precisely produces amylose and amylosic materials, but the problem in practical application is that Glc-1-P is expensive. To overcome it, an attempt has been made to synthesize Glc-1-P by phosphorolysis of an inexpensive starch catalyzed by Thermus caldophilus GK24 phosphorylase [201]. The optimal pH and temperature were 7.0 and 70 °C for the phosphorolytic reaction producing Glc-1-P. Soluble starch (amylopectin, amylose) turned out to be a better substrate giving a higher yield of Glc-1-P than glycogen, potato starch, etc. As a result, Glc-1-P was produced in a good yield (47%) in the reaction containing 5% soluble starch in 0.7 M potassium phosphate at pH 7.0.
Another possible solution to the expensive problem of Glc-1-P is to combine another enzyme that produces Glc-1-P. The combined action by plural enzymes often shows synergistic effect to efficiently produce target products. Sucrose phosphorylase catalyzes phosphorolysis of sucrose in the presence of Pi to produce Glc-1-P and fructose. The Glc-1-P thus produced in situ by this enzyme can be used for the subsequent GP-catalyzed synthesis of amylose (Fig. 3.20). However, the following antagonistic reaction conditions are conceived when these two individual enzymatic reactions should efficiently occur, in which the first reaction should be carried out in high concentration of Pi, while the Pi should be removed as soon as possible from the media of the second reaction. Waldmann et al. have reported in the system for the production of amylose from sucrose, interestingly, Pi produced in the second GP-catalyzed reaction is recycled for the first sucrose phosphorylase-catalyzed reaction. Therefore, the cooperative action by the two phosphorylases takes place continuously with a constant Pi concentration without any inhibition caused by an accumulation of Pi.

Production of amylose by combined used of sucrose or cellobiose phosphorylase and GP
Thermostable sucrose phosphorylase was generated by introducing a random and site-directed mutagenesis on the sucrose phosphorylase gene from Streptococcus mutans to increase and used together with the triple-mutant thermostable GP (F39 L/N135S/T706I) originally from potato for the production of amylose from sucrose. These thermostable variants of sucrose phosphorylase and GP were employed to optimize the conditions for the production of amylose from sucrose. The yields of amylose produced the two enzymatic reactions from sucrose were higher than those from Glc-1-P. The molecular weights of the produced amyloses were strictly controlled by the sucrose/primer feed ratios, and their M w/M n values were close to 1. The amyloses with molecular weights less than 7.1 × 104 were produced as insoluble particles, whereas those with molecular weights more than 3.05 × 105 were produced in the solution. These results indicated that the properties of amylose differ according to the molecular weights.
For the purpose to produce Glc-1-P in situ, the use of cellobiose phosphorylase combined with GP was also examined (Fig. 3.20). Cellobiose phosphorylase catalyzes phosphorolysis of cellobiose in the presence of Pi to produce Glc-1-P and glucose. When a partially purified cellobiose phosphorylase was incubated with cellobiose and GP in the presence of Pi, various sizes of amylose (from 4.2 × 104 to 7.3 × 105) were produced. However, the yield (38.6%) was not as high as that in the system using sucrose phosphorylase. To improve the yield of amylose, mutarotase and glucose oxidase were added to the initial reaction mixture. The role of these enzymes is to remove glucose derived by the cellobiose-catalyzed reaction, leading to shifting the equilibrium state to phosphorolysis. As the result, the yield of amylose increased to 64.8% by the action of these enzymes.
Branched glucan has been prepared by the combined action of GP and BE on Glc-1-P in the presence of an adequate primer (Fig. 3.21) [202,203,204,205,206,207,208,209]. The molecular weight and branching pattern of the product were controlled by the Glc-1-P/ primer feed ratio and by the relative BE/GP activity ratio, respectively. Thus, various branched glucans were produced by using different BE/GP activity ratios. The produced glucans at high BE/GP activity ratios had more frequently branching points than those produced at low BE/GP ratios. Deinococcus geothermalis glycogen-branching enzyme is known to catalyze the redistribution of short α-glucans via inter- and intramolecular chain transfer from α(1→4)-positions to α(1→6)-positions. The combined use of GP and glycogen-branching enzyme, therefore, gave highly branched amylose from Glc-1-P [203].

Synthesis of branched glucan by combined use of GP and BE
Amylose-modified silica gels have been prepared by the GP-catalyzed enzymatic polymerization using maltooligosaccharide primers having appropriate functional groups at the reducing end, followed by immobilization on the surface of silica gels, by two different approaches (Fig. 3.22) [210, 211]. In approach I, 1,3-aminopropyltriethoxysilane-modified maltooligosaccharide was used of the GP-catalyzed enzymatic polymerization. The produced amylose derivative was immobilized by reaction with silica gel. In approach II, amylose lactone was first prepared by the GP-catalyzed enzymatic polymerization using maltooligosaccharide having potassium gluconate at reducing end (precursor of lactone), followed by the lactonization. The produced lactone was reacted with 3-aminopropyl-silanized silica gel. The two modified silica gels were reacted with a large excess of 3,5-dimethylphenyl isocyanate to convert hydroxy groups in glucose residues to carbamate derivatives. The products can be used as chiral stationary phases in high-performance liquid chromatography, owing to their intrinsic chirality. The modified silica gels display excellent enantioseparation of ten different racemates. The systems are thus widely used to separate racemic compounds into their enantiomers.

Preparation of amylose-grafted silica gels
The GP-catalyzed enzymatic polymerization using modified primers has been applied to an enzyme-responsive artificial chaperone system for protein refolding. Glc5-block-alkyl chain surfactants with different numbers of carbon (e.g., Glc5C8, Glc5C12, and Glc5C16) have been used as primers for the GP-catalyzed enzymatic polymerization, where the reducing end of Glc5 is substituted with an alkyl group (C8, C12, and C16). The primer surfactants formed micelles in water, which dissociated upon the GP-catalyzed enzymatic polymerization [212]. By this property, the micelle-to-vesicle transition of the mixed lipid–primer systems was observed during the enzymatic polymerization. Consequently, Glc5C12 micelles behaved as enzyme-responsive molecular assembly systems. Accordingly, an enzyme-responsive artificial chaperone system by means of the behavior of this surfactant was constructed to enable protein refolding (Fig. 3.23) [213]. The effective refolding of carbonic anhydrase B after either treatment with guanidine hydrochloride or heat denaturation was observed by controlled association between the protein molecules and surfactant primer micelles via the GP-catalyzed enzymatic polymerization.

Schematic illustration of artificial chaperone system by GP-catalyzed enzymatic polymerization of Glc5C12 primer. (Reprinted with permission from ref. [213]. Copyright 2009 Elsevier)
3.5 Conclusion
Polysaccharides are important biobased materials with applications spanning the whole range of cheap commodity plastics to advanced medical applications. Phosphorylases are excellent biocatalysts for the synthesis of well-defined oligo- and polysaccharides. They also provide environmentally friendly processes, because the reactions can be operated using unprotected substrates in aqueous media under mild conditions. They are extensively used for the synthesis of oligo- and polysaccharides as standard materials, in hybrid structures, etc.
Advancements in the discovery, production, and characterization of phosphorylases, like high-throughput screening for enzyme evolution and metabolic pathway engineering, have broadened the scope of possibilities and allowed the improved synthesis of oligo- and polysaccharide libraries and hybrid materials thereof. The synthesized hybrid materials show very interesting properties and can be used for various applications.
It can be envisioned that both the discovery of new phosphorylases and the synthesis of new hybrid materials will be the focus of more intensified research in the near future as the possibilities summarized in this review show the great potential of this area of research, based on the viewpoints of several topics, e.g., synthetic method, functional material, biological function, and environmental aspect.
References
Schuerch C (1986) Polysaccharides. In: Mark HF, Bilkales N, Overberger CG (eds) Encyclopedia of polymer science and engineering, vol 13, 2nd edn. John Wiley & Sons, New York, pp 87–162
Berg JM, Tymoczko JL, Stryer L (2012) Biochemistry, 7th edn. W.H. Freeman, New York
Williams R, Galan MC (2017) Recent advances in organocatalytic glycosylations. Eur J Org Chem 2017:6247–6264
Paulsen H (1982) Advances in selective chemical syntheses of complex oligosaccharides. Angew Chem Int Ed Engl 21:155–173
Schmidt RR (1986) New methods for the synthesis of glycosides and oligosaccharides—are there alternatives to the Koenigs-Knorr method? Angew Chem Int Ed Engl 25:212–235
Toshima K, Tatsuta K (1993) Recent progress in O-glycosylation methods and its application to natural products synthesis. Chem Rev 93:1503–1531
Kobayashi S, Shoda S (1996) Enzymatic synthesis of polysaccharides: a new concept in polymerization chemistry. In: Kamachi M, Nakamura A (eds) New macromolecular architecture and functions. Springer, Heidelberg, pp 171–180
Shoda S, Kobayashi S (1997) Recent developments in the use of enzymes in oligo- and polysaccharide synthesis. Trends Polym Sci 5:109–115
Shoda S, Fujita M, Kobayashi S (1998) Glycanase-catalyzed synthesis of non-natural oligosaccharides. Trends Glycosci Glycotechnol 10:279–289
Kobayashi S, Shoda S, Donnelly M et al (1999) Enzymatic synthesis of cellulose. In: Bucke C (ed) Methods in biotechnology 10, Carbohydrate biotechnology protocols. Humana Press, Totowa, NJ, pp 57–69
Kobayashi S, Kimura S (1999) In vitro biosynthesis of natural and unnatural polysaccharides catalyzed by isolated hydrolases. In: Steinbuechel A (ed) Biochemical principles and mechanism of biosynthesis and biodegradation of polymers. Wiley-VCH, Weinheim, Germany, pp 161–167
Kobayashi S, Uyama H, Kimura S (2001) Enzymatic polymerization. Chem Rev 101:3793–3818
Kobayashi S, Sakamoto J, Kimura S (2001) In vitro synthesis of cellulose and related polysaccharides. Prog Polym Sci 26:1525–1560
Shoda S, Izumi R, Fujita M (2003) Green process in glycotechnology. Bull Chem Soc Jpn 76:1–13
Kobayashi S (2007) New developments of polysaccharide synthesis via enzymatic polymerization. Proc Jpn Acad Ser B 83:215–247
Kobayashi S, Makino A (2009) Enzymatic polymer synthesis: an opportunity for green polymer chemistry. Chem Rev 109:5288–5353
Makino A, Kobayashi S (2010) Chemistry of 2-oxazolines: a crossing of cationic ring-opening polymerization and enzymatic ring-opening polyaddition. J Polym Sci Polym Chem 48:1251–1270
Kadokawa J, Kobayashi S (2010) Polymer synthesis by enzymatic catalysis. Curr Opin Chem Biol 14:145–153
Kadokawa J (2011) Precision polysaccharide synthesis catalyzed by enzymes. Chem Rev 111:4308–4345
Kobayashi S (2005) Challenge of synthetic cellulose. J Polym Sci Polym Chem 43:693–710
Shoda S, Uyama H, Kadokawa J et al (2016) Enzymes as green catalysts for precision macromolecular synthesis. Chem Rev 116:2307–2413
Nagel B, Dellweg H, Gierasch LM (1992) Glossary for chemists of terms used in biotechnology. Pure Appl Chem 64:143–168
Lairson LL, Henrissat B, Davies GJ et al (2008) Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem 77:521–555
Qian XP, Sujino K, Palcic MM et al (2002) Glycosyltransferases in oligosaccharide synthesis (Reprinted from Glycochemistry: principles, synthesis, and applications, pg 535-565, 2001). J Carbohydr Chem 21:911–942
Davies GJ, Charnock SJ, Henrissat B (2001) The enzymatic synthesis of glycosidic bonds: “Glycosynthases” and glycosyltransferases. Trends Glycosci Glycotechnol 13:105–120
Blixt O, Razi N (2008) In: Fraser-Reis B, Tatsuta K, Thiem J (eds) Glycoscience. Springer, Berlin
Song J, Zhang HC, Li L et al (2006) Enzymatic biosynthesis of oligosacch a rides and glycoconjugates. Curr Org Synth 3:159–168
Jakeman DL, Withers SG (2002) Glycosynthases: new tools for oligosaccharide synthesis. Trends Glycosci Glycotechnol 14:13–25
Feng J, Zhang P, Cui YL et al (2017) Regio- and stereospecific O-glycosylation of phenolic compounds catalyzed by a fungal glycosyltransferase from Mucor hiemalis. Adv Synth & Catal 359:995–1006
Li YH, Xue MY, Sheng X et al (2016) Donor substrate promiscuity of bacterial beta 1-3-N-acetylglucosaminyltransferases and acceptor substrate flexibility of beta 1-4-galactosyltransferases. Bioorg Med Chem 24:1696–1705
Macdonald SS, Patel A, Larmour VLC et al (2018) Structural and mechanistic analysis of a beta-glycoside phosphorylase identified by screening a metagenomic library. J Biol Chem 293:3451–3467
McArthur JB, Chen X (2016) Glycosyltransferase engineering for carbohydrate synthesis. Biochem Soc Trans 44:129–142
Palcic MM (2011) Glycosyltransferases as biocatalysts. Curr Opin Chem Biol 15:226–233
Kitaoka M, Hayashi K (2002) Carbohydrate-processing phosphorolytic enzymes. Trends Glycosci Glycotechnol 14:35–50
Nakai H, Kitaoka M, Svensson B et al (2013) Recent development of phosphorylases possessing large potential for oligosaccharide synthesis. Curr Opin Chem Biol 17:301–309
Puchart V (2015) Glycoside phosphorylases: structure, catalytic properties and biotechnological potential. Biotechnol Adv 33:261–276
Chaen H, Nishimoto T, Nakada T et al (2001) Enzymatic synthesis of kojioligosaccharides using kojibiose phosphorylase. J Biosci Bioeng 92:177–182
Kitaoka M, Sasaki T, Taniguchi H (1991) Synthesis of laminarioligosaccharides using crude extract of Euglena-gracilis Z-cells. Agric Biol Chem Tokyo 55:1431–1432
Nakajima M, Toyoizumi H, Abe K et al (2014) 1, 2-β-Oligoglucan phosphorylase from Listeria innocua. PLoS One 9:e92353
O'Neill EC, Field RA (2015) Enzymatic synthesis using glycoside phosphorylases. Carbohydr Res 403:23–37
Kadokawa J (2016) Precision synthesis of functional polysaccharide materials by phosphorylase-catalyzed enzymatic reactions. Polymers 8:138. https://doi.org/10.3390/polym8040138
Kadokawa J (2017) α-Glucan phosphorylase: a useful catalyst for precision enzymatic synthesis of oligo- and polysaccharides. Curr Org Chem 21:1192–1204
Iwanow L (1902) Über die umwandlungen des phosphors beim keimen der wicke. Ber Deutsch Bot Ges 20:366–372
Zaleski W (1906) Über die rolle der enzyme bei der umwandlung organischer phosphorverbindungen in keimenden samen. Ber Deutsch Bot Ges 24:285–291
Zaleski W (1911) Über die rolle der nucleoproteide in den pflanzen. Ber Deutsch Bot Ges 29:146–155
Suzuki U, Yoshimura K, Takaishi M (1906) On the occurrence of an enzyme which decomposes anhydrooxymethylenephosphoric acid. Tokyo Kagaku Kaishi 27:1330–1342
Bodńar J (1925) Biochemie des phosphorsäurestoffwechsels der höheren pflanzen. Über die enzymatische Überführung der anorganischen phosphorsäure in organische form. Biochem Z 165:1–15
Cori GT, Cori CF (1936) The formation of hexosephosphate esters in frog muscle. J Biol Chem 116:119–128
Cori GT, Cori CF (1936) An unusual case of esterification in muscle. J Biol Chem 116:129–132
Cori CF, Colowick SP, Cori GT (1937) The isolation and synthesis of glucose-1-phosphoric acid. J Biol Chem 121:465–477
Cori CF, Cori GT (1937) Formation of glucose-1-phosphoric acid in muscle extract. Proc Soc Exp Biol Med 36:119–122
Kiessling W (1938) Preparation in a pure state of glucose 1 phosphoric acid (Cori-ester). Biochem Z 298:421–430
Wolfrom ML, Pletcher DE (1941) The structure of the Cori ester. J Am Chem Soc 63:1050–1053
Palm D, Klein HW, Schinzel R et al (1990) The role of pyridoxal 5′-phosphate in glycogen-phosphorylase catalysis. Biochemistry 29:1099–1107
Schäfner A, Specht H (1938) Über die amylasen der hefe und über die umsetzungen der glukose-1-phosphorsäure durch hefeextrakte. Naturwissenschaften 26:494–495
Kiessling W (1939) Über ein neues fermentprotein der hefe und eine reversible enzymatische synthese des glykogens. Naturwissenschaften 27:129–130
Cori CF, Schmidt G, Cori GT (1939) Synthesis of a polysaccharide from glucose-1-phosphate in muscle extract. Science 89:464–465
Hanes CS (1940) The breakdown and synthesis of starch by an enzyme system from pea seeds. Proc Roy Soc B 128:421–450
Ostern P, Holmes E (1939) Formation and breakdown of glycogen in the liver. Nature 144:34–34
Cori GT, Cori CF, Schmidt G (1939) The role of glucose-1-phosphate in the formation of blood sugar and synthesis of glycogen in the liver. J Biol Chem 129:629–639
Ostern P, Herbert D, Holmes E (1939) Formation and breakdown of glycogen in the liver. Biochem J 33:1858–1878
Imberty A, Chanzy H, Perez S et al (1988) The double-helical nature of the crystalline part of A-starch. J Mol Biol 201:365–378
Imberty A, Perez S (1988) A revisit to the three-dimensional structure of B-type starch. Biopolymers 27:1205–1221
Kadokawa J, Kaneko Y (2013) Engineering of polysaccharide materials – by phosphorylase-catalyzed enzymatic chain-elongation. Pan Stanford Publishing Pte Ltd, Singapore
Kadokawa J (2014) Chemoenzymatic synthesis of functional amylosic materials. Pure Appl Chem 86:701–709
Omagari Y, Kadokawa J (2011) Synthesis of heteropolysaccharides having amylose chains using phosphorylase-catalyzed enzymatic polymerization. Kobunshi Ronbunshu 68:242–249
Kadoakwa J (2012) Synthesis of amylose-grafted polysaccharide materials by phosphorylase-catalyzed enzymatic polymerization. In: Smith PB, Gross RA (eds) Biobased monomers, polymers, and materials. Vol 1043. ACS symposium series 1105. American Chemical Society, Washington, DC, pp 237–255
Kadoakwa J (2013) Synthesis of new polysaccharide materials by phosphorylase-catalyzed enzymatic α-glycosylations using polymeric glycosyl acceptors. In: Cheng HN, Gross RA, Smith PB (eds) Green polymer chemistry: biocatalysis and materials II. Vol 1144. ACS symposium series 1144. American Chemical Society, Washington, DC, pp 141–161
Nishimura T, Akiyoshi K (2016) Amylose engineering: phosphorylase-catalyzed polymerization of functional saccharide primers for glycobiomaterials. Wiley Interdiscip Rev Nanomed Nanobiotechnol 9
Green DE, Stumpf PK (1942) Starch phosphorylase of potato. J Biol Chem 142:355–366
Weibull C, Tiselius A (1945) A study of the starch phosphorylase of potato. Arkiv för Kemi Mineralogi Och Geologi 19:1–25
Whelan WJ, Bailey JM (1954) The action pattern of potato phosphorylase. Biochem J 58:560–569
Pfannemüller B, Burchard W (1969) Difference in course of phosphorolytic synthesis of amylose with maltotriose and higher maltodextrines as initiators. Makromol Chem 121:1
Pfannemüller B (1975) Living polymerization and enzymatic polysaccharide synthesis. Naturwissenschaften 62:231–233
Suganuma T, Kitazono JI, Yoshinaga K et al (1991) Determination of kinetic-parameters for maltotriose and higher maltooligosaccharides in the reactions catalyzed by α-D-glucan phosphorylase from potato. Carbohydr Res 217:213–220
Kadokawa J (2011) Facile synthesis of unnatural oligosaccharides by phosphorylase-catalyzed enzymatic glycosylations using new glycosyl donors. In: Gordon NS (ed) Oligosaccharides: sources, properties and applications. Nova Science Publishers, Inc., Hauppauge, pp 269–281
Kadokawa J (2013) Synthesis of non-natural oligosaccharides by α-glucan phosphorylase-catalyzed enzymatic glycosylations using analogue substrates of α-D-glucose 1-phosphate. Trends Glycosci Glycotechnol 25:57–69
Kadoakwa J (2015) Enzymatic synthesis of non-natural oligo- and polysaccharides by phosphorylase-catalyzed glycosylations using analogue substrates. In: Cheng HN, Gross RA, Smith PB (eds) Green polymer chemistry: biobased materials and biocatalysis. Vol 1192. ACS symposium series 1192. American Chemical Society, Washington, DC, pp 87–99
Percival MD, Withers SG (1988) Applications of enzymes in the synthesis and hydrolytic study of 2-deoxy-α-D-glucopyranosyl phosphate. Can J Chem 66:1970–1972
Evers B, Mischnick P, Thiem J (1994) Synthesis of 2-deoxy-α-D-arabino-hexopyranosyl phosphate and 2-deoxy-maltooligosaccharides with phosphorylase. Carbohydr Res 262:335–341
Evers B, Thiem J (1997) Further syntheses employing phosphorylase. Bioorg Med Chem 5:857–863
Nawaji M, Izawa H, Kaneko Y et al (2008) Enzymatic synthesis of α-D-xylosylated maltooligosaccharides by phosphorylase-catalyzed xylosylation. J Carbohydr Chem 27:214–222
Nawaji M, Izawa H, Kaneko Y et al (2008) Enzymatic α-glucosaminylation of maltooligosaccharides catalyzed by phosphorylase. Carbohydr Res 343:2692–2696
Kawazoe S, Izawa H, Nawaji M et al (2010) Phosphorylase-catalyzed N-formyl-α-glucosaminylation of maltooligosaccharides. Carbohydr Res 345:631–636
Bhuiyan SH, Rus’d AA, Kitaoka M et al (2003) Characterization of a hyperthermostable glycogen phosphorylase from Aquifex aeolicus expressed in Escherichia coli. J Mol Catal 22:173–180
Umegatani Y, Izawa H, Nawaji M et al (2012) Enzymatic α-glucuronylation of maltooligosaccharides using α-glucuronic acid 1-phosphate as glycosyl donor catalyzed by a thermostable phosphorylase from Aquifex aeolicus VF5. Carbohydr Res 350:81–85
Takemoto Y, Izawa H, Umegatani Y et al (2013) Synthesis of highly branched anionic α-glucans by thermostable phosphorylase-catalyzed α-glucuronylation. Carbohydr Res 366:38–44
Takata H, Takaha T, Okada S et al (1996) Cyclization reaction catalyzed by branching enzyme. J Bacteriol 178:1600–1606
Takata H, Takaha T, Okada S et al (1996) Structure of the cyclic glucan produced from amylopectin by Bacillus stearothermophilus branching enzyme. Carbohydr Res 295:91–101
Takata H, Takaha T, Nakamura H et al (1997) Production and some properties of a dextrin with a narrow size distribution by the cyclization reaction of branching enzyme. J Ferment Bioeng 84:119–123
Takata Y, Shimohigoshi R, Yamamoto K et al (2014) Enzymatic synthesis of dendritic amphoteric α-glucans by thermostable phosphorylase catalysis. Macromol Biosci 14:1437–1443
Shimohigoshi R, Takemoto Y, Yamamoto K et al (2013) Thermostable α-glucan phosphorylase-catalyzed successive α-mannosylations. Chem Lett 42:822–824
Kadokawa J, Shimohigoshi R, Yamashita K et al (2015) Synthesis of chitin and chitosan stereoisomers by thermostable α-glucan phosphorylase-catalyzed enzymatic polymerization of α-D-glucosamine 1-phosphate. Org Biomol Chem 13:4336–4343
Borgerding J (1972) Phosphate deposits in digestion systems. J Water Pollut Control Fed 44:813–819
Yui T, Uto T, Nakauchida T et al (2018) Double helix formation from non-natural amylose analog polysaccharides. Carbohydr Polym 189:184–189
Yamashita K, Yamamoto K, Kadoakwa J (2015) Synthesis of non-natural heteroaminopolysaccharides by α-glucan phosphorylase-catalyzed enzymatic copolymerization: α(1->4)-linked glucosaminoglucans. Biomacromolecules 16:3989–3994
Baba R, Yamamoto K, Kadokawa J (2016) Synthesis of α(1-->4)-linked non-natural mannoglucans by alpha-glucan phosphorylase-catalyzed enzymatic copolymerization. Carbohydr Polym 151:1034–1039
Nakauchida T, Takata Y, Yamamoto K et al (2016) Chemoenzymatic synthesis and pH-responsive properties of amphoteric block polysaccharides. Org Biomol Chem 14:6449–6456
Sheth K, Alexander JK (1969) Purification and properties of β-1, 4-oligoglucan – orthophosphate glucosyltransferase from clostridium thermocellum. J Biol Chem 244:457–464
Samain E, Lancelonpin C, Ferigo F et al (1995) Phosphorolytic synthesis of cellodextrins. Carbohydr Res 271:217–226
Nakai H, Hachem MA, Petersen BO et al (2010) Efficient chemoenzymatic oligosaccharide synthesis by reverse phosphorolysis using cellobiose phosphorylase and cellodextrin phosphorylase from Clostridium thermocellum. Biochimie 92:1818–1826
Petrovic DM, Kok I, Woortman AJ et al (2015) Characterization of oligocellulose synthesized by reverse phosphorolysis using different cellodextrin phosphorylases. Anal Chem 87:9639–9646
Hiraishi M, Igarashi K, Kimura S et al (2009) Synthesis of highly ordered cellulose II in vitro using cellodextrin phosphorylase. Carbohydr Res 344:2468–2473
Moreau V, Viladot JL, Samain E et al (1996) Design and chemoenzymatic synthesis of thiooligosaccharide inhibitors of 1,3:1,4-β-D-glucanases. Bioorg Med Chem 4:1849–1855
Hrmova M, Fincher GB, Viladot JL et al (1998) Chemoenzymic synthesis of (1 -> 3,1 -> 4)-β-D-glucooligosaccharides for subsite mapping of (1 ->,3,1 -> 4)-β-D-glucan endohydrolases. J Chem Soc Perk Trans 1:3571–3576
Choudhury AK, Kitaoka M, Hayashi K (2003) Synthesis of a cellobiosylated dimer and trimer and of cellobiose-coated polyamidoamine (PAMAM) dendrimers to study accessibility of an enzyme, cellodextrin phosphorylase. Eur J Org Chem 2003:2462–2470
Tran HG, Desmet T, Saerens K et al (2012) Biocatalytic production of novel glycolipids with cellodextrin phosphorylase. Bioresour Technol 115:84–87
Okada H, Fukushi E, Onodera S et al (2003) Synthesis and structural analysis of five novel oligosaccharides prepared by glucosyltransfer from β-d-glucose 1-phosphate to isokestose and nystose using Thermoanaerobacter brockii kojibiose phosphorylase. Carbohydr Res 338:879–885
Takahashi N, Okada H, Fukushi E et al (2005) Structural analysis of six novel oligosaccharides synthesized by glucosyl transfer from β-d-glucose 1-phosphate to raffinose and stachyose using Thermoanaerobacter brockii kojibiose phosphorylase. Tetrahedron Asymmetry 16:57–63
Watanabe H, Higashiyama T, Aga H et al (2005) Enzymatic synthesis of a 2-O-α-D-glucopyranosyl cyclic tetrasaccharide by kojibiose phosphorylase. Carbohydr Res 340:449–454
Takahashi N, Fukushi E, Onodera S et al (2007) Three novel oligosaccharides synthesized using Thermoanaerobacter brockii kojibiose phosphorylase. Chem Cent J 1:18
Emmerling W, Pfannemüller B (1978) Block copolymers with monosaccharide, disaccharide and oligosaccharide side-chains linked through amide bonds. Chemiker-Zeitung 102:233–233
Emmerling WN, Pfannemüller B (1981) Preparative methods for the preparation of higher maltooligomers and their coupling with aliphatic diamines. Stärke 33:202–208
Müller-Fahrnow A, Hilgenfeld R, Hesse H et al (1988) Amphiphile properties of synthetic glycolipids based on amide linkages 3. Molecular and crystal-structures of N-(normal-heptyl)-D-gluconamide and N-(normal-decyl)-D-gluconamide. Carbohydr Res 176:165–174
Taravel FR, Pfannemüller B (1990) Amphiphilic properties of synthetic glycolipids based on amide linkages 4. C-13 NMR spectroscopic studies on the gelation of N-octyl-D-gluconamide in aqueous-solution. Makromol Chem 191:3097–3106
Tuzov I, Cramer K, Pfannemüller B et al (1995) Molecular-structure of self-organized layers of N-octyl-D-gluconamide. Adv Mater 7:656–659
Pfannemüller B, Schmidt M, Ziegast G et al (1984) Properties of a once-broken wormlike chain based on amylose tricarbanilate – light-scattering, viscosity, and dielectric-relaxation. Macromolecules 17:710–716
Pfannemüller B, Kühn I (1988) Amphiphilic properties of synthetic glycolipids based on amide linkages 3. Temperature and concentration-dependence of the reduced viscosity of gel-forming alkyl gluconamides. Makromol Chem 189:2433–2442
Emmerling WN, Pfannemüller B (1983) Chemical synthesis of branched polysaccharides.10. Polymers with monosaccharide and oligosaccharide side-chains linked by amide bonds. Makromol Chem 184:1441–1458
Biermann M, Schmid K, Schulz P (1993) Alkylpolyglucosides – technology and properties. Stärke 45:281–288
Hill K, Rhode O (1999) Sugar-based surfactants for consumer products and technical applications. Fett-Lipid 101:25–33
von Rybinski W, Hill K (1998) Alkyl polyglycosides – properties and applications of a new class of surfactants. Angew Chem Int Ed Engl 37:1328–1345
Niemann C, Nuck R, Pfannemüller B et al (1990) Phosphorolytic synthesis of low-molecular-weight amyloses with modified terminal groups. Carbohydr Res 197:187–196
Ziegast G, Pfannemüller B (1987) Linear and star-shaped hybrid polymers 4. Phosphorolytic syntheses with di-functional, oligo-functional and multifunctional primers. Carbohydr Res 160:185–204
Loos K, Stadler R (1997) Synthesis of amylose-block-polystyrene rod-coil block copolymers. Macromolecules 30:7641–7643
Loos K, Müller AHE (2002) New routes to the synthesis of amylose-block-polystyrene rod-coil block copolymers. Biomacromolecules 3:368–373
Loos K, Böker A, Zettl H et al (2005) Micellar aggregates of amylose-block-polystyrene rod-coil block copolymers in water and THF. Macromolecules 38:873–879
Rachmawati R, de Gier HD, Woortman AJJ et al (2015) Synthesis of telechelic and three-arm polytetrahydrofuran-block-amylose. Macromol Chem Phys 216:1091–1102
Tanaka T, Sasayama S, Yamamoto K et al (2015) Evaluating relative chain orientation of amylose and poly(L-lactide) in inclusion complexes formed by vine-twining polymerization using primer–guest conjugates. Macromol Chem Phys 216:794–800
Kumar K, Woortman AJJ, Loos K (2015) Synthesis of amylose-b-P2VP block copolymers. Macromol Rapid Commun 36:2097–2101
Akiyoshi K, Kohara M, Ito K et al (1999) Enzymatic synthesis and characterization of amphiphilic block copolymers of poly(ethylene oxide) and amylose. Macromol Rapid Commun 20:112–115
Akiyoshi K, Maruichi N, Kohara M et al (2002) Amphiphilic block copolymer with a molecular recognition site: induction of a novel binding characteristic of amylose by self-assembly of poly(ethylene oxide)-block-amylose in chloroform. Biomacromolecules 3:280–283
Ziegast G, Pfannemüller B (1984) Linear and star-shaped hybrid polymers 3. An improved purification procedure for coupling products of oligosaccharides by amide linkage. Makromol Chem 185:1855–1866
Ziegast G, Pfannemüller B (1984) Linear and star-shaped hybrid polymers 1. A new method for the conversion of hydroxyl end groups of poly(oxyethylene) and other polyols into amino end groups. Makromol Chem Rapid Commun 5:363–371
Ziegast G, Pfannemüller B (1984) Linear and star-shaped hybrid polymers 2. Coupling of monosaccharide and oligosaccharide to alpha, omega-diamino substituted poly(oxyethylene) and multifunctional amines by amide linkage. Makromol Chem Rapid Commun 5:373–379
Calder PC (1991) Glycogen structure and biogenesis. Int J Biochem 23:1335–1352
Manners DJ (1991) Recent developments in our understanding of glycogen structure. Carbohydr Polym 16:37–82
Izawa H, Nawaji M, Kaneko Y et al (2009) Preparation of glycogen-based polysaccharide materials by phosphorylase-catalyzed chain elongation of glycogen. Macromol Biosci 9:1098–1104
Takata Y, Yamamoto K, Kadokawa J (2015) Preparation of pH-responsive amphoteric glycogen hydrogels by α-glucan phosphorylase-catalyzed successive enzymatic reactions. Macromol Chem Phys 216:1415–1420
Kobayashi K, Kamiya S, Enomoto N (1996) Amylose-carrying styrene macromonomer and its homo- and copolymers: synthesis via enzyme-catalyzed polymerization and complex formation with iodine. Macromolecules 29:8670–8676
Narumi A, Kawasaki K, Kaga H et al (2003) Glycoconjugated polymer 6. Synthesis of poly[styrene-block-(styrene-graft-amylose)] via potato phosphorylase-catalyzed polymerization. Polym Bull 49:405–410
von Braunmühl V, Jonas G, Stadler R (1995) Enzymatic grafting of amylose from poly(dimethylsiloxanes). Macromolecules 28:17–24
Sasaki Y, Kaneko Y, Kadokawa J (2009) Chemoenzymatic synthesis of amylose-grafted polyacetylene by polymer reaction manner and its conversion into organogel with DMSO by cross-linking. Polym Bull 62:291–303
Kaneko Y, Matsuda S, Kadokawa J (2010) Chemoenzymatic synthesis of amylose-grafted poly(vinyl alcohol). Polym Chem 1:193–197
Nishimura T, Mukai S, Sawada S et al (2015) Glyco star polymers as helical multivalent host and biofunctional nano-platform. ACS Macro Lett 4:367–371
Mazzocchetti L, Tsoufis T, Rudolf P et al (2014) Enzymatic synthesis of amylose brushes revisited: details from X-ray photoelectron spectroscopy and spectroscopic ellipsometry. Macromol Biosci 14:186–194
Cao Z, Woortman AJJ, Rudolf P et al (2015) Facile synthesis and structural characterization of amylose-fatty acid inclusion complexes. Macromol Biosci 15:691–697
Matsuda S, Kaneko Y, Kadokawa J (2007) Chemoenzymatic synthesis of amylose-grafted chitosan. Macromol Rapid Commun 28:863–867
Kaneko Y, Matsuda S, Kadokawa J (2007) Chemoenzymatic syntheses of amylose-grafted chitin and chitosan. Biomacromolecules 8:3959–3964
Omagari Y, Matsuda S, Kaneko Y et al (2009) Chemoenzymatic synthesis of amylose-grafted cellulose. Macromol Biosci 9:450–455
Egashira N, Yamamoto K, Kadokawa J (2017) Enzymatic grafting of amylose on chitin nanofibers for hierarchical construction of controlled microstructures. In: Polym Chem, vol 8, p 3279
Tanaka K, Yamamoto K, Kadokawa J (2014) Facile nanofibrillation of chitin derivatives by gas bubbling and ultrasonic treatments in water. Carbohydr Res 398:25–30
Omagari Y, Kaneko Y, Kadokawa J (2010) Chemoenzymatic synthesis of amylose-grafted alginate and its formation of enzymatic disintegratable beads. Carbohydr Polym 82:394–400
Arimura T, Omagari Y, Yamamoto K et al (2011) Chemoenzymatic synthesis and hydrogelation of amylose-grafted xanthan gums. Int J Biol Macromol 49:498–503
Kadokawa J, Arimura T, Takemoto Y et al (2012) Self-assembly of amylose-grafted carboxymethyl cellulose. Carbohydr Polym 90:1371–1377
Hatanaka D, Takemoto Y, Yamamoto K et al (2013) Hierarchically self-assembled nanofiber films from amylose-grafted carboxymethyl cellulose. Fibers 2:34–44
Kamiya S, Kobayashi K (1998) Synthesis and helix formation of saccharide-poly(L-glutamic acid) conjugates. Macromol Chem Phys 199:1589–1596
Morimoto N, Yamazaki M, Tamada J et al (2013) Polysaccharide-hair cationic polypeptide nanogels: self-assembly and enzymatic polymerization of amylose primer-modified cholesteryl poly(L-lysine). Langmuir 29:7509–7514
Shouji T, Yamamoto K, Kadokawa J (2017) Chemoenzyamtic synthesis and self-assembling gelation behavior of amylose-grafted poly(γ-glutamic acid). Int J Biol Macromol 97:99–105
Sarko A, Zugenmaier P (1980) Crystal structures of amylose and its derivatives. In: French AD, Gardner KH (eds) Fiber diffraction methods. Vol 141. ACS symposium series 141. American Chemical Society, Washington, DC, pp 459–482
Putseys JA, Lamberts L, Delcour JA (2010) Amylose-inclusion complexes: formation, identity and physico-chemical properties. J Cereal Sci 51:238–247
Shogren RL (1993) Complexes of starch with telechelic poly(epsilon-caprolactone) phosphate. Carbohydr Polym 22:93–98
Shogren RL, Greene RV, Wu YV (1991) Complexes of starch polysaccharides and poly(ethylene coacrylic acid) – structure and stability in solution. J Appl Polym Sci 42:1701–1709
Star A, Steuerman DW, Heath JR et al (2002) Starched carbon nanotubes. Angew Chem Int Ed 41:2508–1512
Ikeda M, Furusho Y, Okoshi K et al (2006) A luminescent poly(phenylenevinylene)-amylose composite with supramolecular liquid crystallinity. Angew Chem Int Ed 45:6491–6495
Kumar K, Woortman AJJ, Loos K (2013) Synthesis of amylose-polystyrene inclusion complexes by a facile preparation route. Biomacromolecules 14:1955–1960
Kaneko Y, Kadokawa J (2005) Vine-twining polymerization: a new preparation method for well-defined supramolecules composed of amylose and synthetic polymers. Chem Rec 5:36–46
Kaneko Y, Kadokawa J (2006) Synthesis of nanostructured bio-related materials by hybridization of synthetic polymers with polysaccharides or saccharide residues. J Biomater Sci Polym Ed 17:1269–1284
Kaneko Y, Beppu K, Kadokawa J (2009) Amylose selectively includes a specific range of molecular weights in poly(tetrahydrofuran)s in vine-twining polymerization. Polym J 41:792–796
Kadokawa J (2012) Preparation and applications of amylose supramolecules by means of phosphorylase-catalyzed enzymatic polymerization. Polymers 4:116–133
Kadokawa J (2013) Architecture of amylose supramolecules in form of inclusion complexes by phosphorylase-catalyzed enzymatic polymerization. Biomolecules 3:369–385
Orio S, Yamamoto K, Kadokawa J (2017) Preparation and material application of amylose-polymer inclusion complexes by enzymatic polymerization approach. Polymers 9:729. https://doi.org/10.3390/polym9120729
Kadokawa J, Kaneko Y, Tagaya H et al (2001) Synthesis of an amylose-polymer inclusion complex by enzymatic polymerization of glucose 1-phosphate catalyzed by phosphorylase enzyme in the presence of polythf: a new method for synthesis of polymer-polymer inclusion complexes. Chem Commun 5:449–450
Kadokawa J, Kaneko Y, Nagase S et al (2002) Vine-twining polymerization: amylose twines around polyethers to form amylose – polyether inclusion complexes. Chem Eur J 8:3321–3326
Kadokawa J, Kaneko Y, Nakaya A et al (2001) Formation of an amylose-polyester inclusion complex by means of phosphorylase-catalyzed enzymatic polymerization of α-D-glucose 1-phosphate monomer in the presence of poly(ε-caprolactone). Macromolecules 34:6536–6538
Kadokawa J, Nakaya A, Kaneko Y et al (2003) Preparation of inclusion complexes between amylose and ester-containing polymers by means of vine-twining polymerization. Macromol Chem Phys 204:1451–1457
Nomura S, Kyutoku T, Shimomura N et al (2011) Preparation of inclusion complexes composed of amylose and biodegradable poly(glycolic acid-co-ε-caprolactone) by vine-twining polymerization and their lipase-catalyzed hydrolysis behavior. Polym J 43:971–977
Kaneko Y, Beppu K, Kadokawa JI (2008) Preparation of amylose/polycarbonate inclusion complexes by means of vine-twining polymerization. Macromol Chem Phys 209:1037–1042
Kaneko Y, Saito Y, Nakaya A et al (2008) Preparation of inclusion complexes composed of amylose and strongly hydrophobic polyesters in parallel enzymatic polymerization system. Macromolecules 41:5665–5670
Kobayashi S, Uyama H, Suda S et al (1997) Dehydration polymerization in aqueous medium catalyzed by lipase. Chem Lett 26:105–105
Suda S, Uyama H, Kobayashi S (1999) Dehydration polycondensation in water for synthesis of polyesters by lipase catalyst. Proc Jpn Acad B Phys 75:201–206
Kaneko Y, Beppu K, Kadokawa JI (2007) Amylose selectively includes one from a mixture of two resemblant polyethers in vine-twining polymerization. Biomacromolecules 8:2983–2985
Kaneko Y, Beppu K, Kyutoku T et al (2009) Selectivity and priority on inclusion of amylose toward guest polyethers and polyesters in vine-twining polymerization. Polym J 41:279–286
Kaneko Y, Ueno K, Yui T et al (2011) Amylose’s recognition of chirality in polylactides on formation of inclusion complexes in vine-twining polymerization. Macromol Biosci 11:1407–1415
Gotanda R, Yamamoto K, Kadokawa J (2016) Amylose stereoselectively includes poly(D-alanine) to form inclusion complex in vine-twining polymerization: a novel saccharide-peptide supramolecular conjugate. Macromol Chem Phys 217:1074
Kaneko Y, Fujisaki K, Kyutoku T et al (2010) Preparation of enzymatically recyclable hydrogels through the formation of inclusion complexes of amylose in a vine-twining polymerization. Chem Asian J 5:1627–1633
Kadokawa J, Nomura S, Hatanaka D et al (2013) Preparation of polysaccharide supramolecular films by vine-twining polymerization approach. Carbohydr Polym 98:611–617
Kadokawa J, Tanaka K, Hatanaka D et al (2015) Preparation of multiformable supramolecular gels through helical complexation by amylose in vine-twining polymerization. Polym Chem 6:6402–6408
Kadokawa J, Shoji T, Yamamoto K (2018) Preparation of supramolecular network materials by means of amylose helical assemblies. Polymer 140:73–79
Tanaka T, Sasayama S, Nomura S et al (2013) An amylose-poly(L-lactide) inclusion supramolecular polymer: enzymatic synthesis by means of vine-twining polymerization using a primer-guest conjugate. Macromol Chem Phys 214:2829–2834
Tanaka T, Tsutsui A, Gotanda R et al (2015) Synthesis of amylose-polyether inclusion supramolecular polymers by vine-twining polymerization using maltoheptaose-functionalized poly(tetrahydrofuran) as a primer-guest conjugate. J Appl Glycosci 62:135–141
Tanaka T, Gotanda R, Tsutsui A et al (2015) Synthesis and gel formation of hyperbranched supramolecular polymer by vine-twining polymerization using branched primer-guest conjugate. Polymer 73:9–16
Yataka Y, Sawada T, Serizawa T (2015) Enzymatic synthesis and post-functionalization of two-dimensional crystalline cellulose oligomers with surface-reactive groups. Chem Commun 51:12525–12528
Serizawa T, Kato M, Okura H et al (2016) Hydrolytic activities of artificial nanocellulose synthesized via phosphorylase-catalyzed enzymatic reactions. Polym J 48:539–544
Wang J, Niu J, Sawada T et al (2017) A bottom-up synthesis of vinyl-cellulose nanosheets and their nanocomposite hydrogels with enhanced strength. Biomacromolecules 18:4196–4205
Serizawa T, Fukaya Y, Sawada T (2017) Self-assembly of cellulose oligomers into nanoribbon network structures based on kinetic control of enzymatic oligomerization. Langmuir 33:13415–13422
Hata Y, Kojima T, Koizumi T et al (2017) Enzymatic synthesis of cellulose oligomer hydrogels composed of crystalline nanoribbon networks under macromolecular crowding conditions. ACS Macro Lett 6:165–170
Hata Y, Sawada T, Serizawa T (2017) Effect of solution viscosity on the production of nanoribbon network hydrogels composed of enzymatically synthesized cellulose oligomers under macromolecular crowding conditions. Polym J 49:575–581
Hata Y, Sawada T, Sakai T et al (2018) Enzyme-catalyzed bottom-up synthesis of mechanically and physicochemically stable cellulose hydrogels for spatial immobilization of functional colloidal particles. Biomacromolecules 19:1269–1275
Nohara T, Sawada T, Tanaka H et al (2017) Enzymatic synthesis and protein adsorption properties of crystalline nanoribbons composed of cellulose oligomer derivatives with primary amino groups. J Biomater Sci Polym Ed 28:925–938
Bae J, Lee D, Kim D et al (2005) Facile synthesis of glucose-1-phosphate from starch by Thermus caldophilus GK24 α-glucan phosphorylase. Process Biochem 40:3707–3713
Fujii K, Takata H, Yanase M et al (2003) Bioengineering and application of novel glucose polymers. Biocatal Biotransformation 21:167–172
van der Vlist J, Palomo Reixach M, van der Maarel M et al (2008) Synthesis of branched polyglucans by the tandem action of potato phosphorylase and Deinococcus geothermalis glycogen branching enzyme. Macromol Rapid Commun 29:1293–1297
Kakutani R, Adachi Y, Kajiura H et al (2008) Stimulation of macrophage by enzymatically synthesized glycogen: the relationship between structure and biological activity. Biocatal Biotransformation 26:152–160
Kajiura H, Kakutani R, Akiyama T et al (2008) A novel enzymatic process for glycogen production. Biocatal Biotransformation 26:133–140
Takata H, Kajiura H, Furuyashiki T et al (2009) Fine structural properties of natural and synthetic glycogens. Carbohydr Res 344:654–659
Takata H, Akiyama T, Kajiura H et al (2010) Application of branching enzyme in starch processing. Biocatal Biotransformation 28:60–63
Kajiura H, Takata H, Akiyama T et al (2011) In vitro synthesis of glycogen: the structure, properties, and physiological function of enzymatically-synthesized glycogen. Biologia 66:387–394
Hernandez JM, Gaborieau M, Castignolles P et al (2008) Mechanistic investigation of a starch-branching enzyme using hydrodynamic volume SEC analysis. Biomacromolecules 9:954–965
Enomoto N, Furukawa S, Ogasawara Y et al (1996) Preparation of silica gel-bonded amylose through enzyme-catalyzed polymerization and chiral recognition ability of its phenylcarbamate derivative in HPLC. Anal Chem 68:2798–2804
Loos K, von Braunmuhl V, Stadler R et al (1997) Saccharide modified silica particles by enzymatic grafting. Macromol Rapid Commun 18:927–938
Morimoto N, Ogino N, Narita T et al (2007) Enzyme-responsive molecular assembly system with amylose-primer surfactants. J Am Chem Soc 129:458–459
Morimoto N, Ogino N, Narita T et al (2009) Enzyme-responsive artificial chaperone system with amphiphilic amylose primer. J Biotechnol 140:246–249
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Loos, K., Kadokawa, Ji. (2019). Synthesis of Polysaccharides II: Phosphorylase as Catalyst. In: Kobayashi, S., Uyama, H., Kadokawa, Ji. (eds) Enzymatic Polymerization towards Green Polymer Chemistry. Green Chemistry and Sustainable Technology. Springer, Singapore. https://doi.org/10.1007/978-981-13-3813-7_3
Download citation
DOI: https://doi.org/10.1007/978-981-13-3813-7_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3812-0
Online ISBN: 978-981-13-3813-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)