
Abstract
Primary open-angle glaucoma (POAG) is an optic nerve disease with elevated IOP as the main risk factor. In recent years, with the development of interdisciplinary in the ophthalmology and neurology, new questions about the essence of POAG have been raised. Is POAG just an ocular disease? Is it a disease which involves the eye first and then the whole visual pathway? Or sometimes is it an ocular manifestation of a particular central nervous system (CNS) disease? Recent experimental and clinical studies have suggested that POAG patients may have an abnormally low cerebrospinal fluid pressure (CSFP). It was thought that trans-lamina cribrosa pressure difference (TLPD) may be associated with pathogenesis of POAG, instead of either IOP or CSFP alone. These questions have caused controversy in the field of ophthalmology. Previous studies showed that POAG does not only involve the optic nerve damage but also affects the lateral geniculate body, the optic radiation, and the visual cortex. It is a multilevel syndrome throughout the visual pathway. The mechanism of glaucomatous lesion is complex, which involves transsynaptic damage, blood supply disorder of the visual pathway, and blood-brain barrier abnormalities. It was hypothesized that glaucoma might be recognized as a CNS disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Primary open-angle glaucoma (POAG) is an optic nerve disease with elevated IOP as the main risk factor. In recent years, with the development of interdisciplinary in the ophthalmology and neurology, new questions about the essence of POAG have been raised. Is POAG just an ocular disease? Is it a disease which involves the eye first and then the whole visual pathway? Or sometimes is it an ocular manifestation of a particular central nervous system (CNS) disease? Recent experimental and clinical studies have suggested that POAG patients may have an abnormally low cerebrospinal fluid pressure (CSFP). It was thought that trans-lamina cribrosa pressure difference (TLPD) may be associated with pathogenesis of POAG, instead of either IOP or CSFP alone. These questions have caused controversy in the field of ophthalmology. Previous studies showed that POAG does not only involve the optic nerve damage but also affects the lateral geniculate body, the optic radiation, and the visual cortex. It is a multilevel syndrome throughout the visual pathway. The mechanism of glaucomatous lesion is complex, which involves transsynaptic damage, blood supply disorder of the visual pathway, and blood-brain barrier abnormalities. It was hypothesized that glaucoma might be recognized as a CNS disease.
POAG is the second leading cause of blindness in the world after cataract. The prevalence rate of POAG is 0.21–1.64%, and the prevalence rate of people over 40 years old is 1–2%. By 2020, the number of POAG patients worldwide will be approximately 80 million [1]. The retinal ganglion cell death is the major cause of retinal nerve fiber layer thinning and visual field defects in POAG, and reducing IOP is the main treatment proven to be effective. The progression of glaucomatous damage can also sometimes be delayed by reducing the IOP in patients with normal tension glaucoma (NTG). However, even if the IOP is well controlled in some patients with advanced glaucoma, the glaucomatous optic neuropathy is still progressing. Recent studies revealed that some CNS degenerative diseases were similar to glaucoma in the characteristics and mechanisms. Animal experiments and human studies showed that POAG was more like a syndrome of the entire visual pathway from the retina to the optic nerve, optic chiasm, optic tract, lateral geniculate body, visual radiation, and visual cortex. Morphological and functional changes existed in all parts above, leading to a fundamental change in the concept of glaucoma – is POAG a CNS disease?
1 The Relationship Between POAG and CNS Diseases
POAG is similar to many other CNS diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and muscular atrophic lateral sclerosis. In general, these diseases start with axonal transport abnormality and are then followed by chronic neuronal degeneration with apoptosis as a common pathway. The relationship and similarity between AD and glaucoma are some of the examples.
Tamura et al. [2] found that 41 (23.8%) of 172 AD patients suffered from POAG, which was significantly higher than that (9.9%) of 176 age-matched controls. It was also found that there was no significant difference in IOP between glaucoma patients and non-glaucoma patients in the Alzheimer’s disease group. A retrospective study of 112 AD patients in Germany by Bayer et al. [3] found that 29 patients of them (25.9%) had glaucomatous visual field defects and/or the cup-to-disc ratio is greater than 0.8. This study showed that the morbidity rate of POAG in AD patients was ten times higher than that in patients with non-AD. Bayer et al. [4] also reported 12 cases (24.5%) with glaucoma visual field defection or disc ratio >0.8 out of 49 patients with AD, which was significantly higher than that in the normal control group (6.5%). The above studies suggest that patients with AD were more possibly to develop glaucoma. Hence some authors proposed that glaucoma was an ocular Alzheimer [5].
This viewpoint was supported by more and more evidences recently. In 2017, Chiara Criscuolo et al. [6] reviewed the similarity between AD and POAG in terms of synaptic dysfunction. Pathologically, the two diseases form chronic neurological damage by characteristic synaptic dysfunction. Both of them have protein aggregates such as the beta amyloid (Aβ) and intracellular microtubule inclusions with hyperphosphorylated tau, which belongs to microtubule-associated protein family. During the early phase of degeneration, the two diseases are characterized by synaptic dysfunction and mitogen-activated protein kinase (MAPK) changes.
Aβ plays an important role in both diseases. Ambra Masuzzo et al. [7] reviewed the amyloidosis in retinal neurodegenerative diseases including age-related macular degeneration (AMD) and POAG comparing to AD. Among the neurodegenerative diseases, which are related to Aβ amyloidosis, AD is undoubtedly the best known and the most studied. Recently, it has been recognized that Aβ-related amyloidosis also exists in POAG and AMD. The Aβ-related amyloidopathies include the increase of intra- and/or extracellular Aβ accumulation and deposition, in the form of insoluble substances, such as amyloid plaques or drusens. Most neurodegenerative diseases are associated with amyloidopathies. The amount of Aβ that exceeds normal physiological level would produce cytotoxicity that might be related to the disorders of Aβ-associated modulation of synaptic excitability in physiological conditions [8]. In addition, Aβ is also thought to be associated with mitochondrial dysfunction and glial activation in both visual pathway and the whole CNS [7].
Li et al. [9] developed a therapy method targeting amyloid β which was used to be a target of AD to treat POAG. They demonstrated that targeting different components of the Aβ formation and aggregation pathway could effectively reduce glaucomatous RGC apoptosis in vivo, and finally, the combined triple treatments were more effective than monotherapy.
There are also some central nervous demyelinating lesions, such as PD, that may be associated with POAG. Bayer et al. [4] found that 9 out of 38 PD patients (23.7%) suffered from POAG, which was significantly higher than the prevalence of glaucoma in the general population, indicating that these two diseases may also have a certain correlation.
In 2015, Fani Tsolaki et al. [10] elucidated the putative association between various forms of dementia, POAG, and Helicobacter pylori (H. pylori) infection in all possible combinations. A total of 156 patients were recruited in this study. Then they were divided into a dementia group, a POAG group, and two control groups. All patients were tested by neuropsychological evaluation, POAG detection, and H. pylori diagnostic testing. After statistical analysis, their results suggested that there were positive correlations between H. pylori infection and dementia, as well as H. pylori infection and POAG. H. pylori infection was more popular in CNS disease (dementia) group and POAG group comparing to the control group (68.33% in patients with dementia vs. 45.16%, p < 0.05, 68.57% in patients with POAG vs. 45.16%, p < 0.05). Furthermore, there was a higher rate of POAG in patients with AD and PD (16.66% dementia vs 0% control), as well as higher frequency of dementia (AD and frontotemporal dementia, FTD) in patients with POAG (16.66% POAG vs. 0% control), comparing to the control group. They concluded that neurodegenerative diseases such as dementia and POAG were linked to each other and to H. pylori infection.
Besides the same pathogenesis factors which can support that CNS diseases and POAG may be a same kind of disease, the similar manifestations can also indicate that POAG is a type of CNS disease. Multiple sclerosis is a chronic, inflammatory, and demyelinating lesion of CNS. Its optic nerve changes are very similar to POAG. It was believed that both of them may share a similar pathogenesis, and both of them express high level of vasoconstrictor cytokines [11].
2 The Relationship Between POAG and TLPD
The two optic nerves are the only cranial nerves which pass through the three sealing containers with pressure. Firstly, the RGCs and its axons belong to the ocular container. Then, the axons are surrounded by cerebrospinal fluid along its pathway until into the third container, the intracranial container. The connection between the first two containers is called the lamina cribrosa, which is the part of the sclera tissue that is pierced by the axons. The intracranial container is a bony structure without space to expand. Therefore, the intracranial pressure change could severely impair the neuro structures. Meanwhile, the ocular container is constructed with compact fiber tissues, also with no space to expand.
Normally, the IOP is a little bit higher than the intracranial pressure, which is 5–11 mmHg. When the pressure changes either in the intracranial or intraocular container, the pressure gradient might impose a sheared force on the site where the two pressures meet. The backward indentation of the lamina cribrosa of sclera is related to the increased IOP. The low intracranial pressure may also show the similar effects. In addition, the changes of pressure and composition of cerebrospinal fluid might also be associated with the glaucomatous optic neuropathy [12].
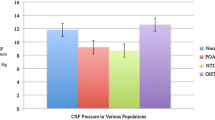
A prospective iCOP study from the Ningli Wang and his colleagues revealed that patients with NTG and/or high-pressure glaucoma as well as patients with ocular hypertension received a neurological examination of a lumbar puncture wit lumbar CSFP measurement. When compared to non-glaucomatous control group, the patients with NTG had significantly lower lumbar CSFP than those with high-pressure glaucoma or those in the control group. Consequently, the TLPD was significantly higher in both glaucoma groups than in the control group. Patients with ocular hypertension had significantly higher CSFP. It also confirmed that the glaucomatous optic nerve damage, evaluated by neuroretinal rim area and visual field defect, was related to the decrease of CSFP and the increase of TLPD [13].
Gallina et al. [14] investigated 22 patients with normal pressure hydrocephalus (NPH) who had undergone ventriculoperitoneal (VP) shunt placement. Interestingly, after more than 6 months follow-up, they found nine of them had NTG. The median follow-up time was 12.0 months in patients with NTG and 18.0 months in those without NTG. This study concluded that changes in the pressure gradient between intraocular and intracranial compartments at the lamina cribrosa level play a role in the pathogenesis of NTG. If this is true, the decreased pressure gradient should be beneficial to prevent the development and progression of NTG. The study from Sawada et al. [15] supported this hypothesis. One hundred and fifty-nine myopic eyes with glaucomatous VFD under treatment and follow-up for 7 years were studied. The results showed that in myopic eyes, there are specific patterns of LC defect, which can function as a pathway to balance the pressure between the IOP and ICP, suggesting an important role of the pressure gradient in the pathogenesis of NTG. Imbalance of the TLPD may play an important role in the pathogenesis of glaucomatous optic nerve damage and other intracranial and intraocular pressure-related diseases.
All these suggest the association between glaucoma and CNS diseases. It is very necessary to have further study in this field. In another chapter of this book, the theory of intracranial and intraocular pressure-related diseases is discussed in detail.
3 DBA/2 Mouse Model of Glaucoma
Calkins et al. [16] pointed out that the initial lesion of POAG was located in the midbrain instead of the eye. The DBA/2 glaucoma mouse was a rodent model which IOP increased along with age. When cholera toxin – cholera toxin B (CTB) – was injected into the vitreous cavity of DBA/2 mouse glaucoma model, by observing the uptake and transport of CTB by RGCs, the early axonal injury could be detected. It was thought that the first sign of glaucomatous damage should be found in the retina. However, the results were unexpected. The earliest site of injury was in the midbrain, where the end of optic nerve fibers project at. His main findings are described below.
3.1 Obstruction of Axoplasmic Transport in Glaucoma Was Developed from the Proximal to the Distal Axon
Some of the optic nerve fibers of the DBA/2 mouse model of glaucoma were projected to the second layer of the superior colliculus (SC) via the third layer of SC, and the others were projected to the optic nerve nucleus, lateral geniculate nucleus, and ovule dome. The transportation of 3-month-old DBA/2 mouse axons was normal, and ordinary CTB signals could be detected in the SC. When these mice grew to 12 months old, their IOPs increased significantly, with the CTB signal reduced in the SC. The CTB signals could not be detected in the SC in the 14 DBA/2 mice aged 10–12 months and could be observed in their lateral geniculate body (43%), optic tract (29%), and optic nerve (21%), respectively. These finds suggested that the early axoplasmic transport disorder and axonal malnutrition in DBA/2 glaucoma mice were in the SC – the area where the RGC cell fibers projected, not in the eye, and the lesion developed slowly from the distal end to the proximal end. This pathological process of peripheral axons is very similar to some CNS diseases, such as AD, PD, and amyotrophic lateral sclerosis [17,18,19].
3.2 Obstruction of Distal Axonal Transport in Glaucoma Owns Characteristics of Age–Dependent and Retinal Localization
Calkins et al. [16] found that the CTB signal in the SC in the 5-month-old DBA/2 mice and the C57BL/6 mice was almost at the same time, while that in the 8-month-old and 10-month-old DBA/2 mice was decreased by 25% and 88%, respectively. And the pattern of CTB signal decrease owned retinal localization. At the same time, the authors also analyzed the correlation between the age, the IOP, and the decrease of CTB signal in the SC. The results were summarized below. In DBA/2 mice aged from 3 to 10 months, the decrease of CTB signal in the SC was significantly correlated with age. Particularly, no significant IOP change was found in 3- to 5-month-old mice, and their CTB signal intensity showed almost no difference. However, in mice aged from 5 to 8 months, the increase of IOP was found to be the most obvious one. But there was no significant correlation between the IOP change and the CTB signal decrease. At last, it was found in 8- to 12-month-old mice that the increase of IOP was relatively slow, while the CTB signal expressed was decreased by 96%. The elevation of IOP in this period was significantly related to the CTB signal change. In order to confirm the relationship among age, IOP, and the axonal terminal transport disorder of ganglion cells, the polystyrene microspheres were injected into the anterior chambers of two groups of DBA/2 mice aged from 3 to 4 months and 7 to 9 months, respectively. The IOP of mice in both groups elevated about 45% drastically. Meanwhile the same volume of saline was injected into the anterior chambers of mice from their corresponding control groups; no IOP change was found. In CTB signal analysis, it was found that there was no obvious SC CTB signal decrease in the 3- to 4-month-old group after the IOP elevated, but in the 7- to 9-month-old group, the same CTB signal was significantly weakened while the IOP was increasing. These results indicated that the glaucomatous distal axonal transport deficits were age related. As a highly risk factor of glaucoma, elevated IOP was more likely to cause distal axonal transport deficits in the elder group, which also suggested that glaucoma was similar to the other age-related neurodegenerative diseases, such as AD and PD.
3.3 The Axonal Structure of RGC Cells in Glaucoma Is Still Preserved After the Obstruction of Axonal Transport
Calkins chose two specific antibodies to mark the estrogen-related receptor and the glutamate vesicle transporter 2, respectively. The former can show the fibers of the whole RGC, and the latter can specifically show the synapse of RGCs. The results showed that the number of distal axons and proximal axons in DBA/2 mice decreased with the increase of age and the number of distal axons always decreased more obviously, which indicated that axonal damage also developed from the distal end to the proximal end. In 18-month-old DBA/2 mice, distal axonal transport was impaired, and CTB signals were not seen in the SC, but the anti-ERR staining results showed that there was no significant difference comparing to the 3-month-old DBA/2 mice. Moreover, in those 17-month-old DBA/2 mice, some of them might not observe the SC CTB signal, but both the anti-ERR and anti-VGluT2 staining results of them showed almost 100% stained, which indicates that the structures of the RGCs axons are still present for a certain period of time after the axonal transport is impaired in glaucoma.
4 The Visual Pathway Damage Induced by POAG
The axons of the retinal ganglion cells converge into the brain and form the optic nerve. Ninety percent of the fibers reach the lateral geniculate body. And the optic radiation projects to the occipital visual cortex. The other 10% of the fibers project to the superior colliculus and the pretectal area. The DBAs/2 glaucoma first showed the axoplasmic transport barrier in the peripheral axles of RGC cells, which is a typical lesion of senile dementia and amyotrophic lateral sclerosis. According to the transsynaptic mechanism of nerve injury, the damage of neurons causes secondary degeneration of neurons that are anatomically or functionally linked to it; in other words, the retrogression of RGCs can lead to damage to the lateral geniculate body, visual radiation, and visual cortex; meanwhile degeneration also retrogrades damage to RGC cells. The mechanisms may be oxidative damage and glutamate excitatory toxicity. Yucel et al. [20] have observed the obvious protective effects of retinal ganglion cells and lateral geniculate cells on glaucoma model of rhesus monkey by blocking N-methyl-d-aspartate receptor.
4.1 The Damage of the Anterior Visual Pathway of POAG
Sasaoka et al. [21] used a monkey laser-induced high intraocular pressure (IOP) model and observed that the diameter of the optic nerve was reduced and the optic chiasm became thin after 16 weeks after laser. In human studies, Kashiwagi et al. [22] performed MRI examination on 31 POAG patients and 23 normal controls. It was found that the optic nerve diameter and the thickness of optic chiasm in glaucoma patients were significantly smaller than those in normal controls and were positively correlated with the degree of visual field defect and the enlargement of cup-disc ratio. Garaci et al. [23] used NMR diffusion tensor imaging (DTI) and showed that the partial anisotropy ratio of optic nerve in POAG patients was reduced, the average diffusion rate was increased, and the range of change was related to the course of POAG. In the autopsy of POAG patients, it was also found that the diameter of the optic nerve was significantly reduced, the intraorbital pial meninges of the optic nerve were thickened, and the number of subarachnoid meningeal cell nests was obviously increased [24, 25].
4.2 The Damage of Posterior Visual Pathway in POAG
The lateral geniculate body is an important transit point for the visual system. It is divided into six layers. The cross fibers from two eyes terminate in the first, fourth, and sixth layer of the opposite side, and the non-cross fibers end in the ipsilateral layer of second, third, and fifth. The monocular glaucoma models of crab monkey and rhesus monkey were both confirmed that the replacement neurons in layers 1, 4, and 6 of the contralateral geniculate body and the ipsilateral layers 2, 3, and 5 of the ipsilateral geniculate body were obviously reduced, the large cell neurons and small cell neurons were both involved, and the denaturation degree of small cell neurons was significantly higher than that of large cell neurons [26]. The degeneration of neurons was accompanied with the proliferation of glial cells [27]. Postmortem [23] and MRI results [28] also verified that the lateral geniculate body of POAG patients was significantly smaller than that of age-matched normal persons. The decrease in the number and function of neurons in the lateral geniculate body caused by POAG is bound to correspond to the changes of visual radiation and the visual cortex. Chan et al. [29] found that the ratio of choline/creatine in the visual cortex metabolites of chronic glaucoma mice was significantly lower than that in the normal control group by using magnetic resonance spectroscopy. Choline was involved in the synthesis of some neurotransmitters and acetylcholine, and it was also involved in cell membrane synthesis and cell regeneration, which may be related to the decreased activity of visual cortex cells. The decrease of the ratio of choline/creatine may be related to the decrease in the activity of visual cortex cells. Magnetic resonance imaging (MRI) on human body showed that visual radiation and injury to the visual cortex were confirmed. By using magnetic resonance diffusion imaging, Garaci et al. [23] found the decreasing of the anisotropy ratio of visual radiation and the increasing of the average diffusion rate in POAG patients and then proved the damage of visual radiation fiber. At the same time, Kitsos et al. [30] found that the white matter of the cerebrum of alive patients with POAG was higher than that of the normal control group in MRI results. Magnetization transfer imaging showed that the magnetization transfer rate of gray matter and white matter in POAG patients was lower than that in normal controls. Human autopsy provides strong evidence too that comparing with normal people, POAG patients manifested an obvious atrophy – visual cortex gray matter was thinner, and cerebral gyrus was shallower [25].
4.3 Other Glaucomatous Damage in CNS
Chiquet et al. [31] found that in the mouse glaucoma model, except the damage of the whole optic pathway, damage of the suprachiasmatic nucleus, which is related to the synchronization of the circadian rhythm, also exists. This may be initiated from the synaptic damage caused by the death of the retinal ganglion cells in the nucleus. Animal experiments showed that both acute and chronic high IOP could damage the melanopsin-containing retinal ganglion cells which were related to the regulation of the circadian rhythm [32], indicating that glaucoma could also damage the non-shape sensitive conduction pathway.
5 Mechanism of Action (MOA) Study of CNS Damage in Glaucoma
Besides the damaged RGCs, high IOP which blocks the axonal transportation and the secondary demyelination of nerve fibers may also contribute to the glaucomatous damage and dysfunction of the optic nerve fiber. Downs et al. [33] using the three-dimensional tissue measurement techniques reconstructed the optic disc of the monkey glaucoma model. It was found that the nerve tube and the subarachnoid structure in the beginning of the optic disc were obviously deformed. The optic nerve is part of the CNS, but the anterior vessels of the optic nerve sieve do not have the function of the blood-brain barrier and thus lack the barrier to the harmful substances. In the glaucoma patients, the barrier function is more obvious, which may also be one of the causes of the optic nerve damage. The CNS lesion of POAG is closely related to extensive cerebral blood supply deficiency. In 15 cases of POAG, Siesky et al. [34] found that the visual acuity, contrast sensitivity, mean central visual field defect, and the amplitude of electroretinogram of these subjects were closely related to the mean blood flow rate of the middle cerebral artery.
Neuronal damage caused secondary degeneration of its anatomic or functional contacted neuron, which is known as transsynaptic damage and is a typical lesion of AD and amyotrophic lateral sclerosis. The lateral geniculate body and visual cortex damage caused by glaucoma belong to the category of transsynaptic damage. Both of the anterograde and retrograde cross-synaptic damages exist in this kind of lesion. The mechanism may be oxidative damage and excitatory toxicity of glutamate. Reactive oxygen species which accumulated in the oxidative stress state reacting with nitrous oxide formed pernitrite. Thereby the intracellular environment was altered, and cell death was conducted. Pernitrite can turn protein into nitrotyrosine, and it is a marker of various neurodegenerative diseases. Some researchers found nitrotyrosine in the lateral geniculate parenchyma and vascular endothelium of glaucomatous animal models which indicates the mechanism of oxidative damage in glaucoma. Glutamate excitatory toxicity is also one of the causes of many central degenerative diseases. Blocking the N-methyl-D-aspartate (NMDA) receptor can effectively protect the retinal ganglion cells and lateral geniculate bodies from lesion which is caused by glaucoma in Macaca rhesus [20]. Sposato et al. [35] found that the expression of nerve growth factor and its receptor in the lateral geniculate body and visual cortex is downregulated significantly in the glaucoma mouse model. It is considered that the regulation of cytokines and its receptors plays a very important role in glaucomatous damage. Neurochemical changes in the lateral geniculate nucleus and primary optic cortex were studied by Crawford et al. [36] in the glaucoma monkey model, which confirmed that the cytochrome oxidase activity in the lateral geniculate nucleus and the visual cortex was significantly reduced, suggesting that the cytochrome system also played a role in the pathogenesis of glaucoma. The ratio of choline/creatine in the visual cortex metabolites of mice with chronic glaucoma was significantly lower than that in the normal control group by using magnetic resonance spectroscopy [25]. It was believed that the magnetic resonance spectroscopy technique has the potential to study the pathogenesis of glaucoma. The spectrum analysis of the ratio of choline/creatine in the cortex can be used to judge the central damage of glaucoma.
Interestingly, the possible MOA of that Helicobacter pylori infection increases the incidence rate of both CNS diseases such as AD, PD, etc., and POAG was discussed by some literatures. They may share same pathogenesis under the same H. pylori infection stress. Sergio C Sacca et al. [37] reviewed 152 literatures which related to the relationship between H. pylori infection and eye diseases, most of which were POAG from 1998 to 2014. In particular, the pathogenetic researches were focused on oxidative damage. H. pylori infection can accumulate the reactive oxygen species (ROS) production, and then, mitochondrial function declined, mitochondrial DNA mutations accumulated under this condition. The level of oxidative damage to DNA, proteins, and lipids increased, while the capacity to degrade oxidatively damaged proteins and other macromolecules decreased. At last, RGC apoptosis is initiated, and glaucoma is formed.
In CNS diseases, ROS increase after H. pylori infection also played a very important role. Giulia Nesi et al. [38] reviewed the relationship between ROS and AD. They showed that an increasing body of evidence reveals that both mitochondrial abnormalities and metal accumulations synergistically act as major producers of ROS, thus contributing to neuronal toxicity.
Additionally, more pathogenesis factors under other conditions such as microglia reaction were reported contributing to both of CNS diseases and POAG. Ana I. Ramirez et al. [39] reviewed the same role of microglial played in the AD, PD, and POAG. After the literature analysis, they found microglial activation had been reported in AD, PD, and POAG in relation to protein aggregates and degenerated neurons. The activated microglia could release pro-inflammatory cytokines which could aggravate and propagate neuro-inflammation, thereby degenerating neurons and impairing brain as well as retinal function.
Furthermore, same therapy which works for both CNS disease and POAG can also indicate they are same kind of disease. Mead et al. [40] introduced a method of dental pulp stem cell therapy, which can repair CNS damage, including recent findings on RGC neuroprotection and regeneration in optic nerve injury and glaucoma.
It was proved that trans-lamina cribrosa pressure difference (TLPD) may be the pathogenesis for glaucoma, not the elevated IOP or the reduced CSFP alone. It is still hard to say whether increase of TLPD is the hypostasis for POAG or just a manifestation. Many results can be provoked beyond the mechanical environment change by an increased trans-lamina cribrosa pressure difference. For example, the increased TLPD may also increase the strain power of the laminar pore and change the ocular perfusion status; in addition, reduced CSF flow in the optic nerve subarachnoid space may cause accumulation of metabolites and conglutination between optic nerve and its sheath. Killer et al. [41] suggested compartment of subarachnoid space and showed CSF composition difference in POAG patients with normal IOP; it is still hard to say whether the lowering of CSFP or CSF flow in the optic nerve subarachnoid space happens first or the optic nerve sheath compartment happens first. In the future, it is important to explore the downstream pathophysiology of the increased trans-lamina cribrosa pressure difference in POAG to find the true hypostasis.
The orbital CSFP as counterpressure against IOP is a determinant of the TLPD and is important for the pathophysiology of the pressure-related diseases originating at the optic nerve head, such as glaucoma. Taking the physiological triangular relationships between IOP, CSFP, and BP into account, glaucoma may be described as a misbalance between IOP, CSFP, and BP, finally leading to an increase in TLPD.
Since the central retinal vein passes through the orbital CSF space, the orbital CSFP is of importance for the pressure in the retinal veins and potentially for the development of retinal vein occlusions. In a similar manner, since the retinal venous pressure influences the retinal capillary pressure, the orbital CSFP may influence the development and severity of diabetic retinopathy. Similarly, since the choroid drains through the vortex veins into the superior ophthalmic vein and cavernous sinus intracranial, higher CSFP may be associated with a thicker choroid. But it remains unexplored about the body position-dependent and time-dependent changes in TLPD. Some experimental studies suggested that dorsomedial and perifornical hypothalamic neurons may be involved in the regulation of both IOP and CSFP [13].
6 Summary
In summary, it is believed that POAG may be a special CNS disease, because it not only shares similarities with some degenerative diseases of the CNS but also exhibits the functional and structural changes of the whole optic pathway. Additionally, imbalance of the TLPD may play an important role in the pathogenesis of glaucomatous optic nerve damage and other intracranial and intraocular pressure-related diseases.
Therefore, POAG should not only be considered as an ocular disease but a CNS disease that begins with the dysfunction of the axonal transport of RGC cells. On the other hand, it should be pointed out that lowering IOP only is not enough to treat POAG; neuroprotective therapy is also very important, for not only protecting the optical nerve but also the optic pathway and, furthermore, the CNS.
References
Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90(3):262–7.
Tamura H, Kawakami H, Kanamoto T, Kato T, Yokoyama T, Sasaki K, Izumi Y, Matsumoto M, Mishima HK. High frequency of open-angle glaucoma in Japanese patients with Alzheimer’s disease. J Neurol Sci. 2006;246(1–2):79–83.
Bayer AU, Ferrari F, Erb C. High occurrence rate of glaucoma among patients with Alzheimer’s disease. Eur Neurol. 2002;47(3):165–8.
Bayer AU, Keller ON, Ferrari F, Maag KP. Association of glaucoma with neurodegenerative diseases with apoptotic cell death: Alzheimer’s disease and Parkinson’s disease. Am J Ophthalmol. 2002;133(1):135–7.
Mckinnon SJ. Glaucoma: ocular Alzheimer’s disease? Front Biosci. 2003;8(1–3):s1140–56.
Criscuolo C, Fabiani C, Cerri E, Domenici L. Synaptic dysfunction in Alzheimer’s disease and glaucoma: from common degenerative mechanisms toward neuroprotection. Front Cell Neurosci. 2017;111(53):1–7.
Masuzzo A, Dinet V, Cavanagh C, Mascarelli F, Krantic S. Amyloidosis in retinal neurodegenerative diseases. Front Neurol. 2016;7(127):1–17.
Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37(6):925–37.
Guo L, Salt TE, Luong V, Wood N, Cheung W, Maass A, Ferrari G, Russo-Marie F, Sillito AM, Cheetham ME, Moss SE, Fitzke FW, Cordeiro MF. Targeting amyloid-β in glaucoma treatment. PNAS. 2007;104(33):13444–9.
Tsolaki F, Kountouras J, Topouzis F, Tsolaki M. Helicobacter pylori infection, dementia and primary open-angle glaucoma: are they connected? BMC Ophthalmol. 2015;15(1):24.
Jankowska-Lech I, Terelak-Borys B, Grabska-Liberek I, Palasik W. Glaucoma neuropathy and neuropathy in multiple sclerosis- -common elements of pathogenesis? Klin Oczna. 2007;109(7–9):317–20.
Ren R, Jonas JB, Tian G, Zhen Y, Ma K, Li S, Wang H, Li B, Zhang X, Wang N. Cerebrospinal fluid pressure in glaucoma: a prospective study. Ophthalmology. 2010;117(2):259–66.
Jonas JB, Wang N, Yang D, Ritch R, Panda-Jonas S. Facts and myths of cerebrospinal fluid pressure for the physiology of the eye. Prog Retin Eye Res. 2015;46(3):67–83.
Gallina P, Savastano A, Becattini E, Orlandini S, Scollato A, Rizzo S, Carreras G, Di Lorenzo N, Porfirio B. Glaucoma in patients with shunt-treated normal pressure hydrocephalus. J Neurosurg. 2017;17:1–7.
Sawada Y, Araie M, Kasuga H, Ishikawa M, Iwata T, Murata K, Yoshitomi T. Focal lamina cribrosa defect in myopic eyes with nonprogressive glaucomatous visual field defect. Am J Ophthalmol. 2018;190:34–49.
Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci U S A. 2010;107(11):5196–201.
Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellanosanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185(2):232–40.
Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307(5713):1282.
Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6(11):889–98.
Yücel YH, Gupta N, Zhang Q, Mizisin AP, Kalichman MW, Weinreb RN. Memantine protects neurons from shrinkage in the lateral geniculate nucleus in experimental glaucoma. Arch Ophthalmol. 2006;124(2):217–27.
Sasaoka M, Nakamura K, Shimazawa M, Ito Y, Araie M, Hara H. Changes in visual fields and lateral geniculate nucleus in monkey laser-induced high intraocular pressure model. Exp Eye Res. 2008;86(5):770–82.
Kashiwagi K, Okubo T, Tsukahara S. Association of magnetic resonance imaging of anterior optic pathway with glaucomatous visual field damage and optic disc cupping. J Glaucoma. 2004;13(3):189–95.
Garaci FG, Bolacchi F, Cerulli A, Melis M, Spanò A, Cedrone C, Floris R, Simonetti G, Nucci C. Optic nerve and optic radiation neurodegeneration in patients with glaucoma: in vivo analysis with 3-T diffusion-tensor MR imaging. Radiology. 2009;252(2):496.
Pache M, Meyer P. Morphological changes of the retrobulbar optic nerve and its meningeal sheaths in glaucoma. Ophthalmologica. 2006;220(6):393–6.
Gupta N, Ang L, Tilly LND, Bidaisee L, Yücel YH. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus, and visual cortex. Br J Ophthalmol. 2006;90(6):674.
Yücel YH, Zhang Q, Weinreb RN, Kaufman PL, Gupta N. Effects of retinal ganglion cell loss on magno-, parvo-, koniocellular pathways in the lateral geniculate nucleus and visual cortex in glaucoma. Prog Retin Eye Res. 2003;22(4):465–81.
Zhang S, Wang H, Lu Q, Qing GP, Wang NL, Wang YD, Li SN, Yang DY, Yan FC. Detection of early neuron degeneration and accompanying glial responses in the visual pathway in a rat model of acute intraocular hypertension. Brain Res. 2009;1303:131–43.
Gupta N, Greenberg G, Noël de Tilly L, Gray B, Polemidiotis M, Yücel YH. Atrophy of the lateral geniculate nucleus in human glaucoma detected by magnetic resonance imaging. Br J Ophthalmol. 2009;93(1):56–60.
Chan KC, So KF, Wuabc EX. Proton magnetic resonance spectroscopy revealed choline reduction in the visual cortex in an experimental model of chronic glaucoma. Exp Eye Res. 2009;88(1):65–70.
Kitsos G, Zikou AK, Bagli E, Kosta P, Argyropoulou MI. Conventional MRI and magnetisation transfer imaging of the brain and optic pathway in primary open-angle glaucoma. Br J Radiol. 2009;82(983):896.
Chiquet C, Drouyer E, Woldemussie E, Ruiz G, Wheeler L, Denis P, Cooper H, Romanet JP. Consequences of glaucoma on circadian and central visual system. J Fr Ophtalmol. 2006;29(7):847–51.
Wang H, Lu Q, Wang N, Liu H, Zhang L, Zhan GL. Loss of melanopsin-containing retinal ganglion cells in a rat glaucoma model. Chin Med J (Engl). 2008;121(11):1015–9.
Downs JC, Yang H, Girkin C, Sakata L, Bellezza A, Thompson H, Burgoyne CF. Three-dimensional histomor-phometry of the normal and early glaucomatous monkey optic nerve head: neural canal and subarachnoid space architecture[J]. Invest Ophthalmol Vis Sci. 2007;48(7):3195–208.
Harris A, Siesky BD, Haine C, Catoira Y, Sines D, Mccranor L, Garzozi H. Relationship of cerebral blood flow and central visual function in primary open-angle glaucoma. J Glaucoma. 2007;16(1):159–63.
Sposato V, Parisi V, Manni L, et al. Glaucoma alters the expression of NGF and NGF receptors in visual cortex and geniculate nucleus of rats: effect of eye NGF application. Vision Res. 2009;49(1):54–63.
Crawford ML, Harwerth RS, Smith EL, Mills S, Ewing B. Experimental glaucoma in primates: changes in cytochrome oxidase blobs in V1 cortex. Invest Ophthalmol Vis Sci. 2001;42(2):358–64.
Sacca SC, Vagge A, Pulliero A, Izzotti A. Helicobacter pylori infection and eye diseases: a systematic review. Medicine. 2014;93(28):1–13.
Nesi G, Sestito S, Digiacomo M, Rapposelli S. Oxidative stress, mitochondrial abnormalities and proteins deposition: multitarget approaches in Alzheimer’s disease. Curr Top Med Chem. 2017;17(27):3062–79.
Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, López-Cuenca I, Rojas P, Triviño A, Ramírez JM. The role of microglia in retinal neurodegeneration: Alzheimer’s disease, Parkinson, and glaucoma. Front Aging Neurosci. 2017;09(214):1–21.
Mead B, Logan A, Berry M, Leadbeater W, Scheven BA. Concise review: dental pulp stem cells: a novel cell therapy for retinal and central nervous system repair. Stem Cells. 2017;35(1):61–7.
Killer HE, Jaggi GP, Flammer J, Miller NR, Huber AR, Mironov A. Cerebrospinal fluid dynamics between the intracranial and the subarachnoid space of the optic nerve. Is it always bidirectional? Brain. 2007;130(Pt 2):514–20.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Fan, N., Liu, G., Zhang, X., Liu, X. (2019). Primary Open-Angle Glaucoma, Trans-Lamina Cribrosa Pressure Difference, and Central Nerve System. In: Wang, N. (eds) Intraocular and Intracranial Pressure Gradient in Glaucoma. Advances in Visual Science and Eye Diseases, vol 1. Springer, Singapore. https://doi.org/10.1007/978-981-13-2137-5_5
Download citation
DOI: https://doi.org/10.1007/978-981-13-2137-5_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-2136-8
Online ISBN: 978-981-13-2137-5
eBook Packages: MedicineMedicine (R0)