
Abstract
Isothermal titration calorimetry (ITC) is a well-established technique that allows the accurate and precise determination of binding equilibrium constants. It is able to provide detailed thermodynamic description of reacting systems without the need for van’t Hoff analysis. ITC plays an important role in biology, biochemistry and medicinal chemistry, providing researchers with important information on the structure, stability and functionality of biological and synthetic molecules. This review demonstrates the power and versatility of ITC in providing accurate, rapid, and label-free measurement of the thermodynamics of molecular interactions. Moreover, this work focuses on recent studies employing ITC to investigate compounds of great biotechnological interest.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Isothermal titration calorimetry
- Molecular recognition
- Binding thermodynamics
- Nanoparticles
- Polyelectrolytes
- Cyclodextrins
- Liposomes
- Rational drug design
1 Introduction
One of the most fundamental processes in biology, defined as molecular recognition, is the specific binding of molecules driven by noncovalent forces such as of hydrogen bonds, hydrophobic, van der Waals and charge-charge interactions. Networks of interacting molecules form intricate biological pathways that regulate all critical aspects of life, including homeostasis, signal transduction, energy production and gene expression.
To gain insight into cellular functions, many key-components of these networks, like proteins, peptides, lipids, carbohydrates and nucleic acids have been identified and studied extensively. Over the past 60 years, rapid developments in genomics and proteomics allowed scientists to collect a large amount of structural information of ever-increasing detail on these biomolecules and their complexes. As of 2018, over 137,000 macromolecular structures are accessible via the Protein Data Bank repository (www.rcsb.org) [1] and new structures are being added every year at an accelerated rate. Knowledge of spatial orientation and surface complementarity of biological assemblies has been crucial for the in-depth study of many important cellular processes, allowing us to make useful connections about specific three-dimensional architectures and functionality. However, biological recognition is not governed by structural features alone; to explore fully the binding mechanisms and expand our understanding of physiological processes at the molecular level, we must be also able to describe how strongly and how fast the binding components interact. Any effort to modulate cellular activity by therapeutic molecules or bio-inspired nanomaterials requires a detailed information on the nature of the molecular forces involved. Thus, a combination of kinetic, structural and thermodynamic data is necessary for a rigorous description of any biological interaction.
Several biophysical techniques, including fluorescence spectroscopy, capillary electrophoresis, surface plasmon resonance and mass spectroscopy have been successfully employed over the years to study noncovalent molecular interactions. The strengths and weaknesses of the most common techniques in terms of sensitivity, sample concentration, throughput and ease of use are presented in Table 1. Isothermal titration calorimetry (ITC) stands out as a versatile and sensitive method that allows the rigorous characterization of molecular interactions without any need for modification or immobilization of the compounds involved.
ITC has near-universal applicability, allowing a non-invasive approach to quantify binding events in solution. Energy exchange with the environment in the form of heat is an inherit property of all binding reactions; as a result, the range of interactions that can be studied by ITC is vast. The most significant advantage of the technique is its unique ability to provide a model-independent measurement of the binding enthalpy change (ΔΗb), without the underlying assumptions of van’t Hoff analysis used by other methods. In addition, with the use of an appropriate reaction model, important binding parameters including the binding constant (Kb), the binding entropy change (ΔSb), the stoichiometry of the interaction (N) and the Gibbs free energy change (ΔGb) can be determined from a limited set of experiments and calculations. This detailed thermodynamic description of the system facilitates the unraveling of the binding mechanism and the molecular forces governing the binding process.
Up until a few decades ago, only a few custom-made titration calorimeters were available worldwide, accessible only to pioneers that painstakingly developed methods and technologies that are still in use today. A prototype model of a titration calorimeter was presented by Christensen and Izatt in 1968. The sensitivity of the instrument was not sufficient to study biological interactions; however, the usefulness of the technique was demonstrated by measurements of binding enthalpies for simpler reactions [2]. In 1979, Langerman and Biltonen described instrumentation, concepts and methods that could be used in the calorimetric study of biological systems. The first commercially available titration calorimeters, designed specifically for biomolecular reactions, appeared a decade later [3]. Along with technological advancements, the sensitivity of titration calorimeters improved drastically. Modern isothermal titration calorimeters are able to detect very small amounts of heat and provide quantitative characterization of noncovalent interactions in conditions resembling those of biological systems.
As any other analytical technique, the use of ITC comes with certain drawbacks. ITC is a laborious and time-consuming method (low throughput). Compared to other techniques, it requires high concentrations of sample that needs to be of high purity (>95%). Despite these limitations, ITC is widely considered as the “gold standard” for the study of binding interaction. The popularity of the technique is evident by the number of publications generated each year (Fig. 1), covering disciplines including biochemistry, biophysics, structural biology, materials science, medicinal chemistry, supramolecular chemistry and food science.

Number of publications containing isothermal titration calorimetry data since 1990, sourced from the Web of Science citation database. Black bars correspond to the total number of available articles while grey bars represent the annual growth
This chapter aims to demonstrate the power of ITC for characterizing noncovalent molecular interactions and to highlight the suitability of the technique for pharmaceutical and biotechnological applications with selected examples from the literature.
2 Basic Concepts, Experimental Procedure and Data Analysis
2.1 Device Design and Experimental Procedure
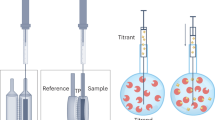
Figure 2 shows the basic parts of a typical ITC instrument. Although significant steps have been made over the years towards scaled-down and ultrasensitive devices, the operating principles and general design remain unchanged.

Schematic representation of a typical isothermal titration calorimeter. Two identical cells are surrounded by a thermostated jacket that acts as an isothermal enclosure. A syringe is used for delivering small aliquots of a binding partner and also serves as a stirring device. Thermal sensors between the cells feed data to a temperature controller that adjusts the power supplied to the heating units to keep the temperature difference between the cells stable
Almost all ITC instruments operate in a differential mode. At the center of the device, symmetrically placed inside a jacket, there are two identical cells made of a heat-conductive corrosion-resistant metal. The sample cell is the vessel where the titration occurs, while the other cell serves throughout the experiment as a reference system with similar thermal behavior. Monitoring thermal fluxes as differential measurements ensures higher signal sensitivity and faster system responsiveness consequently the concept has been incorporated in most of the commercially available titration calorimeters. The temperature inside the jacket is constantly kept identical to that of the cells, functioning as an isothermal enclosure that prohibits any heat exchange with the environment. A series of thermoelectric sensors are attached to the cells and constantly provide temperature data to a temperature controller. The sensors detect temperature changes only in the active cell volume and not in the stem region that is used to collect the excess volume.
Separate heating and cooling systems in close proximity of the cells modulate their temperature. Some devices use a passive cooling system, while in other models it is actively adjusted by the temperature controller. Hence, in steady-state conditions, a certain amount of power needs to be supplied constantly to the cells in order to keep their temperature constant.
The direct observable in an ITC experiment is the differential power supply required to maintain the two cells at the same temperature as a function of time. When the sensors detect a small temperature gradient (ΔΤ) between the two cells, the power supply is properly adjusted to return the temperature back to the desired value (power compensation). The power supply difference between the two cells is often called differential power or cell feedback power (CFB). The CFB signal at the steady state before the injections occur consists the baseline of the titration. The baseline level can be adjusted by maintaining a small constant temperature difference (ΔT ~ 0.005 °C) between the cells. This is important because near-zero CFB values destabilize the feedback circuit.
Two separate solutions of the reacting species are prepared. The solutions must be of high purity, known concentration and aggregate-free. The first reactant (titrand) is placed in the sample cell and the other (titrant) is loaded to the instrument’s syringe. The syringe is then positioned on the top of the sample cell, with its lower tip submerged in the titrand solution. The syringe is computer-controlled and it can be programmed to execute automatically a series of small-volume injections at preset intervals with high accuracy. In addition, during the experiment, the syringe rotates at a constant speed, serving as a stirring device to ensure sample homogeneity and fast thermal equilibration.
For most applications, the heat capacity of the sample cell is effectively matched by filling the reference cell with deionized water. Filling the cell with the titration buffer is often avoided for cleaning purposes, without any measurable effect on data quality. However, in cases where the reacting solutions contain significant amounts of organic compounds, the reference cell should be filled with an identical solvent. The reference cell solution should be frequently replaced (i.e. every week), especially when working at higher-than-room temperatures.
The injection of solution into the sample cell is accompanied by a heat flow that tends to change the temperature of the sample cell. The heat sensors detect the imbalance and the feedback circuit minimizes the temperature difference between the cells by a proper adjustment of the power supply. Heat-generating reactions will reduce the power feed to the cells, while heat-absorbing reactions will result in an increased power feedback, until a new equilibrium is reached. The recorded heat change is proportional to the number of complex molecules formed after the injection. As the titration progresses, the number of binding sites that are available to the titrant is reduced. As a result, the observed heat effect becomes smaller with each subsequent injection until nearly all binding sites are occupied. After this saturation point is reached, only a small heat signal from mixing, dilution and friction phenomena is observed. The raw data of an ITC experiment is the plot of the CFB signal as a function of time, from the initial steady state to the end of the titration. This plot is the so-called thermogram of an ITC measurement (Fig. 3a).

(Upper panel) Change of power supply to the calorimetric cell during the titration of 10 mM of CaCl2 (syringe) into 1 mM of EDTA (cell) at 25 °C in MES buffer (100 mM, pH 6.0). (Lower panel) Integration of the area under each injection, normalized per mol of injectant and plotted as a function of the [Ca2+]:[EDTA] ratio at each point of the titration
ITC measurements are based on the first law of thermodynamics, which states that the change in the internal energy (ΔU) of a reacting system is equal to the difference between the total heat exchanged (Q) with its surroundings and the amount of work done by the system (W):
In an ITC experiment, the reaction takes place at constant pressure (atmospheric pressure) and volume (active cell volume) and as a result, the total work is zero. Hence, for systems analyzed by ITC, it can be concluded that ΔU = Q. For isobaric processes, ΔU is also equal to the total enthalpy change of the system (ΔΗ):
In this fashion, by measuring the total heat exchange for a given titration we can directly measure the enthalpy change of the occurring reaction without prior assumptions for the binding mechanism.
Measuring the binding enthalpy change of noncovalent interactions is not a trivial task. Noncovalent bonds are intrinsically weak, typically associated with total heat changes in the range of 4–20 kJ/mol upon formation. To make things even more challenging, during a titration experiment this small amount of heat is released or absorbed gradually. Nevertheless, modern ITC instruments are sensitive enough to detect changes of heat as small as 100 nJ.
Before the development of ITC, the only approach to calculate ΔHb was to measure the binding constant of an interaction with a convenient method at several temperatures and then use the well-known van’t Hoff equation:
where Kb is the equilibrium constant, T is the absolute temperature and R is the gas constant. Although in theory the calorimetric and van’t Hoff enthalpies are equivalent, there are reports of significant discrepancies for some systems [4,5,6,7,8,9].
2.2 Design of an ITC Experiment
To take full advantage of ITC as a powerful technique to investigate molecular interactions, solid experimental design and careful data analysis is required. There is no universal protocol for ITC measurements; for optimal results, each experimental setup should be based on the thermodynamic characteristics of the investigated system. In this section, we will present some general guidelines for a successful titration.
Selection of concentrations: The shape of the thermogram depends on both the Kb and the concentration of binding sites in the sample cell and can be described by the so-called Wiseman parameter (c) [3]:
where N is the stoichiometry of the reaction (number of titrant molecules bound to one molecule of titrand when full saturation is reached) and [A]tot is the titrand concentration in the sample cell. The parameter c is dimensionless and can be used to determine the ideal concentrations for an ITC experiment. The c value of the reacting system is very important for an accurate determination of the binding constant. Very small c values lead to almost linear binding isotherms with poor ΔΗb or Kb resolution. On the other hand, large c values will result in step-like curves that cannot be used for Kb estimations. Moreover, for system with very high affinity, optimal concentrations will be so low that the heat produced will be lower than the sensitivity threshold of the instruments. For that reason, the largest binding constant that can be determined directly by an ITC experiment is around 109 M−1.
Experience shows that selection of a c value between 5 and 50 leads to more accurate results. For interactions of known Kb and N the following inequality can be used to determine the optimal concentration range of the titrand:
For interactions of unknown Kb and N, an initial titration under the assumptions N = 1 and Kb = 105 M−1 is designed and then the protocol is corrected based on the results of the experiment. Once the concentration of the titrand is selected, the following equation is typically used for calculating the titrant concentration in the syringe ([T]syr):
Frequently, the intrinsic properties of the reacting species (i.e. low solubility, self-association etc.) or sample availability issues are prohibiting factors for working in the optimal range of concentrations. In this case, Eq. 5 is used for determining the concentrations based on the limitations imposed by the system.
The analysis of low c value systems is still possible if certain conditions are met. Turnbull and Daranas [10] demonstrated that it possible to extract meaningful data from systems of c < 1 by using a fixed N value when fitting the data to a binding model. This requires former knowledge of N, which can be provided by other methods or by the nature of the reaction in study. Sophisticated ITC protocols have also been developed for the indirect analysis of systems with c > 1000 or c < 1 [11,12,13,14].
In any case, for reliable measurements, the selected concentrations should produce heat signals of at least 3–5 μcal for the larger peaks of the titration. The accuracy of the ITC-derived parameters directly depends on how well the concentrations of the reacting species are determined. Hence, it is advisable to verify the concentrations used with an appropriate analytical method (i.e. UV/Vis absorbance).
Selection of injection sequence: In a conventional ITC experiment, the titrant solution is added to the reaction vessel in a stepwise manner as a sequence of small-volume injections; this approach ensures a strict control of the reactant concentrations in the cell for each point of the titration. Before an ITC measurement begin, the user must select the total number of injections, the time distance between two consecutive injections as well as the volume and duration of each injection.
The number of injections is equal to the number of points that can be fitted to an appropriate thermodynamic model. A common misconception is that one must have as many points as possible for better fitting results; by increasing the number of injections, one divides the finite amount of reaction heat into smaller and smaller portions, a practice that eventually leads to larger uncertainties and poor reproducibility. Tellinghuisen [15] provided a compelling case and mathematical foundation for using no more than 10 injections for standard ITC titrations. However, 20–25 injections is still the norm for most experiments.
Typical injection volumes range from 1 to 15 μL. Smaller injection volumes are inaccurate and significantly affected from the unavoidable diffusion of the titrant solution in the sample cell [16]. Larger injection volumes must be of very long duration to allow thermal equilibration of the sample cell solution before it is displaced from it as excess volume.
The duration of each injection depends on the injected volume and it should be such that proper mixing is achieved before significant volume displacements. On the other hand, injections with extremely long duration times suffer from titrant diffusion and the measured heat effect is underestimated. Consequently, a balancing act is required; for most reactions, 1–2 s duration for each μL of solution delivered will give reproducible results.
The time distance between two consecutive injections must be long enough to allow the occurring reaction to reach an equilibrium state and for the CFB signal to return to baseline levels. Typical spacing between injections is 300 s, while for reactions with very slow kinetics the waiting time can be longer than 600 s. Special attention is needed for systems with multiple binding sites that exhibit negative cooperativity. In these reactions, the binding process is slower at the middle of the experiment than at the beginning of it and the time distance needs to be adjusted accordingly.
The number of injections and the equilibration time between injections define the duration of the ITC experiment. A typical 20-injections titration with 300 s intervals between injections needs approximately two hours to finish (including the pre-titration equilibration period of the instrument).
A common problem in ITC experiments is that the first injection systematically produces a smaller than expected heat change (see Fig. 3, first injection). The prevailing explanation is that this phenomenon stems from the passive diffusion of the titrant during the long equilibration phase (≥15 min) that precedes the injection sequence; however, it has been suggested that mechanical issues from the automated syringe motor could also give rise to this effect [17]. To circumvent this problem, it is common practice to include a small 1-μL injection at the beginning of the titration and then exclude the first point from data analysis.
An alternative approach that can be used in place of the conventional sequence of injections method is to deliver the syringe solution by a single continuous slow titration. The main advantage of this method is the increase rate of data acquisition, that significantly reduces the duration of the experiment and leads to more points in the binding isotherm for non-linear regression [18].
Selection of buffer and solution preparation: Buffer selection is very important, as it can have a significant effect on the ITC thermogram. Whenever the complex formation is accompanied by protonation or deprotonation of the reacting species, an equivalent number of will have to be taken up or released by the buffer to maintain the pH of the solution stable. This buffer activity is coupled to the binding reaction and will have a measurable heat effect (Qion), which depends only on the number of protons (np) exchanged per mole of titrant bound and the ionization enthalpy of the buffer (ΔHion). The net heat change of the binding reaction (Qbind) can then be calculated from the experimentally determined heat (Qexp) by the following equation:
Due to the complexity of the interactions, the np term is seldom known a priori. However, it can be calculated by repeating the binding experiment a few times in buffers with different ionization enthalpies; the collected Qexp data are then plotted versus the ΔHion of the buffer and fitted linearly to Eq. 7 to estimate both Qbind and np (Fig. 4). Knowledge of np can be very useful for molecular simulations of the binding reaction, especially in cases where no structural information is available. Moreover, buffer effects can be exploited to amplify the heat signal of a weak interaction by selecting a buffer with large ionization enthalpy. Nonetheless, for most titrations, a buffer with small ionization enthalpy (ΔHion ≈ 0) should be selected to avoid this kind of thermal interference. The ionization enthalpies and other important thermodynamic data for many buffer systems are readily available from previous studies [19, 20]. Table 2 contains ionization enthalpies and heat capacity changes for common biological buffers.

Binding enthalpy (ΔΗb) versus buffer ionization enthalpy (ΔΗion) plot (□) for the the titration of 10 mM of CaCl2 (syringe) into 1 mM of EDTA (cell) at 25 °C in the following buffers: Tris (ΔΗion = 47.5 kJ/mol), MOPS (ΔΗion = 21.1 kJ/mol), PIPES (ΔΗion = 11.2 kJ/mol), MES (ΔΗion = 14.8 kJ/mol), acetate (ΔΗion = −0.4 kJ/mol) and HEPES (ΔΗion = 20.4 kJ/mol). The intercept of the linear fit (---) is 0.39 ± 1.7 kJ/mol and the slope is −1.1 ± 0.1 (R2 = 0.98)
Mixing of solutions with very different chemical compositions can generate large heat signals of mixing or dilution that will mask any heat produced by a binding reaction. To minimize such heat signals, the buffer of the titrant and the titrand solutions should be identical in every aspect (pH, ionic strength, concentration, presence of co-solvents etc.). Hence, it is advisable to dialyze exhaustively the samples against large volumes of buffer or to directly dissolve them using a buffer solution of the same batch. To acquire data of high quality, the purity of samples must be higher than 95%. If a reducing agent must be added in the buffer, it is preferable to use low concentrations of mercaptoethanol or tris(2-carboxyethyl)phosphine (TCEP), instead of dithiothreitol (DTT).
Before loading the samples for an ITC experiment, it important to thoroughly degass the solutions using a suitable vacuum chamber. Introduction of small air bubbles into the cell can generate a noisy power baseline and spurious heat signals that will affect the overall quality of the measurement. For the same reason, both solutions should be filtered or centrifuged to remove any precipitated material before filling the cell or the syringe. In case there is some precipitation, after the removal of the aggregates the concentration of the sample must be corrected.
Solution Placement: Another decision that needs to be made when designing an ITC experiment is to select which solution will be loaded in the syringe and which in the sample cell. Routinely, the compound with the lower molecular weight is filled the syringe and the larger compound is placed in the cell. Nevertheless, this is not always the optimal configuration for an ITC experiment; if a compound with significant population of oligomers is placed in the syringe, it will dissociate when diluted in the sample cell and will introduce an additional heat effect to the binding isotherm (dissociation heat). Considering that the cell solution is diluted to a lesser degree (around 20%), loading this compound in the cell is preferable in order to minimize these kind of heat contributions.
Another factor that can force the decision of solution placements is the concentrations needed for the titration. If there are solubility or availability issues with a specific sample, it is better to load it in the cell, where typically the concentration requirements are much lower than those needed for the syringe solution.
Selection of Temperature: The ITC’s temperature operating range is 2–80 °C. The experimental temperature should be selected based on the properties of the system. If there are no system-specific requirements, the most common titration temperatures selected are 25 and ~37 °C. In general, ΔGb, ΔSb, ΔHb and Kb exhibit temperature dependence and caution is required when comparing results from titrations at different experimental temperatures.
2.3 Experimental Results and Data Analysis
2.3.1 Raw Data Treatment
The experimentally determined heats of the binding isotherm (DQtitration) unavoidably include heat contributions from the dilution of both the titrant and sample cell solutions, which need to be separated from the actual heat generated by the interaction (DQreaction). To achieve this, a series of control measurements is required: the dilution heat for the titrant (DQtitrant), usually the most significant contribution, can be determined by replacing the sample cell solution with buffer and repeating the titration following the same protocol with the initial experiment. Likewise, the dilution heat of the titrand (DQtitrand) is estimated by another experiment in which the syringe solution is the one replaced with buffer. For a thorough analysis, a final titration in which buffer is injected into buffer is used for calculating the buffer dilution heat (DQbuffer). The DQbuffer term arises from temperature differences between the syringe and the cell as well as friction heat from stirring and it can be considered as an instrument blank. The heat data from this set of titrations are used to correct the final binding isotherm of the reaction according to the following equation:
The term DQbuffer is added and not subtracted because buffer dilution heat is already included in both DQtitrant and DQtitrand terms and it is removed twice from DQtitration. For most reactions, the dilution heats of the titrand and the buffer are negligible and the corresponding control experiments are often omitted. Then, Eq. 8 is reduced to the equation:
If the titrant is available in limited quantities or is too valuable to be used for a control experiment, it is possible to estimate the DQtitrant term from the final binding isotherm points of a fully saturated system. After the point of saturation is reached, any heat released upon injection is in essence the dilution heat of the titrant (DQreaction ≈ 0). A linear fit of these last few points in the binding isotherm will provide a good approximation of DQtitrant. However, it is strongly recommended to collect all necessary control experiments prior to data analysis for a more accurate calculation of the thermodynamic parameters.
2.3.2 Conversion of Raw Data to a Binding Isotherm
In an ideal situation, where the reacting system is always in thermal and thermodynamic equilibrium without any mechanical or electrical interference, the instrument’s power baseline will always be constant. However, in reality the power baseline will always exhibits some drift and background noise. A very important step for calculating the binding isotherm from raw ITC data is to define the power baseline of the experiment. The baseline trace cannot be determined by a separate experiment and it is only intermittently observed in the thermogram. Hence, the parts of the baseline that are disrupted by the injection sequence must be extrapolated, a process that introduces a certain degree of ambiguity and affects the precision of the measurements [21]. Baseline approximations are generated automatically by the data analysis software provided by the device manufacturer and can be manually corrected if deemed necessary.
The heat change associated with each injection is calculated from the thermogram by integration of the area between the baseline and the corresponding CFB trace with respect to time. The lower time bound for the integration is a few seconds before the injection begins and the upper bound is a few seconds before the next injection is made. Each injection heat change is normalized as heat change per mole of titrant and plotted against the ratio of titrant to titrand concentrations in the active volume (molar ratio). The resulting curve is known as the binding isotherm of the titration (Fig. 3b).
2.3.3 Equilibrium Binding Models for ITC Data Analysis
The binding constant, binding enthalpy and stoichiometry of the interaction are determined by the best fit of the experimental data to an appropriate binding model. The equations describing these models derive from the law of mass action, based on the assumptions made for the binding mechanism and the equilibria involved. The dependent variable (i.e. heat change per injection) is expressed as a function of an independent variable (i.e. moles of titrant added) and the corresponding thermodynamic parameters (e.g. Kb, N and ΔHb). The chosen model should be as simple as possible and compatible with the intrinsic properties of the system in study.
The fitting of a binding isotherm is an iterative process. At first, initial assumptions are made for the value of the fitted parameters. Based on these values, a curve is generated and compared to the experimental data. The deviation between experimental data and the curve is calculated and a suitable algorithm is used to adjust the value of the parameters in order to minimize the error. This procedure is repeated enough times until no further minimization is possible.
The simplest binding reaction is that of an analyte (A) with one available binding site, interacting with free titrant molecules (T) to form a stable complex (AT).
The equilibrium association constant or binding constant (Kb) of the interaction is:
where [A], [T] and [AT] are the concentrations of the corresponding species in the sample cell. Often, in biochemical or pharmacological studies, the reciprocally related dissociation constant (Kd) is used instead of Kb (Kb = 1/Kd). The dimension of Kb is M−1.
If Q is the measured total heat change for the binding reaction (experimental observable), V is the active volume, [A]tot is the total titrand concentration, [A] is the unbound titrand concentration and [AT] is the concentration of the complex in the sample cell, then we have:
and

If the analyte has N identical binding sites, then Eq. 11 can be rewritten in the form:
In cases where the analyte contains N independent binding sites, each with different binding enthalpy \( (\Delta H_{b}^{i} ) \), binding constant \( (K_{b}^{i} ) \) and stoichiometry \( (N^{i} ) \), Eq. 12 is transformed to the following equation:
In order to be able to use these equations for nonlinear regression, we must determine the concentration of the unbound titrant in the active volume [T], at each point of the titration.
For systems with one binding site (N = 1):
where [T]tot is the total concentration of the titrant injected in the sample cell. The combination of Eqs. 10, 11 and 15 leads to:
or equivalently:
where b = −[A]tot − [T]tot − 1/Kb and c = [A]tot ⋅ [T]tot. The solution of Eq. 15 is then:
For systems with multiple identical binding sites (N > 1), Kb is equal to:
Following a similar logic, we get to a solution identical to that of Eq. 17, with the following c and b values:
Parameter c in both cases is equal to the Wiseman parameter of the system. Once [T] is determined, the experimental data can be fitted to Eq. 12 or 13. All concentration-dependent parameters are normalized per mole of injectant. If the thermodynamic data need to be presented in respect to the sample cell compound, then the analysis model must be modified accordingly [22].
The binding models described above are adequate for analyzing simple binding reactions. However, in some cases the nature of the reacting systems requires a different thermodynamic description. Binding models for complex interactions, including three independent set of sites [23], competitive binding [11, 12], multiple interactive binding sites [24], dimer dissociation [25] and combinations of them [26] are available in the literature. Model-free methodologies that utilize binding polynomials for the analysis of ITC data have also been described [27, 28].
Frequently, different binding models produce similar binding isotherms, making difficult to discriminate binding mechanisms. A useful test for the validity of the chosen binding model will be to repeat a second titration with reverse solution placement. If the model is correct, the calculated thermodynamic parameters from the two titrations should be the same [29].
2.3.4 Thermodynamics of Noncovalent Interactions
The measurement of binding affinity allows the calculation of the Gibbs free energy change of the interaction (ΔGb) using the equation:
ΔGb is the result of structural and energy differences between free and bound states for all reacting species. A reaction under thermodynamic control will spontaneously proceed to the direction in which ΔGb < 0, until a thermodynamic equilibrium is reached. When analyzed to enthalpic and entropic contributions (ΔGb = ΔHb − TΔSb), ΔGb can provide additional information for the binding interaction. The binding enthalpy change (ΔHb) reflects variations on the interactions of all the atoms participating in the reacting system [30]. ΔHb is the net result of the formation, disruption and destruction of many individual bonds; when the system gains energy through increased bonding, we have release of heat to the environment and a negative enthalpy change for the interaction (ΔΗb < 0). The reaction is then termed exothermic and the observed ΔHb is considered favorable, since it makes ΔGb more negative. Analogously, when ΔΗb > 0, we have decreased bonding when the complex is formed with a contemporary uptake of energy from the surrounding in the form of heat; the reaction is then described as endothermic and the observed ΔHb is considered unfavorable.
The binding entropy change (ΔSb) is associated with changes in the order of the system. In general, a strongly interacting system tends to be more ordered than a weakly interacting system. Positive ΔSb (increased disorder) leads to even more negative ΔGb and it is favorable for the interaction. Parallel to ΔHb, ΔSb represents the sum of many positive or negative contributions: i.e., a positive solvation entropy change is associated with the burial on non-polar groups from the solvent, while conformational entropy penalties arise from the unavoidable loss of degrees of freedom upon complex formation. Additional contributions to the binding entropy change stem from the burial of polar surfaces and release of organized water molecules to the bulk [31].
Interactions with ΔHb as the most significant contribution towards a negative ΔGb are considered as enthalpy-driven, while interactions with a dominant entropic term are described as entropy-driven.
It is already described how ITC can directly measure the binding enthalpy of a given reaction at a specific temperature. If this process is repeated over a temperature range, the heat capacity change under constant pressure (ΔCp) of the interaction can be determined, based on the following equation:
where T0 is an appropriate reference temperature. Consequently, if ΔHb is plotted against temperature for a series of measurements and fitted using a linear model, the slope of the best fit will correspond to the ΔCp of the system (Fig. 5). The use of Eq. 20 implies that ΔCp is constant within the temperature range of the experiments. ΔCp measures differences in the ability of the solution to absorb heat, reflecting changes in the molecular level on the order and structure of the system in study.

Binding enthalpy versus temperature plot for the binding oxacillin to octakis(6-(2-aminoethylthio)-6-deoxy)-γ-cyclodextrin [123] at 288.15, 298.15 and 309.75 K. Dashed line represents the fit of the data to a simple linear model of the form: ΔHb(T) = ΔH° + ΔCp·T (R = 0.99, ΔCp = 0.11 ± 0.02 kJ/mol K, ΔH° = −45.76 ± 7.56 kJ/mol)
The fact that many reactions are accompanied by large changes in ΔCp may have some practical implications for ITC experiments. For these systems, there is a temperature range where ΔHb will be very small or zero. Consequently, no heat will be detected by the calorimeter. For this reason, in cases where no reaction is observed although it is expected, it is advisable to repeat the experiment at a new experimental temperature that differs at least 10–15 °C to that of the original experiment.
Heat capacity changes are of particular interest in the case of protein-protein and protein-peptide interactions. It has been established that there is a strong correlation between ΔCp and the surface area screened from the solvent upon complex formation [32, 33].
Many noncovalent interactions are accompanied by characteristic changes in enthalpy, entropy and heat capacity that make them distinguishable from other heat-generating phenomena. These “thermodynamic signatures” are extremely useful for the interpretation of ITC data, by providing insight to the driving forces and mechanism of the interaction [34,35,36,37,38,39,40]. The thermodynamic profile of various processes is discussed below.
Hydrogen Bonds: Hydrogen bonds are very important for all processes involving aqueous solutions. Although weaker than covalent bonds, hydrogen bonds play a critical role in the stability, conformation and function of many molecular assemblies [41]. In solution, compounds with solvent-exposed hydrogen bonding groups interact with water molecules in their surroundings. When two molecules interact by forming hydrogen bonds at their binding interface, an equal number of hydrogen bonds with water molecules will have to be broken. Considering that hydrogen bonds are relatively weak and have more or less the same energy (around 20 kJ/mol), only a small change is observed in binding enthalpy. The properties of the hydrogen bonding groups and the solvent before and after the formation of the complex dictate the magnitude of the enthalpy change. Despite their small enthalpic contribution, hydrogen bonds significantly enhance the selectivity and affinity of interactions, with a single bond often leading to a 10- to 1000-fold increase in Kb [42].
Hydrophobic Interactions: By the term hydrophobic interaction, we describe the tendency of non-polar groups to aggregate in aqueous solutions to minimize their contact with the solvent. Non-polar surfaces exposed to the solvent cannot form hydrogen bonds with the surrounding water molecules. Consequently, water molecules near hydrophobic areas tend to make stronger hydrogen bonds among themselves, creating regions of “structured” water around them. Hiding non-polar surfaces from water leads to release of these organized molecules back to bulk solvent, a process characterized by a small enthalpy change (ΔHb ~ 0), a large favorable entropic term (–TΔSb < 0) and a negative contribution to heat capacity change (ΔCp < 0) [43,44,45,46].
Electrostatic Interactions: Electrostatic forces originate from coulombic interactions among polar and charged chemical groups. The strength of these interactions is determined by the dielectric constant of the binding interface and the inverse distance between the charges. Electrostatic interactions in general are entropically driven processes, with a small binding enthalpy change [47]. The entropic gain originates from the release of water molecules solvating the charged groups. Electrostatic interactions have a varying effect on ΔCp. Naturally, electrostatic interactions do not exhibit any binding specificity or selectivity.
Enthalpy-Entropy Compensation: Molecular interactions often yield compensating changes for –TΔSb and ΔHb terms with no significant effect on ΔGb (Fig. 6). This effect, better known as enthalpy-entropy compensation, makes hard to predict the effect of noncovalent interactions on binding affinity. This phenomenon is not limited in aqueous solutions and it appears be a universal property of systems stabilized by interconnecting networks of weak interactions [48,49,50,51,52]. The origin of the enthalpy-entropy compensation effect is a subject of debate for decades. The fact that both enthalpic and entropic contributions are controlled by ΔCp may provide a mechanism for this correlation [53].

Enthalpy-entropy compensation plot (□) for the the titration of 10 mM of CaCl2 (syringe) into 1 mM of EDTA (cell) at 25 °C in Tris, MOPS, PIPES, MES, acetate and HEPES buffers. The intercept of the linear fit (---) is −31.7 ± 1.1 kJ/mol and the slope is −0.70 ± 0.04 (R2 = 0.98)
Thermodynamic parameters and structural changes: Empirical equations connecting important thermodynamic parameters (ΔHb, ΔSb and ΔCp) with structural changes, based on protein folding/unfolding studies have been described in the literature [34, 54,55,56]. It was expected that these equations would have general use since molecular interactions are based on the same noncovalent forces. However, their applicability beyond protein-protein interactions is questionable, especially when the binding surface is relatively small [53]. Therefore, extreme caution is needed for the use of structural data to predict thermodynamic parameters and vice versa [57, 58]. The collection of thermodynamic data for interactions of pharmaceutical interest will eventually allow the development of similar equations for the binding of small ligands, a task of great importance for structure-based rational drug design [29, 59, 60].
3 Applications
ITC was originally developed to study the interactions of proteins and other biological macromolecules with small ligands. The number of protein-protein and protein-ligand interactions that have been investigated by ITC is truly impressive and there is a plethora of excellent reviews that summarize the most important methods and findings [61,62,63,64,65,66,67,68].
Even today, the majority of ITC-related publications still involve the study of protein interactions (Fig. 7). However, new application fields and protocols have emerged over the last decade to include the study of surfactant demicellization [69], membrane partitioning [70], food chemistry reactions [71, 72], antimicrobial agents [73, 74], degradation of pollutants [75] and enzyme kinetics [76,77,78,79]. In this section, we describe the advantages of including ITC in the drug optimization process and demonstrate the power of the technique for the characterization of novel drug delivery systems, nanomaterials and supramolecular assemblies.

Scientific articles containing isothermal titration calorimetry data in 2107, categorized in fields of research. Protein chemistry still dominates the number of publications (362), although the number of articles for synthetic molecules and lipid-based systems is constantly increasing
3.1 The Value of ITC in Rational Drug Design
The development of a new drug is a laborious multi-step process which can take 12–15 years to complete with a total cost that often exceeds the $1,000,000,000 mark [80]. The first step in drug design is to identify ligands that exhibit affinity for a specific target of interest. This task is accomplished by screening compound libraries, often containing thousands of molecules, against a drug target of interest. Typically, initial hits involve compounds that bind with micromolar or even weaker dissociation constants (lead compounds). However, nanomolar and sub-nanomolar range affinities are necessary for the clinical use of a therapeutic compound. In addition, a successful drug should satisfy a wide range of physicochemical requirements, including binding selectivity, low toxicity, high solubility, membrane permeability etc. Therefore, the next step involves chemical optimization of the initial lead candidates for the development of molecules with a suitable pharmacokinetic profile (drug candidates).
Binding affinity is directly related to the free energy change of the interaction (Eq. 19). Therefore, different combinations of ΔHb and ΔSb contributions can result in identical binding affinities (Fig. 8). Enthalpic contributions in general provide a measure of the numerous noncovalent bonds (van der Waals, H-bonds, salt bridges etc.) created or destroyed at the binding interface upon complex formation. In a similar manner, the entropic term is the net effect of various smaller contributions: solvation entropy change is associated with the burial on non-polar groups from the solvent, while conformational entropy penalties arise from the unavoidable loss of degrees of freedom upon complex formation. Another phenomenon linked with the total entropy change is the burial of polar surfaces and release of organized water molecules from the hydration layer to the bulk [31]. Consequently, the thermodynamic signature of a binding event can provide valuable insight to the molecular mechanisms involved in the association. Enthalpy-driven interactions are based on the formation of stabilizing forces between chemical groups and complementary surfaces while entropy-driven interactions are dominated by the hydrophobic effect. Obviously, high binding affinity is achieved only when both enthalpic and entropic contributions are favorable [81]. As a result, the analysis of binding affinity in terms of enthalpy and entropy can be used to identify the best lead compounds among molecules with similar binding constants and to provide useful guidelines for the chemical alterations that would improve their effectiveness.

Thermodynamic profiles for three different interactions with identical binding affinities. Interaction 1 has favorable enthalpic and entropic contributions; interaction 2 is an enthalpy-driven process and interaction 3 is an entropy-driven process. Blue bars represent the Gibbs free energy change (ΔGb); green bars (ΔHb) represent the binding enthalpy and red bars represent the contribution of the entropic term (–TΔSb)
Even when detailed structural and thermodynamic data are available, lead optimization is a common bottleneck in drug development that many projects fail to overcome [81]. For example, the binding affinity of a compound for a hydrophobic binding pocket can be theoretically increased by introducing non-polar groups to its structure. However, this modification would decrease the solubility of compound and could have a negative effect on the surface complementarity and binding orientation of the molecule, leading to sub-optimal affinities. Therefore, a delicate balance of forces must be achieved for fabricating therapeutic agents with the desired set of properties.
ITC can be directly used to identify lead compounds by monitoring their affinity (Kb) for specific binding sites of interest. Furthermore, the thermodynamic signature (N, ΔHb, ΔSb, ΔCp) of these interactions provides additional information on the complexation mechanism, which can be further exploited for the rational design of new compounds with optimized physicochemical characteristics. Describing an interaction not only in terms of affinity but in terms of ΔHb as well is extremely helpful for identifying the most promising positive hits from a large collection of successfully screened compounds. Large favorable ΔHb is a difficult optimization task that includes good shape complementarity, increased number of stabilizing bonds in the binding site interface and proper orientation of interacting groups. The ability to cull compounds that already exhibit large favorable ΔHb can accelerate the optimization process considerably [60].
As mentioned previously, the effectiveness of a therapeutic molecule hinges on more than one properties. Naturally, a good drug should show high affinity for its target, but it should also be able to interact with serum proteins or biological membranes for an effective delivery to the site of interest. ITC can be employed to monitor these interactions as well. Another distinct advantage of the technique is the ability to measure accurately the stoichiometry of a binding interaction. Exact knowledge of N is an excellent method to monitor the effectiveness of a purification process or to assess sample consistency between batches. The ITC-derived stoichiometry is based on the “active” and not the total concentration of the reactants and therefore can be utilized as a sensitive quality indicator.
One of the main disadvantages for the use of ITC in drug development was the low throughput of the technique. Until recently, ITC was able to screen only 5–8 compounds on a daily basis, following protocols that required manual cleaning and loading of the device between experiments. However, technical advancements allowed the development of highly sophisticated instruments and the throughput of the technique was considerably increased. Nowadays, new instruments allow the conduct of 100 experiments per week by automated processes that require even smaller sample volumes [82]. Furthermore, advanced ITC protocols are now available for identifying unknown target proteins from a mixture of biomolecules for a given drug or a lead compound [83]. Although ITC is still a low throughput technique and not suitable as a primary screening method, it provides information-rich data that are invaluable for the identification and optimization of the most promising compounds.
3.2 Nanoparticle-Protein Interactions
The field of biotechnology has broadened considerably over the last decade, to include a wide range of novel nanomaterials to the traditional arsenal of lipid-based and polymer-based systems discuss above [84]. One of the most notable new approaches is the use of nanoparticles (NPs) for the development of bio-reactive assemblies with controlled surface characteristics.
Nanoparticles offer considerable scalability and versatility for the development of novel materials for problem-specific applications. Similar in size (<100 nm) to many biological components, their functionality can be exploited to control processes at the cellular level. However, their ability to penetrate cells and organelles can also be hazardous [85]. Hence, a deeper understanding of nanoparticle interactions with respect to their intrinsic properties is crucial in their design, applications and safe handling.
In principle, when introduced to biological fluids, NPs with the appropriate functionality will be able to readily interact with biomolecules due to their small dimensions and large surface-to-mass ratio. Since the absorbed biomolecules are mainly proteins, the term protein corona is often used to describe such complexes [86]. The corona composition is based on a dynamic equilibrium between the NP and the surroundings. When a NP is introduced to a new environment, the composition of the corona will change until a new equilibrium state is reached. Typical protein coronas in human plasma are consisted of proteins such as albumins, apolipoproteins and immunoglobins [87].
In parallel to many biological interfaces, the absorption of biomolecules to the nanoparticles surface is facilitated by several “weak” noncovalent forces such as van der Waals interactions, solvation effects, hydrogen bonds etc. As a result, the mechanism of the interaction depends on both the physicochemical properties of the reactants and the medium [88]. The release of the absorbed molecules can be achieved by disruption stabilizing forces between the nanoparticle and the compound, i.e. by changes in the pH or the ionic strength of the solution [89].
The absorption of biomolecules to the NP surface can promote the translocation of the assembly across membranes to specific cellular targets. On the other hand, absorption forces can also induce conformation changes to proteins, affecting the overall reactivity of the complex or even triggering immune system responses [90]. These adverse effects highlight the need to for a better understanding of the molecular mechanisms involved in NP interactions in order to engineer bio-reacting nanomaterials for future applications.
Absorbed serum proteins by functionalized NPs are frequently used as a model system to gain insight to the dynamics and energetics of such interactions. ITC is one of the few analytical methods that can provide detail information on NP-protein interactions and it has been used for the study a number of NP-protein systems.
Cedervall et al. [91] employed ITC to investigate the interaction of human serum albumin (HSA) with N-isopropylacrylamide (NIPAM): N-tert-butylacrylamide (BAM) copolymer nanoparticles. The absorption was exothermic with a binding isotherm that was adequately fitted by a single-set-of-sites thermodynamic model, indicating the absence of protein-protein interactions or binding cooperativity. The interactions were very strong (Kb ~ 106 M−1) and the binding affinity was not affected by the size or the composition of the nanoparticles, implying that the interactions are not specific. The stoichiometry of the interactions revealed that a large number of protein molecules could be absorbed to the NP surface. Small size NP (~70 nm) were able to interact with dozens of proteins, while large size NPs (~200 nm) needed more than 1000 protein molecules to reach saturation. Interestingly, the stoichiometry of the interaction increased with the NP hydrophobicity. This finding suggests that the hydrophobic BAM groups are the main protein binding sites and that hydrophobic effects are the driving force for protein absorption by NIPAM/ BAM copolymer nanoparticles.
Another study by Mandal et al. [88] investigated the interaction of BSA and HSA with carbon nanoparticles. The calorimetric profiles of the interactions were almost identical, showing a single binding event driven by equally favorable enthalpic and entropic contributions. The affinity of the NP-BSA and NP-HSA interactions is remarkably high (~2 × 107 M−1) compared to typical affinities for binding of small ligands to BSA and HSA (around 105 M−1). The 40 nm carbon nanoparticles were able to interact with approximately 40 protein molecules, showing the same binding capacity for both BSA and HSA. Strong binding interactions on the highly curved surface of the NPs were expected to have an effect on the protein conformation. Circular dichroism data confirmed partial unfolding and significant alteration of protein secondary structure in the presence of carbon NPs. The authors suggested that the BSA and HSA unfolding on the NP surface would significantly change the transport properties of the complex, affecting in turn all the BSA- and HSA-mediated processes of a biological system.
A later publication by Fleischer and Payne [92] monitored the interactions of bovine serum albumin (BSA) with polystyrene NPs functionalized with either amine or carboxylate groups to provide a cationic or anionic surface charge respectively. ITC thermograms revealed that BSA is able to bind to both cationic and anionic NPs following a binding equilibrium that can be analyzed by a single set-of-sites model. The total enthalpy change of BSA binding was large, favorable and almost identical for both NPs, suggesting the presence of an extensive network of stabilizing forces between the NPs and the protein. However, the other parameters of the interaction were very different for the oppositely charged NPs. The binding constant was almost an order of magnitude greater for BSA adsorbed on anionic NP than that on their cationic counterpart. Moreover, the average number of BSA molecules absorbed on the carboxylate-modified NPs was much larger than that of the amine-modified NPs (871 and 27 respectively), even though they were approximately of the same size. The authors attributed the lower binding capacity of the cationic NPs to the loss of protein secondary structure, resulting in a less energetically favorable packing of BSA upon absorption on the positively charged surface. These differences in corona characteristics were very important for the binding of the BSA-NP complexes to cellular receptors. Although the same protein forms the corona, it was established that anionic NPs and cationic NPs bind to different cellular receptors, showing that the protein secondary structure is a key parameter that controls the NP interactions with the cell.
Eren et al. [93] investigated the interaction of lysozyme with silica NPs. The thermal profile of the interaction revealed the presence of two distinct modes of interaction that lead to lysozyme complexation. The binding isotherm was analyzed with the use of a sequential binding model that allowed the estimation of thermodynamic parameters for both processes. The first binding mode was of higher affinity, with favorable enthalpic and entropic terms and it was succeed by a weaker binding mode with a similar, although less pronounced, thermodynamic signature. The enthalpic change in both processes was attributed to the formation of multiple noncovalent interactions between the protein and the NP, whereas the entropic gain was associated with reorganization of the charged NP surface and release of solvent molecules and counterions from the binding interface to the bulk. Connecting ITC and zeta potential data, the authors proposed an interesting binding mechanism for the interaction of lysozyme with silica NPs. The first thermodynamic equilibrium involves binding of protein molecules to a positively charged NP. As the complexation progresses, more negatively charged protein molecules are bound and as a result the surface charge of NP becomes neutral. The stoichiometry ratio from ITC data confirm that this is the point where the second binding mode takes over, with the second thermodynamic equilibrium corresponding to protein binding events to a negatively charged surface. These energetically different thermodynamic equilibria appear as discrete binding events in the ITC thermogram.
3.3 Polyelectrolyte Interactions
Polyelectrolytes is a fascinating and rapidly growing research domain [94]. Charged polyelectrolytes can serve as model systems to investigate polyelectrolyte complexation events in biological systems, including the wrapping of DNA around histones to form nucleosomes. More importantly, it has been established that interactions between oppositely charged polyelectrolytes can lead to a wide variety of electrostatically assembled macromolecular architectures; for example, when combined with lipids, DNA can form characteristic shapes like rods or toroids [95,96,97]. Therefore, by introducing different chemical groups to the polyelectrolyte structures, we can create self-assembled complexes with many desired properties, conformations and functionalities. Appropriately, polyelectrolytes have many industrial and biotechnological applications including wastewater treatment, microencapsulation, protein separation, enzyme immobilization and drug delivery.
Despite their importance, our understanding of their behavior and functionality is far from complete. ITC is one of the few experimental techniques capable of monitoring their interactions without any significant limitation in respect to their charge, size and internal architecture. In addition, ITC can provide valuable data on the polyelectrolyte’s aggregation state under various solvent conditions, by conducting proper dilution experiments [98,99,100]. However, studying these systems with ITC can be very challenging, considering that the complexation between oppositely charged polyelectrolytes is often dominated by favorable entropic contributions, with a very small enthalpy change. The main source of heat during the complexation process is the release of counterions from the polyelectrolyte’s hydration layer. Fortunately, modern ITC devices have become sensitive enough to follow this type of low-enthalpy reactions. As a result, during the last decade, ITC has been frequently employed to gain further insight into the mechanisms of various polyelectrolyte interactions. Some characteristic examples from the literature are discussed in brief below.
Feng et al. [101] studied the interactions between the strong polyelectrolytes poly(diallyldimethylammonium chloride) and potassium poly(vinyl sulfate) and between the weak polyelectrolytes polyvinylamine and carboxymethyl cellulose by ITC under various pH and ionic strength conditions. Strong polyelectrolytes were found to interact with high affinity through a relatively simple two-step process; the first step corresponds to the exothermic complexation of the two polyelectrolytes. The second step, which is also exothermic, is the result of aggregation and shrinking of the complex structure to a more compact state. Interestingly, the second process occurs near the point where the total charge of the sample cell solution is zero. The enthalpy of the interaction shows a strong dependence on the choice of buffer, a strong indication for the presence of a linked protonation/deprotonation equilibrium. However, ion specific effects from the buffer counterions could also play a significant role in these densely charged colloidal dispersions. The interaction between the two weak electrolytes was found to follow a different binding mechanism. Based on ITC data from experiments in different solution pH, the authors concluded that the formation of the polyelectrolyte complex at the 5.0–5.9 pH range induces the carboxymethyl cellulose to ionize further during the titration. This increased carboxyl ionization leads to more ionic crosslinks between the polyelectrolytes and the interaction appears to be quite strong. Outside this pH window, the ionization effect becomes less important and the interaction is described by a much smaller binding constant.
Another study by Maurstad et al. [102] investigated the interaction between xanthan, a bacterial polysaccharide, and a chitosan polymer. Thermodynamic analysis of ITC data provided a valuable insight to the mechanism of interaction. The small binding constant of the interaction is indicative of a non-specific, entropy-driven complexation process. Increasing the chain length of the chitosan led to a more complicated binding isotherm, suggesting that there was a secondary process after the formation of the complex. Atomic force microscopy topographs confirmed original ITC findings, showing that initial binding events were followed by aggregation when the concentration and chain length of chitosan were increased over a certain limit.
A recent study by Lounis et al. [103] used ITC to systematically investigate the interactions between linear poly(L-lysine) and oppositely charged acrylamide and 2-acrylamido-2-methyl-1-propanesulfonate copolymers at different ionic strength conditions. The ITC experimental results for these interactions were in very good agreement with previously published data obtained by other techniques. The thermodynamic analysis of the binding signature revealed that the complex formation is an entropy-driven process. Furthermore, the logarithm of the measured binding affinities demonstrated a linear dependence with the logarithm of the solution’s ionic strength; taken together, these finding confirm that the binding mechanism is based on stabilizing electrostatic interactions between oppositely charged polyelectrolyte regions. The authors were able to estimate the number of counter ions release by combining ITC affinity data and the Manning theory [104].
One of the most interesting applications of polyelectrolyte systems is their use as drug stabilizing and delivery systems. Sideratou et al. [105] investigated the interaction between functionalized poly(L-lysine) dendrigrafts and insulin at physiological pH. Analysis of ITC data provided a clear picture for the binding mechanism of the dendrigrafts-insulin interaction. The final binding isotherm appears to be the result of two distinct thermodynamic equilibria: an endothermic process that is the major source of heat signals at low insulin concentrations (up to ~0.7 molar ratio) and an exothermic process that is predominant at higher molar ratio. The competition between these two opposite heat signals produces a characteristic V-shaped curve (Fig. 9). The endothermic trace is produced from the dissociation of insulin, which at high concentrations forms dimers or hexamers. When dendrigraft molecules are injected into the sample cell, the presence of the amino and/or guanidinium groups destabilizes the insulin oligomers, causing them to dissociate into monomers. The observed exothermic process results from the binding of insulin monomers to the functionalized dendrigraft by charge-pairing interactions. After a critical monomer concentration is reached in the solution, less and less dissociation events occur and the exothermic binding trace becomes dominant.

Isothermal titration data for the interaction of 1 mM insulin with 0.5 mM poly(L-lysine) dendrigraft bearing 36 amino groups at the external surface at 25 °C in Tris buffer (10 mM, pH = 7.4). The upper panel shows the raw calorimetric data and the lower panel shows the corresponding integrated and normalized heats (d) given per mole of dendrigraft injected. Dashed line represent the non-linear least square fit of the ITC data to a two independent set-of-sites binding model
3.4 Interactions of Lipid-Based Systems
Some compounds do not have all the necessary properties to become efficient drugs. Problems like poor solubility, non-specific targeting or low permeability of biological barriers reduce their effective concentration significantly, leading to less than optimal results. One of the greatest challenges in the pharmaceutical industry is the development of new strategies and technologies to overcome these obstacles and enhance the bioavailability of the molecules in the area of interest.
The interaction of bioactive ligands with lipid bilayers is a critical step for their transportation through biological membranes to the intended target site. Most drugs will have to cross several membranes to reach the interior of the cell. Therefore, a detailed understanding of the interactions between drugs and biological membranes is very important for the formulation of effective therapeutic agents.
The thermodynamics of lipid interactions with other compounds depends on the structure and various physicochemical properties of the reacting species, including lipophilicity, charge, size and hydrogen bonding capacity. Lipids can assemble in a variety of morphologies, depending on their chemical structure, hydration state and solution conditions. Lipid are a very difficult system to work with; usually, the available sample quantities are limited and contain various impurities. Moreover, the resulting suspensions are generally turbid, which complicates analysis by spectroscopic techniques. Opposite to specific binding, where the association requires the presence of well-defined binding sites, lipid interactions does not necessarily lead to the formation of complexes with certain stoichiometries.
In addition to their usefulness as model systems for biological membranes, lipid vesicles hold promise as efficient drug carriers, with numerous clinical applications [106, 107]. ITC is considered the method of choice for monitoring interactions between bioactive molecules (such as surfactants, peptides, proteins, drugs) and lipid membrane analogs like vesicles or micelles. Below we discuss in brief some interesting ITC studies on the interactions of lipid-based systems.
Ikonen et al. [108] investigated the thermodynamics of the binding and partitioning of three β-blockers (alprenolol, labetalol and propranolol) as well as that of the local anaesthetic tetracaine into liposomes composed of 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG). A series of ITC experiments was performed in order to investigate thoroughly the effect of concentration, membrane curvature, temperature, and ionic strength to binding energetics. The interactions of all drugs were exothermic, with a dominant enthalpic term. The favorable enthalpy change can be explained by van der Waals interactions between the non-polar groups of the drugs and the long hydrocarbon chains of the lipids. Repeating the titrations with different concentrations of liposomes and drugs resulted in almost identical estimations for the thermodynamic parameters of the interaction, indicating that the partitioning of the compounds into liposomes is a thermodynamic equilibrium and not a kinetically controlled process. The effect of membrane curvature was examined by direct comparison of ITC titrations with liposomes of different diameter and it was found that there is no significant change to the binding parameters. Increase of the titration temperature resulted in more exothermic interactions, implying negative ΔCp values for the binding processes. Negative ΔCp values are often attributed to burial of hydrophobic areas, providing further evidence for an extended network of van der Waals interactions between the non-polar groups of the reactants. In conclusion, the effect of ionic strength on the affinity of the interactions was examined by carrying out ITC experiments at different salt concentrations. Measurable differences were detected for the binding constants of the interactions in low and high salt concentrations, providing evidence that electrostatic effects play an important role in the binding process. These electrostatic effects may include direct charge—charge interactions between the drugs and the liposomes as well as structural reorganization of the lipids in the negatively charged membrane.
Another publication by Al-Kaddah et al. [109] explored the interaction of the antibiotics gallidermin and vancomycin with bacterial membranes simulated by 1,2-dioleoyl-sn-glycero-3-phosphocholine liposomes (DOPC). To investigate the role of the cell wall precursor lipid II to the antibiotic interactions, pure DOPC liposomes and DOPC liposomes containing 0.1% (mol/mol) were prepared (DOPC + Lipid II). Both compounds were found to bind to DOPC + Lipid II liposomes with high affinity (Kb ~ 3 × 105 M−1), displaying a typical sigmoidal ITC trace of exothermic peaks that can be analyzed by a single set-of -sites binding model. However, only vancomycin was able to interact with pure DOPC liposomes with similar affinity under identical experimental conditions. These results provide a clear indication for a direct interaction between gallidermin and Lipid II that facilitates the membrane insertion of the molecule. On the other hand, the vancomycin insertion is probably mediated by non-specific hydrophobic interactions with the membrane. These differences in binding behavior could also imply a different mechanism of antimicrobial activity for the two peptides.
Another interesting class of lipid-based molecules, which combine the aggregation characteristics of lipids and the specific functionalities of nucleosides, are the nucleolipids [110]. These hybrid moieties, composed of a lipid covalently linked to a nucleoside, show great promise as biocompatible nanocarriers for therapeutic compounds. A recent study [111] explored the interactions of thymidine- and adenine-3′-(1,2-dipalmitoyl-sn-glycero-3-phosphate) nucleolipids (abbreviated as diC16dT and diC16dA respectively) with the RNA analogs polyuridylic acid (polyU) and polyadenylic acid (polyA). Both nucleolipids were able to bind to their base-corresponding RNA analogs, however the interactions were relatively weak (Kb < 103 M−1) and non-specific, driven mainly by favorable entopic contributions (Fig. 10). The stoichiometry of the interactions (N < 1) also revealed that due to steric restrictions, only a fraction of the nucleolipid molecules are able to interact with the nucleic acid analogues. The conclusions of this study were used for an appropriate modification of the diC16dT and diC16dA nucleolipids, in order to increase the selectivity and affinity of the interactions [112]. The results were very encouraging, showing a tenfold increase in affinity for both compounds. Although the optimization of nucleolipids is far from complete, these studies demonstrate the usefulness of ITC in the development of new design strategies.

Calorimetric data for the interaction of poly-A with diC16dT; Upper panel: Raw data for the titration of a 6 mM solution of poly-A [syringe] into 1 mM solution of diC16dT [cell]. Lower panel: Normalized injection heats, corrected for dilution effects and the fitting curve to the single set of sites model (dashed line). The binding constant is Kb = 6660 M−1 and the binding enthalpy is 5.6 kJ/mol
3.5 Cyclodextrin Interactions
For most drugs, a difficult balance needs to be achieved: they must be hydrophilic enough to dissolve in biological fluids and lipophilic enough to be able to penetrate all the biological membranes in their path. However, quite often, the most promising drug candidates exhibit poor water solubility [113] and they are classified as Class II (high permeability but low solubility) or Class IV (low permeability and low solubility) drugs in the FDA’s biopharmaceutical classification system [114]. There are many ways to increase the apparent solubility of a compound, including salt formulation, particle size reduction, microemulsions etc. One very effective strategy to increase drug solubility is to create reversible complexes with a more hydrophilic carrier. Cyclodextrins are increasingly used for drug-delivery purposes, by virtue of their non-toxicity and effectiveness in enhancing the solubility, stability and bioavailability of otherwise poorly soluble drugs. Today, many therapeutic agents that contain natural or modified cyclodextrins are available in markets worldwide [115]. Cyclodextrins are naturally occurring cyclic oligosaccharides formed by six (α-CD), seven (β-CD) or eight (γ-CD) D-glucopyranosyl units organized in a hollow truncated cone architecture with amphiphilic properties; the exterior of the structure is hydrophilic, while the interior cavity surface is relatively hydrophobic. They are known for more than 100 years [116], but it is only recently that highly purified cyclodextrins have been incorporated in pharmaceutical formulations. In aqueous environments, CDs form inclusion complexes by incorporating hydrophobic molecules or hydrophobic parts of the molecule (depending on chemical structure and steric restrictions) into their central cavity, thereby increasing the solubility of the guest molecules and protecting them from their environment.
The rigidity of the structure and the specific dimensions of the hydrophobic cavity arise certain limitations on the potential guests that can be accommodated by native CDs. Chemical modifications of cyclodextrins have been used to enhance their functionality and in some cases to overcome molecular size restrictions by proper orientation of multiple CDs into new geometries that specifically match the structure of the guest. CD functionalization provide new opportunities for their use to complexation, delivery and controlled release of therapeutic molecules. Natural CDs are relatively large and contain a large number of hydrogen donors and acceptors; consequently, they do not readily permeate biological membranes [117]. However, modified cyclodextrins, such as methylated β-cyclodextrins, can enhance drug penetration through lipophilic barriers [118].
Most CD-drug inclusion complexes involve 1:1 stoichiometries (one molecule of drug per CD molecule) and binding constants in the range of 50–2000 M−1 [119]. The driving force for the inclusion can have many origins: inclusion of the drug in the central cavity displaces closely packed water molecules to the bulk with a favorable change in entropy. Electrostatic interactions, van der Waals interactions, hydrophobic effect, hydrogen-bond formation can also contribute to the energetics of the complexation process. Hence, CD-drug interactions are relatively weak interactions, which are often hard to study. ITC is an invaluable tool for studying the binding mechanism and the energetics of CD—drug inclusion, especially when combined with structural information from other techniques like NMR. In the following paragraphs, instructive examples of ITC studies on CD-drug interactions are discussed.
Segura-Sanchez et al. [120] used ITC to study the complexation mechanism of (+)-usnic acid (a molecule with antitumor activity), by native and modified CDs. The stoichiometry of all interactions was found to be 1:1 and the binding constants were in the range between 100 and 1100 M−1, with γ-CD being the most suitable host of all the CDs of the study. The thermodynamic profile of the interactions suggests that the complex formation is enthalpically driven, stabilized by a combination of van der Waals forces and hydrogen bonds. To investigate further the complexation mechanism, the authors repeated the γ-CD experiments in different solution pH. The observed changes in binding energetics were correlated with various possible orientations and a planar configuration was finally proposed for the guest molecule. Molecular modeling simulations confirmed that the indicated binding topology was the most stable for this specific inclusion system.
Mazzaferro et al. [121] investigated the inclusion complexes of docetaxel (a potent anti-cancer drug) with various cyclodextrins. The solubility of the drug was increased over 5000 times when complexed with methyl-β-cyclodextrin (Me-β-CD), while the other cyclodextrins of the study offered less significant solubility enhancements. Two-dimensional 1H NMR, ITC, molecular docking simulations and circular dichroism spectroscopy were employed to investigate the binding of docetaxel to Me-β-CD. Analysis of ITC data revealed an unusual binding mechanism: the stoichiometry of the complex was found to be 2:1 (two Me-β-CDs bind one docetaxel molecule) and the experimental binding isotherm was indicative of an exothermic sequential binding process. The first step of the complex formation involves the exothermic binding of docetaxel to one Me-β-CD with an unfavorable entropic term. The exposed part of docetaxel then interacts with another Me-β-CD to form the final 2:1 complex, this time with a favorable entropy change. Molecular docking simulations explain the opposite entropic contributions of the sequential binding events by differences in the hydrophobic area of the two binding interfaces and the smaller flexibility of the already bound docetaxel molecule.
A study by Ignaczak et al. [122] evaluated the thermodynamic parameters of the complex formation between mianserin hydrochloride, a common antidepressant, with β-CD by ITC and quantum chemical methods. Titration data shown that the interaction is relatively strong (>1300 M−1), with a small negative enthalpic term and a large favorable entropic change that is associated with release of high-ordered water molecules from the hydration layer to the bulk. Interestingly, the stoichiometry of the interaction suggests the formation of 2:1 complexes along with a smaller population of 1:1 β-CD—mianserin complexes. However, the binding isotherm was adequately described by a single step process, indicating that the 2:1 complex involves the synchronous binding of two β-CDs to a mianserin molecule. Quantum molecular dynamics calculations on both 1:1 and 2:1 structures confirmed that 2:1 complexes are energetically more favorable than their 1:1 counterparts.
A recent study by Agnes et al. used ITC to identify the optimal host for delivering penicillins to mammalian cells from a collection of anionic and cationic thioether-substituted-β- and γ-CD derivatives. The strongest interaction of the study was that of the positively charged octakis(6-(2-aminoethylthio)-6-deoxy)-γ-CD with oxacillin (Fig. 11). The binding constant was ~1700 M−1, with almost equally favorable entropic and enthalpic terms. For this system, the binding stoichiometry suggested the presence of 1:1 complexes along with measurable population of 2:1 (host/guest) complexes, a result that was later confirmed by NMR data as well. The authors used this interaction to investigate the role of electrostatic effects in the β-lactam antibiotics inclusion mechanism by studying the effect of ionic strength on the binding of the oppositely charged molecules. No significant change in both enthalpy and binding affinity was detected for biologically relevant salt concentrations and it was concluded that the complex was stabilized though host-guest soft contacts rather than by electrostatic interactions.

(Upper panel) Change of power supply to the calorimetric cell during the titration of 10 mM of octakis(6-(2-aminoethylthio)-6-deoxy)-γ-cyclodextrin into 1 mM of various oxacillin at 25 °C in PBS buffer, after the subtraction of the appropriate reference experiments. (Lower panel) Integration of the area under each injection, normalized per mol of injectant and plotted as a function of the [Guest]:[Host] ratio at each point of the titration. Dashed line represent the non-linear least square fit of the ITC data to a simple 1:1 complexation thermodynamic model
4 Conclusions
Isothermal titration calorimetry is a well-established biophysical technique for studying the formation or dissociation of molecular complexes. Over the past years, ITC has developed from a convenient tool for studying macromolecular interactions with small ligands to a powerful and versatile method that is used in various research fields including biochemistry, biophysics, structural biology, materials science, medicinal chemistry, supramolecular chemistry and food science. The increased popularity of ITC in basic biochemical, biomedical biotechnology-oriented research lies in the ability to provide a complete thermodynamic characterization of noncovalent interactions and valuable insight to the underlying mechanisms that govern molecular recognition. Measuring the small amounts of heat released or absorbed upon binding, ITC is able to monitor molecular events that are invisible to other techniques such as binding to surfactants, cyclodextrin inclusions and nanoparticle corona formation. As titration calorimeters become more sensitive and widespread with time, the spectrum of ITC applications is expected to broaden even more in the following years.
References
Berman, H.M., et al.: The protein data bank. Nucleic Acids Res. 28(1), 235–242 (2000)
Christensen, J.J., Johnston, H.D., Izatt, R.M.: An isothermal titration calorimeter. Rev. Sci. Instrum. 39(9), 1356–1359 (1968)
Wiseman, T., et al.: Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179(1), 131–137 (1989)
Horn, J.R., Brandts, J.F., Murphy, K.P.: van’t Hoff and calorimetric enthalpies II: effects of linked equilibria. Biochemistry 41(23), 7501–7507 (2002)
Horn, J.R., et al.: van’t Hoff and calorimetric enthalpies from isothermal titration calorimetry: are there significant discrepancies? Biochemistry 40(6), 1774–1778 (2001)
Kantonen, S.A., Henriksen, N.M., Gilson, M.K.: Accounting for apparent deviations between calorimetric and van’t Hoff enthalpies. Biochim. Biophys. Acta 1862, 692–704 (2018)
Liu, Y., Sturtevant, J.M.: Significant discrepancies between van’t Hoff and calorimetric enthalpies. II. Protein Sci. 4(12), 2559–2561 (1995)
Liu, Y., Sturtevant, J.M.: Significant discrepancies between van’t Hoff and calorimetric enthalpies. III. Biophys. Chem. 64(1–3), 121–126 (1997)
Naghibi, H., Tamura, A., Sturtevant, J.M.: Significant discrepancies between van’t Hoff and calorimetric enthalpies. Proc. Natl. Acad. Sci. U.S.A. 92(12), 5597–5599 (1995)
Turnbull, W.B., Daranas, A.H.: On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J. Am. Chem. Soc. 125(48), 14859–14866 (2003)
Velazquez-Campoy, A., Freire, E.: Isothermal titration calorimetry to determine association constants for high-affinity ligands. Nat. Protoc. 1(1), 186–191 (2006)
Khalifah, R.G., et al.: Thermodynamics of binding of the carbon dioxide-competitive inhibitor imidazole and related compounds to human carbonic anhydrase I: an isothermal titration calorimetry approach to studying weak binding by displacement with strong inhibitors. Biochemistry 32(12), 3058–3066 (1993)
Zhang, Y.L., Zhang, Z.Y.: Low-affinity binding determined by titration calorimetry using a high-affinity coupling ligand: a thermodynamic study of ligand binding to protein tyrosine phosphatase 1B. Anal. Biochem. 261(2), 139–148 (1998)
Sigurskjold, B.W.: Exact analysis of competition ligand binding by displacement isothermal titration calorimetry. Anal. Biochem. 277(2), 260–266 (2000)
Tellinghuisen, J.: Designing isothermal titration calorimetry experiments for the study of 1:1 binding: problems with the “standard protocol”. Anal. Biochem. 424(2), 211–220 (2012)
Nilsson, S.O., Wadso, I.: A flow-microcalorimetric vessel for solution of small quantities of easily or slightly soluble liquids—solution of benzene in water at 298.15-K. J. Chem. Thermodyn. 16(4), 317–330 (1984)
Mizoue, L.S., Tellinghuisen, J.: The role of backlash in the “first injection anomaly” in isothermal titration calorimetry. Anal. Biochem. 326(1), 125–127 (2004)
Markova, N., Hallen, D.: The development of a continuous isothermal titration calorimetric method for equilibrium studies. Anal. Biochem. 331(1), 77–88 (2004)
Christensen, J.J., Hansen, L.D., Izatt, R.M.: Handbook of Proton Ionization Heats and Related Thermodynamic Quantities. Wiley, New York (1976)
Goldberg, R.N., Kishore, N., Lennen, R.M.: Thermodynamic quantities for the ionization reactions of buffers. J. Phys. Chem. Ref. Data 31(2), 231–370 (2002)
de Rivera, M.R., Socorro, F.: Baseline changes in an isothermal titration microcalorimeter. J. Therm. Anal. Calorim. 80(3), 769–773 (2005)
Bhatnagar, R.S., Gordon, J.I.: Thermodynamic studies of myristoyl-coa—protein N-myristoyltransferase using isothermal titration calorimetry. Lipid Modif. Proteins 250, 467–486 (1995)
Brautigam, C.A.: Fitting two- and three-site binding models to isothermal titration calorimetric data. Methods 76, 124–136 (2015)
Brown, A.: Analysis of cooperativity by isothermal titration calorimetry. Int. J. Mol. Sci. 10(8), 3457–3477 (2009)
McPhail, D., Cooper, A.: Thermodynamics and kinetics of dissociation of ligand-induced dimers of vancomycin antibiotics. J. Chem. Soc. Faraday Trans. 93(13), 2283–2289 (1997)
Buurma, N.J., Haq, I.: Advances in the analysis of isothermal titration calorimetry data for ligand-DNA interactions. Methods 42(2), 162–172 (2007)
Keeler, C., et al.: An explicit formulation approach for the analysis of calcium binding to EF-hand proteins using isothermal titration calorimetry. Biophys. J. 105(12), 2843–2853 (2013)
Freire, E., Schon, A., Velazquez-Campoy, A.: Isothermal titration calorimetry: general formalism using binding polynomials. Methods Enzymol. 455, 127–155 (2009)
Ladbury, J.E.: Calorimetry as a tool for understanding biomolecular interactions and an aid to drug design. Biochem. Soc. Trans. 38(4), 888–893 (2010)
Fisher, H.F., Singh, N.: Calorimetric methods for interpreting protein-ligand interactions. Energ. Biol. Macromol. 259, 194–221 (1995)
Ladbury, J.E., Chowdhry, B.Z.: Sensing the heat: the application of isothermal titration calorimetry to thermodynamic studies of biomolecular interactions. Chem. Biol. 3(10), 791–801 (1996)
Livingstone, J.R., Spolar, R.S., Record Jr., M.T.: Contribution to the thermodynamics of protein folding from the reduction in water-accessible nonpolar surface area. Biochemistry 30(17), 4237–4244 (1991)
Spolar, R.S., Livingstone, J.R., Record, M.T.: Use of liquid-hydrocarbon and amide transfer data to estimate contributions to thermodynamic functions of protein folding from the removal of nonpolar and polar surface from water. Biochemistry 31(16), 3947–3955 (1992)
Sturtevant, J.M.: Heat capacity and entropy changes in processes involving proteins. Proc. Natl. Acad. Sci. U.S.A 74(6), 2236–2240 (1977)
Gomez, J., Freire, E.: Thermodynamic mapping of the inhibitor site of the aspartic protease endothiapepsin. J. Mol. Biol. 252(3), 337–350 (1995)
Haq, I., et al.: Specific binding of hoechst 33258 to the d(CGCAAATTTGCG)2 duplex: calorimetric and spectroscopic studies. J. Mol. Biol. 271(2), 244–257 (1997)
Chaires, J.B.: Energetics of drug-DNA interactions. Biopolymers 44(3), 201–215 (1997)
Cooper, A.: Thermodynamic analysis of biomolecular interactions. Curr. Opin. Chem. Biol. 3(5), 557–563 (1999)
Holdgate, G.A., Ward, W.H.: Measurements of binding thermodynamics in drug discovery. Drug Discov. Today 10(22), 1543–1550 (2005)
Spolar, R.S., Record Jr., M.T.: Coupling of local folding to site-specific binding of proteins to DNA. Science 263(5148), 777–784 (1994)
Jeffrey, G.A., Saenger, W.: Hydrogen Bonding in Biological Structures. Springer, New York (1991)
Fersht, A.: Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, vol. xxi, 631 p. W.H. Freeman, New York (1999)
Scatena, L.F., Brown, M.G., Richmond, G.L.: Water at hydrophobic surfaces: weak hydrogen bonding and strong orientation effects. Science 292(5518), 908–912 (2001)
Matulis, D.: Thermodynamics of the hydrophobic effect. III. Condensation and aggregation of alkanes, alcohols, and alkylamines. Biophys. Chem. 93(1), 67–82 (2001)
Matulis, D., Bloomfield, V.A.: Thermodynamics of the hydrophobic effect. II. Calorimetric measurement of enthalpy, entropy, and heat capacity of aggregation of alkylamines and long aliphatic chains. Biophys. Chem. 93(1), 53–65 (2001)
Matulis, D., Bloomfield, V.A.: Thermodynamics of the hydrophobic effect. I. Coupling of aggregation and pK(a) shifts in solutions of aliphatic amines. Biophys. Chem. 93(1), 37–51 (2001)
Matulis, D., Rouzina, I., Bloomfield, V.A.: Thermodynamics of DNA binding and condensation: isothermal titration calorimetry and electrostatic mechanism. J. Mol. Biol. 296(4), 1053–1063 (2000)
Ahmad, M., et al.: Enthalpy-entropy compensation upon molecular conformational changes. J. Chem. Theory Comput. 11(4), 1410–1418 (2015)
Breiten, B., et al.: Water networks contribute to enthalpy/entropy compensation in protein-ligand binding. J. Am. Chem. Soc. 135(41), 15579–15584 (2013)
Dragan, A.I., Read, C.M., Crane-Robinson, C.: Enthalpy-entropy compensation: the role of solvation. Eur. Biophys. J. 46(4), 301–308 (2017)
Dunitz, J.D.: Win some, lose some: enthalpy-entropy compensation in weak intermolecular interactions. Chem. Biol. 2(11), 709–712 (1995)
Lee, B.: Enthalpy-entropy compensation in the thermodynamics of hydrophobicity. Biophys. Chem. 51(2–3), 271–277; discussion 277–278 (1994)
Holdgate, G.A.: Making cool drugs hot: isothermal titration calorimetry as a tool to study binding energetics. Biotechniques 31(1), 164–166, 168, 170 passim (2001)
Baldwin, R.L.: Temperature dependence of the hydrophobic interaction in protein folding. Proc. Natl. Acad. Sci. U.S.A. 83(21), 8069–8072 (1986)
Privalov, P.L., Gill, S.J.: Stability of protein structure and hydrophobic interaction. Adv. Protein Chem. 39, 191–234 (1988)
Spolar, R.S., Ha, J.H., Record Jr., M.T.: Hydrophobic effect in protein folding and other noncovalent processes involving proteins. Proc. Natl. Acad. Sci. U.S.A. 86(21), 8382–8385 (1989)
Kelley, R.F., O’Connell, M.P.: Thermodynamic analysis of an antibody functional epitope. Biochemistry 32(27), 6828–6835 (1993)
Davies, T.G., Hubbard, R.E., Tame, J.R.: Relating structure to thermodynamics: the crystal structures and binding affinity of eight OppA-peptide complexes. Protein Sci. 8(7), 1432–1444 (1999)
Chaires, J.B.: Calorimetry and thermodynamics in drug design. Ann. Rev. Biophys. 37, 135–151 (2008)
Garbett, N.C., Chaires, J.B.: Thermodynamic studies for drug design and screening. Expert Opin. Drug Discov. 7(4), 299–314 (2012)
Pierce, M.M., Raman, C.S., Nall, B.T.: Isothermal titration calorimetry of protein-protein interactions. Methods 19(2), 213–221 (1999)
Freire, E., Mayorga, O.L., Straume, M.: Isothermal titration calorimetry. Anal. Chem. 62(18), A950–A959 (1990)
Lewis, E.A., Murphy, K.P.: Isothermal titration calorimetry. Methods Mol. Biol. 305, 1–16 (2005)
Leavitt, S., Freire, E.: Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 11(5), 560–566 (2001)
Ababou, A., Ladbury, J.E.: Survey of the year 2004: literature on applications of isothermal titration calorimetry. J. Mol. Recognit. 19(1), 79–89 (2006)
Cliff, M.J., Ladbury, J.E.: A survey of the year 2002 literature on applications of isothermal titration calorimetry. J. Mol. Recogn. 16(6), 383–391 (2003)
Ghai, R., Falconer, R.J., Collins, B.M.: Applications of isothermal titration calorimetry in pure and applied research–survey of the literature from 2010. J. Mol. Recogn. 25(1), 32–52 (2012)
Falconer, R.J.: Applications of isothermal titration calorimetry—the research and technical developments from 2011 to 2015. J. Mol. Recogn. 29(10), 504–515 (2016)
Majhi, P.R., Blume, A.: Thermodynamic characterization of temperature-induced micellization and demicellization of detergents studied by differential scanning calorimetry. Langmuir 17(13), 3844–3851 (2001)
Vargas, C., Klingler, J., Keller, S.: Membrane partitioning and translocation studied by isothermal titration calorimetry. Methods Mol. Biol. 1033, 253–271 (2013)
Poncet-Legrand, C., et al.: Interactions between flavan-3-ols and poly(L-proline) studied by isothermal titration calorimetry: effect of the tannin structure. J. Agric. Food Chem. 55(22), 9235–9240 (2007)
Zheng, Y., et al.: Effect of pH on the complexation of kaempferol-4′-glucoside with three beta-cyclodextrin derivatives: isothermal titration calorimetry and spectroscopy study. J. Agric. Food Chem. 62(1), 244–250 (2014)
Baldoni, D., et al.: Performance of microcalorimetry for early detection of methicillin resistance in clinical isolates of Staphylococcus aureus. J. Clin. Microbiol. 47(3), 774–776 (2009)
Xi, L., et al.: Microcalorimetric study of Staphylococcus aureus growth affected by selenium compounds. Thermochim. Acta 387(1), 57–61 (2002)
Mariana, F., et al.: Isothermal titration calorimetry—a new method for the quantification of microbial degradation of trace pollutants. J. Microbiol. Methods 82(1), 42–48 (2010)
Hansen, L.D., et al.: Enzyme-catalyzed and binding reaction kinetics determined by titration calorimetry. Biochim. Biophys. Acta 1860(5), 957–966 (2016)
Transtrum, M.K., Hansen, L.D., Quinn, C.: Enzyme kinetics determined by single-injection isothermal titration calorimetry. Methods 76, 194–200 (2015)
Demarse, N.A., et al.: Determining enzyme kinetics via isothermal titration calorimetry. Methods Mol. Biol. 978, 21–30 (2013)
Todd, M.J., Gomez, J.: Enzyme kinetics determined using calorimetry: a general assay for enzyme activity? Anal. Biochem. 296(2), 179–187 (2001)
Hughes, J.P., et al.: Principles of early drug discovery. Br. J. Pharmacol. 162(6), 1239–1249 (2011)
Ruben, A.J., Kiso, Y., Freire, E.: Overcoming roadblocks in lead optimization: a thermodynamic perspective. Chem. Biol. Drug Des. 67(1), 2–4 (2006)
Torres, F.E., et al.: Higher throughput calorimetry: opportunities, approaches and challenges. Curr. Opin. Struct. Biol. 20(5), 598–605 (2010)
Zhou, X., Kini, R.M., Sivaraman, J.: Application of isothermal titration calorimetry and column chromatography for identification of biomolecular targets. Nat. Protoc. 6(2), 158–165 (2011)
Malmsten, M.: Soft drug delivery systems. Soft Matter 2(9), 760–769 (2006)
Gwinn, M.R., Vallyathan, V.: Nanoparticles: health effects—pros and cons. Environ. Health Perspect. 114(12), 1818–1825 (2006)
Cedervall, T., et al.: Detailed identification of plasma proteins adsorbed on copolymer nanoparticles. Angew. Chem. Int. Ed. Engl. 46(30), 5754–5756 (2007)
Saptarshi, S.R., Duschl, A., Lopata, A.L.: Interaction of nanoparticles with proteins: relation to bio-reactivity of the nanoparticle. J. Nanobiotechnol. 11, 26 (2013)
Mandal, S., et al.: Interaction of carbon nanoparticles to serum albumin: elucidation of the extent of perturbation of serum albumin conformations and thermodynamical parameters. J. Hazard. Mater. 248–249, 238–245 (2013)
Malmsten, M.: Inorganic nanomaterials as delivery systems for proteins, peptides, DNA, and siRNA. Curr. Opin. Colloid Interface Sci. 18(5), 468–480 (2013)
Cukalevski, R., et al.: Structural changes in apolipoproteins bound to nanoparticles. Langmuir 27(23), 14360–14369 (2011)
Cedervall, T., et al.: Understanding the nanoparticle–protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. 104(7), 2050–2055 (2007)
Fleischer, C.C., Payne, C.K.: Secondary structure of corona proteins determines the cell surface receptors used by nanoparticles. J. Phys. Chem. B 118(49), 14017–14026 (2014)
Eren, N.M., Narsimhan, G., Campanella, O.H.: Protein adsorption induced bridging flocculation: the dominant entropic pathway for nano-bio complexation. Nanoscale 8(6), 3326–3336 (2016)
Decher, G., Schlenoff, J.B.: Multilayer thin Films: Sequential Assembly Of Nanocomposite Materials. 2nd compl. rev. and enl. edn. Wiley, Weinheim (2012)
Dan, N.: The structure of DNA complexes with cationic liposomes-cylindrical or flat bilayers? Biochim. Biophys. Acta 1369(1), 34–38 (1998)
Golan, R., et al.: DNA toroids: stages in condensation. Biochemistry 38(42), 14069–14076 (1999)
Vilfan, I.D., et al.: Time study of DNA condensate morphology: implications regarding the nucleation, growth, and equilibrium populations of toroids and rods. Biochemistry 45(26), 8174–8183 (2006)
Perspicace, S., et al.: Isothermal titration calorimetry with micelles: Thermodynamics of inhibitor binding to carnitine palmitoyltransferase 2 membrane protein. FEBS Open Bio 3, 204–211 (2013)
Loh, W., Brinatti, C., Tam, K.C.: Use of isothermal titration calorimetry to study surfactant aggregation in colloidal systems. Biochim. Biophys. Acta 1860(5), 999–1016 (2016)
Marsh, D.: Thermodynamics of phospholipid self-assembly. Biophys. J. 102(5), 1079–1087 (2012)
Feng, X., Leduc, M., Pelton, R.: Polyelectrolyte complex characterization with isothermal titration calorimetry and colloid titration. Colloids Surf. A 317(1), 535–542 (2008)
Maurstad, G., Kitamura, S., Stokke, B.T.: Isothermal titration calorimetry study of the polyelectrolyte complexation of xanthan and chitosan samples of different degree of polymerization. Biopolymers 97(1), 1–10 (2012)
Lounis, F.M., et al.: Interactions between oppositely charged polyelectrolytes by isothermal titration calorimetry: effect of ionic strength and charge density. J. Phys. Chem. B 121(12), 2684–2694 (2017)
Manning, G.S.: Limiting laws and counterion condensation in polyelectrolyte solutions I. Colligative properties. J. Chem. Phys. 51(3), 924–933 (1969)
Sideratou, Z., et al.: Arginine end-functionalized poly(L-lysine) dendrigrafts for the stabilization and controlled release of insulin. J. Colloid Interface Sci. 351(2), 433–441 (2010)
Bulbake, U., et al.: Liposomal formulations in clinical use: an updated review. Pharmaceutics 9(2), 12 (2017)
Sercombe, L., et al.: Advances and challenges of liposome assisted drug delivery. Front Pharmacol 6, 286 (2015)
Ikonen, M., Murtomaki, L., Kontturi, K.: Microcalorimetric and zeta potential study on binding of drugs on liposomes. Colloids Surf B Biointerfaces 78(2), 275–282 (2010)
Al-Kaddah, S., et al.: Analysis of membrane interactions of antibiotic peptides using ITC and biosensor measurements. Biophys. Chem. 152(1), 145–152 (2010)
Allain, V., Bourgaux, C., Couvreur, P.: Self-assembled nucleolipids: from supramolecular structure to soft nucleic acid and drug delivery devices. Nucleic Acids Res. 40(5), 1891–1903 (2012)
Thanassoulas, A., et al.: From nucleobases to nucleolipids: an ITC approach on the thermodynamics of their interactions in aqueous solutions. J. Phys. Chem. B 118(24), 6570–6585 (2014)
Patwa, A., et al.: Tuning molecular interactions in lipid-oligonucleotides assemblies via locked nucleic acid (LNA)-based lipids. Org. Biomol. Chem. 11(41), 7108–7112 (2013)
Lipinski, C.A.: Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 44(1), 235–249 (2000)
Amidon, G.L., et al.: A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12(3), 413–420 (1995)
Gidwani, B., Vyas, A.: A comprehensive review on cyclodextrin-based carriers for delivery of chemotherapeutic cytotoxic anticancer drugs. Biomed. Res. Int. 2015, 198268 (2015)
Villiers, A.: Sur la fermentation de la fécule par l’action du ferment butyrique. Compt. Rend. Acad. Sci 112, 536–538 (1891)
Lipinski, C.A., et al.: Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46(1–3), 3–26 (2001)
Marttin, E., Verhoef, J.C., Merkus, F.W.: Efficacy, safety and mechanism of cyclodextrins as absorption enhancers in nasal delivery of peptide and protein drugs. J. Drug Target. 6(1), 17–36 (1998)
Connors, K.A.: The stability of cyclodextrin complexes in solution. Chem. Rev. 97(5), 1325–1358 (1997)
Segura-Sanchez, F., et al.: Elucidation of the complexation mechanism between (+)-usnic acid and cyclodextrins studied by isothermal titration calorimetry and phase-solubility diagram experiments. J. Mol. Recogn. 22(3), 232–241 (2009)
Mazzaferro, S., et al.: Bivalent sequential binding of docetaxel to methyl-beta-cyclodextrin. Int. J. Pharm. 416(1), 171–180 (2011)
Ignaczak, A., Palecz, B., Belica-Pacha, S.: Quantum chemical study and isothermal titration calorimetry of beta-cyclodextrin complexes with mianserin in aqueous solution. Org. Biomol. Chem. 15(5), 1209–1216 (2017)
Agnes, M., et al.: Designed positively charged cyclodextrin hosts with enhanced binding of penicillins as carriers for the delivery of antibiotics: the case of oxacillin. Int. J. Pharm. 531(2), 480–491 (2017)
Nguyen, H.H., et al.: Surface plasmon resonance: a versatile technique for biosensor applications. Sens. (Basel) 15(5), 10481–10510 (2015)
Piliarik, M., Vaisocherova, H., Homola, J.: Surface plasmon resonance biosensing. Methods Mol. Biol. 503, 65–88 (2009)
Tyszka, J.M., Fraser, S.E., Jacobs, R.E.: Magnetic resonance microscopy: recent advances and applications. Curr. Opin. Biotechnol. 16(1), 93–99 (2005)
Cala, O., Guilliere, F., Krimm, I.: NMR-based analysis of protein-ligand interactions. Anal. Bioanal. Chem. 406(4), 943–956 (2014)
Lebowitz, J., Lewis, M.S., Schuck, P.: Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11(9), 2067–2079 (2002)
Howlett, G.J., Minton, A.P., Rivas, G.: Analytical ultracentrifugation for the study of protein association and assembly. Curr. Opin. Chem. Biol. 10(5), 430–436 (2006)
Rossi, A.M., Taylor, C.W.: Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc. 6(3), 365–387 (2011)
Hall, M.D., et al.: Fluorescence polarization assays in high-throughput screening and drug discovery: a review. Methods Appl. Fluoresci. 4(2), 022001 (2016)
Heegaard, N.H., Nilsson, S., Guzman, N.A.: Affinity capillary electrophoresis: important application areas and some recent developments. J. Chromatogr. B Biomed. Sci. Appl. 715(1), 29–54 (1998)
Albishri, H.M., et al.: Recent advances in affinity capillary electrophoresis for binding studies. Bioanalysis 6(24), 3369–3392 (2014)
Hofstadler, S.A., Sannes-Lowery, K.A.: Applications of ESI-MS in drug discovery: interrogation of noncovalent complexes. Nat. Rev. Drug Discov. 5(7), 585–595 (2006)
Vivat Hannah, V., et al.: Native MS: an ‘ESI’ way to support structure- and fragment-based drug discovery. Future Med. Chem. 2(1), 35–50 (2010)
Freyer, M.W., Lewis, E.A.: Isothermal titration calorimetry: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 84, 79–113 (2008)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Thanassoulas, A., Nounesis, G. (2019). Isothermal Titration Calorimetry: A Powerful Tool for the Characterization of Molecular Interactions. In: Demetzos, C., Pippa, N. (eds) Thermodynamics and Biophysics of Biomedical Nanosystems. Series in BioEngineering. Springer, Singapore. https://doi.org/10.1007/978-981-13-0989-2_4
Download citation
DOI: https://doi.org/10.1007/978-981-13-0989-2_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-0988-5
Online ISBN: 978-981-13-0989-2
eBook Packages: EngineeringEngineering (R0)