
Abstract
Protein-protein interactions (PPIs) are important to every cellular process, from signaling pathways to protein post-translational modifications, as well as many cellular machines for complex biological functions. These interactions make up the so-called interactomics of the organism, and aberrant PPIs are the cause of multiple diseases, including aggregation-related Alzheimer’s disease and many types of cancers. Therefore, modulating the protein-protein interactions was considered as the precise way for pharmacological interventions. In this chapter, we first briefly introduced the fragment-based drug discovery and summarized the current consideration for constructing the fragment library and commonly adopted fragment screening methods. In the second part, we first classified the PPIs into four categories based on the interaction pattern and size of interfaces. Then we reviewed recent progress of utilizing the fragment-based drug discovery approach to develop antagonists of various interesting PPIs and organized these case studies into a biological function-oriented way.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Protein-protein interface
- Undruggable target
- Fragment-based drug discovery
- Fragment screening
- Apoptosis
- Ras signaling
- Protein post-translational modification
After the completion of human genome sequencing at the beginning of twenty-first century, post-genomic research and systems biology study revolutionized our view of the biology to an extraordinary complex level. The importance of the complex network of direct interactions between proteins—known as the interactome—to both biological systems and the development of disease states is widely recognized [1–3]. Despite its dominance in past drug development, simply targeting regulatory sites of enzymes and receptors is not enough to precisely module the biochemical network. Instead, an increasingly popular approach is to target protein-protein interactions (PPIs) that participate in cell signaling, growth, and survival. Although the exact number of human PPIs is unknown, estimates range from hundreds of thousands to around a million. Several databases have been created to aid the study of PPIs. STRING [4] is a database of predicted and known PPIs, TIMBAL [5] provides small-molecule inhibition data, and 2P2IDB [6], PICCOLO [7] and others provide structural information and analysis. Overall, PPIs provided a substantial amount of drug targets to treat the diseases in the selective and elegant way [8, 9].
However, modulation of PPIs for therapeutic intervention also posed significant challenges to medicinal chemists, as the protein-protein interfaces are generally flat and large (~2000 Å2), making the design of small molecule a daunting task. Therefore, many considered PPIs ‘undruggable.’ It has been recapitulated that the interfaces are predominantly hydrophobic with flanked polar residues, and the interfaces could be classified on the basis of complexity of the binding epitopes. A critical development in the understanding of PPIs was the realization that the interactions driving the affinity of a pair of proteins are not distributed evenly across their surfaces, and with the experiment called alanine scanning analysis some critical residues at the interface could be found to contribute a large portion of binding affinity [10]. Identifying these hot-spot residues is an initial step for designing antagonists of PPIs [11, 12]. Then different approaches can be applied to tackle the challenges of discovering lead compounds, which include peptide-mimics, high-throughput screening, computational virtual screening as well as fragment-based drug discovery.
1 Fragment-Based Drug Discovery
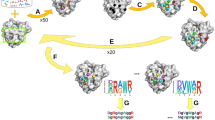
The invention of combinatorial chemistry in the late 1980s dramatically expanded the number of compounds in chemical collections, leading to the advent of high-throughput screening (HTS). This random screening approach is essential to the targets with no or little ligand information, and with serendipity it is perhaps the most venerable way to identify new ligands. In parallel to the development of HTS, computer graphics, macromolecular NMR technology and X-ray crystallography were integrated into drug design field to enable the application of structure-based drug discovery (SBDD) to accelerate the lead optimization phase and therefore speed up and lower the cost of the drug development. These two methods are complementary, as the HTS could be utilized for discovering the hit compounds and SBDD could be applied to the optimization of the identified hits. Although HTS remains an important method for hunting the bioactive compounds, the low efficiency and high cost associated with each HTS campaign hinder its usage. To combine the advantages of random screening and rational design, fragment-based drug discovery was proposed in later 1990s to early 2000s [13–16]. Rather than screening millions of compounds to find drug-sized starting points, fragment-based drug discovery takes a different approach to screen much smaller collections of low molecular weight compounds, which dramatically alleviates the burden on common HTS studies, such as compound collection, library maintenance, screening costs. The origins of FBDD can be traced back to the publication by Jencks [17], in which he proposed that ‘fragment’ can form high-quality interactions that can be optimized into highly potent larger molecules. In 1992, Verlinde et al. also mentioned using a link-fragment approach to discover lead compounds for trypanosomiasis [18]. However, the methodology was firmly established only until the studies at Abbott using ‘SAR by NMR’ [19] and at Astex using directed X-ray crystallography as screening method [20, 21]. Over two decades of development, fragment-based methods are now being used worldwide in many pharmaceutical companies and research laboratories as a very efficient tool for lead discovery. Practically, FBDD involved three components, namely fragment library, fragment screening and fragment optimization. In this chapter, we will first introduce the fragment-based drug discovery, then the case studies of applying FBDD in the development of PPI inhibitors. The overall process of developing PPIs, especially with the FBDD method, is illustrated in Fig. 6.1.

Overview of the current strategies of developing PPI inhibitors
1.1 Fragment Library
Constructing fragment library is the critical step for the successful application of FBDD, as the fragment hits are inevitably coming from the library. Reymond et al. [22] in silico enumerated all possible molecules up to 17 heavy atoms and found that sampling of the chemical space is much more efficient with fragments than with larger molecules. Their analysis indicated that adding one heavy atom roughly increases one order of magnitude to number of possible compounds. If the average fragment has 15 heavy atoms and the average optimized compounds have 28 heavy atoms, it means the chemical space of leads is at least 13 orders of magnitude larger than the chemical space of fragments. And in reality, most fragment libraries contain a few thousand molecules, which are about three orders of magnitude smaller than a common HTS collection. Besides of this sampling advantage, fragments have been considered more druggable in the view of bioactivity and pharmaceutical properties such as pharmacokinetics and toxicity. In 2001, Hann and colleagues [23] in GSK proposed a concept of ‘molecular complexity,’ a term measuring the molecular size and chemical features. They noted that, as molecules become more complex, they have more possible interactions with a protein target, both favorable and unfavorable interactions, which sometimes difficult to separate. Therefore, larger molecules will bind fewer targets and will be difficult to optimize. As a contrast, fragments have few but essential interactions, enabling them to interact with many protein targets and leaving them more direction to optimize the binding affinity as well as the drug-like properties.
To design a valuable fragment library, many have provided guidelines, and good reviews have been written on this topic [24–26]. Generally, most libraries consist of molecules that adhere to the ‘rule of three’: Compounds have a molecular weight below 300 Da, fewer than three hydrogen-bond donors and acceptors, less than three rotatable bonds and the value of ClogP smaller than 3 [27]. Other criteria including solubility and stability are also emphasized in many studies. Molecular weight is a primary property of fragments, as it usually limits other properties. There is a trend in recent years to even use smaller fragments, probably the MW around 200–250 Da. The solubility is critical to fragment screening. Because fragments have few interactions with the targets and showed low to moderate affinity with the IC50 range from 1 mM to 10 μM, the concentration used in these screenings will be high, which requires the fragments with good solubility in aqueous solution. This information can be easily obtained alongside the purity assessment using NMR spectroscopy.
Perhaps the most important issue related to library construction is to avoid compounds known as ‘bad actors,’ which include reactive covalent modifiers, chelators, nonspecific binders, and the compounds fond of aggregation. Baell et al. [28] have compiled a list of substructure to filter such pan assay interference compounds (PAINS), which can be used for excluding bad fragments. The reactive groups such as Michael receptors, alkyl halides or epoxides can be easily detected and removed from the library. Hush et al. also provided an NMR-based method—ALARM NMR—to rapidly and robustly detect reactive fragments [29].
Recently, the 3D features of fragments were considered as a new criterion for assessing the quality of library. Morley et al. argued that current fragment libraries suffered from the limitation of containing too many planar fragments that did not have the capability to bind to difficult targets [30]. They established ‘3D Fragment Consortium’ to enhance the 3D characteristics of fragment libraries and, therefore, to increase the probability of finding fragments interacting with complex binding sites, such as in PPIs. On the contrary, as implied by Hann’s investigation of molecular complexity, increasing 3D features of fragments will lower the hit rates of the library. Then one needs to balance between complexity, size, and diversity of the fragments to ensure the general utility of the fragment library.
1.2 Fragment Screening
Fragment hits make small number but high-quality interactions with the target, and typically, their binding affinities are in the range of 0.01–1 mM; while HTS assays often identify compounds with a stronger affinity (10 μM is a typical lower limit). Thus, the screening methodology used for fragment screening must be compatible with the smaller size, reduced complexity and consequently lower affinity of fragments, needing to provide 100- to 1000-fold higher sensitivity than in HTS. Currently, fragment screening generally utilizes sensitive biophysical technologies [13, 31], including NMR-ligand spectroscopy [32], surface plasmon resonance [33], fluorescence-based thermal shift [34], mass spectrometry [35], NMR-protein spectroscopy [36], and X-ray crystallography [37]. These screening methods are usually used in cascade way: applying the high-throughput methods first and following with more time-consuming techniques to validate the hits.
Thermal shift (TS) Fluorescence-based thermal shift probably is the quickest method for fragment screening [34], as it can be set up in the plate-based format. This technique detects the compounds that increase the melting temperature (Tm) of the target protein by monitoring the unfolding process via a hydrophobic-sensitive fluorescence dye. Because fragment binding is weak, the shift of the melting temperature (ΔTm) is small, usually in the range of 0.5–1.5 °C. And the size of the protein is the important factor of the temperature shift, being the larger the protein is, usually the smaller the change in temperature. Giving the thermal shift experiments are not always reproducible, screening with this method should be seen as the enrichment process. And some more accurate methods, e.g., NMR screen, should be used to assess the fragment hits from TS experiment. Murry et al. [38] at Astex performed a fragment-based screen against DDR1, a unique receptor tyrosine kinase activated by extracellular collagen. Approximately 1500 compounds from the Astex fragment library were screened against DDR1 using a protein thermal shift assay. Then hits from this assay were progressed with crystal soaking to determine the X-ray structures. A unique fragment containing a urea moiety was selected for optimization, as it binds to the back pocket rather than the typical hinge part of ATP site. Finally, a potent and selective DDR1/2 inhibitor was discovered with IC50 value about 3 nM. Laesson and his coworkers used a fragment-based ligand design strategy to find the inhibitors targeting TNKS2 [39]. Initially, a thermal shift assay-based fragment screen method called differential scanning fluorimetry (DSF) was applied to the hit characterization from a 500-compound fragment library. Two compounds gave melting curves distinct from that of the DMSO control, stabilizing TNKS2 and one compound was chosen for the further modification by analyzing the crystal structures of the fragments. As a result, cycles of optimization led a set of compounds with high affinities (IC50 values in the low nanomolar range) and with other favorable properties including good solubility, high PARP-selectivity, and high ligand efficiency.
Surface Plasmon Resonance (SPR) Surface plasmon resonance is the resonant oscillation occurring when polarized light strikes an electrically conducting surface at the interface between two media. This generates electron charge density waves called plasmons, which could reduce the intensity of reflected light at a specific angle (known as the resonance angle) in proportion to the mass on a sensor surface. In SPR fragment screening, usually the target protein is covalently linked to the gold surface of an SPR biosensor chip, and solutions of fragments are sequentially passed over it. If the fragments bind to the immobilized target, the increase in the surface mass is detected with the change of the resonance angle in real time. Since the SPR experiment can measure the time-dependent fragment association–dissociation response, the binding kinetics can be obtained as well as the binding affinity [33, 40, 41]. Mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) is a serine/threonine kinase implicated in the regulation of many biological processes. Crawford conducted a screen of diverse fragment library using SPR technique to identify a suitable hit for MAP4K4 [42]. Detailedly, a screening of 2361 fragments at 100 μM as singletons using SPR led to 225 confirmed hits with LE > 0.35. They followed up on multiple fragment hits initially and settled on two fragments (pyrrolotriazine and oxazole series) for full optimization. By utilizing crystal structures, molecular modeling, and scaffold hopping methods, compound G495 with excellent potency, kinase selectivity, and mouse PK profile was discovered. Navratilova et al. [43] used a novel SPR protocol as the primary fragment screening method to discover novel bromodomain scaffolds by overcome the presence of DMSO interference. They followed with X-ray crystallography to obtain the binding modes of fragments, providing clear clues about how to aid the development of potent and selective inhibitors of PCAF, CREBBP, and BRD4.
Mass Spectrometry (MS) The advances in mass spectrometry techniques enable it to be a complementary fragment screening method. Protein-fragment mixtures are ionized by soft electrospray ionization (ESI), and the binding can be observed by the increase in the mass of the bimolecular ion corresponding to that of a bound fragment. In principle, it is possible to assay the cocktails of fragments, but due to the soft ionization techniques necessary to see non-covalent binding, it is difficult to detect fragment binding directly with MS techniques.
Nevertheless, MS offers a rapid, sensitive, and high-throughput method for fragment screening. And the amount of protein needed depends on its intrinsic ionization efficiency, the type, and concentration of buffer used and the instrumental conditions. Heat shock protein 90 (HSP90) is a molecular chaperone that plays important functional roles in cells and was considered as an anticancer drug target. Fogliatto and coworkers used ESI-MS to investigate the interaction of ligands to the HSP90 [44]. A set of chemically divergent compounds, with a broad range of dissociation constants from 40 pM to 100 μM, were tested to access the reliability of ESI-MS for the study of protein/ligand complexes. A good agreement was found between the values measured with a fluorescence polarization displacement assay and those determined by mass spectrometry. Poulsen et al. use three fragment screening methods (ESI-MS, X-ray crystallography, and SPR) to demonstrate that there is a tremendous opportunity to apply native state mass spectrometry as a complementary fragment screening method to accelerate drug discovery [45]. Specifically, the study was conducted in two stages: Initial screening against a 720-member fragment library was performed by SPR; then the seven identified hits were confirmed by native MS and X-ray crystallography. The screening results showed that three screening methods are in excellent agreement.
NMR Spectroscopy Based on the object of NMR spectra in fragment screening, it can be classified as ligand-detected one-dimensional (1D) or protein-detected two-dimensional (2D) NMR. Ligand-detected NMR methods, including Carr–Purcell–Meiboom–Gill (CPMG), saturation transfer difference (STD), water-ligand-observed via gradient spectroscopy (waterLOGSY), are frequently used in fragment screening. CPMG experiment measures the relaxation dispersion parameter of fragment, and when the fragment bound to the slow tumbling protein, the intensity of proton signals of the fragment will decrease. In the waterLOGSY and STD experiments, an irradiation pulse is applied at either the resonance frequency of bulk water or the specific protons in protein targets. Then due to the transfer of magnetization via the binding of fragment to the target, the resonance signals of binding will be different with the original non-binding signals, which could be used to obtain the fragment hits. The acquisition of 1D spectra is relatively fast (less than 15 min), and the experiments also can apply to the ‘cocktail’ solution—mixing several fragments in one stock solution, making these ligand-detected NMR methods ideal for fragment screening.
Since proposed by scientists in Abbott, 2D bimolecular-detected 1H-15N HSQC experiment was commonly used for fragment screening [19]. This experiment monitors the difference of chemical shifts in the 1H-15N cross-peaks of the 15N-labeled target protein during the process of adding fragments. Although the method is time-consuming, it can provide information about the binding site of fragment/target complex.
The BET family of bromodomain-containing proteins have been potential targets for blocking proliferation in a variety of cancer cell lines. Wang et al. [46] initiated a protein-based NMR-fragment screen carried out against the second bromodomain of BRD4 and found that a phenylpyridazinone fragment showed a strong spectral shift in the NMR screen is a novel small molecule not previously reported. This weak binding fragment was then elaborated by medicinal chemistry efforts and X-ray structure-based design. Several analogs exhibited single-digit nanomolar potency in both biochemical and cell assays and showed high exposures in PK studies and significant tumor growth inhibition efficacy.
Activated factor XI (FXIa) inhibitors are anticipated to combine anticoagulant and profibrinolytic effects with a low bleeding risk. Linda Öster and his coworkers [47] used ligand-detected 1D NMR spectroscopy as the primary fragment screening tool to identify 50 neutral or weakly basic fragment hits from 1800 structures that are selected from the docking study on 65,000 fragments. Finally, two prioritized fragment hits as novel FXIa fragments were chosen for the further study. Sequent work combining the X-ray crystallography and structure-guided linking and expansion work derived a potent and selective FIXa inhibitor with IC50 of 1.0 nM.
X-ray Crystallography X-ray crystallography was heavily used in structure-based drug discovery, as it can provide the details of binding interactions. Along the development of facilities and software in crystallography, the X-ray crystallographic screening gets popular in nowadays because it can simultaneously validate the fragment hits and give the binding information for later optimization [37, 48–50]. To speed up the screening, the cocktails of fragments can be soaked into the crystallized apo-protein. To achieve this, the protein crystal must be robust and have a solvent-exposed binding site in the crystal lattice. For other difficult cases, the co-crystallization with cocktails of fragments will be the better option.
Screening of fragment libraries using X-ray crystallography has been an effective method to sample chemical space, to reveal previously unobserved pockets and to illustrate ligand binding modes. Jhoti et al. reported on the successful application of fragment screening using X-ray crystallography for identifying secondary, allosteric, sites for three separate targets (the viral protease-helicase protein HCV NS3 [51], the p38 MAP kinase [52], and human soluble adenylate cyclase [53]). In 2015, he also reported their analyses of in-house data of 24 previous fragment-based drug discovery campaigns against a wide variety of protein targets, indicating that the majority proteins contain secondary binding sites that could bind with fragments [54]. This provided strong implications of applying FBDD method to generate new chemical tools probing unexploited biological mechanisms.
1.3 Fragment Optimization
Before investing time and resources to elaborate fragments, we should select the optimal fragment based on the potency and synthetic tractability of the fragment. Besides, the optimal fragment may require minor chemical modification to determine the scaffold.
In the guidance of structural binding information and quantitative affinity data, validated fragment hits are elaborated to improve potency in an iterative process of rational design and chemical synthesis. Generally, there are three main approaches to increasing the potency of compounds derived from fragments: fragment merging, linking, and growing (Fig. 6.2).

Three methods of fragment optimization
Fragment merging is to incorporate structural portions of other overlapping molecules into a fragment, based on superimposed complexes of the same target bound with other fragments, substrates, and known ligands. Structure–activity relationship from different series can be used to identify important interactions and pharmacophores, and this information can be used to produce a hybrid series. Nikiforov et al. applied such a fragment merging strategy to develop inhibitors against EthR [55], a novel target in tuberculosis. Initially, multiple techniques were used to screen a 1250-member fragment library. Then, two partially overlapped fragments were optimized with merging approach, giving eightfold increase in binding affinity.
Fragment linking is conceptually the most appealing strategy for fragment optimization. It requires the efficient joining of two fragments that are known to bind at non-overlapping sites. In theory, a compound derived from linking fragments with an ideal linker is expected to have a Gibbs free energy of binding that is better than the sum of the individual fragment binding energies, and such favorable contribution mainly attributes to the entropic factor. Borsi et al. investigated this by using ITC experiments to measure the thermodynamic parameters of two fragments and their linking molecule on MMP-12 protein [56]. Although the binding free energy of two fragments is only −3.01 and −3.85 kcal/mol, the linked inhibitor showed the binding free energy −10.50 kcal/mol. And the difference in entropic contribution is −4.3 kcal/mol, which totally accounting for the improvement in binding. An ideal linker not only allows the linked fragments to recapitulate the conformation of the individual fragments and add the favorable entropic contribution, but also may make additional favorable interactions with the protein and provide little additional enthalpic energy. However, in practice, fragment linking can be challenging, because slight length or geometric deficiencies in the linker can have a dramatically negative effect on binding. Moreover, linked compounds are often larger than ideal.
More frequently, a fragment that binds at a single site is discovered and gradually ‘grown’ through adding chemical groups to explore further interactions. The choice of which direction of optimization to pursue will be influenced not only by potency but also by ligand efficiency that is defined simply as the free energy of binding of a ligand divided by the number of non-hydrogen atoms it has (ligand efficiency LE = ΔG/Nheavy_atoms). In general, optimized ligands with a ligand efficiency of more than 0.3 kcal/mol/atom is considered as promising as it maintains the binding without making fat molecules, which leaves large room for medicinal chemists to simultaneously optimize the potency and the druggability. In discovering inhibitors of acetylcholine-binding protein, Edink et al. reported a study of optimization with fragment growing method [57]. After fragment screening, a fragment with good LE (0.43 kcal/mol/atom) was co-crystallized with Ac-AchBP, a representative acetylcholine-binding protein. Based on the crystal structure of the complex, they modified the fragment by gradually adding ethylbenzene motif and increased the binding affinity up to 150-fold. Pin1 is an emerging oncology target related in Ras and ErbB2-mediated tumorigenesis. Potter et al. from Vernalis use structure-based method to evolve a 5-pyridinyl pyrazole-3-carboxylate fragment into a series of 5-aryl-carbamoyl-3-phenyl-imidazole-4-carboxylates and improve the activity against Pin1 from IC50 about 360 μM of the initial fragment to 0.52 μM of the best-optimized inhibitor [58].
2 FBDD in PPI
Protein-protein interactions are central to all biological processes and are often dysregulated in disease, therefore providing a vast class of therapeutic targets for drug discovery. The last two decades has seen amazing progress in tackling challenging PPI targets with small molecules. Extensive studies have revealed that, although PPIs have many shapes and sizes, most of the inhibitors target PPIs at the essential small binding pockets termed as hot spot. In the mid-1990s, Dagmar Ringe proposed a method of multiple solvent crystal structure determination (MSCS), which is to soak small organic solvent molecules into protein crystals to map out their binding sites experimentally [59]. Similarly, Vajda et al. implemented a computational method called FTMAP to identify the hot spots [60, 61]. In order to design small molecules that interfere with CDk9–cyclin T1 interactions, Randjelovic and colleagues applied the FTMAP to find the low energy binding site and discovered a series of 2-amino-8-hydroxyquinoline inhibitors of CDK9–cyclin T1 interactions [62]. Based on the characteristics of PPI interfaces, Arkin et al. [63] classified the PPIs into four groups, and the classes as well as some examples are listed in Table 6.1. This classification provides insights for designing the small molecular drugs targeting the PPIs. Briefly, the PPI from class I usually is difficult to find potent inhibitors and the interface tends to be much more dynamic than PPIs from other classes. While for PPIs of class II, design approaches have focused on both the binding pocket and the partner peptide. Mapping the hot spot and then focusing on the spot to design small molecules have proved the success with several clinical testing drugs. But the most prevailing drug development of PPIs is coming from class III and class IV, which largely attributes to containing a primary hot spot at the interface. The fragments or pharmacophores bound to this hot spot can dramatically low the binding energy and eventually lead to nanomolar inhibitors. The fragment-based drug discovery has played an important role in the design and developing these drugs, such as ABT199, a drug targeting BCL-2 has been reached to the market for cancer treatment, and several BET bromodomain inhibitors that have entered into clinical trials for various diseases. These successful case studies highlight the importance of FBDD in PPIs.
2.1 FBDD Application
Giving the importance of PPIs in drug discovery and prevalence of FBDD, researchers worldwide, either in big pharmaceutical companies or in small research laboratories, utilize this high efficient technique to find the small molecules blocking the PPIs. In this chapter, we cannot provide the throughout review for enormous applications in this field. Instead, we will divide the examples into several groups according to the biological function of targets, which are most appealing or known to the authors. We hope this particular perspective can accelerate readers to grasp the advance in this territory, and we encourage the readers to survey other journal articles for more case studies.
2.1.1 Targeting PPIs in Apoptosis Process
Apoptosis, also known as type I programmed cell death, is a naturally occurring process that is crucial for tissue homeostasis. As estimated, between 50 and 70 billion cells die each day due to apoptosis in the average human adult. Because apoptosis is so important, it is a highly regulated process. There are at least two broad pathways that lead to apoptosis, an extrinsic pathway and an intrinsic pathway. The molecular mechanisms underlying the intrinsic and extrinsic apoptotic pathways have been extensively investigated and found that the extrinsic pathway is activated in response to the binding of death-inducing ligands to cell-surface death receptors and cell-intrinsic apoptotic stimuli include DNA damage, growth factor deprivation and oxidative stress. Defective apoptosis processes have been implicated in a wide variety of diseases, such as cancer, a phenomenon resulting from an insufficient amount of apoptosis [64, 65]. In the efforts to modulate the dysregulated apoptosis process, three types of protein-protein interactions were identified as essential components in the signaling pathway for designing small molecular drugs (Fig. 6.3), which involves BCL-2 family proteins [66, 67], IAP proteins [68], and p53–Mdm2 interaction [69].

Three important PPIs in the apoptosis signaling pathway
Bcl-2 and Bcl-X L Bcl-2 is located to the out membrane of mitochondria, where it functions as anti-apoptotic factor, and plays an important role in promoting the cellular survival by interacting with pro-apoptosis proteins, such as BAX and BAK. Upregulation of Bcl-2 and its relatives Bcl-XL and Mcl-1 is an important mechanism through which cancer cells avoid cell death in the face of signal dysregulation, radiation, and chemotherapy. Inspecting the structures of Bcl-XL bound with BAX peptide or with BAK peptide elucidated that the binding site of Bcl-XL is an extended hydrophobic groove, of ~20 Å in length, interacts with a critical alpha helix in BH3 domain of the pro-apoptosis proteins. In mid-2000s, Oltersdorf and his coworkers [70] applied a high-throughput NMR-based method called ‘SAR by NMR’—a technology based on the linkage of proximal fragments to achieve high-affinity binding—to screen a chemical library to identify small molecules that bind to the hydrophobic BH3-binding groove of Bcl-XL. Initially, two compounds 6-1 and 6-2 were discovered that bind to distinct but proximal subsites within this cleft (Fig. 6.4a), with affinities of approximately 0.3 mM and 4.0 mM, respectively. Substitution of an acyl-sulfonamide for biphenyl carboxyl group maintained the correct position in site 1 while providing an optimal trajectory to another site. Site-directed parallel synthesis led to compound 6-3 (Ki = 36 nM to Bcl-XL) in which 3-nitro-4-(2-phenylthioethyl) aminophenyl group spans the binding sites and efficiently occupies site 2 (Fig. 6.4b). Later study showed that compound 6-3 tightly bound to domain III of human serum albumin (HSA), which dramatically reduced the binding affinity in vivo study. To reduce binding to HSA, the NMR structure of the thioethylamino-2,4-dimethylphenyl analog of compound 1 complexed with domain III of HSA was compared to the structure of compound 6-3 bound to Bcl-XL and revealed that portions of the ligand at the solvent-exposed part in Bcl-XL can be modified with polar substituents to achieve the selectivity. And their efforts finally resulted ABT-737 that binds with high affinity (Ki ≤ 1 nM) to Bcl-XL and Bcl-2 (Fig. 6.4c). The subsequent optimization aimed to overcome the poor oral absorption created the second-generation analog, navitoclax [67]. However, concomitant on-target thrombocytopenia caused by Bcl-XL inhibition limits the efficacy achievable with navitoclax. Subsequent efforts led to the development of the first highly selective inhibitor of Bcl-2, venetoclax, approved in 2016 for the treatment of patients with chronic lymphocytic leukemia (CLL).

The development of inhibitors of Bcl-2 and Bcl-XL. a Two fragments in two distinct sites of Bcl-XL (PDB code: 1YSG); b co-crystal structure of compound 6-1 bound to Bcl-XL (PDB code: 1YSI); c the development route to marketed drug Venetoclax
Mcl-1 Mcl-1 is a related protein in Bcl-2 family, which is also upregulated in many cancer cells. But Mcl-1 is distinct from Bcl-2 and Bcl-XL as it binds to different BH3 peptides. Friberg et al. described the discovery of potent and selective Mcl-1 inhibitors using fragment-based method [71]. Firstly, 132 hits (93 inhibited Mcl-1 with Ki < 500 μM) was resulted from the screening of their fragment library (>138,000 compounds) by recording SOFAST 1H-15N HMQC spectra of Mcl-1 incubated with mixtures of 12 fragments. On the basis of the affinity and distinct chemical characteristics, two classes of compounds (6-4 and 6-5, Fig. 6.5) were selected for follow-up experiments. Secondly, to determine how the two class of fragments bind to Mcl-1, they performed NMR-based structural studies on Mcl-1/fragment hit complexes. Using double-labeled (15N, 13C) Mcl-1 protein, they acquired NOE-derived distance restraints and used these to dock representative fragments into a previously determined Mcl-1/Bim BH3 peptide complex. The fragments of the two classes exhibited different patterns of protein-ligand NOEs, confirming that they bind to different regions of Mcl-1. Thirdly, they found the attachment of class II aromatic groups with a two- to four-atom linker to the 3-position of the class I 6,5-fused heterocycles would lead to merged compounds that could maintain the favorable hydrophobic contacts of both fragments to Mcl-1 as well as the interaction between the common carboxylic acid and R263 of Mcl-1. With these efforts, they discovered that the four-atom linked compounds as compound 6-6 yielded the most potent Mcl-1 inhibitors in the series, displaying submicromolar dissociation constants. Moreover, these compounds exhibited selectivity for Mcl-1 over Bcl-xL and Bcl-2.

The development of Mcl-1 inhibitor 6-6. Co-crystal structure of compound 6-6 bound to Mcl-1 (PDB code: 4HW3) was depicted as stick and surface model
Sun’s group described using the FBDD method to accelerate the discovery of a novel Mcl-1 inhibitor from two distinct structural classes [72]. An initial NMR-based fragment screen was conducted by employing the protein-based NMR-screening method. In this method, recombinant human Mcl-1 protein labeled with 13C at the methyl groups of isoleucine, leucine, valine, and methionine was expressed in Escherichia coli and purified. By monitoring the changes in the 13C-HSQC spectrum of this methyl-labeled protein upon addition of approximately 17,000 fragments, they found that Mcl-1 is amenable to small-molecule inhibitions demonstrated by a high hit rate of the screening. In this screening method, two compounds (the aryl sulfonamide and salicylic acid as compounds 6-7 and 6-8, shown in Fig. 6.6) were chosen for the further study. Due to the unavailability of co-crystals of Mcl-1 with the fragment hit, NOE-driven docking of the fragment onto the crystal structure of human Mcl-1 was adopted to illustrate the binding mode of the compounds. Based on the NMR-derived model, the optimization of 6-7 and 6-8 fragments yielded compound 6-9 with an IC50 of 30 nM against Mcl-1.

The development of Mcl-1 inhibitor 6-9. Representative binding conformation of aryl sulfonamides (PDB code: 4OQ5)
IAPs-SMAC Inhibitor of apoptosis proteins (IAPs), including cIAP1, cIAP2, and XIAP, are important regulators in the apoptosis. They bind to caspases to prevent the activation of apoptosis and often overexpressed in cancer cells. An endogenous inhibitor of IAPs, called second mitochondrial activator of caspases (Smac), competes with caspase binding to BIR domains of IAPs and thereby stimulates apoptosis. Since Smac peptide motif, Ala-Val-Pro-Ile (AVPI), binds tightly to two adjacent subpockets on BIR domains of XIAP, it provides a template for designing small molecules to mimic the binding features of the tetra-peptide.
Pellecchia and his coworkers [73] reported on the utility of the NMR-based approach by deriving novel and effective SMAC mimetics targeting the anti-apoptotic protein XIAP. Initially, they designed a virtual library of L-alanine derivatives coupled with 578 primary and 815 secondary, low molecular weight, amines. Fifteeen potential fragments predicted by molecular docking were further checked experimentally by protein-observed NMR for their ability to bind to the Bir3 domain of XIAP. By comparing the differences of chemical shift perturbations on Bir3 in the presence of the selected putative SMAC mimics, compound 6-10 stands out as a weak binder (Kd = 200 μM) for the Bir3 domain (Fig. 6.7). Based on the crystal structure, it was predicted that, if compound 1 was modified at position 2 of the 4-phenoxybenzene scaffold, it may provide more interactions with the P2 subpocket, similar to the isoleucine residue of AVPI. Then a second virtual library of derivatives of compound 6-11 (about 900 compounds) was designed, and compound 5 the out from top scoring in the docking study were synthesized and tested by the NMR method with 15 N labeled Bir3. Finally, compound 6-12, an analog of 6-11, was discovered to be the tightest binding affinity among the tested compounds with a Kd value of 1.2 μM to Bir3 domain of XIAP.

The development of XIAP inhibitor 6-12
In 2015, Chessari et al. at Astex, using fragment-based drug discovery method, discovered a non-alanine lead series with dual activity against clAP1 and XIAP [74]. Initial screen with ligand-detected NMR methods, such as LOGSY or STD-NMR, were found to be relatively insensitive for XIAP-BIR3 due to the small size of the protein (11.8 kDa). Therefore, protein 1H NMR was used as a primary screen to detect fragment binding. A protein concentration of 200 µM was used in the protein 1H-NMR spectrum and the chemical shifts and line widths of XIAP-BIR3 1H signals was monitored with threshold δ < 0.4 ppm and the range δ 9.8–10.4 ppm (the ranges were expected to be sensitive to fragments binding in the canonical AVPI pocket). In the second step, an X-ray-based fragment screen of XIAP-BIR3 was applied to the identified fragment hits by soaking crystals of XIAP-BIR3 (residues 250–354) with the fragments for 24–72 h either as singletons (at 50–100 mM fragment, 5–10% DMSO) or doublets (2 × 50 mM, 10% DMSO).Screened from the 1151 fragments with relatively high LE for both XIAP and cIAP1, four fragment hits contained the small primary or secondary aliphatic amines were confirmed, which is consistent with the known binding preference of N-terminal alanine-containing peptides. One compound (6-13) of them was chosen as a starting hit and optimized into a potent non-alanine IAP antagonist (6-14), which is structurally distinct from all IAP antagonists reported previously (Fig. 6.8).

The development of XIAP inhibitor 6-14. a Fragment 6-13 bound to XIAP (PDB: 5C3H); b structure of compound 6-14 bound to XIAP (PDB code: 5C83)
p53-Mdm2 The transcription factor p53 is a master tumor suppressor that regulates cell-fate decisions such as senescence, cell-cycle arrest, and apoptosis. Mutations in p53, resulting in reduction or loss of p53 function, are presented in ~ 50% of human cancers. In other tumors, the p53 pathway is inactivated by upregulation of p53 inhibitors, such as the mouse double minute proteins (Mdm2 and MdmX), or by downregulation of p53 cooperators, such as ARF. In the past decades, extensive efforts of biochemical, structural studies have provided clear picture about function of the p53–Mdm2 interactions. Mdm2 functions as an inhibitor of the N-terminal transactivation domain (TAD) of p53 and promotes p53 degradation through the ubiquitin–proteasome system (E3 ligase activity). On the other hand, MdmX can downregulate p53 by inhibition of the TAD domain and it can upregulate Mdm2. Then the use of Mdm2 antagonists in cancer cells expressing wild-type (WT) p53 should activate p53, resulting in effective antitumor activity. Scrutiny of the Mdm2/p53 complex shows that the p53–Mdm2 interaction can be minimized to an α helix from the TAD domain of p53 that binds into a pocket on the surface of the N-terminal domain of Mdm2 and therefore is druggable by small molecules based on a buried surface area of ~700 Å2. At the interface, the interacted helix of TAD domain is only two turns and contains three critical hot-spot residues (Phe19, Trp23, and Leu26) pointed toward a deep pocket in the center of the peptide-binding groove of Mdm2 [75].
To dissect the interaction features of the binding site of p53-Mdm2, Fry et al. [76] adopted a fragment-based approach to screen a small focused library generated by deconstructing a potent inhibitor RG7112 into 10 small pieces (compounds 6-15 to 6-24) (Fig. 6.9). The results show that the fragment containing three substituents on the core structure, in any combination, is capable of binding to Mdm2. X-ray structures of compounds 6-23 and 6-24 bound to Mdm2 showed that these fragments attain a position as expected based on the RG7112 binding paradigm, exhibiting the same orientation and utilizing the same subpocket-filling strategy as the parent compound. The binding of fragments that retained only two of the four attached groups was more varied, indicating the combination of attached groups have a dramatic influence on activity, causing affinities to range from an undetectable level to a Kd of 26 μM (Fig. 6.9). This study supports the notion that p53–Mdm2 interaction systems should be highly amenable to a fragment-based lead discovery approach, although these systems will likely require some specialized choice of library composition, as exemplified by a report of Boltjes et al. on the library generation for the discovery of p53-Mdm4 inhibitor [77].

The deconstruction of RG7112 and result of binding assay
2.1.2 Targeting PPIs in Ras Signaling Pathway
Mutational activation of the RAS oncogene products (HRAS, NRAS, and KRAS) is a frequent event in human cancers. Ras belongs to the family of small GTPases and functions as molecular switches in controlling of extracellular growth signal transduction in many vital cellular processes such as cell differentiation, proliferation, and survival [78–80]. By conformational transformation from an inactive GDP-bound to an active GTP-bound state, RAS proteins relay the signals from extracellular kinase receptors and interact with various downstream effectors such as Raf, PI3K, and Ral-GDS, therefore triggering further cellular activities (Fig. 6.10). After the discovery of activated RAS genes in cancers in 1982, targeting the mutated RAS remains as a promising anticancer strategy for more than three decades. However, mutated Ras has proven to be an extremely difficult target for drug development. Ras proteins bind GDP and GTP with high affinity in pM range. Moreover, the cellular concentrations of GDP and GTP are at μM level, which further hinder the pharmacological modulation through the development of GDP/GTP competitive inhibitors for Ras. The activities of Ras proteins are regulated by guanine nucleotide exchange factors (GEFs, e.g., SOS1) and GTPase-activating proteins (GAPs, e.g., neurofibromin) [81]. Ras signaling strongly depends on the intracellular localization of Ras at the plasma membrane [82]. After post-translational modified at the C-terminal hypervariable region of Ras protein at endomembrane, Ras needs shuttling factor PDEδ to bind the farnesyl moiety and dissociate it from endomembranes, therefore enhancing its diffusion throughout the cell. With the help of G-protein Arl2, the Ras protein released from PDEδ and relocalized to the plasma membrane through the electrostatic interaction and hydrophobic interaction.

Simplified Ras signaling pathway
SOS1-Ras In order to identify and characterize small-molecule binders to KRas, Maurer and Wang [83] carried out a fragment-based lead discovery campaign. Initially, a ligand-detected NMR screen identified 266 fragments from a library of 3285 diverse compounds. Then protein-detected NMR using isotopically labeled KRas protein was applied for hit validation and binding site characterization. A consensus site of fragment binding was revealed and confirmed by X-ray crystallography study on KRas protein bound with several hits. The consensus site comprises a shallow hydrophobic pocket, which was expanded upon the fragment binding. The binding site is proximal to the protein-protein interface, especially at the SOS1 binding pocket.
In 2012, Maurer et al. [84] reported their effort for the discovery of a small-molecule blocking Ras–SOS interactions. They applied the NMR-based saturation transfer difference (STD) assay to screen a 3000-compound library (using pools of up to six fragments each), resulting in 240 primary hits. These hits were further validated by 2D 1H15N HSQC NMR method. By comparing the HSQC spectra, 25 compounds produced chemical shift perturbations that can be mapped to a contiguous site on the KRasm structure and were thus classified as confirmed hits. The complexes of KRasm with benzamidine (BZDN), benzimidazole (BZIM), or 4,6-dichloro-2-methyl-3-aminoethyl-indole (DCAI) were obtained by soaking the confirmed fragment hits into the KRasm crystals, indicating they all bind to a similar site surrounded by residues K5, L6, V7, I55, L56, and T74 (Fig. 6.11a). Detailed investigation showed that DCAI, binding at RAS-SOS interface, inhibits SOS-mediated nucleotide exchange and release for Ras with IC50 of 342 and 155 μM, respectively.

Fragment hits of RAS-SOS. a Four fragments identified by Maurer et al. [84]; b crystal structure of compound 6-25 bound to SOS (PDB code: 4US2)
Another attempt to discover small molecules binding to Ras/SOS complex was reported by Winter and coworkers in 2015 [85]. A library of 1160 fragments organized into four-compound cocktails was screened with X-ray crystallography method. Three interesting fragments bound to distinct sites on the complex were elucidated from co-crystal structures, but none of these compounds were sufficiently potent to show functional activity in the Ras–Raf HTRF assay. Simple modifications on these fragment hits were unsuccessful to improve the activity. Inspecting the complex found a nearby cysteine residue (Cys118) which could be utilized for designing covalent binding molecules. Therefore, a library of 400 compounds containing potentially reactive functional groups was screened, finding the reactive N-ethylmaleimide (NEM) group as an ideal electrophilic ‘warhead’ to bind to Cys118. Further optimization resulted an irreversible inhibitor 6-25 (shown in Fig. 6.11b) that shows a time-dependent inhibitory activity against both wild-type and mutant KRas, opening the way to a new strategy to target aberrant Ras signaling by intervening in the SOS-mediated activation of Ras.
PDEδ-Ras Despite downstream interactions with GEF, GAP proteins, as mentioned before, Ras protein needs transfer to the plasma membrane to execute its oncogenic activity. The shuttle protein PDEδ was found to act as a solubilizing factor that facilitates the transit of RAS proteins to either the Golgi or the recycling endosomes to the plasma membrane. Waldmann’ group carried out drug discovery based on this RAS–PDEδ interactions, and so far, obtained three chemotypes of PDEδ inhibitors: deltarasin, deltazinone, and deltasonamide [86–88]. Different from their method of high-throughput screening, we applied fragment-based drug discovery to identify several fragment hits from ligand-observed STD and CPMG NMR screening (unpublished work). By solving the co-crystal structure of PDEδ bound with a tetrazole fragment 6-26, the binding mode was revealed as shown below, which supports our structure-based lead optimization and finally obtains a potent PDEδ inhibitor compound 6-27 with IC50 value about 27 nM in the FP assay (Fig. 6.12).

Fragment hit and optimized inhibitor of RAS–PDEδ interactions (unpublished work)
2.1.3 Targeting PPIs Related Histone Post-translational Modifications
Epigenetic regulation of gene expression is currently the focus of intensive research in post-genomic era. At the molecular level, epigenetic regulation involves dynamic and reversible modification of DNA and the proteins that package DNA. Histones, the core proteins within chromatin structure, are subjected to a range of post-translational modifications (PTMs), mainly including acetylation, methylation, phosphorylation, and ubiquitination (Fig. 6.13). The combinatorial characterized covalent modification on the histone tails is coined as ‘histone code,’ which is believed to be key to understanding the gene expression pattern and many heritable changes in phenotype that are not encoded in the underlying DNA sequences. This histone code hypothesis has led to the presumption that there must be protein families for specifically adding, removing, and recognizing the PTM marks. In recent years, all three types of proteins have been identified for histones. Taking acetylation as an example, histone acetyltransferases (HATs) act as a writer to transfer the acetyl group from acetyl CoA to form an ε-N-acetyllysine, whereas histone deacetylases (HDACs) work as an eraser to remove the acetyl group from the histone tail. The proteins containing bromodomains could bind to the acetylated lysine (KAc) and are playing just as readers for signaling transduction of the lysine acetylation states of histone. The aberrant events involving in writer-reader-eraser processes have been associated with many human diseases, especially certain cancers [89–91].

Drug targets in epigenetics
Lysine methyltransferase Histone methyltransferases (HMTs) are a class of enzymes that introduce methyl marks on lysines and arginines of histone proteins using the cofactor S-adenosyl methionine (SAM) as methyl source [92, 93]. DOT1L, especially lysine methyltransferase (KMT) responsible for methylations of lysine K79 on histone H3, distinguishes itself from the other KMTs by the absence of the common SET domain and is more closely related to the protein arginine methyltransferase (PRMT) class. Until 2016, the reported DOT1L inhibitors are all SAM-mimic. And due to its unique structure, the optimized inhibitors can achieve high potency and selectivity among the histone methyltransferases, as represented by the clinical candidate EPZ-5676 that was discovered by researchers at Epizyme. The scientists at Novartis took a different approach by applying the fragment-based drug discovery method [94]. They screened the fragment library using surface plasmon resonance (SPR) with an immobilized DOT1L construct containing the catalytic domain. Compound 6-28 was identified as a weak binder with an estimated equilibrium binding constant of Kd ≈ 50 μM (Fig. 6.14). Further, the compound was confirmed by the biochemical scintillation proximity assay and protein-observed NMR experiments. After solving the co-crystal structure of fragment 1 bound DOT1L, the details of binding mode were revealed. The 2,6-dichlorophenyl moiety of fragment 1 acts as a hydrophobic anchor occupying a hydrophobic cavity formed by side chain movements of Met147, Leu143, Phe239, and Tyr312. The 3-(2-N-methylaminocarbonyl) pyrrolyl moiety is sandwiched between Phe243, Pro130, and Phe131 engaging the flexible lid loop of the SAM pocket in a novel conformation by π–π stacking. Although the initial fragment has a moderate ligand efficiency (LE = 0.25) and binds to an induced plastic pocket, the authors carried on the optimization with fragment growing method. As shown below, after extensive modification and under the guidance of co-crystal structures, they finally discovered a high-potent DOT1L inhibitor 6-29, with IC50 value of 14 nM, increased affinity by more than 4 orders of magnitude.

The development of DOT1L inhibitor
Arginine Methyltransferase Histone arginine methyltransferases using the cofactor S-adenosyl-l-methionine (SAM) transfer a methyl group to the terminal guanidino nitrogens of arginine on histone. Like lysine methyltransferases, they play important roles in histone post-translational modifications and govern essential cellular processes that affect cell growth, proliferation, and differentiation. Therefore, deregulation of these enzymes was implicated in the pathogenesis of several different diseases, including cancer. In mammalian cells total nine protein arginine methyltransferases were identified and can be divided into three types according to the degree and position of methylation. Freitas et al. [95] selected three high-potent PRMT inhibitors and deconstructed them into three fragments containing the essential amine group. From the biochemical assay, all three fragments demonstrated a good ligand efficiency (LE ≥ 0.68). This initial result encouraged them to expand the study to screen a commercial fragment library of 2040 compounds. Finally, they obtained 14 confirmed fragment hits. Among them, fragment 7 was in detail characterized with the ITC experiment and found that this fragment shows a signature of favorable enthalpic free energy (ΔG = −8.2 kcal/mol and ΔH = −17.2 kcal/mol). Further tests indicated that fragment 6-33 is substrate-competitive inhibitor of PRMT6, and it could be served as warheads for developing class I PRMT inhibitors (Fig. 6.15).

The development of PRMT6 inhibitor 6-33. Co-crystal structure of fragment 6-33 bound to PRMT6 (PDB:5EGS)
EED Polycomb repressive complex 2 (PRC2) is a protein complex functioning as a H3K27 methyltransferase and playing a major role in diverse biological processes such as chromatin remodeling and epigenetic silencing. The core of the PRC2 complex is composed of four proteins: EZH2, EED, Suz12, and RBAP48 [96]. The catalytic activity is originated in protein EZH2, which contains the SET domain. Gain-of-function mutations and overexpression of EZH2 are well documented to be involved in various cancer types. In addition to EZH2, EED was shown to be required for enzymatic activity, and its binding mechanism was revealed by structural studies on entire human PRC2 and the minimal core of the PRC2 containing only three proteins: EZH2, EED, and Suz12, visualizing the three proteins forming an intertwined complex in which the catalytic center is formed by EZH2 but is stabilized by domains of EED and Suz12. Moreover, H3K27me3 recognition by EED is essential in stimulating basal PRC2 activity in vitro and propagating H3K27 methylation in repressive chromatin for gene silencing in vivo. Giving the important roles of EED in PRC2 complex, Lingel et al. [97] at Novartis Institutes for BioMedical Research initialized a project of developing EED–histone interaction inhibitor. They conducted a biochemical high-throughput screening campaign and identified compound 1 showing inhibition of PRC2 activity with a single-digit micromolar IC50 (2.5 μM). 2D HSQC NMR spectroscopy was used to assess the binding of 1 to EED protein, and dose-dependent chemical shift perturbations of several 13C-labeled methionine methyl resonances were observed. The binding mode was revealed by the co-crystal structure of 6-34 with EED, showing the compound bound in the trimethyllysine pocket, which is consistent with the biochemical study that is a competitive inhibitor with respect to H3K27Me3 peptide (Fig. 6.16). Since compound 6-34 has large molecular weight and is difficult to perform chemical modification, the authors employed an ingenious approach of ligand deconstruction to identify the minimal fragments (compound 6-35). The co-crystal structure of 6-35 with EED shows that the interactions made with the protein are very similar to the ones observed for the parent compound 6-34. Based on this crystal structure, a series of amino-imidazole compounds were synthesized and characterized. Finally, several EED–histone interaction inhibitors (such as 6-36) with submicromolar activity and favorable drug-like properties were discovered, enabling further optimization and studying the biology of EED-mediated inhibition of PRC2 activity.

The development of EED inhibitors. Co-crystal structure of compounds 6-34, 6-35, and 6-36 (PDB codes: 5U17, 5U5K, 5U5H)
Bromdomain Acetylation of histone lysine residues is one of the most widely studied post-translational modifications (PTMs) that regulate chromatin structure and gene expression in the cell. Acetylated histones are recognized by ‘readers,’ which are typically found in chromatin- and transcription-associated proteins that partake in many protein-protein interactions, facilitating the formation of the protein complexes that drive active transcription. So far, there are three readers (bromodomain, double PHD finger, and pleckstrin homology domain) which recognized acetylated lysine (KAc) and the bromodomain is the most thoroughly characterized of the three. The bromodomain was identified to function as lysine acetylation reader in the early 1990s in the brahma gene from Drosophila melanogaster. After analysis of human genome, it was found that 46 different nuclear and cytoplasmic proteins contain 61 bromodomains. These proteins include HATs and HAT-associated proteins, histone methyltransferases, helicases, ATP-dependent chromatin remodeling complexes, transcriptional co-activators, TBP-associated factors, and nuclear scaffolding proteins. Despite having large sequence variations, bromodomain modules share a conserved fold that comprises a left-handed bundle of four α-helices (namely, αZ, αA, αB, and αC) that are linked by diverse loop regions of variable charge and length (known as ZA and BC loops) which surround a central acetylated lysine binding site [98, 99].
Initial fragment-like bromodomain inhibitors were described in the literature in 2005 [100], but potent inhibitors were not developed until 2009. The discovery of the first potent BET inhibitor (+)-JQ-1 [101] was reported in 2010 (Fig. 6.17). (+)-JQ1 was widely used as chemical probe to study the function of BET proteins in physiology as well as in many diseases. Similar to (+)-JQ1, I-BET-762 is also a selective BET inhibitor and now in a phase II clinical trial for nuclear protein in testis (NUT) midline carcinoma [102]. Since the first reports of BET inhibitors, the field of bromodomain inhibitor development has evolved rapidly, and many new scaffolds were discovered to interact with acetylated lysine binding site, especially through the fragment-based techniques [103–105]. Taking the first such application as an example, in 2012, Chung et al. [103] reported an attempt to discover the bromodomain inhibitor with the FBDD method. Based on the analysis of solved bromodomain co-crystal structures, they assembled a focus fragment library and screened with high-throughput biochemical assay and directed X-ray crystallography. The binding modes of over 40 fragment hits were elucidated, which provided invaluable information for bromodomain inhibitor development. Their follow-up study focused on the optimization of dimethyl isoazole scaffold, and eventually led to several potent inhibitors of BRD2, as representated by well-known inhibitor I-BET151. Besides, the 3,5-dimethyl-isoxazole template was proved to be a warhead of KAc binding pocket, being adopted into several chemical series for diversified bromodomains, including BRD4, CREBBP, and P300 proteins.

The representative BET inhibitors
Recently, a study exploring the druggability of the entire family of bromodomains has paved the way for developing inhibitors for other important bromodomains [106]. For example, Harner et al. [107] from Vanderbilt University reported a fragment-based screening of bromodomain ATAD2, a protein that possesses both AAA+ATPase (ATPases associated with various cellular activities) and bromodomain functionalities, which has been linked to poor prognosis in prostate, lung, and triple-negative breast cancers, as well as in hepatocellular carcinoma. They rely on the use of protein-observed NMR spectroscopy, as this method could not only measure the ligand binding affinity but also detect the binding site. Totally, 13,800 fragments were screened and 1H-15N SOFAST-HMQC NMR spectra were recorded on uniformly 15N-labeled ATAD2 in the presence of mixtures containing 12 fragments. Then, hit mixtures were deconvolved as singletons to isolate the hit fragments, resulting in a total of 65 fragment hits. Based on the chemical features and the novelty of scaffold, the fragment hits were clustered into three groups (compounds from 6-37 60 6-39 shown in Fig. 6.18). Detailed binding modes were characterized with the aid of co-crystal structures. Together with the published data from the SGC, they proposed several strategies to improve ligand binding affinities toward the design of a chemical probe to examine the biological impact of ATAD2 inhibition.

The ATAD2 fragment hits: 6-37, 6-38, and 6-39. Co-crystal structure of compounds bound to ATAD2 bromodomain (PDB: 4TYL,4TZ2,4TZ8)
PHD finger Methyllysine and methylarginine ‘readers’ are proteins with domains that can recognize and bind to methylation marks. They are responsible for conveying the methylation signal downstream, and they do so either by having their own catalytic functions or by recruiting other proteins to sites of action through the formation of multiprotein complexes. The major families of methyl reader domains are the plant homeodomain (PHD) fingers, WD40 repeat domains, chromatin organization modifier domains (chromodomains), Tudor domains, Agenet domains, proline-tryptophan-tryptophan-proline (PWWP) domains, and malignant brain tumor (MBT) domains.
In 2014 Miller et al. [108] reported an application of NMR-fragment screen to identify small molecules that bound to the pocket of the Pygo PHD finger, which participates in the interaction between Pygo-BCL9 complex and oncogenic β-catenin. In detail, the armadillo repeats domain (ARD) of β-catenin interacts with BCL9 adaptor proteins, which in turn interact with the rear of Pygo PHD fingers through a BCL9 adapter domain called HD1. This interaction induces a change in the binding module of the PHD finger, which promotes binding to methylated H3K4 and finally translating the signaling to downstream factors. Since inhibition of β-catenin has proved challenging, inhibitors disrupting the PHD finger–BCL9 interactions will be valuable for testing the hypothesis of cancer treatment.
Interaction between the Pygo PHD finger and H3K4me is mediated by two deep binding pockets: one pocket anchoring the N-terminal alanine residue, and the second pocket binding to the mono-methylated side chain of lysine 4 (H3K4me). Firstly, a library of commercially available compounds was screened in silico, and 313 possible hits were tested by protein-observed 2D NMR spectroscopy with a purified 15N-labeled PHD–HD1 complex. Three fragments (6-40 to 6-42) proved to be positive, and elicited several weak chemical shift perturbations (CSPs) of the same PHD residues, indicating they target to same histone binding pocket (Fig. 6.19). To identify compounds with improved solubility and ligand efficiency, they conducted another three consecutive virtual screens and identified 32 additional hits confirmed by NMR spectra. Finally, a fragment containing benzothiazole scaffold (6-43) was analyzed in co-crystal structure, providing the fragment bound the K4me binding pocket of the PHD–HD1 protein with weak affinity (2.5 mM) but unique interaction mode of making a hydrogen bond to Asp380 of PHD. The fragments discovered demonstrate the ability to inhibit the Pygo PHD finger and could lead to future small-molecule inhibitors of the PHD/HD1 complex and other PHD-containing proteins.

The development of PHD finger inhibitors. Co-crystal structure of benzothiazole scaffold 6-43 (PDB:4UP5)
2.2 Targeting PPIs for Antibacterial and Antiviral Treatment
The treatment of bacterial infections through the administration of chemotherapeutic agents, which began in the 1930s, was one of the most profound medical advances of the twentieth century. Unfortunately, the emergence of antibiotic-resistant bacteria has presented a great challenge to humanity. In the past 40 years only two new structural types, daptomycin and linezolid, have been introduced to the clinic following their discovery using empirical screening methods. However, the determination of complete bacterial genome sequences and the parallel development of other techniques such as proteomics inspired a new genomics-based approach to antibacterial drug discovery. Therefore, many new targets were proposed and structure-based or fragment-based drug discovery methods were applied in developing antibiotics. Here we illustrated the fragment-based approach with two recent examples.
FtsZ–ZipA Cell wall division in E. coli involves a series of events, in which the first step involves the generation of the Z-ring, a circular polymeric structure formed by the cytosolic protein FtsZ. FtsZ is required for cell division in both Gram− and Gram+ bacteria and is highly conserved among different species. The assembly dynamics of FtsZ is regulated in cooperation with other proteins, such as ZipA. Impeding the FtsZ–ZipA interaction was found to block the cell division leading to long filamentous bacteria and consequently bacterial cell death. Thus, the inhibition of interaction between FtsZ and ZipA has become an important strategy for finding a new class of antibacterial drugs. Mosyaket al. [109] attempted to use the NMR-fragment screening method to discover the novel inhibitors of the ZipA/FtsZ protein-protein interaction. Firstly, an NMR library composed of 825 compounds was built up based on the structurally diversity and promising physical properties. The screen was initially conducted by monitoring the ligand binding against ZipA with the 1H-15N HSQC experiment and resulted in 16 compounds bound to ZipA. Subsequent research found that seven of the compounds showed perturbations in the 1H-15N HSQC peaks corresponding to residues that are involved in interaction with FtsZ and compound 6-44 showing the largest perturbations in the 1H-15N HSQC spectrum of ZipA was selected as the hit for further optimization (Fig. 6.20).

The development of ZipA inhibitors
The further exploration for the SAR of compound 6-44 was started with a high-quality X-ray crystal structure of ZipA. Eighty-seven compounds structurally similar to compound 6-17 were chosen for testing in the FP assay and evaluation by 1H-15N HSQC experiments. Six compounds with possessed better solubility displayed the largest HSQC perturbations, suggesting that they were the better binding analogs. X-ray co-crystallization experiments for the six compounds and structure-based design led to compound 6-45.
Sliding clamp The bacterial sliding clamp (SC), also known as the DNA polymerase III (Pol III) β subunit, is a torus-shaped homo-dimeric protein that is conserved across bacterial species. The SC serves as a scaffold protein to form large DNA/protein complex, playing a pivotal role in bacterial DNA replication and repair. Proteins that interact with the SC recognize a surface binding pocket that consists of two subsites (I and II), with one pocket located on each of the SC monomers. A consensus LM sequence, QLx1Lx2F/L (S/D preferred at x1; x2 may be absent), has been identified that interacts with the SC LM-binding pocket. In 2014, Yin et al. [110] applied X-ray crystallography as a primary screen to identify fragment hits against the E. coli SC. A total of 352 fragment compounds from the First Pass Screen (Zenobia Fragment Libraries) were soaked into E. coli SC crystals as 4-in-1 cocktails and screened using X-ray crystallography, resulting in four fragments, 3,4-difluorobenzamide(6-46), 5-chloroisatin(6-47), 6-nitrobenzopyrazole (6-48), and 5-nitroindole (6-49), bound in subsite I of the binding pocket of SC (Fig. 6.21). Compound 6-50, an analog of 6-46, was measured using the FP assay (280 µM). And a co-crystal structure of 6-50 bound SC was solved, showing compound 6-50 fully occupied subsite I, with its fluoroaryl ring adopting a binding pose similar to that of 6-46. Then the ZINC library was subsequently searched for compounds displaying structural similarity to fragments, which led to the identification of additional moderate-active SC inhibitors, providing novel scaffolds for further drug development.

Fragment hits of the sliding clamp protein
HIV-Integrase Antiviral drugs, like antibiotics, face similar situation of resistance due to fast evolution of viruses. Therefore, constant needs for new antiviral drugs are posed to the whole drug discovery community, especially for treating the infections caused by HIV, HBV, and HCV. As summarized by Latham et al. [111], there are many drug discovery programs aimed at finding novel HIV-1 reverse transcriptase and integrase inhibitors by using the fragment-based approach, such as the work reported by Serrao and his coworkers [112]. Based on an initial molecular modeling study, they found 8-hydroxyquinoline (6-51) bound to HIV-1 integrase (IN) at the IN–lens epithelium-derived growth factor/p75 (LEDGF/p75) interface (Fig. 6.22). Then they developed a set of modified 8-hydroxyquinoline fragments demonstrating micromolar IC50 values for inhibition of the IN–LEDGF/p75 interaction. Further modifications at the C5 and C7 carbons of the 8-hydroxyquinoline core improved potency, while only modifications at the C5 position ultimately yielded potent inhibitors with low cytotoxicity. Two of these particular compounds, 5-((p-tolylamino) methyl) quinolin-8-ol (6-52) and 5-(((3,4-dimethylphenyl) amino) methyl) quinolin-8-ol (6-53), inhibited viral replication in MT-4 cells with low micromolar EC50, providing evidence for 8-hydroxyquinolines as novel inhibitors of the IN–LEDGF/p75 interaction.

The development of HIV IN inhibitors
HCV NS3 In 2012, the researchers at Astex reported a potent HCV NS3 allosteric inhibitor identified by fragment screening and structure-guided design [51]. They performed a fragment-based screen using crystals of the HCV NS3–NS4a full-length genotype 1b holoenzyme. From the co-crystal structures of a series of fragment hits, they identified a new binding site at the interface of the two domains. This novel binding site is relatively hydrophobic, with residues Met485, Val524, Cys525, Gln526, and Val630 from the helicase domain and His57, Val78, Asp79, Asp81, and Arg155 from the protease domain making key van der Waals contacts with the fragments. One of the fragment hits (6-54), having a half-maximal inhibitory concentration (IC50) ~ 500 µM, was selected for further elaboration with the structure-based approach (Fig. 6.23). By introducing an ethylamino side chain, the ligand (6-55) achieved additional affinity by forming further interactions with the pocket formed by residues Asp79, Thr519, and Leu522, finally leading to an improvement in potency over four orders of magnitude (Kd = 0.022 μM, IC50 < 0.01 μM, LE ~ 0.39 kcal per heavy atom).

HCV NS3 inhibitors. Co-crystal structure of 6-54 bound to NS3 (PDB code: 4B6F); co-crystal structure of 6-55 bound to NS3 (PDB code: 4B74)
2.3 Other PPIs
CXCL12-CXCR4 Chemokines are small soluble proteins that activate G-protein–coupled receptors (GPCRs) and are involved in many physiological processes including cell trafficking, angiogenesis, and embryogenesis. Of the chemokine receptors, CXCR4 is the only one that is expressed by the majority of cancer types (at least 23 different cancers), functions as a sensor to chemokine ligand 12 (CXCL12) gradients in distant tissues to promoting the migration of the cancer cell. Ziareket al. [113] adopted a fragment-based approach to find a better starting molecule without using traditional hit-to-lead optimization strategy. A5-substituted-1H-tetrazole group, a bioisostere of the carboxylic acid, was chosen to bind the sY21 subpocket of CXCL12. Based on the tetrazole group, a fragment library contains only 9 compounds through altering the length of the linker and the substitution pattern of the phenyl. 2D protein-observed NMR was employed to study the fragment-induced CXCL12 chemical shift perturbations and determine the binding affinity of the synthesized fragments. The result showed that all molecules produced a subset of chemical shift changes indicative of a specific binding interaction. Three compounds were docked against total 21 structure models in order to account for protein flexibility and capture the best conformation. With these efforts, they found a compound as a novel hit binding to the sY21site of CXCL12, and subsequent research led a lead compound 6-56 with improvement in affinity, efficiency, and potency (Fig. 6.24).

The deconstruction of CXCR4 inhibitors
pVHL–HIF1α Ciulli et al. [114] dissected small molecular inhibitors (6-57 to 6-60) targeting the pVHL–HIF-1α interaction to fragments and analyzed the implication of fragment-based approaches in the discovery of PPIs inhibitors (Fig. 6.25). Firstly, they deconstructed three targeting molecules into nine fragments (6-61 to 6-69) as a small library. To monitor the fragment binding, they initially selected DSF, NMR spectroscopy, and FP for the first-line screening, but they were unable to unambiguously detect fragment binding directly in any of these experiments except compound 6-69, which showed a very weak signal in the CPMG and Water LOGSY NMR spectra. To address the question of which size and structural complexity would be required to detect and characterize fragment binding, the promising fragment derivatives (6-70 to 6-73) were synthesized. Evidence of binding of 6-71 was only observed by NMR but not by DSF and FP. In contrast, it was able to detect binding of fragment 6-72 and fragment 6-73 in DSF and NMR and to characterize their binding affinities by FP and ITC. This result indicated of the requirement of fragments of the starting inhibitors to form favorable interactions in at least two subsites of the pVHL-HIF1αinterface. This work contrasted with the previous study that fragments had higher ligand efficiency than the larger compounds they were part of and the results pointed to the possibility that current fragment screens against PPIs may be missing interesting fragments because of the inherently low ligand efficiencies associated with binding small molecules at protein surfaces.

The deconstruction of pVHL–HIF1α inhibitors
NRF2-KEAP1 The nuclear factor erythroid 2-related factor 2 (NRF2) has emerged as a master regulator of cellular resistance to oxidants, mediating the upregulation of multiple phase 2 and cytoprotective enzymes and proteins. The protein Kelch-like ECH-associated protein 1 (KEAP1) plays a key role in the regulation of NRF2. Davies and his colleagues [115] applied a crystallographic screening of approximately 330 fragments, then identified three discrete hot spots within the NRF2 site and found the three novel fragments bound to three hot spots independently (Fig. 6.26). Fragment 1 with a carboxylic acid group were identified as a promising anchor fragment engaged in a key electrostatic interaction with Arg483 on the KEAP1 Kelch domain. Starting from fragment 6-74, fragment growth from the benzylic carbon would add more interactions with the hot spot occupied by fragment 6-75, while growth from the phenyl ring, at the direction ortho to the chlorine, would allow access to the subsite bound by the sulfonamide fragment 6-76. The fragment linking method was then adopted for the subsequent modification. The benzotriazole moiety was chosen for attachment directly to the benzylic carbon of the phenyl acetic acid and showed a dramatic increase in affinity to allow detection using the FP assay and ITC (IC50 = 61 μM; ITC Kd = 59 μM), growth from the 3-position of the chlorophenyl ring with a sulfonamide group, and replacement of the para-chloro-phenyl with methyl led to the identification of a very potent compound 6-77 (ITC Kd = 1.3 nM). This compound fulfills the three-point pharmacophore identified from the fragment screen, suggesting the utility of their fragment-based approach.

The development of NRF2 inhibitors. Fragment 6-74 (PDB code: 5FNQ); fragment 6-75 (PDB code:5FZJ); fragment 6-76 (PDB code:5FZN); co-crystal structure of compound 6-77 (PDB code:5FNU)
RAD51-BRCA2 Scott et al. [116] described a fragment-based approach targeting the interaction between the tumor suppressor BRCA2 and the recombination enzyme RAD51. The first step in the biophysical screening of the fragment library was a thermal-shift screen against the MAYSAM mutant of RadA by monitoring the thermal unfolding temperature (Tm) of the protein by using a sensitive dye that fluoresces when the protein unfolds. Two compounds (6-78 and 6-79), which gave thermal shifts of +2 and +1.5 °C, respectively, were selected for the saturation transfer difference (STD) NMR spectroscopy experiments to confirm the ability of ligand binding (Fig. 6.27). And these two compounds were found to bind with a Kd of 2.1 mM with ligand efficiencies of 0.36, consistent with the competition experiment performed by ITC method. Based on the evidences for the binding site of the fragments, the indole scaffold was chosen as the starting point for SAR study, and finally compound 6-80 was discovered as the most potent compound with KD of 1.3 μM, demonstrating a successful optimization with 1000-fold improvement over the original fragment.

The development of RAD51 inhibitors
3 Conclusion
The approval of the first Bcl2 inhibitor venetoclax has been an important milestone for developing PPIs with fragment-based methodology. Currently, there are a large number of drugs in clinical trials that were discovered using fragment-based methods, reinforcing the utility of this efficient approach. Although the protein-protein interaction placed a big obstacle to develop small molecular drugs, over several decades of evolution, the fragment-based drug discovery can not only aid to identify the hot spots at the interface of PPIs, also provide novel starting points for elaboration into drug candidates. In order to integrate the FBDD in developing drugs of PPIs, the researchers need to incorporate more 3D-type molecules in the fragment library to meet the complex surface of PPIs. The sensitivity of biophysical tools in fragment assay has been improved constantly, empowering us to find more fragment hits even for difficult targets. As interface of PPIs is usually containing several hot spots, the fragment linking approach will be increased in use for optimization. Giving many successful case studies we have introduced, we expect the combination of PPI with FBDD will expand our ability to tackle difficult targets and provide novel bioactive compounds for the next generation of therapeutics.
References
Yamada T, Bork P (2009) Evolution of biomolecular networks: lessons from metabolic and protein interactions. Nat Rev Mol Cell Biol 10(11):791–803. https://doi.org/10.1038/nrm2787
Ryan CJ, Cimermancic P, Szpiech ZA, Sali A, Hernandez RD, Krogan NJ (2013) High-resolution network biology: connecting sequence with function. Nat Rev Genet 14(12):865–879. https://doi.org/10.1038/nrg3574
Chakraborty C, Doss CG, Chen L, Zhu H (2014) Evaluating protein-protein interaction (PPI) networks for diseases pathway, target discovery, and drug-design using ‘in silico pharmacology’. Curr Protein Pept Sci 15(6):561–571
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43(Database issue):D447–D452. https://doi.org/10.1093/nar/gku1003
Higueruelo AP, Jubb H, Blundell TL (2013) TIMBAL v2: update of a database holding small molecules modulating protein-protein interactions. Database: J Biol Databases Curation 2013:bat039. https://doi.org/10.1093/database/bat039
Basse MJ, Betzi S, Morelli X, Roche P (2016) 2P2Idb v2: update of a structural database dedicated to orthosteric modulation of protein-protein interactions. Database: J Biol Databases Curation 2016. https://doi.org/10.1093/database/baw007
Bickerton GR, Higueruelo AP, Blundell TL (2011) Comprehensive, atomic-level characterization of structurally characterized protein-protein interactions: the PICCOLO database. BMC Bioinform 12:313. https://doi.org/10.1186/1471-2105-12-313
Coelho ED, Arrais JP, Oliveira JL (2013) From protein-protein interactions to rational drug design: are computational methods up to the challenge? Curr Top Med Chem 13(5):602–618
Kar G, Kuzu G, Keskin O, Gursoy A (2012) Protein-protein interfaces integrated into interaction networks: implications on drug design. Curr Pharm Des 18(30):4697–4705
DeLano WL (2002) Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol 12(1):14–20
Moreira IS, Fernandes PA, Ramos MJ (2007) Hot spots–a review of the protein-protein interface determinant amino-acid residues. Proteins 68(4):803–812. https://doi.org/10.1002/prot.21396
Chen J, Ma X, Yuan Y, Pei J, Lai L (2014) Protein-protein interface analysis and hot spots identification for chemical ligand design. Curr Pharm Des 20(8):1192–1200
Winter A, Higueruelo AP, Marsh M, Sigurdardottir A, Pitt WR, Blundell TL (2012) Biophysical and computational fragment-based approaches to targeting protein-protein interactions: applications in structure-guided drug discovery. Q Rev Biophys 45(4):383–426. https://doi.org/10.1017/s0033583512000108
Scott DE, Coyne AG, Hudson SA, Abell C (2012) Fragment-based approaches in drug discovery and chemical biology. Biochemistry 51(25):4990–5003. https://doi.org/10.1021/bi3005126
Joseph-McCarthy D, Campbell AJ, Kern G, Moustakas D (2014) Fragment-based lead discovery and design. J Chem Inf Model 54(3):693–704. https://doi.org/10.1021/ci400731w
Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H (2016) Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discovery 15(9):605–619. https://doi.org/10.1038/nrd.2016.109
Jencks WP (1981) On the attribution and additivity of binding energies. Proc Natl Acad Sci USA 78(7):4046–4050
Verlinde CL, Rudenko G, Hol WG (1992) In search of new lead compounds for trypanosomiasis drug design: a protein structure-based linked-fragment approach. J Comput Aided Mol Des 6(2):131–147
Shuker SB, Hajduk PJ, Meadows RP, Fesik SW (1996) Discovering high-affinity ligands for proteins: SAR by NMR. Science (New York, NY) 274(5292):1531–1534
Rees DC, Congreve M, Murray CW, Carr R (2004) Fragment-based lead discovery. Nat Rev Drug Discov 3(8):660–672. https://doi.org/10.1038/nrd1467
Carr R, Jhoti H (2002) Structure-based screening of low-affinity compounds. Drug Discov Today 7(9):522–527
van Deursen R, Reymond JL (2007) Chemical space travel. ChemMedChem 2(5):636–640. https://doi.org/10.1002/cmdc.200700021
Hann MM, Leach AR, Harper G (2001) Molecular complexity and its impact on the probability of finding leads for drug discovery. J Chem Inf Comput Sci 41(3):856–864
Wilde F, Link A (2013) Advances in the design of a multipurpose fragment screening library. Expert Opin Drug Discov 8(5):597–606. https://doi.org/10.1517/17460441.2013.780022
Ray PC, Kiczun M, Huggett M, Lim A, Prati F, Gilbert IH, Wyatt PG (2017) Fragment library design, synthesis and expansion: nurturing a synthesis and training platform. Drug Discov Today 22(1):43–56. https://doi.org/10.1016/j.drudis.2016.10.005
Keseru GM, Erlanson DA, Ferenczy GG, Hann MM, Murray CW, Pickett SD (2016) Design principles for fragment libraries: maximizing the value of learnings from pharma fragment-based drug discovery (FBDD) programs for use in academia. J Med Chem 59(18):8189–8206. https://doi.org/10.1021/acs.jmedchem.6b00197
Congreve M, Carr R, Murray C, Jhoti H (2003) A ‘rule of three’ for fragment-based lead discovery? Drug Discov Today 8(19):876–877
Baell JB, Holloway GA (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53(7):2719–2740. https://doi.org/10.1021/jm901137j
Huth JR, Mendoza R, Olejniczak ET, Johnson RW, Cothron DA, Liu Y, Lerner CG, Chen J, Hajduk PJ (2005) ALARM NMR: a rapid and robust experimental method to detect reactive false positives in biochemical screens. J Am Chem Soc 127(1):217–224. https://doi.org/10.1021/ja0455547
Morley AD, Pugliese A, Birchall K, Bower J, Brennan P, Brown N, Chapman T, Drysdale M, Gilbert IH, Hoelder S, Jordan A, Ley SV, Merritt A, Miller D, Swarbrick ME, Wyatt PG (2013) Fragment-based hit identification: thinking in 3D. Drug Discov Today 18(23–24):1221–1227. https://doi.org/10.1016/j.drudis.2013.07.011
Hennig M, Ruf A, Huber W (2012) Combining biophysical screening and X-ray crystallography for fragment-based drug discovery. Top Curr Chem 317:115–143. https://doi.org/10.1007/128_2011_225
Harner MJ, Frank AO, Fesik SW (2013) Fragment-based drug discovery using NMR spectroscopy. J Biomol NMR 56(2):65–75. https://doi.org/10.1007/s10858-013-9740-z
Navratilova I, Hopkins AL (2011) Emerging role of surface plasmon resonance in fragment-based drug discovery. Future Med Chem 3(14):1809–1820. https://doi.org/10.4155/fmc.11.128
Zhang R, Monsma F (2010) Fluorescence-based thermal shift assays. Curr Opin Drug Discov Devel 13(4):389–402
Pedro L, Quinn RJ (2016) Native mass spectrometry in fragment-based drug discovery. Molecules (Basel, Switzerland) 21(8). https://doi.org/10.3390/molecules21080984
Gossert AD, Jahnke W (2016) NMR in drug discovery: a practical guide to identification and validation of ligands interacting with biological macromolecules. Prog Nucl Magn Reson Spectrosc 97:82–125. https://doi.org/10.1016/j.pnmrs.2016.09.001
Davies TG, Tickle IJ (2012) Fragment screening using X-ray crystallography. Top Curr Chem 317:33–59. https://doi.org/10.1007/128_2011_179
Murray CW, Berdini V, Buck IM, Carr ME, Cleasby A, Coyle JE, Curry JE, Day JE, Day PJ, Hearn K, Iqbal A, Lee LY, Martins V, Mortenson PN, Munck JM, Page LW, Patel S, Roomans S, Smith K, Tamanini E, Saxty G (2015) Fragment-based discovery of potent and selective DDR1/2 inhibitors. ACS Med Chem Lett 6(7):798–803. https://doi.org/10.1021/acsmedchemlett.5b00143
Larsson EA, Jansson A, Ng FM, Then SW, Panicker R, Liu B, Sangthongpitag K, Pendharkar V, Tai SJ, Hill J, Dan C, Ho SY, Cheong WW, Poulsen A, Blanchard S, Lin GR, Alam J, Keller TH, Nordlund P (2013) Fragment-based ligand design of novel potent inhibitors of tankyrases. J Med Chem 56(11):4497–4508. https://doi.org/10.1021/jm400211f
Chavanieu A, Pugniere M (2016) Developments in SPR Fragment Screening. Expert Opin Drug Discov 11(5):489–499. https://doi.org/10.1517/17460441.2016.1160888
Neumann T, Junker HD, Schmidt K, Sekul R (2007) SPR-based fragment screening: advantages and applications. Curr Top Med Chem 7(16):1630–1642
Crawford TD, Ndubaku CO, Chen H, Boggs JW, Bravo BJ, Delatorre K, Giannetti AM, Gould SE, Harris SF, Magnuson SR, McNamara E, Murray LJ, Nonomiya J, Sambrone A, Schmidt S, Smyczek T, Stanley M, Vitorino P, Wang L, West K, Wu P, Ye W (2014) Discovery of selective 4-Amino-pyridopyrimidine inhibitors of MAP4K4 using fragment-based lead identification and optimization. J Med Chem 57(8):3484–3493. https://doi.org/10.1021/jm500155b
Navratilova I, Aristotelous T, Picaud S, Chaikuad A, Knapp S, Filappakopoulos P, Hopkins AL (2016) Discovery of New Bromodomain Scaffolds by Biosensor Fragment Screening. ACS Med Chem Letters 7(12):1213–1218. https://doi.org/10.1021/acsmedchemlett.6b00154
Riccardi Sirtori F, Caronni D, Colombo M, Dalvit C, Paolucci M, Regazzoni L, Visco C, Fogliatto G (2015) Establish an automated flow injection ESI-MS method for the screening of fragment based libraries: Application to Hsp90. Eur J Pharm Sci: Off J Eur Fed Pharm Sci 76:83–94. https://doi.org/10.1016/j.ejps.2015.05.001
Drinkwater N, Vu H, Lovell KM, Criscione KR, Collins BM, Prisinzano TE, Poulsen SA, McLeish MJ, Grunewald GL, Martin JL (2010) Fragment-based screening by X-ray crystallography, MS and isothermal titration calorimetry to identify PNMT (phenylethanolamine N-methyltransferase) inhibitors. Biochem J 431(1):51–61. https://doi.org/10.1042/bj20100651
Wang L, Pratt JK, Soltwedel T, Sheppard GS, Fidanze SD, Liu D, Hasvold LA, Mantei RA, Holms JH, McClellan WJ, Wendt MD, Wada C, Frey R, Hansen TM, Hubbard R, Park CH, Li L, Magoc TJ, Albert DH, Lin X, Warder SE, Kovar P, Huang X, Wilcox D, Wang R, Rajaraman G, Petros AM, Hutchins CW, Panchal SC, Sun C, Elmore SW, Shen Y, Kati WM, McDaniel KF (2017) Fragment-Based structure-enabled discovery of novel pyridones and pyridone macrocycles as potent bromodomain and extra-terminal domain (BET) family bromodomain inhibitors. J Med Chem 60(9):3828–3850. https://doi.org/10.1021/acs.jmedchem.7b00017
Fjellström O, Akkaya S, Beisel HG, Eriksson PO, Erixon K, Gustafsson D, Jurva U, Kang D, Karis D, Knecht W, Nerme V, Nilsson I, Olsson T, Redzic A, Roth R, Sandmark J, Tigerström A, Öster L (2015) Creating novel activated factor XI inhibitors through fragment based lead generation and structure aided drug design. PLoS One 10(1):e0113705. https://doi.org/10.1371/journal.pone.0113705
Chilingaryan Z, Yin Z, Oakley AJ (2012) Fragment-based screening by protein crystallography: successes and pitfalls. Int J Mol Sci 13(10):12857–12879. https://doi.org/10.3390/ijms131012857
Sharff A, Jhoti H (2003) High-throughput crystallography to enhance drug discovery. Curr Opin Chem Biol 7(3):340–345
Caliandro R, Belviso DB, Aresta BM, de Candia M, Altomare CD (2013) Protein crystallography and fragment-based drug design. Future Med Chem 5(10):1121–1140. https://doi.org/10.4155/fmc.13.84
Saalau-Bethell SM, Woodhead AJ, Chessari G, Carr MG, Coyle J, Graham B, Hiscock SD, Murray CW, Pathuri P, Rich SJ, Richardson CJ, Williams PA, Jhoti H (2012) Discovery of an allosteric mechanism for the regulation of HCV NS3 protein function. Nat Chem Biol 8(11):920–925. https://doi.org/10.1038/nchembio.1081
Gill A, Cleasby A, Jhoti H (2005) The discovery of novel protein kinase inhibitors by using fragment-based high-throughput x-ray crystallography. Chembiochem: Eur J Chem Biol 6(3):506–512. https://doi.org/10.1002/cbic.200400188
Saalau-Bethell SM, Berdini V, Cleasby A, Congreve M, Coyle JE, Lock V, Murray CW, O’Brien MA, Rich SJ, Sambrook T, Vinkovic M, Yon JR, Jhoti H (2014) Crystal structure of human soluble adenylate cyclase reveals a distinct, highly flexible allosteric bicarbonate binding pocket. ChemMedChem 9(4):823–832. https://doi.org/10.1002/cmdc.201300480
Ludlow RF, Verdonk ML, Saini HK, Tickle IJ, Jhoti H (2015) Detection of secondary binding sites in proteins using fragment screening. Proc Natl Acad Sci USA 112(52):15910–15915. https://doi.org/10.1073/pnas.1518946112
Nikiforov PO, Surade S, Blaszczyk M, Delorme V, Brodin P, Baulard AR, Blundell TL, Abell C (2016) A fragment merging approach towards the development of small molecule inhibitors of Mycobacterium tuberculosis EthR for use as ethionamide boosters. Org Biomol Chem 14(7):2318–2326. https://doi.org/10.1039/c5ob02630j
Borsi V, Calderone V, Fragai M, Luchinat C, Sarti N (2010) Entropic contribution to the linking coefficient in fragment based drug design: a case study. J Med Chem 53(10):4285–4289. https://doi.org/10.1021/jm901723z
Edink E, Rucktooa P, Retra K, Akdemir A, Nahar T, Zuiderveld O, van Elk R, Janssen E, van Nierop P, van Muijlwijk-Koezen J, Smit AB, Sixma TK, Leurs R, de Esch IJ (2011) Fragment growing induces conformational changes in acetylcholine-binding protein: a structural and thermodynamic analysis. J Am Chem Soc 133(14):5363–5371. https://doi.org/10.1021/ja110571r
Potter A, Oldfield V, Nunns C, Fromont C, Ray S, Northfield CJ, Bryant CJ, Scrace SF, Robinson D, Matossova N, Baker L, Dokurno P, Surgenor AE, Davis B, Richardson CM, Murray JB, Moore JD (2010) Discovery of cell-active phenyl-imidazole Pin1 inhibitors by structure-guided fragment evolution. Bioorg Med Chem Lett 20(22):6483–6488. https://doi.org/10.1016/j.bmcl.2010.09.063
Mattos C, Bellamacina CR, Peisach E, Pereira A, Vitkup D, Petsko GA, Ringe D (2006) Multiple solvent crystal structures: probing binding sites, plasticity and hydration. J Mol Biol 357(5):1471–1482. https://doi.org/10.1016/j.jmb.2006.01.039
Brenke R, Kozakov D, Chuang GY, Beglov D, Hall D, Landon MR, Mattos C, Vajda S (2009) Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics (Oxford, England) 25(5):621–627. https://doi.org/10.1093/bioinformatics/btp036
Kozakov D, Grove LE, Hall DR, Bohnuud T, Mottarella SE, Luo L, Xia B, Beglov D, Vajda S (2015) The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc 10(5):733–755. https://doi.org/10.1038/nprot.2015.043
Randjelovic J, Eric S, Savic V (2014) In silico design of small molecule inhibitors of CDK9/cyclin T1 interaction. J Mol Graph Model 50:100–112. https://doi.org/10.1016/j.jmgm.2014.04.002
Arkin MR, Tang Y, Wells JA (2014) Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol 21(9):1102–1114. https://doi.org/10.1016/j.chembiol.2014.09.001
Kim I, Xu W, Reed JC (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discovery 7(12):1013–1030. https://doi.org/10.1038/nrd2755
Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G (2011) Cell death assays for drug discovery. Nat Rev Drug Discov 10(3):221–237. https://doi.org/10.1038/nrd3373
Lessene G, Czabotar PE, Colman PM (2008) BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov 7(12):989–1000. https://doi.org/10.1038/nrd2658
Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ (2017) From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov 16(4):273–284. https://doi.org/10.1038/nrd.2016.253
Fulda S, Vucic D (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov 11(2):109–124. https://doi.org/10.1038/nrd3627
Vazquez A, Bond EE, Levine AJ, Bond GL (2008) The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov 7(12):979–987. https://doi.org/10.1038/nrd2656
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435(7042):677–681. https://doi.org/10.1038/nature03579
Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, Fesik SW (2013) Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem 56(1):15–30. https://doi.org/10.1021/jm301448p
Petros AM, Swann SL, Song D, Swinger K, Park C, Zhang H, Wendt MD, Kunzer AR, Souers AJ, Sun C (2014) Fragment-based discovery of potent inhibitors of the anti-apoptotic MCL-1 protein. Bioorg Med Chem Lett 24(6):1484–1488. https://doi.org/10.1016/j.bmcl.2014.02.010
Huang JW, Zhang Z, Wu B, Cellitti JF, Zhang X, Dahl R, Shiau CW, Welsh K, Emdadi A, Stebbins JL, Reed JC, Pellecchia M (2008) Fragment-based design of small molecule X-linked inhibitor of apoptosis protein inhibitors. J Med Chem 51(22):7111–7118. https://doi.org/10.1021/jm8006992
Chessari G, Buck IM, Day JE, Day PJ, Iqbal A, Johnson CN, Lewis EJ, Martins V, Miller D, Reader M, Rees DC, Rich SJ, Tamanini E, Vitorino M, Ward GA, Williams PA, Williams G, Wilsher NE, Woolford AJ (2015) Fragment-based drug discovery targeting inhibitor of apoptosis proteins: discovery of a non-alanine lead series with dual activity against cIAP1 and XIAP. J Med Chem 58(16):6574–6588. https://doi.org/10.1021/acs.jmedchem.5b00706
Estrada-Ortiz N, Neochoritis CG, Domling A (2016) How to design a successful p53-MDM2/X interaction inhibitor: a thorough overview based on crystal structures. ChemMedChem 11(8):757–772. https://doi.org/10.1002/cmdc.201500487
Fry DC, Wartchow C, Graves B, Janson C, Lukacs C, Kammlott U, Belunis C, Palme S, Klein C, Vu B (2013) Deconstruction of a nutlin: dissecting the binding determinants of a potent protein-protein interaction inhibitor. ACS Med Chem Lett 4(7):660–665. https://doi.org/10.1021/ml400062c
Boltjes A, Huang Y, van de Velde R, Rijkee L, Wolf S, Gaugler J, Lesniak K, Guzik K, Holak TA, Domling A (2014) Fragment-based library generation for the discovery of a peptidomimetic p53-Mdm4 inhibitor. ACS Comb Sci 16(8):393–396. https://doi.org/10.1021/co500026b
Wilson CY, Tolias P (2016) Recent advances in cancer drug discovery targeting RAS. Drug Discov Today 21(12):1915–1919. https://doi.org/10.1016/j.drudis.2016.08.002
Papke B, Der CJ (2017) Drugging RAS: know the enemy. Science 355(6330):1158–1163. https://doi.org/10.1126/science.aam7622
Spiegel J, Cromm PM, Zimmermann G, Grossmann TN, Waldmann H (2014) Small-molecule modulation of Ras signaling. Nat Chem Biol 10(8):613–622. https://doi.org/10.1038/nchembio.1560
Keeton AB, Salter EA, Piazza GA (2017) The RAS-Effector Interaction as a Drug Target. Science (New York, NY) 77 (2):221-226. https://doi.org/10.1126/science.aam7622. https://doi.org/10.1158/0008-5472.can-16-0938
Martin-Gago P, Fansa EK, Wittinghofer A, Waldmann H (2017) Structure-based development of PDEdelta inhibitors. Biol Chem 398(5–6):535–545. https://doi.org/10.1515/hsz-2016-0272
Maurer T, Wang W (2013) NMR study to identify a ligand-binding pocket in Ras. The Enzymes 33 Pt A:15–39. https://doi.org/10.1016/b978-0-12-416749-0.00002-6
Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, Ludlam MJ, Bowman KK, Wu J, Giannetti AM, Starovasnik MA, Mellman I, Jackson PK, Rudolph J, Wang W, Fang G (2012) Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci USA 109(14):5299–5304. https://doi.org/10.1073/pnas.1116510109
Winter JJ, Anderson M, Blades K, Brassington C, Breeze AL, Chresta C, Embrey K, Fairley G, Faulder P, Finlay MR, Kettle JG, Nowak T, Overman R, Patel SJ, Perkins P, Spadola L, Tart J, Tucker JA, Wrigley G (2015) Small molecule binding sites on the Ras:SOS complex can be exploited for inhibition of Ras activation. J Med Chem 58(5):2265–2274. https://doi.org/10.1021/jm501660t
Wittinghofer A, Waldmann H, Bastiaens PI, Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, Waldmann H (2013) Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nat Commun 497(7451):638–642. https://doi.org/10.1038/ncomms11360. https://doi.org/10.1038/nature12205
Papke B, Murarka S, Vogel HA, Martin-Gago P, Kovacevic M, Truxius DC, Fansa EK, Ismail S, Zimmermann G, Heinelt K, Schultz-Fademrecht C, Al Saabi A, Baumann M, Nussbaumer P (2016) Identification of pyrazolopyridazinones as PDEdelta inhibitors. Identification of pyrazolopyridazinones as PDEdelta inhibitors 7:11360. https://doi.org/10.1038/ncomms11360
Martin-Gago P, Fansa EK, Klein CH, Murarka S, Janning P, Schurmann M, Metz M, Ismail S, Schultz-Fademrecht C, Baumann M, Bastiaens PI, Wittinghofer A, Waldmann H (2017) A PDE6delta-KRas inhibitor chemotype with up to seven H-bonds and picomolar affinity that prevents efficient inhibitor release by Arl2. Angew Chem Int Ed Engl 56(9):2423–2428. https://doi.org/10.1002/anie.201610957
Tough DF, Tak PP, Tarakhovsky A, Prinjha RK (2016) Epigenetic drug discovery: breaking through the immune barrier. Nat Rev Drug Discov 15(12):835–853. https://doi.org/10.1038/nrd.2016.185
Shortt J, Ott CJ, Johnstone RW, Bradner JE (2017) A chemical probe toolbox for dissecting the cancer epigenome. Nat Rev Cancer 17(3):160–183. https://doi.org/10.1038/nrc.2016.148
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M (2012) Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discovery 11(5):384–400. https://doi.org/10.1038/nrd3674
Kaniskan HU, Jin J (2015) Chemical probes of histone lysine methyltransferases. ACS Chem Biol 10(1):40–50. https://doi.org/10.1021/cb500785t
He Y, Korboukh I, Jin J, Huang J (2012) Targeting protein lysine methylation and demethylation in cancers. Acta Biochim Biophys Sin 44(1):70–79. https://doi.org/10.1093/abbs/gmr109
Scheufler C, Mobitz H, Gaul C, Ragot C, Be C, Fernandez C, Beyer KS, Tiedt R, Stauffer F (2016) Optimization of a fragment-based screening hit toward potent DOT1L inhibitors interacting in an induced binding pocket. ACS Med Chem Lett 7(8):730–734. https://doi.org/10.1021/acsmedchemlett.6b00168
Ferreira de Freitas R, Eram MS, Szewczyk MM, Steuber H, Smil D, Wu H, Li F, Senisterra G, Dong A, Brown PJ, Hitchcock M, Moosmayer D, Stegmann CM, Egner U, Arrowsmith C, Barsyte-Lovejoy D, Vedadi M, Schapira M (2016) Discovery of a potent Class I protein arginine methyltransferase fragment inhibitor. J Med Chem 59(3):1176–1183. https://doi.org/10.1021/acs.jmedchem.5b01772
Jiao L, Liu X (2015) Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science (New York, NY) 350 (6258):aac4383. https://doi.org/10.1126/science.aac4383
Lingel A, Sendzik M, Huang Y (2017) Structure-guided design of EED binders allosterically inhibiting the epigenetic polycomb repressive complex 2 (PRC2) methyltransferase. 60(1):415–427. https://doi.org/10.1021/acs.jmedchem.6b01473
Fujisawa T, Filippakopoulos P (2017) Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol 18(4):246–262. https://doi.org/10.1038/nrm.2016.143
Filippakopoulos P, Knapp S (2014) Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discovery 13(5):337–356. https://doi.org/10.1038/nrd4286
Zeng L, Li J, Muller M, Yan S, Mujtaba S, Pan C, Wang Z, Zhou MM (2005) Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J Am Chem Soc 127(8):2376–2377. https://doi.org/10.1021/ja044885g
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE (2010) Selective inhibition of BET bromodomains. Nature 468(7327):1067–1073. https://doi.org/10.1038/nature09504
Mirguet O, Gosmini R, Toum J, Clement CA, Barnathan M, Brusq JM, Mordaunt JE, Grimes RM, Crowe M, Pineau O, Ajakane M, Daugan A, Jeffrey P, Cutler L, Haynes AC, Smithers NN, Chung CW, Bamborough P, Uings IJ, Lewis A, Witherington J, Parr N, Prinjha RK, Nicodeme E (2013) Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J Med Chem 56(19):7501–7515. https://doi.org/10.1021/jm401088k
Chung CW, Dean AW, Woolven JM, Bamborough P (2012) Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. J Med Chem 55(2):576–586. https://doi.org/10.1021/jm201320w
Zhao L, Cao D, Chen T, Wang Y, Miao Z, Xu Y, Chen W, Wang X, Li Y, Du Z, Xiong B, Li J, Xu C, Zhang N, He J, Shen J (2013) Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain. J Med Chem 56(10):3833–3851. https://doi.org/10.1021/jm301793a
Spiliotopoulos D, Caflisch A (2016) Fragment-based in silico screening of bromodomain ligands. Drug Discov Today Technol 19:81–90. https://doi.org/10.1016/j.ddtec.2016.06.003
Vidler LR, Brown N, Knapp S, Hoelder S (2012) Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J Med Chem 55(17):7346–7359. https://doi.org/10.1021/jm300346w
Harner MJ, Chauder BA, Phan J, Fesik SW (2014) Fragment-based screening of the bromodomain of ATAD2. J Med Chem 57(22):9687–9692. https://doi.org/10.1021/jm501035j
Miller TC, Rutherford TJ, Birchall K, Chugh J, Fiedler M, Bienz M (2014) Competitive binding of a benzimidazole to the histone-binding pocket of the Pygo PHD finger. ACS Chem Biol 9(12):2864–2874. https://doi.org/10.1021/cb500585s
Tsao DH, Sutherland AG, Jennings LD, Li Y, Rush TS 3rd, Alvarez JC, Ding W, Dushin EG, Dushin RG, Haney SA, Kenny CH, Malakian AK, Nilakantan R, Mosyak L (2006) Discovery of novel inhibitors of the ZipA/FtsZ complex by NMR fragment screening coupled with structure-based design. Bioorg Med Chem 14(23):7953–7961. https://doi.org/10.1016/j.bmc.2006.07.050
Yin Z, Whittell LR, Wang Y, Jergic S, Liu M, Harry EJ, Dixon NE, Beck JL, Kelso MJ, Oakley AJ (2014) Discovery of lead compounds targeting the bacterial sliding clamp using a fragment-based approach. J Med Chem 57(6):2799–2806. https://doi.org/10.1021/jm500122r
Latham CF, La J, Tinetti RN, Chalmers DK, Tachedjian G (2016) Fragment based strategies for discovery of novel HIV-1 reverse transcriptase and integrase inhibitors. Curr Top Med Chem 16(10):1135–1153
Serrao E, Debnath B, Otake H, Kuang Y, Christ F, Debyser Z, Neamati N (2013) Fragment-based discovery of 8-hydroxyquinoline inhibitors of the HIV-1 integrase-lens epithelium-derived growth factor/p75 (IN-LEDGF/p75) interaction. J Med Chem 56(6):2311–2322. https://doi.org/10.1021/jm301632e
Ziarek JJ, Liu Y, Smith E, Zhang G, Peterson FC, Chen J, Yu Y, Chen Y, Volkman BF, Li R (2012) Fragment-based optimization of small molecule CXCL12 inhibitors for antagonizing the CXCL12/CXCR4 interaction. Curr Top Med Chem 12(24):2727–2740
Van Molle I, Thomann A, Buckley DL, So EC, Lang S, Crews CM, Ciulli A (2012) Dissecting fragment-based lead discovery at the von Hippel-Lindau protein:hypoxia inducible factor 1alpha protein-protein interface. Chem Biol 19(10):1300–1312. https://doi.org/10.1016/j.chembiol.2012.08.015
Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, Norton D, Rich SJ, Richardson C, Saxty G, Willems HM, Woolford AJ, Cottom JE, Kou JP, Yonchuk JG, Feldser HG, Sanchez Y, Foley JP, Bolognese BJ, Logan G, Podolin PL, Yan H, Callahan JF, Heightman TD, Kerns JK (2016) Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein-protein interaction with high cell potency identified by fragment-based discovery. J Med Chem 59(8):3991–4006. https://doi.org/10.1021/acs.jmedchem.6b00228
Scott DE, Ehebauer MT, Pukala T, Marsh M, Blundell TL, Venkitaraman AR, Abell C, Hyvonen M (2013) Using a fragment-based approach to target protein-protein interactions. Chembiochem: Eur J Chem Biol 14(3):332–342. https://doi.org/10.1002/cbic.201200521
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Xiong, B., Wang, Q., Shen, J. (2018). Fragment-Based Drug Discovery for Developing Inhibitors of Protein-Protein Interactions. In: Sheng, C., Georg, G. (eds) Targeting Protein-Protein Interactions by Small Molecules. Springer, Singapore. https://doi.org/10.1007/978-981-13-0773-7_6
Download citation
DOI: https://doi.org/10.1007/978-981-13-0773-7_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-0772-0
Online ISBN: 978-981-13-0773-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)