
Abstract
In this chapter, we will focus on the isotropic (or rather less anisotropic) two-dimensional layered materials, including the layered transition metal dichalcogenides, the topologically insulating Bi2X3 (X = Se, Te) etc.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


In this chapter, we will focus on the isotropic (or rather less anisotropic) two-dimensional layered materials, including the layered transition metal dichalcogenides, the topologically insulating Bi2X3 (X = Se, Te) etc.
The layered transition metal dichalcogenides (TMDs), such as MoS2 and WSe2, have been attracting lots of interests in the past decade, because of their unique properties and potential applications [1,2,3,4,5]. Monolayer TMDs are excellent candidates for the study of valley pseudospin [5]. Valley polarization [6,7,8] and valley Hall effect [9] have both been observed and reported. Later, electrical control of valley polarization and valley Hall effect has been realized as well [10, 11], which increases the possibility of potential applications in valleytronics. The observation of valley polarization on spectroscopy, to some extent, benefits from the strong photoluminescence in monolayer. The bulk TMD crystals are indirect-gap semiconductors. As thickness decreases, there is an indirect-to-direct gap transition. Monolayer TMDs possess a direct gap, which gives an emerging photoluminescence [12, 13].
Among all the TMDs, the semiconducting group VI ones (MX2, M = Mo, W; X = S, Se, Te) are widely studied, especially the 2H phase MoS2 [6,7,8,9,10,11,12,13, 16, 17]. Besides the stable 2H stacking, 1T and 3R phases are also common in the S- and Se-based MX2 (M = Mo, W; X = S, Se) [1, 18]. We note that the monolayer counterparts of 2H and 3R phases are the same, also referred to as 1H. The spatial inversion symmetry is broken in the 1H stacking, but preserved in the 1T structure, as shown in Fig. 2.1a. The difference between 2H and 3R starts from the bilayer (2 L). Besides the layer shift, which also occurs in the 3R phase, the Nth layer of the 2H stacking is rotated by 180° with respect to the (N−1)th layer. The 3R stacking is noncentrosymmetric from monolayer (1L) to the bulk, giving rise to the valley polarization regardless of the thickness [19]. In contrast, the 2H stacking with an even number of layers possesses an inversion symmetry point, while that with an odd number of layers is noncentrosymmetric. In Te-based TMDs (MTe2, M = Mo, W), 1T’ phase is also thermodynamically stable, as the large size of the telluride atom distorts the structure [15, 20]. The Raman spectra of MoTe2 have shown dependence on excitation lasers [21,22,23], which will be discussed in Chap. 5. On the other hand, the phonon properties in WTe2 are more complicated than other compounds [24, 25]. Here we will focus on the S- and Se-based TMDs.

Structural polytypism in MX2 (M = Mo, W; X = S, Se, Te). (a) Trilayer (3 L) MX2 (M = Mo, W; X = S, Se) with 1T, 2H, and 3R stacking configurations. (Reproduced with permission from Ref. [14], © Tsinghua University Press and Springer-Verlag Berlin Heidelberg 2016). (b) Comparison of 2H, 1T, and 1T’ structures of monolayer MoTe2. (Reproduced with permission from Ref. [15], © American Association for the Advancement of Science 2014)
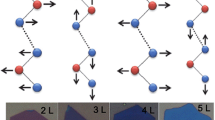
Each layer of 2H-MX2 (M = Mo, W; X = S, Se) consists of X–M–X tri-atomic planes, with the plane of Mo/W atoms sandwiched in two hexagonal planes of chalcogenide atoms. The primitive unit cell of bulk 2H-MX2 has two layers, i.e., 6 inequivalent atoms. Thus, there are 18 Brillouin zone center (Γ) phonons (3 * number of atoms), with 15 optical phonons and 3 acoustic phonons. The irreducible representation of the zone center phonons in bulk 2H-MX2 (M = Mo, W; X = S, Se) is Γ = A 1g + 2A 2u + B 1u + 2B 2g + E 1g + 2E 1u + E 2u + 2E 2g. As the thickness decreases from bulk to few- and mono-layer, we have to consider the difference in symmetry between films with even and odd number of layers (even N and odd N). The irreducible representations in odd N and even N are \( \Gamma =\frac{3N-1}{2}\left({A}_1^{\prime }+{E}^{{\prime\prime}}\right)+\frac{3N+1}{2}\left({A}_2^{{\prime\prime} }+{E}^{\prime}\right) \) (N = 1, 3, 5…) and \( \Gamma =\frac{3N}{2}\left({A}_{1g}+{A}_{2u}+{E}_g+{E}_u\right) \) (N = 2, 4, 6…), respectively. Here E denotes the doubly degenerate in-plane modes, while A and B represent the out-of-plane vibration modes. Among these modes, only A 1g, E 1g, and E 2g are Raman-active. Note that the A 1g mode for bulk 2H-MX2 (∼408 cm−1 for MoS2) is \( {A}_1^{\hbox{'}} \) in 1L due to the change of symmetry, though the corresponding mode is still A 1g in 2L. Modes that are neither Raman active nor infrared (IR) active are called the silent modes, such as B 2g mode in bulk MoS2. The normal mode displacements for 1L, 2L, and bulk 2H-MX2 (M = Mo, W; X = S, Se) are shown in Fig. 2.2a, b.

(a and b) Normal displacements of vibrational modes for 1 L (a) and 2 L & bulk (b) 2H-MX2 (M = Mo, W; X = S, Se). (Reproduced with permission from Ref. [26], © American Chemical Society 2013). (c and d) Phonon dispersion curves of single-layer (c) and bulk MoS2 (d). Points in (d) are experimental data extracted from Ref. [27]. (Reproduced with permission from Ref. [28], © American Physical Society 2011)
In the following, we will take 2H-MoS2 as an example to further discuss the phonon modes. The calculated phonon dispersion curves for 1L and bulk 2H-MoS2 are displayed in Fig. 2.2c, d. There are 3 acoustic branches in 1L: the in-plane longitudinal acoustic (LA), the transverse acoustic (TA), and the out-of-plane acoustic (ZA) modes. The 6 high frequency optical branches are separated from the acoustic branches by a gap of ∼50 cm−1 for MoS2 [28]. The optical phonon dispersion curves for bulk are similar to those for 1L MoS2, as a result of the weak van der Waals interlayer interactions. Meanwhile, we can observe the Davydov splitting of optical modes from Γ point in Fig. 2.2d, e.g. the mode at ∼384 cm−1 (with symmetry of \( {E}_{2g}^1 \) in bulk) and the mode at ∼385.5 cm−1 (with symmetry of E 1u in bulk). Davydov splitting occurs when there is more than one interacting layer from the system [29,30,31]. The difference within the Davydov pair, \( {E}_{2g}^1 \)/E 1u, lies in the displacement between the adjacent sulfur atoms. The sulfur atoms of different layers move in opposite direction for \( {E}_{2g}^1 \) mode, while sulfur atoms of neighbouring sheets are moving in phase for E 1u mode. Due to the weak interlayer coupling, the frequency difference between the two modes is ought to be small, but the out-of-phase displacement for \( {E}_{2g}^1 \) mode is supposed to slightly increase the frequency, compared to that for E 1u mode. The acoustic branches for bulk, which are related to interlayer interaction, differ from those in 1L. Chap. 9 will discuss the interlayer shear and breathing modes in detail.
Raman spectra of bulk MoS2 were collected at different excitation wavelengths, shown in Fig. 2.3a. Only two Raman-active modes (\( {E}_{2g}^1 \) and A 1g) can be seen with 488 nm and 532 nm excitations. Many additional modes, mostly overtones and combinations, appear in the Raman spectra when 594 nm (2.09 eV) and 633 nm (1.96 eV) lasers are used. Resonant Raman spectra of MX2 (M = Mo, W; X = S, Se, Te) will be further discussed in Chap. 5. Here we focus on the \( {E}_{2g}^1 \) and A 1g modes.

(a) Raman spectra of bulk MoS2 crystal under different excitation wavelengths at room temperature. (Reproduced with permission from Ref. [38], © AIP Publishing LLC 2014). (b) Raman spectra of few-layer and bulk MoS2 films (excitation laser: 514.5 nm). The solid line for the 2 L spectrum is a double Voigt fit through data (circles for 2L, solid lines for the rest). (c) Frequencies of \( {E}_{2g}^1 \) and A 1g Raman modes (left vertical axis) and their difference (right vertical axis) as a function of layer thickness. (Reproduced with permission from Ref. [32], © American Chemical Society 2010)
The Raman tensors for \( {E}_{2g}^1 \) and A 1g modes in bulk 2H-MX2 (M = Mo, W; X = S, Se) can be written as:
Correspondingly, A 1g mode has \( {A}_1^{\prime } \) symmetry in odd N, while it is still A 1g in even N. E 2g mode has E '' symmetry and E g symmetry in odd N and even N, respectively. Polarization dependent Raman spectra can distinguish the in-plane E mode and the out-of-plane A mode. As shown in Fig. 2.3b, c, A 1g mode is suppressed under the cross-polarized configuration; In contrast, \( {E}_{2g}^1 \) mode appears under both cross- and parallel-polarized configurations. Reason for the difference is related to the quantity \( {e}_i\cdot \tilde{R}\cdot {e}_s \), where e i is the polarization vector of the incident light, e s is that of the scattered light and \( \tilde{R} \) is the Raman tensor. \( {e}_i\cdot \tilde{R}\cdot {e}_s \) is given by:
For the parallel-polarization \( \overline{z}(xx)z \), y = z = 0, and x is non-zero; For the cross-polarization \( \overline{z}(xy)z \), the incident light is polarized along x direction, while the outgoing scattering light is polarized along y direction. Take the Raman tensor of A 1g mode into calculation. It shows a nonzero value when it is \( \overline{z}(xx)z \) configuration. However, the calculational result is zero when \( \overline{z}(xy)z \) configuration is applied. Therefore, the intensity of A 1g mode at ∼408 cm−1 depends on the polarization of excitation light.
Layer-dependent Raman spectra of MoS2 is displayed in Fig. 2.3d. As thickness increases, A 1g mode shifts to higher frequency (blue shift, stiffening), and \( {E}_{2g}^1 \) mode is shifted towards lower frequency (red shift, softening) [32,33,34,35]. The frequency difference is shown in Fig. 2.3e. In general, the difference increases in thicker sample, and it is around 19 cm−1 for the mechanically exfoliated monolayer MoS2 [32]. While MoS2 is not an exception, similar results have also been found in WS2, WSe2, and MoSe2 [18, 36, 37]. The frequency difference between \( {E}_{2g}^1 \) and A 1g modes provides a novel and rapid way in the identification of layer number in thin film (<6L) TMDs. However, the opposite thickness evolutional trend between \( {E}_{2g}^1 \) and A 1g modes deserves further investigation and discussion.
The phonon dispersion curves in Fig. 2.2c, d show that the A 1g mode is softened away from the Γ point, but the \( {E}_{2g}^1 \) mode is dispersionless. Therefore, a change in the frequency of the \( {E}_{2g}^1 \) mode cannot be explained by the Richter-Wang-Ley (RWL) theory [39], which was widely used in explaining the size dependence of the asymmetric broadening and frequency shift in nanomaterials [39,40,41,42]. In MoS2, the relative displacements of sulfur atoms from adjacent layers are out-of-phase, as shown in Fig. 2.2b. According to the intuitive harmonic oscillator model, the frequencies of both \( {E}_{2g}^1 \) and A 1g modes should be blue-shifted when additional layers are added, owing to the larger accumulated restoring force. This effect will be referred to “the thickness effect”. The frequency-shift for the A 1g mode is consistent with the thickness effect. On the other hand, the opposite frequency trend for the \( {E}_{2g}^1 \) mode is not consistent with the thickness effect.
The differences in lattice constant and interatomic distances as thickness decreases might be a possible reason for the anomalous shift of \( {E}_{2g}^1 \) mode, but it has been ruled out by Molina-Sanchez et al. [28]. In the Raman community, it was widely believed that the anomalous shift of the \( {E}_{2g}^1 \) mode in 2H-MoS2 is related to a thickness dependence of the dielectric screening, a conclusion based on early calculations on dielectric screening by Molina-Sanchez et al. [28]. However, Lin et al. later showed experimentally that the frequencies of the \( {E}_{2g}^1 \) and A 1g modes have negligible dependence on the dielectric constant of the environment [43], consistent with Ref. [33], as well as a recent review by Molina-Sanchez et al. [44]. Here we think that it is the effect of larger M-X (M = Mo, W; X = S, Se) force at the surface of the thin film that makes a difference, which contributes to the anomalous red shift of \( {E}_{2g}^1 \) mode with increasing thickness. Details on surface effect will be discussed in Chap. 7.
Besides the 2H-stacked MX2 (M = Mo, W; X = S, Se), it will also be interesting to investigate how stacking order affects the phonon properties. As 1T phase is metastable, most previous studies just focus on the 2H and 3R stacking configurations in MX2 (M = Mo, W; X = S, Se). It is shown that in MoS2, MoSe2, and WSe2, the intalayer vibrational modes do not show observable dependent on stacking orders; while the low frequency interlayer shear modes are very sensitive to the phase difference [18, 45,46,47]. Similarly, polytypism in the isotropic layered material ReS2 shows distinguishable spectral features on the low-frequency Raman spectra, but there is not much difference on the intralayer vibrations [48, 49]. Stacking-dependent low-frequency interlayer modes will be further discussed in Chap. 9.
However, MoTe2, different from the S- and Se-based TMDs, demonstrates stacking-dependent intralayer vibrational modes [20, 50], as shown in Fig. 2.4. Take the intralayer vibration A 1g mode as an example. It locates at 162–164 cm−1 for the 1T’ phase, and 170–172 cm−1 for the 2H stacking. As a comparison, the high frequency A 1g mode has the same energy in 2H- and 3R- stacked MoS2. It red-shifts by 4 cm−1 in the 1T phase [51]. As we have discussed in Fig. 2.1, the monolayer counterparts of 2H and 3R phases are the same, and the difference between the two phases lies in the interlayer stacking and rotation, which hardly affects the intralayer A 1g mode. Compare 1T and 2H monolayers; we can see that one of the S atoms has 180° rotation (Fig. 2.1a), which would certainly influence the Mo-S force and shift the energy of A 1g mode. Besides having one chalcogenide atom rotated, 1T’ MoTe2 possesses a distorted structure. The combined effects render the energy difference between 2H and 1T’ phases to reach almost 10 cm−1 in A 1g mode.

(a) Layer-dependent Raman signals for 1T’-MoTe2. The normal displacements of vibrational modes are illustrated in the insets. (b) Peak positions in 1T’-MoTe2 with various numbers of layers. (c) Layer-dependent Raman signals for 2H-MoTe2. The normal displacements of vibrational modes are illustrated in the insets. (d) Peak positions in 2H-MoTe2 with various numbers of layers. (Reproduced with permission from Ref. [20], © Nature Publishing Group 2015)
In addition to thickness and stacking, the phonon properties of TMDs can also be influenced by the excitation laser [52, 53], strain [54], doping [55], and temperature [56,57,58,59]. Temperature-dependent Raman scattering can be used to estimate the thermal conductivity in MoS2, thanks to the strong Raman intensities of \( {E}_{2g}^1 \) and A 1g modes [56,57,58,59]. Similarly, Raman spectroscopy has been used to measure the thermal conductivity in graphene [60], while it is not suitable for BN due to the weak Raman signal.
A moderate power of laser is indispensable for the accurate estimation of thermal conductivity, as laser spot can act as a heat source and cause local heating [56,57,58,59, 61]. The measurements on monolayer, few-layer, and bulk MoS2 have been conducted by different groups [56,57,58,59], using both suspended and (silicon-,sapphire-, glass-) supported samples. Most groups observed linear softening of \( {E}_{2g}^1 \) and A 1g modes as temperature increases, and the change of frequencies are within (1.1–1.7) × 10−2 cm−1 K−1 regardless of thickness and substrate. The temperature-dependent Raman shift is mainly attributed to the thermal expansion (volume contribution) and the anharmonic contributions to the ionic interaction potential in the two-dimensional system. It is worth noting that the expansion of (substrate-supported) MoS2 lattice due to increased temperature sometimes is hindered by the cooler substrate. Strain would appear and influence the in-plane \( {E}_{2g}^1 \) mode, while A 1g mode is less sensitive to strain [54, 62, 63]. Thus it will be more accurate to use A 1g mode to obtain thermal conductivity. Figure 2.5 shows an example of temperature-dependent Raman shift in monolayer MoS2. In general, nonlinear coefficients are not considered, given that they are only supposed to impact at higher temperatures [57, 60]. However, two groups have observed the nonlinear temperature-dependent Raman shift at low temperature (<100 K) from MoS2 [59] and WS2 [64]. Taube et al. believe the nonlinearity is related with the optical phonon decay and lattice potential anharmonicity in the monolayer samples [59]. On the other hand, Yan et al. observed the nonlinearity from power-dependent Raman scattering measurements at room temperature. They consider that the appearance of nonlinear effects results either from the nonlinearity of absorption or higher orders of the temperature-dependent coefficients [56]. Combing the linear temperature (Fig. 2.5) and power-dependent coefficients of the A 1g mode, Yan et al. extracted the thermal conductivity κ = (34.5 ± 4) W mK−2 from suspended monolayer MoS2 [56], which is in accordance with theoretical prediction [65].

(a) Four example Raman spectra of suspended, monolayer MoS2 collected at 100, 180, 260, and 320 K. Spectra offset vertically for clarity. (b) Raman peak frequencies of both A 1g (squares) and \( {E}_{2g}^1 \) (circles) modes as a function of temperature. Fit lines and resulting linear temperature coefficients χ T are shown. (Reproduced with permission from Ref. [56], © American Chemical Society 2014)
The new class of quantum matter topological insulators (TIs) have been in the research highlight during the past decade. The bulk phases of TIs are ordinary insulators but their surface states are gapless with nondegenerate spins [66,67,68]. Many of the TIs are layered materials, including bismuth chalcogenides Bi2X3 (X = Se, Te) and 2D HgTe/CdTe quantum wells. Among all, Bi2Te3, Bi2Se3, and Sb2Te3 are the most widely studied [69]. Here we will focus on Bi2X3 (X = Se, Te) in this chapter.
Compared to TMDs, graphene, and phosphorene which are relatively simple in terms of crystal structure, the rhombohedral Bi2X3 (X = Se, Te) has five atomic planes within a single layer. Thus, one layer Bi2X3 (X = Se, Te) can also be called one quintuple layer (1 QL) [69, 70]. Here we take Bi2Se3 as an example. In fact, Bi2Se3 has been known for its large thermoelectric effect before the topological surface states have attracted lots of interests [71]. Layered Bi2Se3 (with crystal symmetry \( R\overline{3}m \)) has a rhombohedral crystal structure belonging to the space group \( {D}_{3d}^5 \). The unit cell consists of five atomic layers, Se-Bi-Se-Bi-Se. The hexagonal close-packed Se-Bi-Se-Bi-Se atomic layers periodically arranged along the c axis, as shown in Fig. 2.6a [72]. Similar to other layered materials, the weakly bound van der Waals forces connect different QLs, but the van der Waals interaction in Bi2Se3 has a slightly covalent nature.

(a) Crystalline structure (\( R\overline{3}m \)) of Bi2Se3. The lattice vectors and the quintuple layer of the rhombohedral unit cell are indicated. (b) Phonon dispersion curves of Bi2Se3 along the Γ−Z direction of the Brillion zone. (c) The vibrational normal modes (only Raman active) of a quintuple layer Bi2Se3. (Reproduced with permission from Ref. [72], © American Chemical Society 2011)
There are 15 dynamical modes at the center of Brillouin zone, among which 12 are the optical modes and other 3 are the acoustic modes. The irreducible representations of the zone center phonons in bulk Bi2Se3 is Γ = 3E u + 3A 2u + 2E g + 2A 1g. A 1g and E g are Raman-active, while E u and A 2u are Raman-inactive but infrared-active. As thickness reduces from bulk to few- and mono-layer, the irreducible representations in odd and even number of layer are \( \Gamma =\frac{5N-1}{2}\left({A}_{1g}+{E}_g\right)+\frac{5N+1}{2}\left({A}_{2u}+{E}_u\right) \) (N = 1, 3, 5…) and \( \Gamma =\frac{5N}{2}\left({A}_{1g}+{A}_{2u}+{E}_g+{E}_u\right) \) (N = 2, 4, 6…), respectively. Phonon dispersion curves of Bi2Se3 along the Γ–Z (where Z corresponds to (0, 0, 3π/c) in reciprocal space) direction of the Brillouin zone is shown in Fig. 2.5b. Only \( {E}_g^2 \) mode exhibits a dispersionless trend among the four Raman-active modes. \( {A}_{1g}^1 \), \( {A}_{1g}^2 \), and \( {E}_g^1 \) all red-shift from Γ to Z. The corresponding vibration patterns of the Raman-active modes are displayed in Fig. 2.6c. According to the outlined normal modes, we remark that the \( {A}_{1g}^1 \) mode at ∼72 cm−1 is supposed to very sensitive to thickness because it reflects the out-of-plane vibrations of the Se and Bi atoms, and the interlayer van der Waals interactions influence the effective restoring forces acting on these atoms. Detailed discussion will be presented later in this chapter.
Figure 2.7a displays the Stokes and anti-Stokes Raman spectra taken from few-QL and bulk Bi2Se3 crystals. \( {A}_{1g}^1 \), \( {A}_{1g}^2 \), and \( {E}_g^1 \) modes red-shift with decreasing thickness, while there is almost no change in the energy of \( {E}_g^2 \) mode. As a further comparison, zoom-in views of \( {A}_{1g}^1 \) and \( {E}_g^2 \) modes are shown in Fig. 2.7b, c, respectively. The slight red-shift in Fig. 2.7c is probably due to systematic error. In contrast, the red-shift is more pronounced in the \( {A}_{1g}^1 \) mode (Fig. 2.7b). This observation is in good agreement with the phonon dispersion curve shown in Fig. 2.6b, and it also suggests that the phonon confinement effect may take a critical role in determining the frequency of phonon modes when thickness decreases.

(a) Stokes and anti-Stokes Raman shifts of Bi2Se3 nanoplatelets for different thicknesses excited by a 632.8 nm laser. (b and c) Zoom-in view of the \( {A}_{1g}^1 \) (b) and \( {E}_g^2 \) (c) modes for various thicknesses of QLs excited by 532 nm (left panel) and 632.8 nm (right panel) lasers. (Reproduced with permission from Ref. [72], © American Chemical Society 2011)
The phonon confinement effect is a type of quantum size effect that occurs when the phonon wave function is confined into a small volume with one or more dimensions of the same magnitude as its wavelength. In Raman spectroscopy, the direct sign of this effect is an asymmetrically broadened Raman peak which shifts to lower frequency in nanomaterials. In 1981, Richter et al. proposed a model to explain this effect in microcrystalline silicon (RWL theory) [39]. Later, the RWL theory was applied to explain the nanowire diameter dependence of the asymmetric broadening and frequency shift of the one phonon band in Si nanowires observed at ∼520 cm−1 [40,41,42]. Confinement effect on phonons has also been observed in nanoparticles [73, 74].
The RWL theory [39] can be understood from the Heisenberg uncertainty principle. Take Bi2Se3 as an example. Following the relation Δp ⋅ Δx ≥ ℏ/2 with Δp = ℏ · Δk and Δx is equal to the crystal size or thickness Δd. The uncertainty of the phonon momentum (Δp) increases with decreased thickness Δd. It is an indication that phonon modes with finite non-zero momentum can be involved in the Raman scattering for small delta x. Based on this principle, we can obtain the frequency shift ∆ω once we know the corresponding phonon dispersion ω(k). This explains why the frequency of \( {E}_g^2 \) mode is insensitive to thickness, while the \( {A}_{1g}^1 \) mode softens with decreasing thickness. The phonon confinement effect has also been suggested to explain the shifts of \( {A}_{1g}^1 \) and \( {A}_{1g}^2 \) modes in Bi2Te3 [75].
We also note that the \( {A}_{1g}^1 \) mode of Bi2Se3 exhibits distinct asymmetry towards higher frequency, which is different from that described in the RWL theory. Such asymmetry towards the higher frequency has been well captured by the Breit-Wigner-Fano (BWF) lineshape as investigated by Zhang et al. in wet chemistry synthesized Bi2Se3 nanoplates [72]. The BWF Raman lineshape originates from the quantum interference between a discrete phonon state and a continuum of electronic states. The constructive or destructive interferences at different frequencies result in the frequency and lineshape renormalization of a Lorentzian Raman peak:
where A, ω 0, W, and q are the peak intensity, frequency, linewidth, and asymmetry parameter, respectively. |1/q| is often used to depict the strength of electron-phonon coupling, and when |1/q| approaches zero, the above equation is reduced to a Lorentzian lineshape.
The asymmetry is more distinct in thinner nanoplates, as shown by the clear contrast between 2 QL and 7 QL (Fig. 2.8). The thickness-dependent asymmetry is shown in Fig. 2.8c, where the electron phonon coupling strength 1/q monotonically increases with decreasing thickness [76]. The detailed information on the continuum states that are coupled with the \( {A}_{1g}^1 \) mode and the thickness-dependent coupling strength are unclear yet and require further investigations. Similar reports on the strong electron-phonon interactions in Bi2Se3 have also been found through other methods such as infrared reflection and transmission [77, 78], angle-resolved photoemission spectroscopy [79], and electron transport measurements [80].

(a and b) Fano fitting in 2 QL (a) and 7 QL (b) Bi2Se3. (c) The asymmetric parameter |1/q| versus crystal thickness. (Reproduced with permission from Ref [14], © Tsinghua University Press and Springer-Verlag Berlin Heidelberg 2016)
It will also be interesting to explore the alloy layered materials on Raman spectroscopy. Figure 2.9 shows the Raman spectra of alloy nanoplates Bi2Te3 − xSex (different anion) and BixSb2 − xTe3 (different cation). Figure 2.8b displays the extracted phonon frequencies versus Se composition for Bi2Te3 − xSex. In general, the phonon modes should blue-shift with increasing concentration of Se, as Se is lighter than Te. Compared to \( {A}_{1g}^2 \) and \( {E}_g^1 \) modes which locate at higher frequency, the lower energy peaks \( {A}_{1g}^1 \) and \( {E}_g^2 \) are less sensitive to Se doping because both modes arises due to the in-phase vibrations of Bi−Te/Se. From the schematic in Fig. 2.6c, it is clear that the central Te/Se atoms are the center of mass of the Raman active vibrations. Consequently, for small doping of Se atoms, which preferentially replaces the central Te atoms, only a slight shifting in the Raman modes is anticipated for Bi2Te2.9Se0.1, Bi2Te2.84Se0.16, and Bi2Te2.7Se0.3 [71]. Likewise, \( {A}_{1g}^2 \) mode is the most sensitive to cation (Bi) doping, since \( {A}_{1g}^2 \) mode involves the Bi/Sb-Te out-of-phase vibrations along the c axis.

(a) Raman spectra of pure and doped Bi2Te3 − xSex nanoplates. (b) Phonon frequencies versus Se composition. (Reproduced with permission from Ref. [71], © American Chemical Society 2012). (c) Raman spectra of BixSb2 − xTe3 nanoplatelets with different Bi composition. The nanoplatelets are deposited on SiO2/Si substrate. (d) Phonon frequencies plotted versus the composition parameter x of BixSb2 − xTe3. (Reproduced with permission from Ref. [81], © ScienceDirect 2015)
There also are plenty of studies on phonon modes in TMD alloy [82,83,84,85,86,87,88,89], with samples including either cation or anion doping such as Mo1 − xWxSe2 and MoS2(1 − x)Se2x. Figure 2.10 demonstrates an example on monolayer and bilayer Mo1 − xWxSe2 alloy. As the Tungsten (W) composition x increases from 0 to 1, the MoSe2-like \( {E}_{2g}^1 \) shifts to lower frequency (\( {E}_{2g}^1 \) mode locates at 287 cm−1 and ∼250 cm−1 in monolayer MoSe2 and WSe2, respectively [90, 91]) and the A 1g mode continuously shifts from 240.1 cm−1 to 249.3 cm−1 in the monolayer alloy (A 1g mode locates at 241 cm−1 and ∼250 cm−1 in monolayer MoSe2 and WSe2, respectively [90, 91]), shown in Fig. 2.10a, b. In addition, the second order Raman peaks also shift and showed two-mode behaviors as the composition of W changes; that is, there are two branches of \( {E}_{2g}^1 \)+ LA(M) mode: one branch is related to WSe2 (at ∼373.6 cm−1 in WSe2) and the other branch is from MoSe2 (at∼432.7 cm−1 in MoSe2). Similar phenomena can be observed in the bilayer alloy as well (Fig. 2.10c, d). The \( {B}_{2g}^1 \) mode, which is optical inactive in bulk and Raman inactive in monolayer (with symmetry of \( {A}_2^{{\prime\prime} } \)), is Raman active in bilayer with symmetry of A 1g due to lower symmetry in few-layers [91]. When the W composition increases, the MoSe2-like \( {B}_{2g}^1 \) mode, which can be observed for x in the range 0–0.81, shifts to lower frequency. While the WSe2-like \( {B}_{2g}^1 \) mode can be observed only for the last two composition alloys (x = 0.94 and 1.00). The \( {B}_{2g}^1 \) mode is at 352 cm−1 and 311 cm−1 in bilayer MoSe2 and WSe2, respectively [90, 91].

(a) Raman spectra of Mo1 − xWxSe2 monolayers with different W composition x in the range 80–480 cm−1. (b) Composition-dependent Raman frequencies of Mo1 − xWxSe2 monolayer alloys. (c) Raman spectra of Mo1 − xWxSe2 bilayers with different W composition x. (d) Composition-dependent Raman frequencies of Mo1 − xWxSe2 bilayer alloys. All the spectra are calibrated with the 520 cm−1 Raman peak from the Si substrate. The black dashed lines in (a) and (c), guides for the eye, show the frequency shift of the first-order Raman modes with different W compositions x. For clarity, the intensity of spectra in the range 220–270 cm−1 is magnified by a factor labelled on the corresponding spectra. The excitation laser wavelength was 514.5 nm. (Reproduced with permission from Ref. [82], © American Chemical Society 2015)
The modified random element isodisplacement (MREI) model [89]can be used to fit the composition-dependent Raman frequencies of the A 1g mode for Mo1 − xWxSe2 monolayers and bilayers. The composition-dependent A 1g frequency, analysed using the MREI model, can be expressed as:
Where m Se is the atomic mass of Se; F 1 and F 2 are the force constants of Mo-Se and W-Se interactions, respectively; F 10 and F 20 are limiting values of F 1 and F 2 as x is reduced to 0; and θ is a parameter that is associated with the variation of force constant in the alloys. The fitting result gives a more accurate composition-dependent frequency shift of the A 1g mode [82],
Besides the group VI TMDs and Bi2X3 (X = Se, Te), there are other isotropic layered materials attracting research focus, such as niobium diselenide (NbSe2) and hexagonal boron nitride (h-BN). NbSe2 is one of the most studied van der Waals layered materials that exhibit both superconductivity (SC) and charge density wave (CDW) at low temperatures [92, 93]. Raman studies on NbSe2 [93,94,95,96,97] mostly focus on the two low-energy broad peaks. One of them is at ultralow frequency (<50 cm−1). As temperature decreases from 8 K to 2 K, the spectral weight from the broad CDW amplitude mode (40 cm−1 at 2 K) evolves to the sharp SC mode (19 cm−1 at 2 K). In the meantime, The SC mode blue-shifts with decreasing temperature whereas the energy of the CDW mode stays almost constant in parallel polarization and slightly shifts to lower energy (∼3 cm−1) in cross polarization. Crucially, the SC and CDW modes develop in an opposite way: When superconductivity is gradually destroyed, the SC mode intensity collapses while the CDW mode intensity recovers [97]. The CDW amplitude mode is found to be layer-sensitive: It blue-shifts by >10 cm−1 from bulk to monolayer, suggesting the increasing electron–phonon coupling strength with decreasing thickness. This finding probably provides the major reason for stronger CDW in atomically thin NbSe2 [93].
The other broad peak is centred at 180 cm−1 with the full width at half-maximum of 50 cm−1 at room temperature, which is much bigger than that of the other phonons (Fig. 2.11a). This peak at 180 cm−1 is called the soft mode, which just appears above the CDW phase transition temperature. Below the transition temperature, the soften mode is frozen [93]. It is considered that the soften mode is not a simple one-phonon mode. As there is no magnetic order in NbSe2, it rules out the possibility of magnetic origin for the peak [94]. Tsang et al. suggested that the broad peak may be associated with the scattering by two branches of LA soft phonons with opposite momentum [95].

(a) Raman spectra of the 2H–NbSe2 single crystal measured from 15 K to 300 K with xx and xy polarization. The spectra have a systematic offset to arrange one above another to give a legible comparison at different temperatures. The peaks are marked by an arrow in the spectra measured at 300 K. (b and c) Temperature dependence of the soft mode. The solid line in (b) corresponds to Eq. (2.5) and the solid line in (b) corresponds to Eq. (2.6). The error bar = 5%. (Reproduced with permission from Ref. [94], © IOP PUBLISHING 2008)
Raman spectroscopy measurements show that the temperature-dependent Raman shift of the soft mode is different from other phonon modes. The A 1g and \( {E}_{2g}^1 \) modes stiffens with decreasing temperature, while the soft mode softens, as shown in Fig. 2.11b, c. Based on mean field theory, the energy change in the second-order soft mode can be described as [98]:
where B is related to the electron-phonon coupling constant, ω 0 is the high-temperature phonon frequency and T c is the CDW transition temperature. However, the obtained T c of 69.7 K from fitting is much higher than the measured CDW transition temperature of 33.5 K. Generally, the two-phonon Raman process is associated with scattering by two phonons from the opposite sides of the BZ, and the k-space vectors’ relationship of the phonons is \( \overrightarrow{k_1}+\overrightarrow{k_2}\approx 0 \). Under the constraint of momentum conservation, it is reasonable to assume that the soft mode is contributed by a soft phonon mode plus a normal one. As reported by neutron diffraction studies [99], the soft mode can be considered as a weak first order phonon. Then the energy of the soft mode can be described as [94]:
where ω 0 is the low-temperature phonon frequency of the normal mode and \( {\omega}_0^{\prime } \) is the high-temperature phonon frequency of the soft mode. The experimental results fit well with the calculation. When the CDW transition temperature of the sample is 33.5 K, one can get the ω 0 of the normal phonon to be 120 cm−1. This mode is not scattered from the centre of the Brillouin zone and cannot be detected from normal Raman scattering [94].
The emerging possibilities to use BN as an ultra-thin insulator separating graphene layers, a spacer layer between TMDs and metal film (to prevent quenched photoluminescence), and an intermediate layer between SiO2/Si and the TMD material (to observe intrinsic linewidth on spectroscopy) have drawn lots of interests to the mono- and few-layer BN insulator. Figure 2.11 shows the Raman spectra taken from atomically thin and bulk BN. BN exhibits a characteristic peak at ∼1366 cm−1 that is due to the vibration of E 2g phonon mode. This peak is analogous to the G peak in graphene [100, 102]. As shown in Fig. 2.11a, the peak at ∼1366 cm−1 becomes weaker as N decreases. In monolayer BN, the intensity of this peak is ∼50 times smaller compared to the G peak in graphene under the same measurement conditions. The integrated intensity for the BN peak is proportional to N with high accuracy from 1 L to 6 L (inset in Fig. 2.11a), which can be exploited to distinguish one, two and more BN layers. However, Fig. 2.12b shows that mono- and bilayers exhibit unexpectedly strong variations in peak position, whereas these are essentially absent for crystals thicker than 5L. The maximum blue shift of monolayers (∼4 cm−1) is in agreement with the theory [100], and the typical shift is between 2 cm to 4 cm−1. Gorbachev et al. suggests that it is strain that causes the sample variation on Raman shift.

(a) Raman spectra of atomically thin BN. The left inset show changes in integrated intensity with the number of layers N. The right picture illustrates the phonon mode responsible for the Raman peak. (b) Position of the Raman peak for different values of N. In mono- and bilayer BN, the peak position is sample-dependent and varies by as much as ±2 cm−1. The dashed line is the Raman shift predicted for monolayer BN, compared to the bulk value (horizontal bar) [100]. The error bar indicates the typical accuracy of determining the peak position. (Reproduced with permission from Ref. [101], © John Wiley & Sons, Inc. 2011)
Recently, 2D magnetic layered materials have attracted lots of interests. Ferromagnetism has been discovered in mono- and few-layer samples [103, 104]. Meanwhile, through the observation of spin-order-induced Raman modes, Wang et al. [105] and Lee et al. [106] reported that iron phosphorus trisulfide (FePS3) crystal exhibits an Ising-type antiferromagnetic ordering down to the monolayer limit.
Figure 2.13a shows the temperature-dependent Raman spectra on bulk FePS3 crystal around the Néel temperature. The Néel temperature of 118 K was obtained from the magnetic susceptibility carried out in the physical property measurement system (Fig. 2.13b). At 77 K, two strong and symmetric peaks located at 88 and 95 cm−1 were resolved in the antiferromagnetic (AFM) phase whereas when the temperature was increased to 150 K, the two peaks disappeared accompanied by the appearance of a broad and asymmetric peak at around 100 cm−1 in the paramagnetic (PM) phase. First-principle calculations did not show any Raman-active modes in the frequency range of 0–130 cm−1 at the Γ point, which suggests that the peaks (88, 95, and 100 cm−1) are spin-dependent. Temperature-dependent measurements show that the spin-order-induced modes at 88 and 95 cm−1 are still robust down to monolayer (Fig. 2.13c). This result confirms that the spin-order-induced Raman modes are attributed to the Brillouin-zone folding in the ab plane [107], because monolayer FePS3 only has a layer of iron atoms, which is impossible to have double-sized magnetic cell along the c axis. The systematic layer-dependent and temperature-dependent measurements show that the Néel temperature decreases from 118 K in bulk to 104 K in monolayers.

(a) Temperature-dependent Raman spectra of bulk FePS3 sample obtained with an excitation wavelength of 532 nm and power∼0.5 mW around Néel temperature. The line shapes of fitting curves are Fano asymmetric and two-Lorentzian peaks above and below Néel temperature, respectively. (b) Plot of magnetic susceptibility of FePS3 bulk crystal versus temperature measured in (filled squares) cooling and (filled circles) heating process. The first derivation of susceptibility was shown in the inset whose inflection points are both around 118 K. (c) Temperature-dependent Raman spectra of mono-, tri- and five-layer FePS3 obtained with an excitation wavelength of 532 nm and power∼0.5 mW. (Reproduced with permission from Ref. [105], © IOP Publishing 2016)
In conclusion, with the focus on isotropic two-dimensional layered materials, we have reviewed the phonon modes in TMDs MX2 (M = Mo, W; X = S, Te, Te), Bi2X3 (X = Se, Te), alloy, NbSe2, h-BN, and magnetic FePS3 crystals in this chapter. Dependence on thickness and the mechanism which leads to the thickness-evolutional trend have been discussed in detail. While temperature dependence, composition (doping) dependence, and the soft mode which is related to CDW have also been presented in this chapter.
References
Q.H. Wang, K. Kalantar-Zadeh, A. Kis, J.N. Coleman, M.S. Strano, Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotech. 7, 699–712 (2012)
M. Chhowalla, H.S. Shin, G. Eda, L.J. Li, K.P. Loh, H.Z. Zhang, The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets. Nat. Chem. 5, 263–275 (2013)
A.K. Geim, I.V. Van der Grigorieva, Waals heterostructures. Nature 499, 419–425 (2013)
K.S. Novoselov, D. Jiang, F. Schedin, T.J. Booth, V.V. Khotkevich, S.V. Morozov, A.K. Geim, Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. U. S. A. 102, 10451–10453 (2005)
X. Xu, W. Yao, D. Xiao, T.F. Heinz, Spin and pseudospins in layered transition metal dichalcogenides. Nat. Phys. 10, 343–350 (2014)
K.F. Mak, K. He, J. Shan, T.F. Heinz, Control of valley polarization in monolayer MoS2 by optical helicity. Nat. Nanotech. 7, 494–498 (2012)
H. Zeng, J. Dai, W. Yao, D. Xiao, X. Cui, Valley polarization in MoS2 monolayers by optical pumping. Nat. Nanotech. 7, 490–493 (2012)
T. Cao, G. Wang, W. Han, H. Ye, C. Zhu, J. Shi, Q. Niu, P. Tan, E. Wang, B. Liu, J. Feng, Valley-selective circular dichroism of monolayer molybdenum disulphide. Nat. Commun. 3, 887 (2012)
K.F. Mak, K.L. McGill, J. Park, P.L. McEuen, The valley hall effect in MoS2 transistors. Science 344, 1489–1492 (2014)
S.F. Wu, J.S. Ross, G.B. Liu, G. Aivazian, A. Jones, Z.Y. Fei, W.G. Zhu, D. Xiao, W. Yao, D. Cobden, X.D. Xu, Electrical tuning of valley magnetic moment through symmetry control in bilayer MoS2. Nat. Phys. 9, 149–153 (2013)
J. Lee, K.F. Mak, J. Shan, Electrical control of the valley hall effect in bilayer MoS2 transistors. Nat. Nanotech. 11, 421–425 (2016)
K.F. Mak, C. Lee, J. Hone, J. Shan, T.F. Heinz, Atomically thin MoS2: a new direct-gap semiconductor. Phys. Rev. Lett. 105, 136805 (2010)
A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C.-Y. Chim, G. Galli, F. Wang, Emerging photoluminescence in monolayer MoS2. Nano Lett. 10, 1271–1275 (2010)
X. Lu, X. Luo, J. Zhang, S.Y. Quek, Q.H. Xiong, Lattice vibrations and Raman scattering in two-dimensional layered materials beyond graphene. Nano Res. 9, 3559–3597 (2016)
X. Qian, J. Liu, L. Fu, J. Li, Quantum spin hall effect in two-dimensional transition metal dichalcogenides. Science 346, 1344–1347 (2014)
B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti, A. Kis, Single-layer MoS2 transistors. Nat. Nanotech. 6, 147–150 (2011)
X. Zhang, X.-F. Qiao, W. Shi, J.-B. Wu, D.-S. Jiang, P.-H. Tan, Phonon and Raman scattering of two-dimensional transition metal dichalcogenides from monolayer, multilayer to bulk material. Chem. Soc. Rev. 44, 2757–2785 (2015)
X. Lu, M.I.B. Utama, J. Lin, X. Luo, Y. Zhao, J. Zhang, S.T. Pantelides, W. Zhou, S.Y. Quek, Q.H. Xiong, Rapid and nondestructive identification of polytypism and stacking sequences in few-layer molybdenum diselenide by Raman spectroscopy. Adv. Mater. 27, 4502–4508 (2015)
R. Suzuki, M. Sakano, Y.J. Zhang, R. Akashi, D. Morikawa, A. Harasawa, K. Yaji, K. Kuroda, K. Miyamoto, T. Okuda, K. Ishizaka, R. Arita, Y. Iwasa, Valley-dependent spin polarization in bulk MoS2 with broken inversion symmetry. Nat. Nanotech. 9, 611–617 (2014)
D.H. Keum, S. Cho, J.H. Kim, D.-H. Choe, H.-J. Sung, M. Kan, H. Kang, J.-Y. Hwang, S.W. Kim, H. Yang, K.J. Chang, Y.H. Lee, Bandgap opening in few-layered monoclinic MoTe2. Nat. Phys. 11, 482–U144 (2015)
Q.J. Song, Q.H. Tan, X. Zhang, J.B. Wu, B.W. Sheng, Y. Wan, X.Q. Wang, L. Dai, P.H. Tan, Physical origin of Davydov splitting and resonant Raman spectroscopy of Davydov components in multilayer MoTe2. Phys. Rev. B 93, 115409 (2016)
G. Froehlicher, E. Lorchat, F. Fernique, C. Joshi, A. Molina-Sanchez, L. Wirtz, S. Berciaud, Unified description of the optical phonon modes in N-layer MoTe2. Nano Lett. 15, 6481–6489 (2015)
C. Ruppert, O.B. Aslan, T.F. Heinz, Optical properties and band gap of single- and few-layer MoTe2 crystals. Nano Lett. 14, 6231–6236 (2014)
Y. Kim, Y.I. Jhon, J. Park, J.H. Kim, S. Lee, Y.M. Jhon, Anomalous Raman scattering and lattice dynamics in mono- and few-layer WTe2. Nanoscale 8, 2309–2316 (2016)
Y.C. Jiang, J. Gao, L. Wang, Raman fingerprint for semi-metal WTe2 evolving from bulk to monolayer. Sci. Rep. 6, 19624 (2016)
Y. Zhao, X. Luo, H. Li, J. Zhang, P.T. Araujo, C.K. Gan, J. Wu, H. Zhang, S.Y. Quek, M.S. Dresselhaus, Q.H. Xiong, Interlayer breathing and shear modes in few-trilayer MoS2 and WSe2. Nano Lett. 13, 1007–1015 (2013)
N. Wakabayashi, H.G. Smith, R.M. Nicklow, Lattice-dynamics of hexagonal MoS2 studied by neutron-scattering. Phys. Rev. B 12, 659–663 (1975)
A. Molina-Sanchez, L. Wirtz, Phonons in single-layer and few-layer MoS2 and WS2. Phys. Rev. B 84, 155413 (2011)
J.L. Verble, T.J. Wieting, Lattice mode degeneracy in MoS2 and other layer compounds. Phys. Rev. Lett. 25, 362 (1970)
P.N. Ghosh, C.R. Maiti, Interlayer force and Davydov splitting in 2H-MoS2. Phys. Rev. B 28, 2237–2239 (1983)
T.J. Wieting, J.L. Verble, Infrared and Raman studies of long-wavelength optical phonons in hexagonal MoS2. Phys. Rev. B 3, 4286 (1971)
C. Lee, H. Yan, L.E. Brus, T.F. Heinz, J. Hone, S. Ryu, Anomalous lattice vibrations of single- and few-layer MoS2. ACS Nano 4, 2695–2700 (2010)
X. Luo, Y. Zhao, J. Zhang, Q. Xiong, S.Y. Quek, Anomalous frequency trends in MoS2 thin films attributed to surface effects. Phys. Rev. B 88, 075320 (2013)
H. Zeng, B. Zhu, K. Liu, J. Fan, X. Cui, Q.M. Zhang, Low-frequency Raman modes and electronic excitations in atomically thin MoS2 films. Phys. Rev. B 86, 241301(R) (2012)
H. Li, Q. Zhang, C.C.R. Yap, B.K. Tay, T.H.T. Edwin, A. Olivier, D. Baillargeat, From bulk to monolayer MoS2: evolution of Raman scattering. Adv. Funct. Mater. 22, 1385–1390 (2012)
W. Zhao, Z. Ghorannevis, K.K. Amara, J.R. Pang, M. Toh, X. Zhang, C. Kloc, P.H. Tan, G. Eda, Lattice dynamics in mono- and few-layer sheets of WS2 and WSe2. Nanoscale 5, 9677–9683 (2013)
P. Tonndorf, R. Schmidt, P. Boettger, X. Zhang, J. Boerner, A. Liebig, M. Albrecht, C. Kloc, O. Gordan, D.R.T. Zahn, S.M. de Vasconcellos, R. Bratschitsch, Photoluminescence emission and Raman response of monolayer MoS2, MoSe2, and WSe2. Opt. Express 21, 4908–4916 (2013)
J.-H. Fan, P. Gao, A.-M. Zhang, B.-R. Zhu, H.-L. Zeng, X.-D. Cui, R. He, Q.-M. Zhang, Resonance Raman scattering in bulk 2H-MX2 (M = Mo, W; X = S, Se) and monolayer MoS2. J. Appl. Phys. 115, 053527 (2014)
H. Richter, Z.P. Wang, L. Ley, The one phonon Raman-spectrum in microcrystalline silicon. Solid State Commun. 39, 625–629 (1981)
R.P. Wang, G.W. Zhou, Y.L. Liu, S.H. Pan, H.Z. Zhang, D.P. Yu, Z. Zhang, Raman spectral study of silicon nanowires: high-order scattering and phonon confinement effects. Phys. Rev. B 61, 16827–16832 (2000)
K.W. Adu, H.R. Gutierrez, U.J. Kim, P.C. Eklund, Inhomogeneous laser heating and phonon confinement in silicon nanowires: a micro-Raman scattering study. Phys. Rev. B 73, 155333 (2006)
K.W. Adu, Q. Xiong, H.R. Gutierrez, G. Chen, P.C. Eklund, Raman scattering as a probe of phonon confinement and surface optical modes in semiconducting nanowires. Appl. Phys. A Mater. Sci. Process. 85, 287–297 (2006)
Y. Lin, X. Ling, L. Yu, S. Huang, A.L. Hsu, Y.-H. Lee, J. Kong, M.S. Dressehaus, T. Palacios, Dielectric screening of excitons and trions in single-layer MoS2. Nano Lett. 14, 5569–5576 (2014)
A. Molina-Sanchez, K. Hummer, L. Wirtz, Vibrational and optical properties of MoS2: from monolayer to bulk. Surf. Sci. Rep. 70, 554–586 (2015)
J. Yan, J. Xia, X. Wang, L. Liu, J.-L. Kuo, B.K. Tay, S. Chen, W. Zhou, Z. Liu, Z.X. Shen, Stacking-dependent interlayer coupling in Trilayer MoS2 with broken inversion symmetry. Nano Lett. 15, 8155–8161 (2015)
J.-U. Lee, K. Kim, S. Han, G.H. Ryu, Z. Lee, H. Cheong, Raman signatures of polytypism in molybdenum disulfide. ACS Nano 10, 1948–1953 (2016)
A.A. Puretzky, L. Liang, X. Li, K. Xiao, K. Wang, M. Mahjouri-Samani, L. Basile, J.C. Idrobo, B.G. Sumpter, V. Meunier, D.B. Geohegan, Low-frequency Raman fingerprints of two-dimensional metal dichalcogenide layer stacking configurations. ACS Nano 9, 6333–6342 (2015)
X.-F. Qiao, J.-B. Wu, L. Zhou, J. Qiao, W. Shi, T. Chen, X. Zhang, J. Zhang, W. Ji, P.-H. Tan, Polytypism and unexpected strong interlayer coupling in two-dimensional layered ReS2. Nanoscale 8, 8324–8332 (2016)
R. He, J.-A. Yan, Z. Yin, Z. Ye, G. Ye, J. Cheng, J. Li, C.H. Lui, Coupling and stacking order of ReS2 atomic layers revealed by ultralow-frequency Raman spectroscopy. Nano Lett. 16, 1404–1409 (2016)
Y. Yoo, Z.P. Degregorio, Y. Su, S.J. Koester, J.E. Johns, In-plane 2H-1T’ MoTe2 homojunctions synthesized by flux-controlled phase engineering. Adv. Mater. 29, 1605461 (2017)
J.J. Wu, M.J. Liu, K. Chatterjee, K.P. Hackenberg, J.F. Shen, X.L. Zou, Y. Yan, J. Gu, Y.C. Yang, J. Lou, P.M. Ajayan, Exfoliated 2D transition metal disulfides for enhanced electrocatalysis of oxygen evolution reaction in acidic medium. Adv. Mater. Interfaces 3, 1500669 (2016)
B.R. Carvalho, L.M. Malard, J.M. Alves, C. Fantini, M.A. Pimenta, Symmetry-dependent exciton-phonon coupling in 2D and bulk MoS2 observed by resonance Raman scattering. Phys. Rev. Lett. 114, 136403 (2015)
L. Sun, J. Yan, D. Zhan, L. Liu, H. Hu, H. Li, B.K. Tay, J.-L. Kuo, C.-C. Huang, D.W. Hewak, P.S. Lee, Z.X. Shen, Spin-orbit splitting in single-layer MoS2 revealed by triply resonant Raman scattering. Phys. Rev. Lett. 111, 126801 (2013)
Y. Wang, C. Cong, C. Qiu, T. Yu, Raman spectroscopy study of lattice vibration and crystallographic orientation of monolayer MoS2 under uniaxial strain. Small 9, 2857–2861 (2013)
B. Chakraborty, A. Bera, D.V.S. Muthu, S. Bhowmick, U.V. Waghmare, A.K. Sood, Symmetry-dependent phonon renormalization in monolayer MoS2 transistor. Phys. Rev. B 85, 161403(R) (2012)
R.S. Yan, J.R. Simpson, S. Bertolazzi, J. Brivio, M. Watson, X.F. Wu, A. Kis, T.F. Luo, A.R.H. Walker, H.G. Xing, Thermal conductivity of monolayer molybdenum disulfide obtained from temperature-dependent Raman spectroscopy. ACS Nano 8, 986–993 (2014)
S. Sahoo, A.P.S. Gaur, M. Ahmadi, M.J.F. Guinel, R.S. Katiyar, Temperature-dependent Raman studies and thermal conductivity of few-layer MoS2. J. Phys. Chem. C 117, 9042–9047 (2013)
N.A. Lanzillo, A.G. Birdwell, M. Amani, F.J. Crowne, P.B. Shah, S. Najmaei, Z. Liu, P.M. Ajayan, J. Lou, M. Dubey, S.K. Nayak, T.P. O’Regan, Temperature-dependent phonon shifts in monolayer MoS2. Appl. Phys. Lett. 103, 093102 (2013)
A. Taube, J. Judek, C. Jastrzebski, A. Duzynska, K. Switkowski, M. Zdrojek, Temperature-dependent nonlinear phonon shifts in a supported MoS2 monolayer. ACS Appl. Mater. Interfaces 6, 8959–8963 (2014)
A.A. Balandin, S. Ghosh, W.Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao, C.N. Lau, Superior thermal conductivity of single-layer graphene. Nano Lett. 8, 902–907 (2008)
S. Najmaei, Z. Liu, P.M. Ajayan, J. Lou, Thermal effects on the characteristic Raman spectrum of molybdenum disulfide (MoS2) of varying thicknesses. Appl. Phys. Lett. 100, 013106 (2012)
C.R. Zhu, G. Wang, B.L. Liu, X. Marie, X.F. Qiao, X. Zhang, X.X. Wu, H. Fan, P.H. Tan, T. Amand, B. Urbaszek, Strain tuning of optical emission energy and polarization in monolayer and bilayer MoS2. Phys. Rev. B 88, 121301(R) (2013)
C.-H. Chang, X. Fan, S.-H. Lin, J.-L. Kuo, Orbital analysis of electronic structure and phonon dispersion in MoS2, MoSe2, WS2, and WSe2 monolayers under strain. Phys. Rev. B 88, 125120 (2013)
M. Thripuranthaka, D.J. Late, Temperature dependent phonon shifts in single-layer WS2. ACS Appl. Mater. Interfaces 6, 1158–1163 (2014)
Y.Q. Cai, J.H. Lan, G. Zhang, Y.W. Zhang, Lattice vibrational modes and phonon thermal conductivity of monolayer MoS2. Phys. Rev. B 89, 035438 (2014)
X.-L. Qi, S.-C. Zhang, Topological insulators and superconductors. Rev. Mod. Phys. 83, 1057–1110 (2011)
M.Z. Hasan, C.L. Kane, Colloquium: topological insulators. Rev. Mod. Phys. 82, 3045–3067 (2010)
J.E. Moore, The birth of topological insulators. Nature 464, 194–198 (2010)
H. Zhang, C.-X. Liu, X.-L. Qi, X. Dai, Z. Fang, S.-C. Zhang, Topological insulators in Bi2Se3, Bi2Te3 and Sb2Te3 with a single Dirac cone on the surface. Nat. Phys. 5, 438–442 (2009)
Y.L. Chen, J.G. Analytis, J.H. Chu, Z.K. Liu, S.K. Mo, X.L. Qi, H.J. Zhang, D.H. Lu, X. Dai, Z. Fang, S.C. Zhang, I.R. Fisher, Z. Hussain, Z.X. Shen, Experimental realization of a three-dimensional topological insulator, Bi2Te3. Science 325, 178–181 (2009)
A. Soni, Y. Zhao, L. Yu, M.K.K. Aik, M.S. Dresselhaus, Q. Xiong, Enhanced thermoelectric properties of solution grown Bi2Te3 −xSex Nanoplatelet composites. Nano Lett. 12, 1203–1209 (2012)
J. Zhang, Z. Peng, A. Soni, Y. Zhao, Y. Xiong, B. Peng, J. Wang, M.S. Dresselhaus, Q.H. Xiong, Raman spectroscopy of few-quintuple layer topological insulator Bi2Se3 nanoplatelets. Nano Lett. 11, 2407–2414 (2011)
G.L. Frey, R. Tenne, M.J. Matthews, M.S. Dresselhaus, G. Dresselhaus, Raman and resonance Raman investigation of MoS2 nanoparticles. Phys. Rev. B 60, 2883–2892 (1999)
Q.-C. Sun, D. Mazumdar, L. Yadgarov, R. Rosentsveig, R. Tenne, J.L. Musfeldt, Spectroscopic determination of phonon lifetimes in rhenium-doped MoS2 nanoparticles. Nano Lett. 13, 2803–2808 (2013)
C. Wang, X. Zhu, L. Nilsson, J. Wen, G. Wang, X. Shan, Q. Zhang, S. Zhang, J. Jia, Q. Xue, In situ Raman spectroscopy of topological insulator Bi2Te3 films with varying thickness. Nano Res. 6, 688–692 (2013)
Y. Zhao, X. Luo, J. Zhang, J. Wu, X. Bai, M. Wang, J. Jia, H. Peng, Z. Liu, S.Y. Quek, Q. Xiong, Interlayer vibrational modes in few-quintuple-layer Bi2Te3 and Bi2Se3 two-dimensional crystals: Raman spectroscopy and first-principles studies. Phys. Rev. B 90, 245428 (2014)
A.D. Laforge, A. Frenzel, B.C. Pursley, T. Lin, X. Liu, J. Shi, D.N. Basov, Optical characterization of Bi2Se3 in a magnetic field: Infrared evidence for magnetoelectric coupling in a topological insulator material. Phys. Rev. B 81, 125120 (2010)
P. Di Pietro, M. Ortolani, O. Limaj, A. Di Gaspare, V. Giliberti, F. Giorgianni, M. Brahlek, N. Bansal, N. Koirala, S. Oh, P. Calvani, S. Lupi, Observation of Dirac plasmons in a topological insulator. Nat. Nanotech. 8, 556–560 (2013)
C. Chen, Z. Xie, Y. Feng, H. Yi, A. Liang, S. He, D. Mou, J. He, Y. Peng, X. Liu, Y. Liu, L. Zhao, G. Liu, X. Dong, J. Zhang, L. Yu, X. Wang, Q. Peng, Z. Wang, S. Zhang, F. Yang, C. Chen, Z. Xu, X.J. Zhou, Tunable Dirac fermion dynamics in topological insulators. Sci. Rep. 3, 2411 (2013)
M.V. Costache, I. Neumann, J.F. Sierra, V. Marinova, M.M. Gospodinov, S. Roche, S.O. Valenzuela, Fingerprints of inelastic transport at the surface of the topological insulator Bi2Se3: role of Electron-phonon coupling. Phys. Rev. Lett. 112, 086601 (2014)
C.H. Zhang, Z.P. Peng, Z. Li, L.G. Yu, K.A. Khor, Q.H. Xiong, Controlled growth of bismuth antimony telluride BixSb2 − xTe3 nanoplatelets and their bulk thermoelectric nanocomposites. Nano Energy 15, 688–696 (2015)
M. Zhang, J.X. Wu, Y.M. Zhu, D.O. Dumcenco, J.H. Hong, N.N. Mao, S.B. Deng, Y.F. Chen, Y.L. Yang, C.H. Jin, S.H. Chaki, Y.S. Huang, J. Zhang, L.M. Xie, Two-dimensional molybdenum tungsten diselenide alloys: photoluminescence, Raman scattering, and electrical transport. ACS Nano 8, 7130–7137 (2014)
S. Tongay, D.S. Narang, J. Kang, W. Fan, C. Ko, A.V. Luce, K.X. Wang, J. Suh, K.D. Patel, V.M. Pathak, J. Li, J. Wu, Two-dimensional semiconductor alloys: monolayer Mo1 − xWxSe2. Appl. Phys. Lett. 104, 012101 (2014)
Y.F. Chen, W. Wen, Y.M. Zhu, N.N. Mao, Q.L. Feng, M. Zhang, H.P. Hsu, J. Zhang, Y.S. Huang, L.M. Xie, Temperature-dependent photoluminescence emission and Raman scattering from Mo1 − xWxS2 monolayers. Nanotechnology 27, 445705 (2016)
X.F. Qiao, X.L. Li, X. Zhang, W. Shi, J.B. Wu, T. Chen, P.H. Tan, Substrate-free layer-number identification of two-dimensional materials: a case of Mo0.5W0.5S2 alloy. Appl. Phys. Lett. 106, 223102 (2015)
S. Zheng, L. Sun, T. Yin, A.M. Dubrovkin, F. Liu, Z. Liu, Z.X. Shen, H.J. Fan, Monolayers of WxMo1 − xS2 alloy heterostructure with in-plane composition variations. Appl. Phys. Lett. 106, 063113 (2015)
Q.L. Feng, N.N. Mao, J.X. Wu, H. Xu, C.M. Wang, J. Zhang, L.M. Xie, Growth of MoS2(1 − x)Se2x (x = 0.41–1.00) monolayer alloys with controlled morphology by physical vapor deposition. ACS Nano 9, 7450–7455 (2015)
W.T. Zhang, X.D. Li, T.T. Jiang, J.L.Q. Song, Y. Lin, L.X. Zhu, X.L. Xu, CVD synthesis of Mo(1 − x)WxS2 and MoS2(1 − x)Se2x alloy monolayers aimed at tuning the bandgap of molybdenum disulfide. Nanoscale 7, 13554–13560 (2015)
Y.F. Chen, D.O. Dumcenco, Y.M. Zhu, X. Zhang, N.N. Mao, Q.L. Feng, M. Zhang, J. Zhang, P.H. Tan, Y.S. Huang, L.M. Xie, Composition-dependent Raman modes of Mo1 − xWxS2 monolayer alloys. Nanoscale 6, 2833–2839 (2014)
X. Lu, M.I.B. Utama, J. Lin, X. Gong, J. Zhang, Y. Zhao, S.T. Pantelides, J. Wang, Z. Dong, Z. Liu, W. Zhou, Q.H. Xiong, Large-area synthesis of monolayer and few-layer MoSe2 films on silicon substrates. Nano Lett. 14, 2419–2425 (2014)
X. Luo, Y. Zhao, J. Zhang, M. Toh, C. Kloc, Q. Xiong, S.Y. Quek, Effects of lower symmetry and dimensionality on Raman spectra in two-dimensional WSe2. Phys. Rev. B 88, 195313 (2013)
J.A. Wilson, F.J. Di Salvo, S. Mahajan, Charge-density waves and superlattices in the metallic layered transition metal dichalcogenides (reprinted from Adv. Phys. 1974, 32, 882). Adv. Phys. 50, 1171–1248 (2001)
X.X. Xi, L. Zhao, Z.F. Wang, H. Berger, L. Forro, J. Shan, K.F. Mak, Strongly enhanced charge-density-wave order in monolayer NbSe2. Nat. Nanotech. 10, 765–770 (2015)
Y. Wu, M. An, R. Xiong, J. Shi, Q.M. Zhang, Raman scattering spectra in the normal phase of 2H-NbSe2. J. Phys. D. Appl. Phys. 41, 175408 (2008)
J.C. Tsang, J.E. Smith, M.W. Shafer, Raman-spectroscopy of soft modes at charge-density-wave phase-transition in 2h-NbSe2. Phys. Rev. Lett. 37, 1407–1410 (1976)
A. Mialitsin, Fano line shape and anti-crossing of Raman active E2g peaks in the charge density wave state of NbSe2. J. Phys. Chem. Solids 72, 568–571 (2011)
M.A. Measson, Y. Gallais, M. Cazayous, B. Clair, P. Rodiere, L. Cario, A. Sacuto, Amplitude Higgs mode in the 2H-NbSe2 superconductor. Phys. Rev. B 89, 060503(R) (2014)
J.F. Scott, Soft-mode spectroscopy: experimental studies of structural phase-transitions. Rev. Mod. Phys. 46, 83–128 (1974)
D.E. Moncton, J.D. Axe, F.J. Disalvo, Study of superlattice formation in 2H-NbSe2 and 2H-TaSe2 by neutron-scattering. Phys. Rev. Lett. 34, 734–737 (1975)
R. Arenal, A.C. Ferrari, S. Reich, L. Wirtz, J.Y. Mevellec, S. Lefrant, A. Rubio, A. Loiseau, Raman spectroscopy of single-wall boron nitride nanotubes. Nano Lett. 6, 1812–1816 (2006)
R.V. Gorbachev, I. Riaz, R.R. Nair, R. Jalil, L. Britnell, B.D. Belle, E.W. Hill, K.S. Novoselov, K. Watanabe, T. Taniguchi, A.K. Geim, P. Blake, Hunting for monolayer boron nitride: optical and Raman signatures. Small 7, 465–468 (2011)
D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C.C. Tang, C.Y. Zhi, Boron nitride nanotubes and nanosheets. ACS Nano 4, 2979–2993 (2010)
C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z.Q. Qiu, R.J. Cava, S.G. Louie, J. Xia, X. Zhang, Discovery of intrinsic ferromagnetism in two-dimensional van der Waals crystals. Nature 546, 265–269 (2017)
B. Huang, G. Clark, E. Navarro-Moratalla, D.R. Klein, R. Cheng, K.L. Seyler, D. Zhong, E. Schmidgall, M.A. McGuire, D.H. Cobden, W. Yao, D. Xiao, P. Jarillo-Herrero, X.D. Xu, Layer-dependent ferromagnetism in a van der Waals crystal down to the monolayer limit. Nature 546, 270–273 (2017)
X.Z. Wang, K.Z. Du, Y.Y.F. Liu, P. Hu, J. Zhang, Q. Zhang, M.H.S. Owen, X. Lu, C.K. Gan, P. Sengupta, C. Kloc, Q.H. Xiong, Raman spectroscopy of atomically thin two-dimensional magnetic iron phosphorus trisulfide (FePS3) crystals. 2D Materials 3, 031009 (2016)
J.U. Lee, S. Lee, J.H. Ryoo, S. Kang, T.Y. Kim, P. Kim, C.H. Park, J.G. Park, H. Cheong, Ising-type magnetic ordering in atomically thin FePS3. Nano Lett. 16, 7433–7438 (2016)
T. Sekine, M. Jouanne, C. Julien, M. Balkanski, Light-scattering study of dynamic behavior of antiferromagnetic spins in the layered magnetic semiconductor FePS3. Phys. Rev. B 42, 8382–8393 (1990)
Acknowledgments
This work is supported by National Basic Research Program of China (2017YFA0303401), Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB28000000) LU JIAXI International team program. J. Z. also acknowledges support from National Young 1000 Talent Plan of China.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Lu, X., Tan, QH., Xiong, Q., Zhang, J. (2019). Raman Spectroscopy of Isotropic Two-Dimensional Materials Beyond Graphene. In: Tan, PH. (eds) Raman Spectroscopy of Two-Dimensional Materials. Springer Series in Materials Science, vol 276. Springer, Singapore. https://doi.org/10.1007/978-981-13-1828-3_2
Download citation
DOI: https://doi.org/10.1007/978-981-13-1828-3_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1827-6
Online ISBN: 978-981-13-1828-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)