
Abstract
Myotonic dystrophy (dystrophia myotonica, DM) is the commonest form of muscular dystrophy affecting adults. This multisystem disorder typically affects the skeletal muscle and is characterized by weakness, wasting, and myotonia; other systemic involvement includes ocular, cardiac, endocrine, and central nervous system dysfunction. DM is classified into two main subtypes: type 1 (DM1) and type 2 (DM2) based on mutations in the dystrophia myotonica protein kinase (DMPK) gene and CCHC-type zinc-finger cellular nucleic acid-binding protein (CNBP) formerly known as the zinc finger 9 (ZNF9) gene, respectively. The multisystem phenotype of DM1 and DM2 is due to the presence of expanded repeats and the attendant effects. DM1 occurs due to the persistence of harmful effects of untranslated RNA transcripts of CTG trinucleotide repeat, which are located in the 3′-untranslated region of the DMPK gene on 19q13. DM2 results from the toxic effects of the untranslated RNA transcripts of CCTG tetranucleotide repeat, which are located in the primary intron of the CNBP gene, on chromosome 3q 21.3. A diagnosis of myotonic dystrophy can be made clinically based on presentation with characteristic features and a positive family history. However, molecular genetic testing for an expanded CTG repeat in the DMPK gene is the gold standard for definitive diagnosis of DM1. If DM1 testing is negative, testing for the CCTG repeat in the CNBP gene is then considered appropriate to establish a diagnosis of DM2.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1.1 Introduction
Myotonic dystrophy (dystrophia myotonica, DM) is an autosomal dominant multisystem disorder and is the commonest form of muscular dystrophy in adults. The condition is clinically and genetically heterogeneous, typically affecting the skeletal muscle with characteristic paradoxical weakness, wasting, and myotonia [1]. Features of multisystem involvement include ocular, cardiac, endocrine, and central nervous system manifestations. And thus, affected individuals may present with early cataract, cardiac conduction abnormalities, insulin resistance, infertility, sleep disorders, and cognitive dysfunction.
Also, severe developmental disability has been reported in a severe congenital form of DM1 [2]. These multiorgan clinical features account for the initial presentation of such individuals to various medical subspecialties including internal medicine, cardiology, ophthalmology, endocrinology, and even pediatrics, before eventually undergoing specialist neurology review. DM is classified into two main forms, myotonic dystrophy type 1 (DM1) and myotonic dystrophy type 2 (DM2), based on the genetic alteration and clinical features. Showing specifically wide variability in terms of severity and age of onset, the disease can also broadly be divided into congenital and adult forms.
The genetic defect in DM1 is a result of mutation in the dystrophia myotonica protein kinase (DMPK) gene with amplified trinucleotide CTG repeats in the 3' untranslated region. Disease severity varies with the number of repeats. In DM2 the defect is in the CCHC-type zinc-finger cellular nucleic acid-binding protein (CNBP) formerly known as the zinc-finger 9 (ZNF9) gene, with tetranucleotide CCTG repeats. Both of them are caused by microsatellite repeat expansions in noncoding regions of the genome giving rise to RNAs having a toxic gain of function among microsatellite expansion diseases encompassing more than 20 neurological disorders, including Huntington’s disease and various spinocerebellar ataxias (Fig. 1.1). The multiorgan phenotype of DM1 and DM2 results from the presence of these expanded repeats and the attendant effects. DM1 occurs due to the persistence of harmful effects of untranslated RNA transcripts of CTG-trinucleotide repeats, which are located in the 3′-untranslated region of the DMPK gene on 19q13. DM2 results from the toxic effects of the untranslated RNA transcripts of CCTG-tetranucleotide repeats, which are located in the primary intron of the CNBP gene, on chromosome 3q 21.3.

Diseases caused by expanded microsatellite repeats. Abbreviations: SCA spinocerebellar ataxia, FXS fragile X syndrome, FXTAS fragile X-associated tremor/ataxia syndrome, SBMA spinal and bulbar muscular atrophy, HD Huntington’s disease, DRPLA dentatorubral-pallidoluysian atrophy, DM myotonic dystrophy, ALS/FTD amyotrophic lateral sclerosis and frontotemporal dementia
The gene mapping, molecular genetics, genotype-phenotype correlation, and population genetics/epidemiology of DM are further expanded below, with discussions on the various phenotypes of DM1 and DM2 and the genetic basis for investigation and diagnosis.
1.2 Genetics
DM1 and DM2 are genetically distinct and are both inherited in an autosomal dominant manner and present with somewhat overlapping phenotypic abnormalities. The diseases may show variable penetrance, with characteristic increasing severity of phenotype over subsequent generations.
The underlying mechanism of the pathogenesis of DM is still unclear. However, studies show a role for the proliferation of transcription products in mutant forms of defective genes. The two genetically distinct subtypes, DM1 and DM2, are caused by similar noncoding repeat expansions in different genes.
In DM1, expanded repeats occur in CTG in the 3′-UTR of DMPK gene. In DM2, expanded repeats occur in the CCTG region in the primary intron of the CNBP gene (formerly known as zinc-finger protein 9; ZNF9) [3,4,5]. The DM1 and DM2 phenotypes result from abnormalities in the transcribed RNA equivalents, CTG-to-CUG for DM1 and CCTG-to-CCUG for DM2.
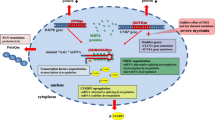
In DM1 and DM2, the abnormal RNA transcripts remain untranslated. They exert a harmful effect on other genes not unique to the DM1 or DM2 locus and aggregate other proteins involved in alternative RNA splicing. These proteins are known as muscle blind-like (MBNL) proteins and CUG-BP and ETR-3-like factors (CELF) [4, 6,7,8]. MBNL proteins are highly conserved across species including man.
The MBNL gene encodes MBNL proteins, responsible for muscle development and photoreceptor neuron differentiation in the eye. Thus, absence of the MBNL gene results in blindness and muscle defects. In patients with DM1 and DM2, MBNL proteins accumulate in the RNA foci due to the activity of the mutated RNA transcripts of CUG or CCUG repeats. Because of this sequestration, the amount of MBNL proteins available for proper function is reduced. A fetal pattern of target transcripts results due to the mutant RNA-induced shift in splicing from normal to abnormal. These mutant RNA transcripts further exert an adverse effect on RNA-binding protein activity, leading to errors in transcript splicing and defects in the function of several genes such as the bridging integrator 1 gene (BIN1) [9], cardiac troponin T [10], insulin receptor [11], and the skeletal muscle chloride channel [12]. These could result in the typical features of abnormalities in cardiac conduction, myotonia, and insulin resistance seen in individuals with DM [4, 6, 7].
1.3 DM1
The genetic defect in DM 1 results from a heterozygous trinucleotide repeat expansion (CTG)n in the 3′ UTR of the DMPK gene on chromosome 19q13. Repeat lengths in excess of 50 CTG repeats are considered pathogenic [13].
DM1 phenotypes can be classified as congenital, childhood-onset, adult-onset “classic DM1,” late-onset/asymptomatic, and premutation DM. Table 1.1 presents an overview of these phenotypes, clinical findings, and CTG length [14].
Clinical features are slowly progressive, and clinical severity varies broadly ranging from asymptomatic to severe. Symptoms usually appear in the second and third decades of life for the most common classic form of DM. Molecular pathways are still unclear.
1.3.1 Mapping
The myotonic dystrophy locus was among the earliest human disease loci to be assigned a chromosome by linkage analysis. Linkage was first suspected between the Lutheran blood group (Lu) and the secretor (Se) loci by Mohr [15]. It was later discovered that complement component 3 (C3) was linked to Lu-Se-DM [16, 17]. C3 had earlier been assigned to chromosome 19 by somatic hybrid studies, and thus this linkage indicated that DM is also on chromosome 19. Subsequently, positive LOD scores were discovered for serum C3 and another chromosome 19 locus, peptidase D (PEPD). PEPD was also assigned to chromosome 19 by somatic cell hybrid studies, and a close linkage was demonstrated between PEPD and DM locus with a LOD score of 3.51 at a recombination fraction of 0.00 [16]. This provided regional assignment and further confirmed the Lu-Se-DM assignment to chromosome 19. Restriction fragment length polymorphism analysis with an associated C3 probe showed evidence of linkage with DM with a LOD score of +3.36 at a recombination fraction of 0.05 in males [17]. Further gene mapping of chromosome 19 with regard to DM revealed suppression of recombination near the centromere, and linkage studies assigned DM a location in the centromere region of chromosome 19 [18].
RFLPs were designated to the D19S19 locus that is linked to DM (maximum LOD score of 11.04 at a recombination fraction of 0.0). The genomic clone LDR152 (D19S19) was reported to be tightly linked to DM with a maximum LOD score of 15.4 at a recombination fraction of 0.0 (95% confidence limits 0.0–0.03) [19, 20].
Friedrich et al. conducted linkage studies in three large kindreds, using RFLPs related to C3 and the chromosome 19 centromeric heteromorphism as genetic markers, and excluded DM from the 19cen-C3 segment by three-point linkage analysis, thus strongly reinforcing the assignment to the proximal long arm of chromosome 19 [21].
It was conclusively established that the DM gene lies in region 19q13.2–q13.3. The APOC2 and CKM were identified as the closest proximal markers located approximately 3 cM and 2 cM from DM, respectively, in the order cen–APOC2–CKMM–DM. Furthermore, among 12 polymorphic markers on 19q, 10 were revealed to be proximal to the DM gene, and 2 were distal to DM, PRKCG (176980), and D19S22 (located at a distance of approximately 25 cM and 15 cM, respectively, from DM) [22].
1.3.2 Molecular Genetics of DM1
Previously known as Steinert disease, DM1 is inherited in the non-Mendelian autosomal dominant pattern, with variable penetrance and transmission between mother and child. A low frequency of about 5–34 copies of the trinucleotide repeats is typical in the general population. In patients with DM1, a greater number of repeats, ranging from upward of 50 copies, are reported [6, 23]. This may explain the parallel seen between the varying severity of DM with the age of onset and the number of repeats. These expanded repeats are unstable and exhibit intergenerational expansion; further expansion may occur during meiosis with an increase in the repeat size during parent-to-child transmission within successive generations [24]. Thus, at-risk offspring may inherit sizably longer repeat lengths than those present in the transmitting parent. Based on these, DM1 is described as showing a characteristic anticipation and exhibiting parental gender effect/maternal bias.
Anticipation is a phenomenon where the increase in repeat size during parent-to-child transmission within successive generations results in increasing disease severity and decreasing age of onset in successive generations. This anticipation results because DMPK alleles of over 34 CTG repeats in length are unstable. Most often a child with early-onset, severe DM1 (i.e., congenital DM1) has inherited the expanded DMPK allele from the mother [25, 26, 27]. Although anticipation typically occurs in maternal transmission of the disease, anticipation with paternal transmission is also possible [25, 28]. Disease severity is directly proportional to the number of repeats: normal individuals have 5–34 repeats, individuals with mild disease have 50–150 repeats, patients with classic DM have 100–1000 repeats, and patients with congenital DM exhibiting over 2000 repeats. The size of the CTG repeat appears to increase over time in the same individual. Somatic instability of the repeat is also recognized as intra-tissue, inter-tissue, and cell-type variability over a patient’s life time [29, 30].
Congenital DM is seen solely when the affected parent is the mother. This maternal bias occurs because expansion of alleles with 40–80 repeats typically occurs in paternal transmissions; however, expansion is seen only in alleles longer than 80 repeats when transmission is maternal. The frequency of repeat contractions is approximately 4.2–6.4% [13].
In congenital DM cases, the CTG-repeat lengths are unusually high, at >1000, and as high as 4000 expanded repeats can occur [6]. Fewer repeat lengths (730–1000) have been reported, but the affected infants may present with infantile hypotonia, mental retardation, and respiratory dysfunction. A decrease in the CTG-repeat size (intergenerational contraction) has been also reported in about 6.4% of transmissions [31]. A paternal factor may contribute to the dynamics of intergenerational contraction, observed in the expanded CTG repeats in DM1. Among the French-Canadian DM1 population, about 7.4% display intergenerational contractions, during transmission. All the cases in this cohort were transmitted from father to child [31]. The intergenerational contraction seen in DM1 could occur within some [32] or all [31] related siblings in a family.
Historically, the isolation of a human genomic clone that detected novel restriction fragments specific to persons with myotonic dystrophy was reported in 1992 [33]. An EcoRI polymorphism with two alleles was seen in normal persons, but in most affected individuals, one of the normal alleles was replaced by a larger unstable fragment. Fragment length showed significant variation within families as well as between unrelated affected individuals. The unstable nature of this region was thought to explain the characteristic variation in severity and age at onset of DM.
Subsequently, a DNA fragment was detected that was larger-sized in DM1 affected persons compared with normal sibs or controls. The sequence containing this fragment was located in chromosome 19 and was flanked by two tightly linked markers, ERCC1 (126380) proximally and D19S51 distally [34]. The essential region between the above-mentioned markers was cloned in a 700-kb contig comprising overlapping cosmids and yeast artificial chromosomes. The central part of the contig bridged an area of about 350 kb between two flanking crossover borders. This segment, which presumably contained the DM gene, has been extensively characterized. Two genomic probes and two homologous cDNA probes were situated within approximately 10 kb of genomic DNA and detected an unstable genomic segment in myotonic dystrophy patients. The length variation in this segment showed similarities to the instability seen in the fragile X locus (300624). The discovery of these changes in repeat expansion in families with fragile X syndrome strongly suggested the possibility that a similar mutation with unstable microsatellite expansion might be involved in the pathogenesis of DM. Subsequently, the CTG expansion was identified [5]. The authors proposed that the length variation was consistent with a direct role in the pathogenesis of DM.
Typically, the size of the pathognomonic CTG triplet repeat is larger in DM patients than in unaffected individuals [22]. The sequence shows high variability among normal populations. Unaffected individuals carry 5–27 copies. Patients with mild disease have 50 or more repeats, and patients with the phenotypically more severe classic DM typically carry repeat expansions of up to several kilobase pairs.
The length of the CTG repeat correlates with the incidence of severe congenital DM. Furthermore, mothers of individuals with congenital DM were found to have higher than average CTG repeat lengths [35].
Recently, using triplet-primed PCR (TP-PCR) of both DNA strands followed by direct sequencing, Musova et al. identified interruptions within expanded DM1 CTG repeats in almost 5% (3 of 63) of Czech DM1 families and in 2 of 2 intermediate alleles [13]. None of 261 normal Czech alleles tested carried interruptions. The expanded alleles contained either regular runs of a (CCGCTG)n hexamer or showed a much higher complexity; they were always located at the 3-prime end of the repeat. The number and location of the interruptions were very unstable within families and subject to substantial change during transmission. However, four of five transmissions of the interrupted expanded allele in one family were accompanied by repeat contraction, suggesting that the interruptions render the DMPK CTG repeat more stable or could even predispose it to contractions. Overall, the contribution of the interrupted alleles to the phenotype was uncertain. Musova et al. suggested that the occurrence of interruptions may be missed by routine testing using PCR or Southern blotting.
1.3.3 Genotype-Phenotype Correlations
1.3.3.1 Congenital DM
Clinically, congenital DM presents with infantile hypotonia, respiratory failure, learning disability, and cardiorespiratory complications. For congenital DM1, an estimated incidence of 2.1–28.6 per 100,000 live births has been reported based on accumulated studies [6]. Onset of typically symptoms present at birth. The majority of congenital DM cases show a maternal transmission pattern, due to a higher probability for expanded CTG repeats in mothers compared with fathers [3, 6, 8]. The mean length of maternal trinucleotide CTG repeats is greater in congenital DM cases compared with adult-onset DM with CTG expansions in excess of 1000 repeats [32]. This is in contrast with paternal trinucleotide repeats, which are smaller and/or show no symptoms at childbirth [6]. Maternal DM can go undetected for most of adult life, with a diagnosis made only after the birth of a neonate with congenital DM [5, 6, 24].
Evidence of congenital DM1 may be detected in utero, with features of clubfoot, polyhydramnios (common in mostly severe cases with swallowing problems in infancy), cardiomyopathy (severe cases), and reduced fetal movement. The mother may experience prolonged labor, likely due to abnormalities in the uterine muscle [3, 5]. Preterm (<34 weeks) and prolonged labor is likely in DM1-affected women, with a resultant increase in the rate of cesarean delivery [6]. Other complications of pregnancy have been reported in DM1. The occurrence of preterm labor was attributed in part to the effects of congenital DM on affected fetuses. Other pregnancy complications have been reported in both DM1 and DM2. In one study of pregnancies in women, ectopic pregnancy (4%), placenta previa (9%), and postpartum hemorrhage (rare) were reported. Uterine atony due to myotonia and muscle weakness may account for the postpartum hemorrhage observed in this cohort, although it is a rare occurrence in DM1-affected individuals [6].
Facial diplegia (bilateral facial paralysis) with a typical V-shaped upper lip, marked hypotonia, poor feeding, respiratory failure, and joint contractures (mainly in the legs) are all typical features of congenital DM1. Muscle weakness and facial diplegia persist into early childhood with a slow and progressive increase in motor function.
Feeding difficulties may necessitate nasogastric feeding and intensive management. Death may occur in the neonatal period due to respiratory abnormalities or in infancy due to abnormalities in cardiac function and feeding [4, 6]. Affected neonates and infants may have to be managed with a feeding tube and mechanical ventilation, in a significant number of cases [6].
Abnormalities of the foot as well as mental and behavioral disorders are common clinical presentations in early childhood (aged 3–5 years). Marked developmental delays in mental and motor development occur in half or more children at this stage. Survival into early childhood is fraught with marked morbidities in the cardiovascular and respiratory systems with a high likelihood of deaths.
The severity of complications in congenital DM1 shows no association with the range of abnormalities that may be observed in teens presenting with the classic, non-congenital DM phenotype. In these cases, abnormalities in muscular and cardiac function present with symptoms and signs of myotonia, muscle weakness (mainly in the lower parts of the body), and cardiac deficits.
Childhood-Onset/Juvenile DM1
In childhood DM1, the CTG-repeat lengths are usually greater (>800), with muscle weakness and physical disability developing later in life, usually before age 10 years [1, 5, 6, 32]. Cases in which the repeat lengths were lower than 800 have been reported [6]. In addition to musculoskeletal abnormalities, behavioral, cardiovascular, and mental complications are typical in childhood DM. Children with cardiovascular complications can go undiagnosed. Behavioral problems can manifest as attentional deficit, anxiety, executive dysfunction, low IQ, and mood disorder.
Adult-Onset Classic DM1
Individuals with classic DM1 have CTG-repeat sizes of 50–1000. Classic DM develops between the first and fourth decades of life, but in the adult-onset classic type of DM, muscle weakness develops later in life [1, 6]. Cataracts, cardiac arrhythmia, excessive daytime sleepiness, and myotonia are presenting features in individuals with classic DM1. Affected individuals have a lower average life expectancy.
Late-Onset/Asymptomatic DM1
Individuals with mild DM also display a CTG-repeat size of 50–150. The onset of DM usually occurs in later years (20–70 years), and individuals have a normal life expectancy. Features common to DM are typical of individuals with mild DM, including cataracts, mild weakness, and myotonia [6]. In a multicenter study, a high sensitivity for detection and reporting of expanded repeats was observed [36]. Also, a high specificity to within two repeats of the consensus was reported in allelic examinations [36]. The ability of genetic testing to accurately define the presence or absence of DM is central to DM management, as a cure is yet to be developed.
More than half of asymptomatic DM cases go undetected throughout childhood, only to have an arrhythmia induced in adolescence due to participation in sports and physical activities. Children of individuals with asymptomatic DM have a high risk of intergenerational expansion or activation, with larger repeat sizes and probability of having symptoms.
Premutation DM1
Generally, the presence of 5–37 repeats is considered normal.
Individuals with premutation DM1 display repeat sizes ranging between 38 and 49. Symptoms begin to manifest in individuals with greater than 50 repeats. Individuals with CTG expansions in the premutation range have been reported to be asymptomatic but are at increased risk of having their children inherit further expanded repeats and thus having symptomatic disease.
1.3.4 Population Genetics and Epidemiology
In general, the prevalence of DM1 ranges from approximately 1:100,000 in certain parts of Japan to 1:10,000 in Iceland; the global estimated prevalence of DM1 is reported to be 1:20,000 [37]. Prevalence may increase in specific regions, such as Quebec, and this is thought to be as a result of founder effects [38, 39]
1.4 DM2
After the discovery of DM1 gene defect, DNA testing identified a group of patients lacking this defect but showing dominantly inherited myotonia, proximal greater than distal weakness, and cataracts. In Europe, this disease was termed proximal myotonic myopathy (PROMM, OMIM 602668) [40, 41] or proximal myotonic dystrophy (PDM) [42], while in the United States, it was termed myotonic dystrophy with no CTG-repeat expansion or myotonic dystrophy type 2 (DM2) [43, 44, 45]. Subsequently, it was demonstrated that many of the families identified as having PROMM, PDM, or DM2 had the same disease that results from an unstable tetranucleotide CCTG-repeat expansion in intron 1 of the nucleic acid-binding protein (CNBP) gene (previously known as zinc-finger 9 gene, ZNF9; OMIM 116955) on chromosome 3q21 [46].
1.4.1 Mapping
The locus for DM2 is located on the 3q 21.3region. In one study of nine German families, linkage analysis based on DNA markers (D3S1541 and D3S1589) showed a close genetic link or allelism between PROMM and DM2, both located on the long arm of chromosome 3 [40].
In study of a five-generation family with myotonic dystrophy [44, 45], Ranum et al. found that the disease locus, DM2, mapped to a 10-cM region of 3q. In addition to excluding the DM1 locus on chromosome 19 in the large family reported by Ranum et al. [44], Day et al. subsequently excluded the chromosomal regions containing the genes for muscle sodium and chloride channels that are involved in other myotonic disorders [45].
Another report described a Norwegian PROMM family in which the proband was clinically diagnosed with myotonic dystrophy but lacked the pathognomonic (CTG)n expansion [41]. Haplotype analysis suggested exclusion of the DM2 locus as well, perhaps indicating further genetic heterogeneity. Interestingly, all family members, affected and unaffected, were heterozygous for the arg894-to-ter (R894X) mutation in the CLCN1 gene [47]. The authors noted that Mastaglia et al.[48] had reported the R894X mutation in only one of two children with PROMM, indicating that it was not the disease-causing mutation in that family: they had termed it an incidental finding. Furthermore, Sun et al. suggested that their findings, combined with those of Mastaglia et al., likely reflected a fairly high carrier frequency in the population, and they presented preliminary data indicating an R894X allele frequency of 0.87% (4/460) in northern Scandinavia [41].
1.4.2 Molecular Genetics
DM2 is caused by expansion of a tetranucleotide CCTG repeat in intron 1 of the CNBP gene on chromosome 3q21 [46]. Patients with DM2 exhibit a wide range of phenotypes that include myotonia, muscle weakness, cardiac anomalies, cataracts, diabetes mellitus, and testicular failure [49]. In a normal allele, the repeat shows a complex motif with an overall configuration of (TG)n(TCTG)n(CCTG)n. The number of CCTG tracts is less than 30, with repeat interruptions of GCTG and/or TCTG motifs [46], and is stably transmitted from one generation to the next. However, in the expanded allele, only the CCTG tract elongates, and the GCTG and TCTG interruptions disappear from the repeat tract. The sizes of expanded alleles are extremely variable, ranging from 75 to 11,000 repeats, with a mean of 5000 repeats. The expanded DM2 alleles show marked somatic instability, with significant increases in length over time [46]. Over 25% of affected individuals have two or more CCTG expansion sizes that can be detected in peripheral blood. Because of this somatic heterogeneity of the repeat size, it can be difficult to establish a pathogenic threshold, and thus affected individuals with the shortest identified CCTG-repeat expansion on one allele (approximately 75 CCTG repeats or 300 bp) would also have an allele with an extremely sizeable CCTG expansion with over 11,000 CCTG repeats (or 44,000 bp); any or both of the expanded alleles could be pathogenic. In DM2, the CCTG expansion usually contracts in the next generation, with no significant differences determined by the gender of the transmitting parent. This may explain the missing of congenital form and the lack of genetic anticipation in DM2.
Three classes of large non-DM2 alleles have been identified [50]. They include short interrupted alleles of up to 24 repeats in CCTG (with up to 2 interruptions), long interrupted alleles of up to 32 repeats in CCTG (with up to 4 interruptions), and uninterrupted alleles of 23–33 repeats in CCTG (with lengths of 92–132 base pairs). The instability common to these large, non-DM2 repeat alleles was higher in the uninterrupted alleles compared to interrupted alleles [50].
Analyses based on single-nucleotide polymorphisms showed a similar haplotype for expanded DM2 repeats and the three classes of large non-DM2 repeat alleles. Thus, a premutation allele pool from the unstable interrupted CCTG alleles may explain the full mutations seen in individuals with DM2. Large non-DM2 classes are more common among African-Americans (8.5%) than European-Caucasians (<2%) [50].
The DM2 CCTG repeat was originally reported to be derived from the Alu element insertion, similar to two other repeat expansion disorders, Friedreich’s ataxia and spinocerebellar ataxia type 10, posing challenges regarding the mechanisms underlying development of Alu-mediated repeats into large, highly unstable expansions common to all three disorders [51].
Although the mechanisms responsible for this unique instability are mostly unknown, the uninterrupted CCTG repeat tends to form a stable hairpin/dumbbell DNA structure with expansion due to an error in the recombination-repair mechanism [52, 53] DM2 CCTG*CAGG repeats are crossover hotspots that are more prone to expansions in comparison with the DM1 CTG*CAG repeats in Escherichia coli [52, 56].
1.4.3 Genotype-Phenotype Correlations
Different from DM1, no significant correlation exists between CCTG repeat size and age of onset of muscle weakness or other indices of disease severity (such as age at cataract extraction). The fact that phenotype in individuals with CCTG-repeat expansions in both CNBP alleles is as severe as those seen in their heterozygous sibs and parents goes to underscore that CCTG repeat number does not modify the clinical course [54].
In DM2, there is no clear correlation between repeat size and the age of the individual at the time that repeat size is measured, demonstrating that repeat length increases with age [46, 49, 55].
Among families that participated in the original characterization of DM2, the severity of clinical features were reported to have increased with successive generations [49, 55]. Data suggested that this was caused by the phenomenon earlier described phenomenon of anticipation (where individuals in successive generations tend to present at an earlier age and/or with more severe phenotype or clinical features) rather than bias constituted ascertainment (inadvertent inclusion of more severely affected younger-generation family members in the study).
However, molecular genetic testing for the CNBP gene showed that there was no obvious congenital form of DM2 analogous to the congenital form of DM1, which established the role of anticipation in that disease. Furthermore, the lack of correlation between disease severity and CCTG repeat length underscores the observation that intergenerational changes in repeat length in successive generations are unlikely to be associated with a definitive increase in disease severity [56].
1.4.4 Population Genetics and Epidemiology
Myotonic dystrophy is the most common adult form of muscular dystrophy, with an estimated incidence of approximately 1 in 8000 in the general population. The exact proportion of myotonic dystrophy represented by DM1 and the milder version DM2 are as yet unknown. Also, the varying range of severity of clinical features obscures the incidence of DM2. An incidence similar to DM1 has been reported in Europe, although it is likely lower in the United States [6].
Prevalence apparently differs in different populations, but comprehensive demographic studies in this regard are limited. The prevalence of DM2 has been reported to be higher in Germany and Poland and in individuals of either German or Polish descent [57] and is reported to be 1:1830 in Finland where the incidence of DM2 is markedly higher than that of DM1 [58]. There have been reports of cases of DM2 in Afghanistan and Sri Lanka, but the condition has not been observed in China, or sub-Saharan Africa. The majority of DM2 mutations have been identified in Caucasians of European descent that are known to have descended from a single common founder and share an identical haplotype [59, 60, 61]. Nevertheless, a previous study has identified the first Japanese DM2 pedigree harboring a distinctive haplotype different from that shared by Caucasians, suggesting the occurrence of DM2 in non-Caucasian populations as well, and that this likely has separate founders [62, 63]. Thus, it would be beneficial to examine whether there is phenotypic haplotypic comparability among DM2 patients from different ethnicities compared with the predominantly European patients that share a common haplotype.
1.5 Genetic Basis for Diagnosis
DM1 and DM2 are clinically and genetically similar but distinct disorders requiring different diagnostic and treatment strategies [3], and thus diagnosis is based on suggestive clinical features and testing, which comprises genetic and nongenetic tests. As is the case with all genetic disease, the key to diagnosis is confirming the presence of the causative mutation by genetic testing. Molecular genetic testing is the gold standard for establishing a diagnosis of DM.
The clinical diagnosis of DM1 can be made where characteristic clinical features such as muscle weakness, demonstrable progressive distal and bulbar dystrophy with myotonia, frontal balding, and cataracts in addition to a positive family history are present. However, clinical diagnosis can be difficult in mild cases, but a high index of suspicion should be maintained in atypical cases of DM, where muscle weakness is absent or a family history is lacking [6]. Clinical diagnosis can be further confirmed by demonstration of depressed serum IgG and elevated CPK as well as other ancillary tests.
For DM2, disease onset is characteristically in adulthood with generally milder symptoms than DM1, and the phenotype clinical manifestations are variable, with early-onset cataracts (before 50 years of age), proximal weakness, varying grip myotonia, as well as demonstrable autosomal dominant inheritance.
1.5.1 Genetic Testing
The major indications for genetic testing for DM1 are as recommended by the International Myotonic Dystrophy Consortium (IDMC) [64].
Genetic testing for DM can be confirmatory/symptomatic, preclinical/asymptomatic, prenatal testing, and preimplantation testing [65]. The procedure must be accompanied by genetic counseling.
1.5.2 Molecular Genetic Analysis
This is the gold standard for the diagnosis of DM [6]. Targeted analysis for pathogenic variants for the presence of expanded repeats for CTG in the DMPK gene (DM1) confirms a diagnosis of DM1. The number of DMPK CTG trinucleotide repeats can be quantified by PCR analysis for expanded alleles with about 100–150 CTG repeats. Southern blot analysis is used for detection of larger CTG expansions. The absence of the CTG-repeat expansion warrants tests for expanded repeats for CCTG in the CNBP gene, a characteristic of DM2.
However, it is requisite to identify the presence of CNBP, which is the only gene in which mutation is known to cause DM2. The CNBP intron 1 carries a complex repeat motif, (TG)n(TCTG)n(CCTG)n, and expansion of the CCTG repeat is causative of DM2 [46]. Because the extremely large size of the expansions in DM2 and marked somatic instability render Southern blot analysis and its interpretation difficult, a repeat-primed PCR assay was developed by Day et al. [49] and was found to increase molecular detection rate of DM2 to 99%. In comparison to laborious and time-consuming Southern blot, this simple PCR assay cannot determine the size of the expansion but can readily and speedily detect the mutational status (the existence of expansion). Recently, a simultaneous repeat assay for both DMPK CTG and CNBP CCTG expansion has also been developed [66].
1.5.3 Prenatal Testing
Individuals at risk of DM should be tested and advised of the options available to them. They should be allowed to make an informed decision concerning the outcome of prenatal tests [1, 10].
1.5.4 Preimplantation Genetic Diagnosis
Where available and in cases where the pathologic expanded repeats have been confirmed, evaluation of the genetic makeup of the fetus at implantation could further assist the parents in making an informed decision [1, 10].
1.6 Conclusion
DM is the most common adult muscular dystrophy, characterized by autosomal dominant progressive myopathy, myotonia, and multiorgan involvement. To date, two distinct forms caused by similar mutations in two different genes have been identified: DM1 and DM2. Aberrant transcription and mRNA processing of multiple genes due to RNA-mediated toxic gain of function have been suggested to cause the complex phenotype in DM1 and DM2. Although the size of the respective repeated DNA sequence was thought to correlate with disease severity and age of onset, the size of the repeats alone does not explain all the differences in DM phenotypes. This shows the importance of considering other factors to regulate disease phenotype in DM.
Abbreviations
- CNBP:
-
Cellular nucleic acid-binding protein
- DM:
-
Myotonic dystrophy
- DMPK:
-
Dystrophia myotonica protein kinase
- MBNL:
-
Muscle blind-like
- PROMM:
-
Proximal myotonic myopathy
- RNA:
-
Ribonucleic acid
- ZNF9:
-
Zinc-finger nuclease 9
References
Ranum LPW, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet. 2004;74(5):793–804. https://doi.org/10.1086/383590.
Ho G, Cardamone M, Farrar M. Congenital and childhood myotonic dystrophy: current aspects of disease and future directions. World J Clin Pediatr. 2015;4(4):66–80. https://doi.org/10.5409/wjcp.v4.i4.66.
Udd B, Krahe R, Sarma S, et al. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11(10):891–905. https://doi.org/10.1016/S1474-4422(12)70204-1.
Yenigun VB, Sirito M, Amcheslavky A, et al. (CCUG)n RNA toxicity in a Drosophila model of myotonic dystrophy type 2 (DM2) activates apoptosis. Dis Model Mech. 2017;10(8):993–1003. https://doi.org/10.1242/dmm.026179.
Thornton CA. Myotonic dystrophy. Neurol Clin. 2014;32(3):705–19., , viii. https://doi.org/10.1016/j.ncl.2014.04.011.
Basil TD Chad DA. Myotonic dystrophy: etiology, clinical features, and diagnosis. https://www.uptodate.com/contents/myotonic-dystrophy-etiology-clinical-features-and-diagnosis. Accessed 24 Aug 2017.
Zhang F, Bodycombe NE, Haskell KM, et al. A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum Mol Genet. 2017;36(16):e24. https://doi.org/10.1093/hmg/ddx190.
Dalton JC, Ranum LP, Day JW. Myotonic dystrophy type 2. Seattle: University of Washington; 1993. http://www.ncbi.nlm.nih.gov/pubmed/20301639. Accessed 24 Aug 2017.
Fugier C, Klein AF, Hammer C, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011;17(6):720–5. https://doi.org/10.1038/nm.2374.
Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280(5364):737–41. http://www.ncbi.nlm.nih.gov/pubmed/9563950. Accessed 24 Aug 2017.
Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29(1):40–7. https://doi.org/10.1038/ng704.
Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10(1):45–53. https://doi.org/10.1016/S1097-2765(02)00572-5.
Musova Z, Mazanec R, Krepelova A, et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am J Med Genet Part A. 2009;149A(7):1365–74. https://doi.org/10.1002/ajmg.a.32987.
Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta. 2015;1852(4):594–606. https://doi.org/10.1016/j.bbadis.2014.05.019.
Mohr J. A study of linkage in man. Copenhagen: Munksgaard; 1954.
O’Brien T, Ball S, Sarfarazi M, Harper PS, Robson EB. Genetic linkage between the loci for myotonic dystrophy and peptidase D. Ann Hum Genet. 1983;47(Pt 2):117–21. http://www.ncbi.nlm.nih.gov/pubmed/6881909. Accessed 24 Aug 2017
Davies KE, Jackson J, Williamson R, et al. Linkage analysis of myotonic dystrophy and sequences on chromosome 19 using a cloned complement 3 gene probe. J Med Genet. 1983;20:259–63. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1049116/pdf/jmedgene00108-0021.pdf. Accessed 25 Aug 2017
Shaw DJ, Brook JD, Meredith AL, Harley HG, Sarfarazi M, Harper PS. Gene mapping and chromosome 19. J Med Genet. 1986;23(1):2–10. http://www.ncbi.nlm.nih.gov/pubmed/3081724. Accessed 24 Aug 2017
Bartlett R, Pericak-Vance M, Yamaoka L, et al. A new probe for the diagnosis of myotonic muscular dystrophy. Science. 1987;235(4796):1648–50. http://science.sciencemag.org/content/235/4796/1648. Accessed 24 Aug 2017
Roses AD, Pericak-Vance MA, Ross DA, Yamaoka L, Bartlett RJ. RFLPs at the D19S19 locus of human chromosome 19 linked to myotonic dystrophy (DM). Nucleic Acids Res. 1986;14(13):5569. http://www.ncbi.nlm.nih.gov/pubmed/3016653. Accessed 24 Aug 2017
Friedrich U, Brunner H, Smeets D, Lambermon E, Ropers H-H. Three-point linkage analysis employing C3 and 19cen markers assigns the myotonic dystrophy gene to 19q. Hum Genet. 1987;75(3):291–3. https://doi.org/10.1007/BF00281077.
Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799–808. https://doi.org/10.1016/0092-8674(92)90154-5.
Yamagata H, Nakagawa M, Johnson K, Miki T. Further evidence for a major ancient mutation underlying myotonic dystrophy from linkage disequilibrium studies in the Japanese population. J Hum Genet. 1998;43(4):246–9. https://doi.org/10.1007/s100380050082.
De Temmerman N, Sermon K, Seneca S, et al. Intergenerational instability of the expanded CTG repeat in the DMPK gene: studies in human gametes and preimplantation embryos. Am J Hum Genet. 2004;75(2):325–9. https://doi.org/10.1086/422762.
Harper PS. Major problems in neurology: myotonic dystrophy. London, UK: WB Saunders; 2001.
Rakocevic-Stojanovic V, Savic D, Pavlovic S, et al. Intergenerational changes of CTG repeat depending on the sex of the transmitting parent in myotonic dystrophy type 1. Eur J Neurol. 2005;12(3):236–7. https://doi.org/10.1111/j.1468-1331.2004.01075.x.
Martorell L, Cobo AM, Baiget M, Naudó M, Poza JJ, Parra J. Prenatal diagnosis in myotonic dystrophy type 1. Thirteen years of experience: implications for reproductive counselling in DM1 families. Prenat Diagn. 2007;27(1):68–72. https://doi.org/10.1002/pd.1627.
Moxley RT. The myotonic dystrophies. In: Rosenberg RN, DiMauro S, Paulson HL, Ptacek L NE, editors. The Molecular and Genetic Basis of Neurologic and Psychiatric Disease. Boston, MA: Wolters Kluwer; 2008, pp. 532–541.
Wong LJ, Ashizawa T, Monckton DG, Caskey CT, Richards CS. Somatic heterogeneity of the CTG repeat in myotonic dystrophy is age and size dependent. Am J Hum Genet. 1995;56:114–22.
Monckton DG, Wong LI, Ashizawa T, Caskey CT. Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum Mol Genet. 1995;4:1–8.
Puymirat J, Giguere Y, Mathieu J, Bouchard J-P. Intergenerational contraction of the CTG repeats in 2 families with myotonic dystrophy type 1. Neurology. 2009;73(24):2126–7. https://doi.org/10.1212/WNL.0b013e3181c677e1.
Ashizawa T, Anvret M, Baiget M, et al. Characteristics of intergenerational contractions of the CTG repeat in myotonic dystrophy. Am J Hum Genet. 1994;54(3):414–23. http://www.ncbi.nlm.nih.gov/pubmed/8116611. Accessed 24 Aug 2017
Harley HG, Brook JD, Rundle SA, et al. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature. 1992;355(6360):545–6. https://doi.org/10.1038/355545a0.
Aslanidis C, Jansen G, Amemiya C, et al. Cloning of the essential myotonic dystrophy region and mapping of the putative defect. Nature. 1992;355(6360):548–51. https://doi.org/10.1038/355548a0.
Tsilfidis C, MacKenzie AE, Mettler G, Barceló J, Korneluk RG. Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat Genet. 1992;1(3):192–5. https://doi.org/10.1038/ng0692-192.
Richards CS, Palomaki GE, Hegde M. Results from an external proficiency testing program: 11 years of molecular genetics testing for myotonic dystrophy type 1. Genet Med. 2016;18(12):1290–4. https://doi.org/10.1038/gim.2016.59.
Theadom A, Rodrigues M, Roxburgh R, et al. Prevalence of muscular dystrophies: a systematic literature review. Neuroepidemiology. 2014;43(3–4):259–68. https://doi.org/10.1159/000369343.
Yotova V, Labuda D, Zietkiewicz E, et al. Anatomy of a founder effect: myotonic dystrophy in Northeastern Quebec. Hum Genet. 2005;117(2-3):177–87. https://doi.org/10.1007/s00439-005-1298-8.
Pratte A, Prévost C, Puymirat J, Mathieu J. Anticipation in myotonic dystrophy type 1 parents with small CTG expansions. Am J Med Genet Part A. 2015;167(4):708–14. https://doi.org/10.1002/ajmg.a.36950.
Ricker K, Grimm T, Koch MC, et al. Linkage of proximal myotonic myopathy to chromosome 3q. Neurology. 1999;52(1):170–1. http://www.ncbi.nlm.nih.gov/pubmed/9921867. Accessed 24 Aug 2017
Sun C, Henriksen OA, Tranebjaerg L. Proximal myotonic myopathy: clinical and molecular investigation of a Norwegian family with PROMM. Clin Genet. 1999;56(6):457–61. https://doi.org/10.1034/j.1399-0004.1999.560609.x.
Udd B, Krahe R, Wallgren-Petterson C, Falck B, Kalimo H. Proximal myotonic dystrophy: a family with autosomal dominant muscular dystrophy, cataracts, hearing loss and hypogonadism: heterogeneity of proximal myotonic syndromes? Neuromuscul Disord. 1997;7:217–28.
Thornton CA, Griggs RC, Moxley RT. Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol. 1994;35(3):269–72. https://doi.org/10.1002/ana.410350305.
Ranum LPW, Rasmussen PF, Benzow KA, Koob MD, Day JW. Genetic mapping of a second myotonic dystrophy locus. Nat Genet. 1998;19(2):196–8. https://doi.org/10.1038/570.
Day JW, Roelofs R, Leroy B, Pech I, Benzow K, Ranum LP. Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2). Neuromuscul Disord. 1999;9(1):19–27. http://www.ncbi.nlm.nih.gov/pubmed/10063831. Accessed 4 Sep 2017
Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293(5531):864–7. https://doi.org/10.1126/science.1062125.
* 118425 CHLORIDE CHANNEL 1, SKELETAL MUSCLE; CLCN1. http://omim.org/entry/118425#0010.
Mastaglia FL, Harker N, Phillips BA, et al. Dominantly inherited proximal myotonic myopathy and leukoencephalopathy in a family with an incidental CLCN1 mutation. J Neurol Neurosurg Psychiatry. 1998;64(4):543–7. http://www.ncbi.nlm.nih.gov/pubmed/9576553. Accessed 24 Aug 2017
Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60(4):657–64. http://www.ncbi.nlm.nih.gov/pubmed/12601109. Accessed 25 Aug 2017
Bachinski LL, Czernuszewicz T, Ramagli LS, et al. Premutation allele pool in myotonic dystrophy type 2. Neurology. 2009;72(6):490–7. https://doi.org/10.1212/01.wnl.0000333665.01888.33.
Kurosaki T, Ueda S, Ishida T, Abe K, Ohno K, Matsuura T. The Unstable CCTG Repeat Responsible for Myotonic Dystrophy Type 2 Originates from an AluSx Element Insertion into an Early Primate Genome. PLoS One. 2012;7(6):e38379. https://doi.org/10.1371/journal.pone.0038379.
Dere R, Wells RD. DM2 CCTG•CAGG repeats are crossover hotspots that are more prone to expansions than the DM1 CTG•CAG repeats in Escherichia coli. J Mol Biol. 2006;360(1):21–36. https://doi.org/10.1016/j.jmb.2006.05.012.
Lam SL, Wu F, Yang H, Chi LM. The origin of genetic instability in CCTG repeats. Nucleic Acids Res. 2011;39(14):6260–8. https://doi.org/10.1093/nar/gkr185.
Schoser BG, Kress W, Walter MC, Halliger-Keller B, Lochmüller H, Ricker K. Homozygosity for CCTG mutation in myotonic dystrophy type 2. Brain. 2004;127(Pt 8):1868–77. Epub 2004 Jul 1
Schneider C, Ziegler A, Ricker K, et al. Proximal myotonic myopathy: evidence for anticipation in families with linkage to chromosome 3q. Neurology. 2000;55(3):383–8. http://www.ncbi.nlm.nih.gov/pubmed/10932272. Accessed 4 Sep 2017
Dalton JC, Ranum LPW, Day JW. Myotonic dystrophy type 2. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle WA: University of Washington, Seattle; 2006. https://www.ncbi.nlm.nih.gov/books/NBK1466/.
Udd B, Meola G, Krahe R, et al. Report of the 115th ENMC workshop: DM2/PROMM and other myotonic dystrophies. 3rd Workshop, 14-16 February 2003, Naarden, The Netherlands. Neuromuscul Disord. 2003;13(7-8):589–96. https://doi.org/10.1016/S0960-8966(03)00092-0.
Suominen T, Bachinski LL, Auvinen S, et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet. 2011;19(7):776–82. https://doi.org/10.1038/ejhg.2011.23.
Liquori CL, Ikeda Y, Weatherspoon M, et al. Myotonic dystrophy type 2: human founder haplotype and evolutionary conservation of the repeat tract. Am J Hum Genet. 2003;73(4):849–62. https://doi.org/10.1086/378720.
Bachinski LL, Udd B, Meola G, et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet. 2003;73(4):835–48. https://doi.org/10.1086/378566.
Coenen MJH, Tieleman AA, Schijvenaars MMVAP, et al. Dutch myotonic dystrophy type 2 patients and a North-African DM2 family carry the common European founder haplotype. Eur J Hum Genet. 2011;19(5):567–70. https://doi.org/10.1038/ejhg.2010.233.
Saito T, Amakusa Y, Kimura T, et al. Myotonic dystrophy type 2 in Japan: ancestral origin distinct from Caucasian families. Neurogenetics. 2008;9(1):61–3. https://doi.org/10.1007/s10048-007-0110-4.
Nakayama T, Nakamura H, Oya Y, et al. Clinical and genetic analysis of the first known Asian family with myotonic dystrophy type 2. J Hum Genet. 2014;59(3):129–33. https://doi.org/10.1038/jhg.2013.133.
New nomenclature and DNA testing guidelines for myotonic dystrophy type 1 (DM1). The International Myotonic Dystrophy Consortium (IDMC). Neurology. 2000;54(6):1218–1221. http://www.ncbi.nlm.nih.gov/pubmed/10746587. Accessed 25 Aug 2017.
Savić Pavićević D, Miladinović J, Brkušanin M, et al. Molecular genetics and genetic testing in myotonic dystrophy type 1. Biomed Res Int. 2013;2013:1–13. https://doi.org/10.1155/2013/391821.
Radvansky J, Ficek A, Kadasi L. Upgrading molecular diagnostics of myotonic dystrophies: Multiplexing for simultaneous characterization of the DMPK and ZNF9 repeat motifs. Mol Cell Probes. 2011;25(4):182–5. https://doi.org/10.1016/j.mcp.2011.04.006.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Matsuura, T. (2018). Genetics of Myotonic Dystrophy. In: Takahashi, M., Matsumura, T. (eds) Myotonic Dystrophy. Springer, Singapore. https://doi.org/10.1007/978-981-13-0508-5_1
Download citation
DOI: https://doi.org/10.1007/978-981-13-0508-5_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-0507-8
Online ISBN: 978-981-13-0508-5
eBook Packages: MedicineMedicine (R0)