
Abstract
Mitochondria supply energy to cells by generating ATP; thus it can be considered as one of the essential organelles of the cell. For the efficient working of cells, a good quality of mitochondria is essential; thus the elimination of injured or nonfunctional mitochondria by means of mitophagy is a very important process for cell function. Mitophagy showed a neuroprotective property in cerebral ischemia by accurate labeling and entrapment of defective mitochondria into isolation membranes. Then the entrapped mitochondria were digested by lysosomes. Therefore, the regulation of mitophagy in ischemic brain injury may be used as a therapeutic strategy to protect the neuron by the efficient removal of injured mitochondria.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keyword
6.1 Introduction
Stroke main outcomes are sudden death or adult disability. Stroke may be ischemic or hemorrhagic. Ischemic stroke alone accounts for 80% of the stroke. Inadequate blood supply to the brain leads to ischemic stroke [1]. Based on the region of the brain affected, ischemic stroke is of two types: global ischemic stroke and focal ischemic stroke. In global ischemic stroke, the blood supply of the entire brain was significantly reduced. However in focal ischemic stroke, the blood supply was reduced in a particular region of the brain blood vessel. Loss of consciousness, impaired voice, blurred vision, and numbness are the principal symptoms of cerebral ischemia. Due to the suppression of the blood flow during the ischemic condition in the brain, the level of the ATP was significantly reduced for a short period of time. That leads to enormous cell death in susceptible regions. Therefore cerebral blood flow is one of the crucial factors for regular functioning of the brain. The damaged region of the ischemic brain which cannot be restored is known as the ischemic core region; however we can restore the region surrounding the core known as the penumbra if treatment is available at the right time. If we are able to recover cerebral blood flow during cerebral ischemia, it limits the nerve cell death in the penumbra region. Therapeutic strategy for ischemic brain injury involves reperfusion and neuroprotection. Usually to restore blood flow in cerebral ischemia, antithrombotic drug was administered as soon as possible. The antithrombotic drugs are of three types based on their mechanism of action. Thrombolytic drugs (recombinant tissue plasminogen activator, streptokinase, etc.) break down an already formed clot. Antiplatelet drugs (aspirin, clopidogrel, etc.) prevent the aggregation of the platelet and limit the formation of a secondary clot. Anticoagulant drugs (heparin, warfarin, dabigatran, etc.) prevent the coagulation of the blood and prevent secondary clot formation. The return of blood flow in the ischemic region leads to a secondary injury known as reperfusion injury. To protect against reperfusion injury and secure the penumbral region from further damage, neuroprotection is another therapeutic approach [2]. But still, the use of recognized neuroprotective agents is unclear in clinical studies.
During ischemic brain damage, the death of nerve cells is reported to have three routes, i.e., necrotic, apoptotic, and autophagic cell death [3, 4]. In this chapter, we have discussed the regulation of mitochondrial autophagy in cerebral ischemia. Mitochondria supply energy to cells by generating ATP; thus it can be considered as one of the essential organelles of the cell. For the efficient working of cells, a good quality of mitochondria is essential; thus the elimination of injured or nonfunctional mitochondria by means of mitophagy is a very important process for cell function. A publication report stated that mitophagy protects nerve cell during stroke condition.
6.2 Autophagy
The autophagy word was given by Christian De Duve [5, 6]. Autophagy is a destructive process which allows the degradation of old, damaged, or nonfunctional cytoplasmic material including proteins and organelle through the liposome’s catalytic enzymes. Stress condition like starvation or ischemic event induces autophagy. Autophagy regulates both cell survival and cell death [7]. Mitochondrial autophagy is also known as mitophagy; it involves the selective removal of the mitochondria from the cytoplasm of the cell. The mitophagy process in yeast resembles with mammals [8]. The maintenance of good quality mitochondria is very important for the survival of the brain cell as mitochondria have an extremely shorter life span compared with neurons. Mitophagy regulation is found to be disturbed in nervous system disorder including ischemic brain injury [9,10,11,12,13,14]. Upregulation of mitophagy enhances elimination of defected mitochondria and has a protective role in cerebral ischemia [11].
6.3 Nonselective Autophagy and Mitophagy
Nonselective autophagy involves the hydrolysis of cytoplasmic contents, while mitophagy involves selective mitochondrial content hydrolysis by liposome through autolysosomes. Nonselective autophagy mainly occurs at the time of nutrient deficiency. Nonselective autophagy involves the degradation of cytosolic material including proteins and organelle to supply ATP. However mitophagy involves selective degradation of damaged or nonfunctional mitochondria from the cytoplasm [15].
6.4 Mitochondria Dynamics: Fission and Fusion of Mitochondria
Mitochondria are the powerhouse of the cell as they are responsible for the generation of energy. Further mitochondria also supervise the programmed death of the cell [16]. Since brain cells cannot store enough energy sources to perform their function, therefore they carry numerous mitochondria in their cytoplasm. The shape of the mitochondria changes according to the fission or fusion. Mitofusins control mitochondrial fusion through the outer mitochondrial membranes. Mitofusins are of two types: mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2). Optic atrophy 1 (OPA1) controls the fusion of the mitochondria through the inner mitochondria membranes [16, 17]. Any impairment in the Mfn1, Mfn2, and OPA1 is responsible for the wide-ranging mitochondrial fragmentation and loss of mitochondrial DNA [18, 19]. To identify the role of fusion mediators in the early development of the embryo, a study has been performed. This study showed that mice lacking these mediators of the fusion die at an initial stage of fetus development, indicating that mitofusins and optic atrophy 1 are very important in the early stage of embryo formation [18, 20]. Similarly another published report stated that a point mutation in mitofusins and OPA1 leads to severe nervous system disorder [21, 22].
Mitochondrial fission results in the splitting of mitochondria into two daughter mitochondria. Regulation of mitochondria splitting is done by dynamin-related protein 1 (Drp1) and fission 1 protein (Fis1) [23]. During the period of the fission, the Drp1 is enrolled to the mitochondria to form the complex with the Fis1. This complex is responsible for the split of the mitochondria [24, 25]. Mitochondrial fission significantly decreases in Drp1-deficient cell; studies have shown lengthened mitochondria in the cell lacking Drp1 [26]. Mitochondrial fusion and fission are greatly affected by disease states. Reports are available showing that in pathological condition such as neurodegenerative disorders and brain trauma, there are imbalances between fusion and fission events [27]. Furthermore, mitochondrial fission mediates mitochondria-dependent cell death [26]. The discharge of pro-apoptotic factors, such as cytochrome c from depolarized mitochondria into the cytosol, is associated with Drp1-mediated fragmentation of the mitochondria which induced apoptosis [23].
6.5 Molecular Mechanisms of Mitophagy
Mitophagy involves the removal of the selective mitochondria from the cytoplasm of the cell. To remove the selective mitochondria from the cytoplasm, damaged or nonfunctional mitochondria are tagged followed by the entrapment into the isolation membrane and formation of autolysosomes.
The autophagy-related gene (Atg) regulates mitophagy through the autophagy-related protein as per the metabolic demands of the cell. For example, Atg 32 protein helps in the initiation of the mitophagy, and Atg 8 protein is necessary for the sealing of the mitochondria through a membrane in yeast. Another protein known as Atg11 is helpful in the sealing of the mitochondria by isolation membrane. Once the mitochondria are sealed completely by the membrane in autophagosome, the sealed mitochondria are fused with the liposomes which digest the defected mitochondria.
During the differentiation of the red blood cell, mitophagy is responsible for the removal of the red blood cell mitochondria. At the time of mitochondrial removal from the red blood cell, the BNIP3L (NIX) expression increases on the outer mitochondrial membrane and has a WXXL-like motif which forms a linkage to LC3. This complex generates the isolation membranes and forms autophagosomes and fuses liposome to form autolysosome. The catalytic enzymes of the liposome hydrolyzed the mitochondrial content.
When mitochondria are injured and lose membrane potential, the PINK1 accumulates and recruits the E3 ubiquitin ligase parkin from the cytosol particularly to the damaged mitochondrion. Parkin promotes the binding of ubiquitinated substrates such as MARF, Mfn1, Mfn2, and VDAC1 to the outer mitochondrial membrane; p62 is recruited to the mitochondria, binding with the ubiquitinated substrates and linking to LC3 to form an isolated membrane and then fuse with lysosomes [15].
6.6 Mitophagy in Cerebral Ischemia
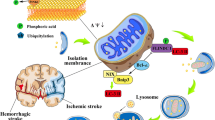
Adenosine monophosphate-activated protein kinase (AMPK), autophagy proteins 5–12 (Atg5–Atg12), dynamin-related protein 1(DRP1), light chain 3 (LC3), mammalian target of rapamycin (mTOR), mitochondrial assembly regulatory factor (MARF), mitofusins 1 and 2 (Mfn1, Mfn2), mycophenolate mofetil (MMF), p62/SQSTM1/sequestosome-1 (p62), PTEN-induced putative kinase 1 (PINK1), serine/threonine-protein kinase (ULK1), voltage-dependent anion-channel 1 (VDAC1) (Fig. 6.1).

Showing the role of mitophagy mediators in cerebral ischemia
Mitophagy involves selective removal of mitochondria from the cytoplasm by liposomes. During ischemic event, there is shortage of blood supply to the brain. Blood supplies oxygen and nutrient to the brain, and shortage of blood to the brain event for a short period of 1–2 min causes severe shortage of ATP in the brain. This causes formation of reactive oxygen species and leads to the mitochondrial membrane’s potential depolarization. Reduction in ATP levels and accumulation of AMP activate the AMPK. AMPK induces mitophagy by activating ULK1 (mTOR inhibit ULK1). Further AMPK activates MFF to recruit DRP1 to induce mitochondrial fission and mitophagy. PINK1 is accumulated in the defective mitochondria triggering the recruitment of the parkin. Parkin promotes binding of ubiquitinated substrates such as MARF, Mfn1, Mfn2, and VDAC1 to the outer mitochondrial membrane. p62 is recruited to the mitochondria, binding with ubiquitinated substrates and linking to LC3 for the autophagic degradation. LC3 with Atg5–Atg12 forms autophagosomes. Autophagosomes fuse with lysosomes to form the autolysosomes and hydrolyze the mitochondrial contents. Rapamycin suppresses mTOR, defending the rat brain from ischemic injury by upregulating mitophagy. Similarly Parkin and Atg5 knockdown aggravates ischemia-induced neuronal cell death, indicating that upregulation of mitophagy is responsible for the protection of the brain cell from ischemic brain injury [28, 30, 31].
6.7 Effect of Cerebral Ischemia in Mitochondrial Dynamic Mediators
Ischemic event in the brain shifts the balance of the mitochondrial fission and fusion toward the fission. Dynamin-related GTPases, i.e., Mfn1, Mfn2, and Opa1, regulate the mitochondrial fusion, while Fis1 and Drp1 regulate mitochondrial fission [12, 17, 24, 32]. Two daughter mitochondria are generated after fission. The daughter mitochondria have altered membrane potential. These daughter mitochondria cannot be fused and are more likely to be taken up by autophagosome due to their smaller size and facilitate mitophagy [33]. Both mitochondrial fission and mitophagy are beneficial, working together to eliminate damaged and depolarized mitochondria. A study revealed that the level of the mitochondrial fusion proteins Opa1 and Mfn2 was significantly reduced in the condition of cerebral ischemia [34]. Further another recent study showed that Drp1 protects against ischemic injury by facilitating mitophagy [35] (Fig. 6.2).

Showing the role of mitochondrial dynamic mediators in cerebral ischemia
Mitofusins Mfn1 and Mfn2 (Mfn1, Mfn2), optic atrophy 1 (Opa1), fission 1 protein (Fis1), dynamin-related protein 1 (Drp1)
6.8 Mitophagy Regulation: An Anti-inflammation Approach in Cerebral Ischemia
Defected mitochondria lead to the release of mitochondrial DNA and reactive oxygen species (ROS) which in turn activate inflammasome [36]. The inflammasome is responsible for the release of the pro-inflammatory marker such as cytokines interleukin 1β and interleukin 18 which leads to programmed cell death due to inflammation. The published report suggests that nuclear factor-κB avoids inflammasome activation by regulating mitophagy to prevent inflammation [37]. Further, studies confirmed that compound that sustains mitochondrial membrane potential intact and inhibited NOD-like receptor (NLRP3) are found to decrease the injury related to the ischemia in the brain [38].
6.9 Mitophagy Regulation: A Neuroprotective Approach in Cerebral Ischemia
Rapamycin shows neuroprotection against cerebral ischemic reperfusion injury through the activation of autophagy [39]. Similarly another recent study reported that rapamycin promotes the L3-II and Beclin-1 that attenuate cerebral ischemia-induced brain injury [14]. The proposed mechanism of action of rapamycin during ischemia is through the increased expression of p62 in mitochondria [14]. Further another compound, methylene blue, shows neuroprotection against cerebral ischemia-reperfusion injury [40]. The exact mechanism of methylene blue is still not known, but it is thought to augment mitophagy [41, 42].
6.10 Conclusion
Mitophagy involves the specific degradation of the damaged or nonfunctional mitochondria. Starvation, oxidative stress, or specific signals including mitochondrial targeting of signaling proteins or modification of mitochondrial proteins induce mitophagy. Since the clearance of the damaged or nonfunctional mitochondria is essential to maintain the homeostasis of cells, the dysfunction of mitophagy is closely related to cerebral diseases. Cerebral ischemic reperfusion injury activates mitophagy. Therefore the regulation of the mitophagy pathway in cerebral ischemic reperfusion injury to protect the nerve cells might be an effective strategy for the treatment of stroke.
References
Paliwal, P., Dash, D., & Krishnamurthy, S. (2017). Pharmacokinetic study of piracetam in focal cerebral ischemic rats. European Journal of Drug Metabolism and Pharmacokinetics, 1–9.
Paliwal, P., Chauhan, G., Gautam, D., Dash, D., Patne, S. C. U., & Krishnamurthy, S. (2018). Indole-3-Carbinol improves neurobehavioral symptoms in a cerebral ischemic stroke model. Naunyn-Schmiedeberg’s Archives of Pharmacology, 391, 613–625.
Lipton, P. (1999). Ischemic cell death in brain neurons. Physiological Reviews, 79, 1431–1568.
Carloni, S., Girelli, S., Scopa, C., Buonocore, G., Longini, M., & Balduini, W. (2010). Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy, 6, 366–377.
Deter, R. L., & De Duve, C. (1967). Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. The Journal of Cell Biology, 33, 437–449.
Deter, R. L., Baudhuin, P., & De Duve, C. (1967). Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. The Journal of Cell Biology, 35, C11–C16.
Yu, L., Alva, A., Su, H., Dutt, P., Freundt, E., Welsh, S., Baehrecke, E. H., & Lenardo, M. J. (2004). Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science, 304(5676), 1500–1502.
Liu, L., Sakakibar, a. K., Chen, Q., & Okamoto, K. (2014). Receptor-mediated mitophagy in yeast and mammalian systems. Cell Research, 24, 787–795.
Santos, R. X., SC, C. a., Wang, X., Perry, G., Smith, M. A., Moreira, P. I., et al. (2010). A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. Journal of Alzheimer’s Disease, 20(2), 401–412.
Vives-Bauza, C., & Przedborski, S. (2011). Mitophagy: The latest problem for Parkinson’s disease. Trends in Molecular Medicine, 17(3), 158–165.
Zhang, X., Yan, H., Yuan, Y., Gao, J., Shen, Z., Cheng, Y., Shen, Y., Wang, R. R., Wang, X., Hu, W. W., & Wang, G. (2013). Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy, 9(9), 1321–1333.
Zuo, W., Zhang, S., Xia, C. Y., Guo, X. F., He, W. B., & Chen, N. H. (2014). Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: The role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology, 86, 103–115.
Huang, C., Andres, A. M., Ratliff, E. P., Hernandez, G., Lee, P., & Gottlieb, R. A. (2011). Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One, 6(6), e20975.
Li, Q., Zhang, T., Wang, J., Zhang, Z., Zhai, Y., Yang, G. Y., & Sun, X. (2014). Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochemical and Biophysical Research Communications, 444, 182–188.
Youle, R. J., & Narendra, D. P. (2011). Mechanisms of mitophagy. Nature Reviews Molecular Cell Biology, 12(1), 9.
Kroemer, G., Dallaporta, B., & Resche-Rigon, M. (1998). The mitochondrial death/life regulator in apoptosis and necrosis. Annual Review of Physiology, 60(1), 619–642.
Chen, H., & Chan, D. C. (2010). Physiological functions of mitochondrial fusion. Annals of the New York Academy of Sciences, 1201, 21–25.
Chen, H., Chomyn, A., & Chan, D. C. (2005). Disruption of fusion results in mitochondrial heterogeneity and dysfunction. The Journal of Biological Chemistry, 280, 26185–26192.
Detmer, S. A., & Chan, D. C. (2007). Functions and dysfunctions of mitochondrial dynamics. Nature Reviews Molecular Cell Biology, 8, 870–879.
Cipolat, S., Rudka, T., Hartmann, D., Costa, V., Serneels, L., Craessaerts, K., Metzger, K., Frezza, C., Annaert, W., D'Adamio, L., & Derks, C. (2006). Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell, 126(1), 163–175.
Züchner, S., Mersiyanova, I. V., Muglia, M., Bissar-Tadmouri, N., Rochelle, J., Dadali, E. L., Zappia, M., Nelis, E., Patitucci, A., Senderek, J., & Parman, Y. (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nature Genetics, 36(5), 449.
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., Rodriguez, M., Kellner, U., Leo-Kottler, B., Auburger, G., & Bhattacharya, S. S. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nature Genetics, 26(2), 211.
Chang, C. R., Manlandro, C. M., Arnoult, D., Stadler, J., Posey, A. E., Hill, R. B., & Blackstone, C. (2010). A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. Journal of Biological Chemistry, 285(42), 32494–32503.
James, D. I., Parone, P. A., Mattenberger, Y., & Martinou, J. C. (2003). hFis1, a novel component of the mammalian mitochondrial fission machinery. The Journal of Biological Chemistry, 278, 36373–36379.
Smirnova, E., Shurland, D. L., Ryazantsev, S. N., & van der Bliek, A. M. (1998). A human dynamin-related protein controls the distribution of mitochondria. The Journal of Cell Biology, 143, 351–358.
Ishihara, N., Nomura, M., & Jofuku, A. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nature Cell Biology, 11, 958–966.
Hoppins, S., Lackner, L., & Nunnari, J. (2007). The machines that divide and fuse mitochondria. Annual Review of Biochemistry, 76, 751–780.
Takagi, H., Matsui, Y., Hirotani, S., Sakoda, H., Asano, T., & Sadoshima, J. (2007). AMPK mediates autophagy during myocardial ischemia in vivo. Autophagy, 3, 405–407.
Mengesdorf, T., Jensen, P. H., Mies, G., Aufenberg, C., & Paschen, W. (2002). Down-regulation of parkin protein in transient focal cerebral ischemia: A link between stroke and degenerative disease? Proceedings of the National Academy of Sciences of the United States of America, 99, 15042–15047.
Tang, Y. C., Tian, H. X., Yi, T., & Chen, H. B. (2016). The critical roles of mitophagy in cerebral ischemia. Protein & Cell, 7(10), 699–713.
Wang, P., Guan, Y. F., Du, H., Zhai, Q. W., Su, D. F., & Miao, C. Y. (2012). Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy, 8(1), 77–87.
Yamamori, T., Ike, S., Bo, T., Sasagawa, T., Sakai, Y., Suzuki, M., Yamamoto, K., Nagane, M., Yasui, H., & Inanami, O. (2015). Inhibition of the mitochondrial fission protein dynamin-related protein 1 (Drp1) impairs mitochondrial fission and mitotic catastrophe after x-irradiation. Molecular Biology of the Cell, 26(25), 4607–4617.
Gomes, L. C., Di Benedetto, G., & Scorrano, L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature Cell Biology, 13, 589–598.
Kumari, S., Anderson, L., Farmer, S., Mehta, S. L., & Li, P. A. (2012). Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Translational Stroke Research, 3, 296–304.
Zuo, W., Yang, P. F., Chen, J., Zhang, Z., & Chen, N. H. (2016). Drp-1, a potential therapeutic target for brain ischaemic stroke. British Journal of Pharmacology, 173(10), 1665–1677.
Gurung, P., Lukens, J. R., & Kanneganti, T. D. (2015). Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends in Molecular Medicine, 21, 193–201.
Zhong, Z., Umemura, A., Sanchez-Lopez, E., Liang, S., Shalapour, S., Wong, J., He, F., Boassa, D., Perkins, G., Ali, S. R., & McGeough, M. D. (2016). NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell, 164(5), 896–910.
Zhao, J., Mou, Y., Bernstock, J. D., Klimanis, D., Wang, S., Spatz, M., Maric, D., Johnson, K., Klinman, D. M., Li, X., & Li, X. (2015). Synthetic oligodeoxynucleotides containing multiple telemeric TTAGGG motifs suppress inflammasome activity in macrophages subjected to oxygen and glucose deprivation and reduce ischemic brain injury in stroke-prone spontaneously hypertensive rats. PLoS One, 10(10), e0140772.
Malagelada, C., Jin, Z. H., Jackson-Lewis, V., Przedborski, S., & Greene, L. A. (2010). Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. The Journal of Neuroscience, 30, 1166–1175.
Miclescu, A., Sharma, H. S., Martijn, C., & Wiklund, L. (2010). Methylene blue protects the cortical blood–brain barrier against ischemia/reperfusion-induced disruptions. Critical Care Medicine, 38, 2199–2206.
Di, Y., He, Y. L., Zhao, T., Huang, X., Wu, K. W., Liu, S. H., Zhao, Y. Q., Fan, M., Wu, L. Y., & Zhu, L. L. (2015). Methylene blue reduces acute cerebral ischemic injury via the induction of mitophagy. Molecular Medicine, 21, 420–429.
Jin, R., Yang, G., & Li, G. (2010). Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. Journal of Leukocyte Biology, 87, 779–789.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Paliwal, P., Krishnamurthy, S., Kumar, G., Patnaik, R. (2019). Critical Role of Mitochondrial Autophagy in Cerebral Stroke. In: Patnaik, R., Tripathi, A., Dwivedi, A. (eds) Advancement in the Pathophysiology of Cerebral Stroke. Springer, Singapore. https://doi.org/10.1007/978-981-13-1453-7_6
Download citation
DOI: https://doi.org/10.1007/978-981-13-1453-7_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1452-0
Online ISBN: 978-981-13-1453-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)