
Abstract
The structural nuclear proteins known as “lamins” (A-type and B-type) provide a scaffold for the compartmentalization of genome function that is important to maintain genome stability. Mutations in the LMNA gene -encoding for A-type lamins- are associated with over a dozen of degenerative disorders termed laminopathies, which include muscular dystrophies, lipodystrophies, neuropathies, and premature ageing diseases such as Hutchinson Gilford Progeria Syndrome (HGPS). This devastating disease is caused by the expression of a truncated lamin A protein named “progerin”. To date, there is no effective treatment for HGPS patients, who die in their teens from cardiovascular disease. At a cellular level, progerin expression impacts nuclear architecture, chromatin organization, response to mechanical stress, and DNA transactions such as transcription, replication and repair. However, the current view is that key mechanisms behind progerin toxicity still remain to be discovered. Here, we discuss new findings about pathological mechanisms in HGPS, especially the contribution of replication stress to cellular decline, and therapeutic strategies to ameliorate progerin toxicity. In particular, we present evidence for retinoids and calcitriol (hormonal vitamin D metabolite) being among the most potent compounds to ameliorate HGPS cellular phenotypes in vitro, providing the rationale for testing these compounds in preclinical models of the disease in the near term, and in patients in the future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
To the study of normal ageing, Hutchinson-Gilford Progeria Syndrome (HGPS) is certainly an outlier. As a severe premature ageing disease, patients develop alopecia, bone and joint abnormalities, subcutaneous fat loss, and severe atherosclerosis, all before their teenage years. Patients ultimately die at an average age of 14.6 years from myocardial infarction or stroke as a result of rapidly progressive atherosclerosis (Ullrich and Gordon 2015). Fortunately, this disease is extremely rare, with an estimated 350–400 children worldwide.Yet, since the mutation driving its pathophysiology was discovered in 2003, it has been the subject of an ever-growing volume of research. This is not only because treatment is desperately needed to help these patients, but it is thought that studying the complexities of this fascinating disease might reveal new insights into the normal ageing process. This is corroborated by the finding that progerin, the toxic protein driving disease pathology, is also found in the fibroblasts and vascular smooth muscle cells from old individuals (Dahl et al. 2006, McClintock et al. 2007). Importantly, progerin is upregulated in the hearts of dilated cardiomyopathy patients, where its expression correlates with left ventricular remodeling (Messner et al. 2018). This suggests that progerin might contribute to the progression of cardiovascular disease with age. Here, we review the clinical manifestations of HGPS, underlying cellular drivers of this disease, and emerging therapies for treating patients.
Hutchinson-Gilford Progeria Syndrome
HGPS results from the disruption of the nuclear lamina, a key nuclear structure for innumerate cellular processes, by a de novo single-base substitution within the LMNA exon 11 (c.1824C>T) (De Sandre-Giovannoli et al. 2003, Eriksson et al. 2003). This mutation activates a cryptic splice site, leading to an in-frame deletion of 50 amino acids near the C-terminus of prelamin A. This prevents proper post-translational processing of prelamin A to lamin A and leaves a permanently farnesylated and carboxymethylated toxic product called “progerin.” This mutant form of lamin A acts in a dominant fashion to induce a variety of abnormalities in nuclear processes, which eventually lead to cellular and organismal decline. Although c.1824C>T remains the most frequent mutation in HGPS patients, other mutations in the LMNA gene have also been reported that result in increased usage of the cryptic splice site.
HGPS patients are seemingly normal at birth, but quickly begin showing symptoms of their underlying disease. Skin alterations are often among the first manifestations of HGPS. Though manifestations can present with differing degrees of severity, typical alterations include areas of discoloration, stippled pigmentation, and tightened areas that restrict movement. Sclerodermoid changes, which give the skin a dimpled appearance with varying pigmentation, frequently appear over the abdomen and lower extremities (Rork et al. 2014). By 1 year of age, patients often present with failure to thrive, alopecia, circumoral cyanosis, prominent scalp veins, and decreased range of motion (Merideth et al. 2008, Ullrich and Gordon 2015). They develop a distinct progeroid appearance. Often remaining less than four feet tall and 30 kg, a decreased and linear rate of weight gain prevents growth comparable to age matched peers (Gordon et al. 2007; Kieran et al. 2007). Patients begin to lose cranial hair around 10 months of age, with progression to almost complete alopecia with time (Rork et al. 2014). HGPS patients also have distinct craniofacial characteristics, developing micrognathia, prominent eyes, and a beaked nose (Kieran et al. 2007, Domingo et al. 2009). Prominent forehead scalp veins and perioral cyanosis become evident, both likely the result of decreased subcutaneous fat (Rork et al. 2014). Patients also have multiple dental abnormalities, including both lack of teeth as well as dental crowding, which can manifest as double rows of teeth (Gordon et al. 2007, Domingo et al. 2009). Middle ear abnormalities and aberrations in the ear canal also lead to low-frequency hearing loss in many patients (Guardiani et al. 2011).
HGPS is a “segmental ageing disease,” since some features of normal ageing are present, whereas other features are notably absent. The liver, kidneys, lungs, and gastrointestinal tract are relatively spared in these patients (Kieran et al. 2007; Ullrich and Gordon 2015). However, others cell and tissue types, such as those of mesenchymal origin, are particularly susceptible to progerin-induced cellular defects, causing notable fat and bone abnormalities in HGPS patients (McClintock et al. 2007; Merideth et al. 2008; Zhang et al. 2011). HGPS patients develop lipodystrophy as well as bone and joint abnormalities consistent with skeletal dysplasia (Gordon et al. 2011). A profound loss of subcutaneous fat is readily apparent in examining these patients. Loss of fat in some body areas, such as the feet, can lead to discomfort and often requires supportive therapies (Gordon et al. 2014b; Ullrich and Gordon 2015). Interestingly, levels of body fat did not correlate with onset of menarche, which girls with this condition often experience in spite of lack of other pubertal features (Greer et al. 2017). Bone problems for these patients include small clavicles, thin ribs, and acroosteolysis. Patients exhibit reduced bone mineral density with accentuated demineralization at the end of long bones. Avascular necrosis is also present, including at the femoral head, likely resulting from vascular compromise (Cleveland et al. 2012). Interestingly, fracture incidence among HGPS patients is not increased compared to the general population, though HGPS patients are more susceptible to skull fractures. This is likely the result of disrupted bone formation in the skull. Patent anterior and posterior fontanels can persist in patients as old as 9 years of age, and these patients often also have widened calvarial sutures and a thin calvarium (Ullrich and Gordon 2015).
The most significant problems in HGPS are the cardiovascular complications, which underlie patient death. Patients develop severe and progressive atherosclerosis, eventually leading to myocardial ischemia, infarction, and stroke (Stehbens et al. 1999). Patients also develop readily evident left ventricular diastolic dysfunction, which increases with age (Prakash et al. 2018). Left ventricular hypertrophy and systolic dysfunction are also observed, which are more evident in older patients. Cardiac manifestations include increased afterload and angina (Ullrich and Gordon 2015). Remarkably, it is estimated that 50% of children have radiographically detectable strokes by the age of eight, and infarcts were common on imageing studies of patients between 5 and 10 years of age (Silvera et al. 2013). Most of these strokes are often clinically silent. This suggests that cardiovascular problems are present well before the end of life contributing to both morbidity and mortality.
The atherosclerosis that develops in HGPS has some important differences from the normal ageing population, although calcification, inflammation, and plaque rupture are present in both HGPS and normal ageing. Interestingly, HGPS patients do not develop hypercholesterolemia or increased serum high-sensitivity C-reactive protein, which are often seen with cardiovascular disease in the normal population (Stehbens et al. 1999; Olive et al. 2010). Additionally, vessels have a more complete fibrosis throughout the vessel wall, as arteries and veins show marked adventitial fibrosis with a dense rim of collagen. This complete stiffening of the wall leads to many measurable changes in the vasculature. Patients can become hypertensive, and some patients also have elongated QT intervals by EKG (Merideth et al. 2008; Gerhard-Herman et al. 2012). Carotid-femoral pulse wave velocity is dramatically elevated, indicating an increase in arterial stiffness. Patients also have abnormally echodense vascular walls by ultrasound, thought to correspond to a dramatically thickened fibrotic matrix. In these patients, as well as mouse models of disease, there is a striking depletion of vascular smooth muscle cells from the media, even in the outermost lamellar units adjacent to the adventitia, that is replaced by proteoglycans and collagen (Varga et al. 2006; Osorio et al. 2011; Gerhard-Herman et al. 2012; Villa-Bellosta et al. 2013). This is likely due to the extreme sensitivity of vascular smooth muscle cells to progerin expression.
The vascular abnormalities present in HGPS can compromise the nervous system as well. In the absence of vascular disease, the nervous system is relatively spared due to the fact that both lamin A and progerin expression is limited in the nervous system by miR-9 (Jung et al. 2012). HGPS patients show no evidence of memory or cognitive challenges often associated with the normal ageing process and have normal cognition. However, many HGPS patients experience neurological symptoms such as headaches of migraine-type quality, muscular weakness, or seizures as a result of impaired blood flow and diseased vasculature (Ullrich and Gordon 2015).
Cellular Disruptions
For the plethora of pathologies comprising this disease, it is surprising that it all results from a single nucleotide substitution and a deleterious protein. Progerin’s toxic cellular effects are substantial and caused primarily by alterations in genome function and integrity. Hallmarks of progerin-expressing cells include nuclear morphological abnormalities, changes in chromatin organization, DNA damage, telomere shortening, and premature senescence (Goldman et al. 2004; Prokocimer et al. 2013; Gonzalo and Kreienkamp 2015; Gonzalo et al. 2017). Despite enormous progress in recent years identifying cellular processes altered by progerin, we still lack a clear picture of the molecular mechanisms whereby progerin expression causes all these cellular phenotypes.
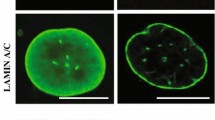
Nuclear morphological abnormalities are probably the most robust marker of HGPS patient-derived fibroblasts, and the phenotype that is most often ameliorated by therapeutic strategies (Capell et al. 2005) (Fig. 15.1). HGPS nuclei appear big, dysmorphic, and full of protrusions throughout that are accompanied by nuclear lamina thickening and disorganization of nuclear pore complexes and chromatin (Goldman et al. 2004; Kubben et al. 2015; Kreienkamp et al. 2016). A dosage-dependent effect of progerin inducing morphological nuclear defects has been reported, which is exacerbated with continuous proliferation (Chojnowski et al. 2015). Thus, strategies that lower levels of progerin have shown improvement of HGPS cells in vitro and in mouse models of disease in vivo. This is the case of antisense oligonucleotides (ASO) targeting lamin A/progerin production (Scaffidi and Misteli 2005; Osorio et al. 2011; Bridgeman et al. 2017), compounds such as rapamycin and everolimus that increase clearance of progerin via autophagy (DuBose et al. 2018), and MG132 that induces progerin nucleocytoplasmic translocation and progerin clearance through macroautophagy (Harhouri et al. 2017). In addition, the E3 ubiquitin ligase Smurf2 directly binds, ubiquitinates, and negatively regulates expression of lamin A/progerin, which in HGPS cells reduces nuclear deformability (Borroni et al. 2018). Furthermore, nuclei from HGPS cells exhibit increased nuclear stiffness and impaired mechanotransduction (Dahl et al. 2006; Verstraeten et al. 2008); phenotypes that are thought to have a big impact on tissues such as bone, skeletal muscle, heart, and vessels that are subjected to significant mechanical stress (Prokocimer et al. 2013; Dobrzynska et al. 2016).

Progerin expression elicits profound alterations in nuclear morphology and genome integrity and function. Compared to normal fibroblasts, HGPS patient-derived fibroblasts exhibit increased nuclear size and nuclear envelope morphological abnormalities characterized by invaginations, protrusions and blebbing. Progerin-expressing cells lose heterochromatin from the nuclear periphery and accumulate DNA damage. In particular, progerin causes increased levels of γΗ2AX (marker of DNA DSBs), and phosphorylated-RPA (marker of replication stress). The structure of the nuclear lamina is impacted by progerin expression. A-type and B-type lamins form independent networks, and progerin seems to be able to intercalate in both types of networks, eliciting structural alterations that affect nuclear stiffness and stability. In fact, nuclear rupture is common in progerin-expressing cells, with leakage of DNA fragments, chromatin, and other nucleoplasmic proteins into the cytoplasm. Similarly, mitochondrial integrity and function is often compromised in HGPS patient-derived cells. Moreover, progerin causes broad changes in gene expression. Recently, we showed that the transcription factor STAT1 is activated by phosphorylation in progerin-expressing cells, leading to its translocation to the nucleus and the activation of target genes in the interferon (IFN) response. (Graphic illustrations generated by Michael Andrus, BS, St Louis University)
Epigenetic changes are characteristic of HGPS cells, including alterations in DNA methylation (Osorio et al. 2010; Heyn et al. 2013), post-translational modifications of histones -mainly H3K9me3, H3K27me3, and H4K20me3- (Columbaro et al. 2005; Scaffidi and Misteli 2005; Shumaker et al. 2006), expression levels of chromatin-modifying activities such as the NURD complex (Pegoraro et al. 2009), and miRNAs (Arancio et al. 2014; Frankel et al. 2018). Interestingly, these chromatin changes and expression of progerin are also observed in cells from old individuals, suggesting their implication in physiological ageing (Dahl et al. 2006). Special attention has been given lately to the consequences of loss of function of SIRT6 in progerin-expressing cells. This sirtuin family member has histone deacetylation and mono-ADP ribosylation activities and has been associated with genomic instability and accelerated ageing similar to HGPS (Ghosh et al. 2015).
Deregulated gene expression is another hallmark of HGPS, which resembles the gene expression profile of disorders affecting mesodermal and mesenchymal cell lineages. Functional categories more often found differentially expressed in HGPS cells include transcription factors (Duband-Goulet et al. 2011) and extracellular matrix (ECM) components (Csoka et al. 2004), in addition to signaling cascades such as the Wnt pathway (Hernandez et al. 2010; Vidak and Foisner 2016), the retinoblastoma pathway (Marji et al. 2010), Notch signalling (Scaffidi and Misteli 2008), NFκB inflammatory pathway, and the recently identified, interferon (IFN)-related innate immune responses (Kreienkamp et al. 2018).
DNA repair defects, telomere dysfunction, and genomic instability are amongst the more potent drivers of ageing and malignancy. In HGPS, there is strong evidence for deficiencies in DNA repair, which are characterized by delayed recruitment of DNA repair factors such as 53BP1 and RAD51 to γH2AX-labeled DNA repair foci (Liu et al. 2005) or anomalous accumulation of Xeroderma Pigmentosum group A (XPA) (Liu et al. 2005, 2008), a protein with an important function in nucleotide excision repair (NER), among others. HGPS cells also develop telomere dysfunction, with faster telomere attrition during proliferation that elicits DNA damage and premature senescence (Gonzalo and Kreienkamp 2015). In addition, there is accumulation of ROS due to mitochondrial dysfunction, which was has been linked to impaired NRF2 pathway activity (Kubben et al. 2016). An interesting study has recently found that inhibition of ROCK (rho-associated protein kinase) activity recovers mitochondrial function in HGPS fibroblasts, ameliorating nuclear morphological abnormalities and genomic instability phenotypes (Kang et al. 2017). Many reviews have previously described the phenotypes of genomic instability in progerin-expressing cells (Gonzalo and Kreienkamp 2015; Dobrzynska et al. 2016; Gonzalo et al. 2017). Here, we will focus on newly identified mechanisms underlying genomic instability and their contribution to premature ageing.
Recently, special emphasis has been placed on understanding how progerin affects DNA replication, given that most of the DNA damage that accumulates in cells is generated during replication. Early replication studies using Xenopus extracts showed that nuclear lamina disruption causes a marked reduction in DNA replication, concomitant with alteration in the distribution of Proliferating Cell Nuclear Antigen (PCNA) and the Replication Factor Complex (RFC), key factors in the elongation phase of DNA replication (Spann et al. 1997). Another study revealed that altered organization of the nuclear lamina inhibits chain elongation in a dose-dependent manner (Moir et al. 2000). In addition, PCNA has been found to co-localize with A- and B-type lamins in early and late sites of DNA replication, respectively (Moir et al. 1994; Goldberg et al. 1995; Jenkins et al. 1995; Kennedy et al. 2000; Dechat et al. 2008), suggesting a role for lamins in the spatial/temporal organization of replication.
More recently, expression of pre-lamin-A was associated with mono-ubiquitination of PCNA and induction of Pol η, two hallmarks of replication fork stalling (Cobb et al. 2016). It was suggested that pre-lamin-A mitigates the interaction of PCNA with mature lamin-A, eliciting replication fork stalling. In HGPS cells, RFC1 is aberrantly degraded by a serine protease, and the cleavage causes defects in the loading of PCNA and Pol δ onto DNA for replication (Tang et al. 2012). In an unbiased screen of lamin-A- and progerin-interacting proteins by mass spectrometry, progerin was also shown to interact with PCNA more robustly than lamin A (Kubben et al. 2010), and also reported to sequester PCNA away from replicating DNA (Wheaton et al. 2017). These findings support the idea that expression of pre-lamin-A and progerin elicit replication stress by sequestering PCNA away from the replication fork. Consistent with this idea, progerin-expressing cells accumulate XPA at stalled or collapsed replication forks, concomitant with a significant loss of PCNA at the forks (Hilton et al. 2017). Depletion of XPA or progerin restores PCNA at replication forks, while reducing the extent of progerin-induced apoptosis. Therefore, progerin expression seems to alter the binding of key factors to the replication fork, including PCNA and proteins such as XPA that participate in the repair of DNA lesions. Altogether, these findings suggest that alterations in nuclear lamins impact DNA replication and that replication stress could play a major role in the proliferation defects and genomic instability that characterize lamins-defective cells. Despite these findings, our mechanistic understanding of how mutant lamins hinder DNA replication is limited.
Mutant lamins such as progerin could hinder the progression of the replication fork by inducing mis-localization of factors that associate with the replisome -PCNA and RFC-. Lamin dysfunction could also hinder the proper recruitment of replication fork protective factors upon fork stalling, causing replication stress-induced genomic instability. To understand mechanistically how lamin dysfunction affects DNA replication requires the utilization of newly developed techniques such as genome-wide single-molecule replication assays (DNA fiber assays), iPOND (Isolation of Proteins On Nascent DNA) (Sirbu et al. 2011), and electron microscopy (Vindigni and Lopes 2017). Our recent studies performing DNA fiber assays have revealed that progerin expression, but not overexpression of lamin-A, causes a robust phenotype hindering replication (Kreienkamp et al. 2018). Progerin elicits replication stress, characterized by increased replication fork stalling in the absence of drugs that inhibit replication. In addition, we find that stalled replication forks are deprotected and susceptible to MRE11 nuclease-mediated fork degradation. As such, inhibition of MRE11 nuclease rescues replication defects in progerin-expressing cells. Moreover, we find that a variety of compounds known to ameliorate phenotypes of genomic instability in progerin-expressing cells, including vitamin D (Gonzalez-Suarez et al. 2011), all-trans retinoic acid (ATRA) (Swift et al. 2013; Kubben et al. 2015; Pellegrini et al. 2015), remodelin (Larrieu et al. 2014), and the combination of a farnesyltransferase inhibitor (FTI) and rapamycin (Cao et al. 2011; Pellegrini et al. 2015; Gordon et al. 2016) markedly reduce replication stress in progerin-expressing cells (Kreienkamp et al. 2018). Despite the fact that molecular mechanisms underlying the beneficial effects of these drugs rescuing replication stress and genomic instability in progerin-expressing cells remain to be identified, this finding has important implications from a therapeutic perspective and for defining the importance of replication stress to the progeria phenotype.
Interestingly, our recent studies demonstrate that replication stress in progerin-expressing cells not only contribute to genomic instability, but also activate inflammatory responses that contribute to cellular ageing. Replication stress in HGPS patient-derived fibroblasts and progerin-expressing normal fibroblasts is accompanied by accumulation of chromatin at the cytoplasm, upregulation of cytosolic sensors of nucleic acids -cGAS, STING, RIG-I, MDA5, and OASs-, and robust activation of a cell intrinsic interferon (IFN)-like response (Kreienkamp et al. 2018). This IFN-like response, which is regulated by STAT1, contributes to cellular ageing phenotypes such as reduced proliferation and migration capabilities. This finding is important because STAT1 is a notorious regulator of inflammation in immune and vascular cells during atherosclerosis, and an important therapeutic target for cardiovascular disease (Szelag et al. 2016), the main cause of death of HGPS patients. We hypothesize that progerin expression in vascular cells from HGPS patients could recapitulate the STAT1 pathway activation observed in fibroblasts, being a contributor to the decline of vascular cells characteristic of this disease.
Importantly, we showed that the same treatments that ameliorate replication stress -vitamin D, ATRA, remodelin, FTI and rapamycin-, markedly repress the STAT1/IFN-like response (Kreienkamp et al. 2018). We propose that in progerin-expressing cells, DNA damage and replication stress, together with disruption of nuclear integrity, results in accumulation of immunogenic nucleic acids in the cytoplasm, where they activate cytosolic sensors of foreign nucleic acids. This in turn leads to activation of inflammatory pathways such as NFκB and STAT1 that drive an IFN-like response. Defining the causes of this cell-intrinsic IFN response and its consequences for organismal decline in HGPS, as well as the mechanisms whereby different compounds ameliorate this response might reveal ways to reduce its pathological impact in HGPS and in normal ageing, as progerin is expressed in cells from old individuals (Dahl et al. 2006).
Current and Future Therapies
With such a multitude of cellular processes and organ systems affected, developing therapies for HGPS has proven challenging. However, since progerin was identified as the driver behind disease phenotypes, researchers have searched for ways to combat its detrimental effects (Harhouri et al. 2018). Among the first of the drugs that emerged was lonafarnib, a farnesyltransferase inhibitor (FTI) designed to prevent processing of prelamin A to progerin. FTIs reduced nuclear blebbing, nuclear stiffness, rescued heterochromatin organization in HGPS cells, decreasing onset of premature senescence and improving proliferation (Capell et al. 2005; Columbaro et al. 2005; Yang et al. 2005; Verstraeten et al. 2008). Further, FTIs had a remarkable effect in mouse models of disease (Fong et al. 2006; Yang et al. 2006; Varela et al. 2008). Based on these promising results, a clinical trial was initiated. There, the results were equally compelling. Administration of the FTI lonafarnib for 2 years in HGPS patients improved pulse-wave velocity, carotid artery wall echodensity, and incidence of stroke, headaches, and seizures (Gordon et al. 2012). FTIs increased mean survival by 1.6 years (Gordon et al. 2014a). Recently, it was demonstrated that lonafarnib monotherapy was associated with a lower mortality rate after 2.2 years of follow-up (Gordon et al. 2018). While this drug certainly is the first treatment for HGPS patients, it can be hard for patients to tolerate due to a number of side effects. Therefore, the next wave of therapies, and the possibility for combination therapies, are desperately needed to allow for synergy or even lower dosages for effectiveness.
The next compound that has made its way to patients is everolimus. Everolimus is an analog of rapamycin, which promotes the removal of toxic, insoluble aggregates like progerin by enhanced autophagy (Cao et al. 2011). Everolimus increased proliferative ability and delayed cellular senescence in cell lines, including those without the classical HGPS mutation (DuBose et al. 2018). Based on these studies, a phase I/II dose-escalation clinical trial of everolimus in combination with lonafarnib was initiated in 2015, with results from these studies expected by 2020 (https://clinicaltrials.gov/ct2/show/study/NCT02579044).
Sulforaphane acts in a similar manner to everolimus, and, as such, has shown benefit in vitro by increasing progerin clearance by autophagy (Gabriel et al. 2015). More recent studies have demonstrated that combination of sulforaphane with lonafarnib is toxic, but intermittent treatment of sulforaphane with lonafarnib separately and in repeated cycles rescues HGPS cellular phenotype (Gabriel et al. 2017).
Other therapies are likely next for testing in patients. Remodelin, an inhibitor of N-acetyltransferase-10 (NAT10), increases chromatin compaction while rescuing nuclear morphological abnormalities, proliferation defects, and accumulation of DNA damage characteristic of progerin-expressing cells (Larrieu et al. 2014). A preclinical study recently performed with remodelin treatment revealed an improvement in healthspan in the progeria mice (Balmus et al. 2018), and similar effects by chemical inhibition of NAT10.
The retinoids are among other treatment strategies that now await testing in vivo. The LMNA gene promoter contains retinoic acid responsive elements (L-RARE) that downregulate LMNA gene expression with all trans retinoic acid (ATRA) treatment (Swift et al. 2013). In HGPS patient-derived fibroblasts, ATRA treatment reduces significantly progerin expression and actually synergizes with rapamycin in downregulating progerin levels, ameliorating a variety of progerin-induced phenotypes (Pellegrini et al. 2015). Retinoids were also identified in a high-throughput, high-content based screening of a library of FDA approved drugs as a class of compounds able to revert cellular HGPS phenotypes (Kubben et al. 2016). Similarly, our studies show that activation of vitamin D receptor signaling by ligand (1,25α-dihydroxy-vitamin D3) binding ameliorates a broad repertoire of phenotypes of HGPS patient-derived cells (Kreienkamp et al. 2016, 2018). Other therapeutic strategies of benefit have included inhibitors of the enzyme responsible for carboxymethylation of the farnesylcysteine of progerin (Ibrahim et al. 2013); the ROS scavenger N-acetyl cysteine, which reduces the amount of unrepairable DNA damage caused by the increased generation of ROS (Pekovic et al. 2011; Richards et al. 2011; Lattanzi et al. 2012; Sieprath et al. 2012); methylene blue, a mitochondrial-targeting antioxidant (Xiong et al. 2016); or resveratrol, an enhancer of SIRT1 deacetylase activity that alleviates progeroid features (Liu et al. 2012). The plethora of potential therapeutic options is encourageing, since it is likely that the best treatment options for these patients will consist of combination therapy. Combination therapy might allow for synergy among compounds, reducing toxicity owed to lowering the doses of each single compound.
Concluding Remarks
HGPS, with its severity and time-course, is certainly an outlier to the ageing process. As an outlier, though, its study portends value not only for these patients, but also for the normal ageing population. Since 2003, our understanding of both normal and abnormal ageing has greatly increased. In the last few years, the fruits of these studies have become tangible for HGPS patients with the first wave of therapies to help them. It is hoped that the next years will yield further therapies of benefit to these children.
Much remains to be learned. Our understanding of the cardiovascular disease that drives patient death is still quite limited. Our studies are also challenged by the fact that we still lack a suitable animal model for recapitulating well the cardiovascular disease driving human death. The coming years seem destined for advances in our understanding of these critical disease processes. Finding targeted biomarkers for disease remains an important goal, as is identifying other genes that impact disease phenotype. With the increased utilization of next generation sequencing, it is hoped that this technology will also benefit HGPS, particularly with identifying other genetic traits that either potentiate or reduce progerin’s toxic effect. Other new technologies, like CRISPR-Cas9, have obvious applications in diseases like HGPS, which now just wait application. Time will certainly reveal new mysteries for disease. And, as we develop ways to help these children live longer, maybe some of these findings might also have relevance for the rest of us in the normal ageing population.
References
Arancio W, Pizzolanti G, Genovese SI, Pitrone M, Giordano C (2014) Epigenetic involvement in Hutchinson-Gilford progeria syndrome: a mini-review. Gerontology 60(3):197–203
Balmus G, Larrieu D, Barros AC, Collins C, Abrudan M, Demir M, Geisler NJ, Lelliott CJ, White JK, Karp NA, Atkinson J, Kirton A, Jacobsen M, Clift D, Rodriguez R, Sanger Mouse Genetics P, Adams DJ, Jackson SP (2018) Targeting of NAT10 enhances healthspan in a mouse model of human accelerated ageing syndrome. Nat Commun 9(1):1700
Borroni AP, Emanuelli A, Shah PA, Ilic N, Apel-Sarid L, Paolini B, Manikoth Ayyathan D, Koganti P, Levy-Cohen G, Blank M (2018) Smurf2 regulates stability and the autophagic-lysosomal turnover of lamin A and its disease-associated form progerin. Ageing Cell 17(2):e12732
Bridgeman VL, Vermeulen PB, Foo S, Bilecz A, Daley F, Kostaras E, Nathan MR, Wan E, Frentzas S, Schweiger T, Hegedus B, Hoetzenecker K, Renyi-Vamos F, Kuczynski EA, Vasudev NS, Larkin J, Gore M, Dvorak HF, Paku S, Kerbel RS, Dome B, Reynolds AR (2017) Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J Pathol 241(3):362–374
Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS (2011) Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med 3(89):89ra58
Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS (2005) Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 102(36):12879–12884
Chojnowski A, Ong PF, Dreesen O (2015) Nuclear lamina remodelling and its implications for human disease. Cell Tissue Res 360(3):621–631
Cleveland RH, Gordon LB, Kleinman ME, Miller DT, Gordon CM, Snyder BD, Nazarian A, Giobbie-Hurder A, Neuberg D, Kieran MW (2012) A prospective study of radiographic manifestations in Hutchinson-Gilford progeria syndrome. Pediatr Radiol 42(9):1089–1098
Cobb AM, Murray TV, Warren DT, Liu Y, Shanahan CM (2016) Disruption of PCNA-lamins A/C interactions by prelamin A induces DNA replication fork stalling. Nucleus 7(5):498–511
Columbaro M, Capanni C, Mattioli E, Novelli G, Parnaik VK, Squarzoni S, Maraldi NM, Lattanzi G (2005) Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell Mol Life Sci 62(22):2669–2678
Csoka AB, English SB, Simkevich CP, Ginzinger DG, Butte AJ, Schatten GP, Rothman FG, Sedivy JM (2004) Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Ageing Cell 3(4):235–243
Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T (2006) Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 103(27):10271–10276
De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N (2003) Lamin a truncation in Hutchinson-Gilford progeria. Science 300(5628):2055
Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD (2008) Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 22(7):832–853
Dobrzynska A, Gonzalo S, Shanahan C, Askjaer P (2016) The nuclear Lamina in health and disease. Nucleus 7:233–248
Domingo DL, Trujillo MI, Council SE, Merideth MA, Gordon LB, Wu T, Introne WJ, Gahl WA, Hart TC (2009) Hutchinson-Gilford progeria syndrome: oral and craniofacial phenotypes. Oral Dis 15(3):187–195
Duband-Goulet I, Woerner S, Gasparini S, Attanda W, Konde E, Tellier-Lebegue C, Craescu CT, Gombault A, Roussel P, Vadrot N, Vicart P, Ostlund C, Worman HJ, Zinn-Justin S, Buendia B (2011) Subcellular localization of SREBP1 depends on its interaction with the C-terminal region of wild-type and disease related A-type lamins. Exp Cell Res 317(20):2800–2813
DuBose AJ, Lichtenstein ST, Petrash NM, Erdos MR, Gordon LB, Collins FS (2018) Everolimus rescues multiple cellular defects in laminopathy-patient fibroblasts. Proc Natl Acad Sci U S A 115(16):4206–4211
Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS (2003) Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423(6937):293–298
Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG (2006) A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 311(5767):1621–1623
Frankel D, Delecourt V, Harhouri K, De Sandre-Giovannoli A, Levy N, Kaspi E, Roll P (2018) MicroRNAs in hereditary and sporadic premature ageing syndromes and other laminopathies. Aging Cell. 2018 Apr 25:e12766. https://doi.org/10.1111/acel.12766
Gabriel D, Roedl D, Gordon LB, Djabali K (2015) Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Ageing Cell 14(1):78–91
Gabriel D, Shafry DD, Gordon LB, Djabali K (2017) Intermittent treatment with farnesyltransferase inhibitor and sulforaphane improves cellular homeostasis in Hutchinson-Gilford progeria fibroblasts. Oncotarget 8(39):64809–64826
Gerhard-Herman M, Smoot LB, Wake N, Kieran MW, Kleinman ME, Miller DT, Schwartzman A, Giobbie-Hurder A, Neuberg D, Gordon LB (2012) Mechanisms of premature vascular ageing in children with Hutchinson-Gilford progeria syndrome. Hypertension 59(1):92–97
Ghosh S, Liu B, Wang Y, Hao Q, Zhou Z (2015) Lamin A Is an Endogenous SIRT6 Activator and Promotes SIRT6-Mediated DNA Repair. Cell Rep 13(7):1396–1406
Goldberg M, Jenkins H, Allen T, Whitfield WG, Hutchison CJ (1995) Xenopus lamin B3 has a direct role in the assembly of a replication competent nucleus: evidence from cell-free egg extracts. J Cell Sci 108(Pt 11):3451–3461
Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS (2004) Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 101(24):8963–8968
Gonzalez-Suarez I, Redwood AB, Grotsky DA, Neumann MA, Cheng EH, Stewart CL, Dusso A, Gonzalo S (2011) A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair. EMBO J 30(16):3383–3396
Gonzalo S, Kreienkamp R (2015) DNA repair defects and genome instability in Hutchinson-Gilford progeria syndrome. Curr Opin Cell Biol 34:75–83
Gonzalo S, Kreienkamp R, Askjaer P (2017) Hutchinson-Gilford progeria syndrome: a premature ageing disease caused by LMNA gene mutations. Ageing Res Rev 33:18–29
Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, Kieran MW (2007) Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics 120(4):824–833
Gordon CM, Gordon LB, Snyder BD, Nazarian A, Quinn N, Huh S, Giobbie-Hurder A, Neuberg D, Cleveland R, Kleinman M, Miller DT, Kieran MW (2011) Hutchinson-Gilford progeria is a skeletal dysplasia. J Bone Miner Res 26(7):1670–1679
Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW (2012) Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 109(41):16666–16671
Gordon LB, Massaro J, D'Agostino RB Sr, Campbell SE, Brazier J, Brown WT, Kleinman ME, Kieran MW, C. Progeria Clinical Trials (2014a) Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 130(1):27–34
Gordon LB, Rothman FG, Lopez-Otin C, Misteli T (2014b) Progeria: a paradigm for translational medicine. Cell 156(3):400–407
Gordon LB, Kleinman ME, Massaro J, D'Agostino RB Sr, Shappell H, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland RH, Nazarian A, Snyder BD, Ullrich NJ, Silvera VM, Liang MG, Quinn N, Miller DT, Huh SY, Dowton AA, Littlefield K, Greer MM, Kieran MW (2016) Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation 134(2):114–125
Gordon LB, Shappell H, Massaro J, D'Agostino RB Sr, Brazier J, Campbell SE, Kleinman ME, Kieran MW (2018) Association of lonafarnib treatment vs no treatment with mortality rate in patients with Hutchinson-Gilford progeria syndrome. JAMA 319(16):1687–1695
Greer MM, Kleinman ME, Gordon LB, Massaro J, D’Agostino RB Sr, Baltrusaitis K, Kieran MW, Gordon CM (2017) Pubertal progression in female adolescents with progeria. J Pediatr Adolesc Gynecol. 2018 Jun 31(3):238–241
Guardiani E, Zalewski C, Brewer C, Merideth M, Introne W, Smith AC, Gordon L, Gahl W, Kim HJ (2011) Otologic and audiologic manifestations of Hutchinson-Gilford progeria syndrome. Laryngoscope 121(10):2250–2255
Harhouri K, Navarro C, Depetris D, Mattei MG, Nissan X, Cau P, De Sandre-Giovannoli A, Levy N (2017) MG132-induced progerin clearance is mediated by autophagy activation and splicing regulation. EMBO Mol Med 9(9):1294–1313
Harhouri K, Frankel D, Bartoli C, Roll P, De Sandre-Giovannoli A, Levy N (2018) An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome. Nucleus 9(1):246–257
Hernandez L, Roux KJ, Wong ES, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong JW, Wang H, Lee HS, Kozlov S, Grunert M, Keeble T, Jones CM, Meta MD, Young SG, Daar IO, Burke B, Perantoni AO, Stewart CL (2010) Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell 19(3):413–425
Heyn H, Moran S, Esteller M (2013) Aberrant DNA methylation profiles in the premature ageing disorders Hutchinson-Gilford Progeria and Werner syndrome. Epigenetics 8(1):28–33
Hilton BA, Liu J, Cartwright BM, Liu Y, Breitman M, Wang Y, Jones R, Tang H, Rusinol A, Musich PR, Zou Y (2017) Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. FASEB J 31(9):3882–3893
Ibrahim MX, Sayin VI, Akula MK, Liu M, Fong LG, Young SG, Bergo MO (2013) Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 340(6138):1330–1333
Jenkins H, Whitfield WG, Goldberg MW, Allen TD, Hutchison CJ (1995) Evidence for the direct involvement of lamins in the assembly of a replication competent nucleus. Acta Biochim Pol 42(2):133–143
Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BS, Yang SH, Barnes RH 2nd, Hong J, Sun T, Pleasure SJ, Young SG, Fong LG (2012) Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A 109(7):E423–E431
Kang HT, Park JT, Choi K, Choi HJC, Jung CW, Kim GR, Lee YS, Park SC (2017) Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Ageing Cell 16(3):541–550
Kennedy BK, Barbie DA, Classon M, Dyson N, Harlow E (2000) Nuclear organization of DNA replication in primary mammalian cells. Genes Dev 14(22):2855–2868
Kieran MW, Gordon L, Kleinman M (2007) New approaches to progeria. Pediatrics 120(4):834–841
Kreienkamp R, Croke M, Neumann MA, Bedia-Diaz G, Graziano S, Dusso A, Dorsett D, Carlberg C, Gonzalo S (2016) Vitamin D receptor signaling improves Hutchinson-Gilford progeria syndrome cellular phenotypes. Oncotarget 7:30018–30031
Kreienkamp R, Graziano S, Coll-Bonfill N, Bedia-Diaz G, Cybulla E, Vindigni A, Dorsett D, Kubben N, Batista LFZ, Gonzalo S (2018) A cell-intrinsic interferon-like response links replication stress to cellular ageing caused by progerin. Cell Rep 22(8):2006–2015
Kubben N, Voncken JW, Demmers J, Calis C, van Almen G, Pinto Y, Misteli T (2010) Identification of differential protein interactors of lamin A and progerin. Nucleus 1(6):513–525
Kubben N, Brimacombe KR, Donegan M, Li Z, Misteli T (2015) A high-content imageing-based screening pipeline for the systematic identification of anti-progeroid compounds. Methods. 2016 Mar 1:96:46–58. https://doi.org/10.1016/j.ymeth.2015.08.024. Epub 2015 Sep 1.
Kubben N, Zhang W, Wang L, Voss TC, Yang J, Qu J, Liu GH, Misteli T (2016) Repression of the antioxidant NRF2 pathway in premature ageing. Cell 165(6):1361–1374
Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP (2014) Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 344(6183):527–532
Lattanzi G, Marmiroli S, Facchini A, Maraldi NM (2012) Nuclear damages and oxidative stress: new perspectives for laminopathies. Eur J Histochem 56(4):e45
Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadinanos J, Lopez-Otin C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z (2005) Genomic instability in laminopathy-based premature ageing. Nat Med 11(7):780–785
Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, Zou Y (2008) Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. FASEB J 22(2):603–611
Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, Kaeberlein M, Tryggvason K, Zhou Z (2012) Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab 16(6):738–750
Marji J, O’Donoghue SI, McClintock D, Satagopam VP, Schneider R, Ratner D, Worman HJ, Gordon LB, Djabali K (2010) Defective lamin A-Rb signaling in Hutchinson-Gilford progeria syndrome and reversal by farnesyltransferase inhibition. PLoS One 5(6):e11132
McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K (2007) The mutant form of lamin A that causes Hutchinson-Gilford progeria is a biomarker of cellular ageing in human skin. PLoS One 2(12):e1269
Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, Brewer CC, Zalewski C, Kim HJ, Solomon B, Brooks BP, Gerber LH, Turner ML, Domingo DL, Hart TC, Graf J, Reynolds JC, Gropman A, Yanovski JA, Gerhard-Herman M, Collins FS, Nabel EG, Cannon RO 3rd, Gahl WA, Introne WJ (2008) Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med 358(6):592–604
Messner M, Ghadge SK, Goetsch V, Wimmer A, Dorler J, Polzl G, Zaruba MM (2018) Upregulation of the ageing related LMNA splice variant progerin in dilated cardiomyopathy. PLoS One 13(4):e0196739
Moir RD, Montag-Lowy M, Goldman RD (1994) Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. J Cell Biol 125(6):1201–1212
Moir RD, Spann TP, Herrmann H, Goldman RD (2000) Disruption of nuclear lamin organization blocks the elongation phase of DNA replication. J Cell Biol 149(6):1179–1192
Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB (2010) Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of ageing. Arterioscler Thromb Vasc Biol 30(11):2301–2309
Osorio FG, Varela I, Lara E, Puente XS, Espada J, Santoro R, Freije JM, Fraga MF, Lopez-Otin C (2010) Nuclear envelope alterations generate an ageing-like epigenetic pattern in mice deficient in Zmpste24 metalloprotease. Ageing Cell 9(6):947–957
Osorio FG, Navarro CL, Cadinanos J, Lopez-Mejia IC, Quiros PM, Bartoli C, Rivera J, Tazi J, Guzman G, Varela I, Depetris D, de Carlos F, Cobo J, Andres V, De Sandre-Giovannoli A, Freije JM, Levy N, Lopez-Otin C (2011) Splicing-directed therapy in a new mouse model of human accelerated ageing. Sci Transl Med 3(106):106ra107
Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T (2009) Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol 11(10):1261–1267
Pekovic V, Gibbs-Seymour I, Markiewicz E, Alzoghaibi F, Benham AM, Edwards R, Wenhert M, von Zglinicki T, Hutchison CJ (2011) Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Ageing Cell 10(6):1067–1079
Pellegrini C, Columbaro M, Capanni C, D'Apice MR, Cavallo C, Murdocca M, Lattanzi G, Squarzoni S (2015) All-trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget 6(30):29914–29928
Prakash A, Gordon LB, Kleinman ME, Gurary EB, Massaro J, D’Agostino R Sr, Kieran MW, Gerhard-Herman M, Smoot L (2018) Cardiac abnormalities in patients with Hutchinson-Gilford progeria syndrome. JAMA Cardiol 3(4):326–334
Prokocimer M, Barkan R, Gruenbaum Y (2013) Hutchinson-Gilford progeria syndrome through the lens of transcription. Ageing Cell 12(4):533–543
Richards SA, Muter J, Ritchie P, Lattanzi G, Hutchison CJ (2011) The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum Mol Genet 20(20):3997–4004
Rork, J. F., J. T. Huang, L. B. Gordon, M. Kleinman, M. W. Kieran¸ M. G. Liang (2014). Initial cutaneous manifestations of Hutchinson-Gilford progeria syndrome. Pediatr Dermatol 31(2): 196–202
Scaffidi P, Misteli T (2005) Reversal of the cellular phenotype in the premature ageing disease Hutchinson-Gilford progeria syndrome. Nat Med 11(4):440–445
Scaffidi P, Misteli T (2008) Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol 10(4):452–459
Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD (2006) Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature ageing. Proc Natl Acad Sci U S A 103(23):8703–8708
Sieprath T, Darwiche R, De Vos WH (2012) Lamins as mediators of oxidative stress. Biochem Biophys Res Commun 421(4):635–639
Silvera VM, Gordon LB, Orbach DB, Campbell SE, Machan JT, Ullrich NJ (2013) Imageing characteristics of cerebrovascular arteriopathy and stroke in Hutchinson-Gilford progeria syndrome. AJNR Am J Neuroradiol 34(5):1091–1097
Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25(12):1320–1327
Spann TP, Moir RD, Goldman AE, Stick R, Goldman RD (1997) Disruption of nuclear lamin organization alters the distribution of replication factors and inhibits DNA synthesis. J Cell Biol 136(6):1201–1212
Stehbens WE, Wakefield SJ, Gilbert-Barness E, Olson RE, Ackerman J (1999) Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol 8(1):29–39
Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW, Discher DE (2013) Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341(6149):1240104
Szelag M, Piaszyk-Borychowska A, Plens-Galaska M, Wesoly J, Bluyssen HA (2016) Targeted inhibition of STATs and IRFs as a potential treatment strategy in cardiovascular disease. Oncotarget 7:48788–48812
Tang H, Hilton B, Musich PR, Fang DZ, Zou Y (2012) Replication factor C1, the large subunit of replication factor C, is proteolytically truncated in Hutchinson-Gilford progeria syndrome. Ageing Cell 11(2):363–365
Ullrich NJ, Gordon LB (2015) Hutchinson-Gilford progeria syndrome. Handb Clin Neurol 132:249–264
Varela I, Pereira S, Ugalde AP, Navarro CL, Suarez MF, Cau P, Cadinanos J, Osorio FG, Foray N, Cobo J, de Carlos F, Levy N, Freije JM, Lopez-Otin C (2008) Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature ageing. Nat Med 14(7):767–772
Varga R, Eriksson M, Erdos MR, Olive M, Harten I, Kolodgie F, Capell BC, Cheng J, Faddah D, Perkins S, Avallone H, San H, Qu X, Ganesh S, Gordon LB, Virmani R, Wight TN, Nabel EG, Collins FS (2006) Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 103(9):3250–3255
Verstraeten VL, Ji JY, Cummings KS, Lee RT, Lammerding J (2008) Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: effects of farnesyltransferase inhibitors. Ageing Cell 7(3):383–393
Vidak S, Foisner R (2016) Molecular insights into the premature ageing disease progeria. Histochem Cell Biol 145(4):401–417
Villa-Bellosta R, Rivera-Torres J, Osorio FG, Acin-Perez R, Enriquez JA, Lopez-Otin C, Andres V (2013) Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 127(24):2442–2451
Vindigni A, Lopes M (2017) Combining electron microscopy with single molecule DNA fiber approaches to study DNA replication dynamics. Biophys Chem 225:3–9
Wheaton, K., D. Campuzano, W. Ma, M. Sheinis, B. Ho, G. W. Brown, S. Benchimol (2017) Progerin-induced replication stress facilitates premature senescence in Hutchinson-Gilford progeria syndrome. Mol Cell Biol 37(14)
Xiong ZM, Choi JY, Wang K, Zhang H, Tariq Z, Wu D, Ko E, LaDana C, Sesaki H, Cao K (2016) Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Ageing Cell 15(2):279–290
Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG (2005) Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc Natl Acad Sci U S A 102(29):10291–10296
Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG (2006) A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest 116(8):2115–2121
Zhang J, Lian Q, Zhu G, Zhou F, Sui L, Tan C, Mutalif RA, Navasankari R, Zhang Y, Tse HF, Stewart CL, Colman A (2011) A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 8(1):31–45
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kreienkamp, R., Gonzalo, S. (2019). Hutchinson-Gilford Progeria Syndrome: Challenges at Bench and Bedside. In: Harris, J., Korolchuk, V. (eds) Biochemistry and Cell Biology of Ageing: Part II Clinical Science. Subcellular Biochemistry, vol 91. Springer, Singapore. https://doi.org/10.1007/978-981-13-3681-2_15
Download citation
DOI: https://doi.org/10.1007/978-981-13-3681-2_15
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3680-5
Online ISBN: 978-981-13-3681-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)