
Abstract
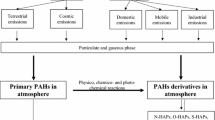
Polycyclic aromatic compounds are ubiquitous atmospheric pollutants with toxic, mutagenic, and carcinogenic properties. They are produced from the chemical reactions of their parent or related compounds in the atmosphere as well as from a wide variety of anthropogenic sources, such as fuel combustion. In this chapter, chemical reaction pathways for the atmospheric secondary formation of several polycyclic aromatic hydrocarbon (PAH) derivatives, i.e., gas-phase formation of mutagenic 1- and 2-nitrotriphenylene via OH or NO3 radical-initiated reactions of the parent triphenylene, formation of carcinogenic 1-nitropyrene from heterogeneous nitration of pyrene on mineral dust aerosols, atmospheric formation of hydroxynitropyrenes from a photochemical reaction of 1-nitropyrene, and photochemical degradation of selected nitrated and oxygenated PAHs on airborne particles under simulated solar UV irradiation, are addressed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Polycyclic aromatic compounds
- Secondary formation
- Photoreaction
- Heterogeneous reaction
- Gas-phase reaction
1 Introduction
Polycyclic aromatic hydrocarbons (PAHs ) and their derivatives (e.g., nitrated, oxygenated, and hydroxylated derivatives: NPAHs, OPAHs, and OHPAHs) are ubiquitous atmospheric pollutants with toxic, mutagenic, and carcinogenic properties (Salmeen et al. 1984; Schuetzle 1983). These compounds are produced from a wide variety of anthropogenic sources such as the incomplete combustion of fossil fuels used in industrial plants, through heating, and in diesel-powered vehicles (Schuetzle et al. 1982; Bamford et al. 2003; Bezabeh et al. 2003). NPAHs, which generally exhibit higher mutagenicity and carcinogenicity than their parent PAHs, are also generated from atmospheric reactions of PAHs released in the gas phase with radical species such as OH and NO3 radicals and nitrogen oxides (Atkinson and Arey 1994; Ciccioli et al. 1996; Reisen and Arey 2005; Sasaki et al. 1997; Zielinska et al. 1989). OPAHs and OHPAHs, which may show biological effects beyond mutagenicity and carcinogenicity, are also known to be formed secondarily via oxidation of PAHs and NPAH s in the atmosphere (Cvrčková et al. 2006; Warner et al. 2004). These PAH derivatives generally have lower volatility than the parent PAHs. As a result, they are more likely to be deposited on airborne particles, especially fine particles which are easily suspended in the atmosphere and can easily be inhaled into the human body. Thus, it is necessary to understand the secondary formation processes of PAH derivatives in order to fully understand their effects on humans. In this chapter, chemical reaction pathways for the secondary formation of PAH derivatives are addressed.
2 Formation of Nitrotriphenylenes by Radical-Initiated Reactions
As described above, several types of NPAHs are formed via gas-phase reactions of semi-volatile PAHs. For example, 2-nitropyrene (2-NP) is formed by the gas-phase reaction of pyrene with OH radicals in the presence of NO2, and 2-nitrofluoranthene (2-NFR) is formed by OH or NO3 radical-initiated reactions of fluoranthene in the gas phase (Atkinson and Arey 1994). Benzanthrone, which has very low vapor pressure, was also found to react with radical species and NO2 to yield a nitrated compound in the gas phase (Phousongphouang and Arey 2003). Nitrotriphenylenes (NTPs), which include the strongly mutagenic isomer 2-nitrotriphenylene (2-NTP), have been found in airborne particles as well (Ishii, et al. 2000, 2001; Kameda et al. 2004; Kawanaka et al. 2005). The possibility of the atmospheric formation of NTPs via reactions of triphenylene was based on seasonal and diurnal changes in the concentration of ambient NTPs (Ishii et al. 2001; Kameda et al. 2004). Because the strongly mutagenic 2-NTP has been found in air at concentrations comparable to those of 1-nitropyrene (1-NP) and 2-NFR, which are the most abundant airborne NPAH s (Ishii, et al. 2000, 2001; Kameda et al. 2004; Kawanaka et al. 2005), the contribution of 2-NTP to the mutagenicity of airborne particles may be significant. From the point of view of public hygiene, it is important to acquire more detailed data about the environmental occurrence of NTPs. Thus, the formation of 1-nitrotriphenylene (1-NTP) and 2-NTP by gas-phase OH or NO3 radical-initiated reactions of triphenylene was demonstrated using a flow reaction system (Kameda et al. 2006). Nitration of triphenylene with N2O5 in CCl4 was also examined in order to determine the isomer distributions of NTPs formed via the NO3 radical-initiated nitration of triphenylene and in order to predict the rate constants of the gas-phase OH or NO3 radical-initiated reactions of triphenylene.
The formation of 1- and 2-NTP was clearly shown by HPLC analysis of the products of the gas-phase reaction of triphenylene initiated by OH radicals. A 2-NTP/1-NTP ratio of 1.22 was obtained for OH radical-initiated nitration. In contrast, the NO3 radical-initiated reaction predominantly gave 2-NTP, with traces of 1-NTP. Because the amount of 1-NTP formed in the NO3 radical-initiated reaction was too small to determine, a precise 2-NTP/1-NTP value could not be calculated and was thus assumed to be a value greater than 1.5. The preferential production of 2-NTP was also observed in the nitration of triphenylene with N2O5 in CCl4, for which the yields of 1- and 2-NTP were 6 (±2) % and 35 (±2) %, respectively. It has been reported that the reactions of several kinds of PAH with N2O5 in CCl4 are similar in terms of nitro-isomer distribution to gas-phase NO3 radical-initiated nitration (Phousongphouang and Arey 2003; Zielinska et al. 1986). Thus, the analogous isomer distribution of NTP (the larger yield of 2-NTP over 1-NTP) in the gas-phase reaction to that in CCl4 liquid-phase nitration does not contradict previous findings regarding the nitration of PAHs. The mean 2-NTP/1-NTP ratio in samples of airborne particles was >1.55. This value was similar to the ratios from the radical-initiated reactions and was much higher than that of the diesel exhaust particulate (DEP) samples (2-NTP/1-NTP = 0.37). This indicates that the atmospheric radical-initiated reactions significantly contribute to the formation of airborne NTPs, especially 2-NTP. The gas-phase formation of NPAHs via OH or NO3 radical-initiated reactions involves the addition of an OH or NO3 radical to the PAH at the carbon atom with the highest electron density, followed by ortho-addition of NO2. This is followed by a loss of water or nitric acid. For triphenylene, the carbon at the 1-position is the most electron-rich (Barker et al. 1955; Radner 1983); therefore, preferential formation of 2-NTP over 1-NTP is expected in the gas-phase radical-initiated reaction (Fig. 7.1).

A schematic for the formation of 2-NTP from OH or NO3 radical-initiated reactions of triphenylene (Kameda 2011). (Reproduced with permission from Journal of Health Science Vol. 57 No. 6. Copyright 2011 The Pharmaceutical Society of Japan )
The rate constants of gas-phase reactions of triphenylene with OH and NO3 radicals at 298 K were predicted to be (8.6 ± 1.2) × 10−12 cm3molecule−1 s−1 and (6.6 ± 1.5) × 10−29 [NO2] cm3 molecule−1 s−1, respectively, using a relative-rate method in a CCl4 liquid-phase system (Kameda et al. 2013). Based on the ambient concentrations of 2-NTP and the rate constant obtained for the reaction of triphenylene with the radicals, the atmospheric loss rate of 2-NTP relative to 2-NFR (which is the most abundant NPAH and is also produced from radical reactions) was successfully estimated. That is to say, 2-NTP is less susceptible to decomposition than 2-NFR, under ambient conditions.
3 Secondary Formation of 1-Nitropyrene Promoted on Mineral Dust Aerosols
One of the most abundant NPAHs is 1-NP , which is formed through the combustion of fossil fuels such as coal and diesel fuel (Schuetzle 1983; Yang et al. 2010). 1-NP is likely a carcinogen (IARC 2013) and can also be formed from gas-particle phase heterogeneous reactions (Esteve et al. 2004; Finlayson-Pitts and Pitts 2000; Inazu et al. 2000; Miet et al. 2009; Nguyen et al. 2009; Ramdahl et al. 1984; Wang et al. 2000). It is formed by the reaction of pyrene (Py) with gaseous NO2 on various substrates such as graphite, as a model for soot (Esteve et al. 2004), and a variety of metal oxides, as models for mineral aerosols (Inazu et al. 2000; Miet et al. 2009; Ramdahl et al. 1984; Wang et al. 2000). However, the heterogeneous formation of atmospheric 1-NP has been previously thought to be negligible because the reaction rate and the yield of 1-NP through this process are not sufficient to account for ambient 1-NP concentration (Finlayson-Pitts and Pitts 2000; Nguyen et al. 2009; Ramdahl et al. 1984; Shiraiwa et al. 2009). Previous studies of heterogeneous NPAH formation used simple inorganic oxides such as SiO2, Al2O3, and TiO2 as models of mineral dust aerosols (Inazu et al. 2000; Ma et al. 2011; Wang et al. 2000), but these substances lack the complexity of real mineral dust aerosols and thus may not be good models for investigating heterogeneous NPAH formation. Mineral dust is a major component of airborne particulates on a global scale (Cwiertny et al. 2008). It is transported by wind from deserts or semiarid regions (Tanaka and Chiba 2006), which account for 40% of the total world land area (Fernández 2002). Organic compounds adsorbed on the surface of mineral dust can have important health implications (Falkovich et al. 2004). Thus, the formation of 1-NP from Py and NO2 on authentic mineral dust was examined (Kameda et al. 2016).
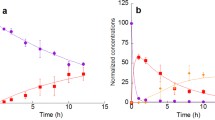
In the NO2 exposure experiments of particle-bound Py, degradation of Py was measured under 3 ppmv NO2 air in the dark. On quartz (SiO2) particles, Py was slowly converted to 1-NP, reaching a yield of ~40% in 12 h (Fig. 7.2a). On Chinese desert dust (CDD) particles, more than 90% of the initial amount of Py was degraded, and the maximum yield of 1-NP was attained after a reaction time of 1 h (Fig. 7.2b). 1-NP was then gradually converted to dinitropyrenes (DNPs) (Fig. 7.2b). Other mononitropyrene isomers were not detected. Desert dust is generally composed of various minerals including quartz, corundum (α-Al2O3), clay minerals, carbonates, feldspars, and hematite (Fe2O3) (Usher et al. 2003). To determine which components contribute to rapid nitration, the percentage of degraded Py (D Py) and the yield of 1-NP (Y 1-NP) were compared during a reaction time of 2 h on various substrates that generally constitute desert dust. The most active components were natural montmorillonites, kaolin, and saponite, as well as Arizona test dust (ATD; standard test dust made from Arizona desert sand) and CDD (Table 7.1). In most of these cases, the conversion of Py to 1-NP was completed within 2 h (Table 7.1). DNP formation was observed except on saponite. Kaolin, montmorillonites A and B, and saponite are types of clay minerals. For the other mineral substrates, such as quartz, carbonates (limestone and dolomite), and feldspars, D Py and Y 1-NP were less than 20%, and no DNP was formed during the NO2 exposure (Table 7.1). To quantify the rate of degradation of Py on each substrate , the kinetics of the heterogeneous reaction between NO2 and Py adsorbed on the substrates tested in this study were determined by following the consumption of Py as a function of NO2 exposure time. The apparent rate constants of the pseudo-first-order reaction, k obs, were (2.9 × 10−4 to 2.5 × 10−3) s−1 on CDD, ATD, and clay minerals and (2.5 × 10−6 to 9.0 × 10−5) s−1 on the other substrates when the concentration of NO2 was 3 ppmv.

Concentrations of Py and nitropyrenes (1-NP and DNPs) on quartz (a) and CDD (b) (expressed as a percent of the initial Py concentration) after exposure to 3 ppmv NO2 for the indicated times (Reproduced from Kameda et al. 2016). The data points represent mean values (± 1 SD) of triplicate experiments: circles, Py; squares, 1-NP; diamonds, DNPs (= 1,3-DNP + 1,6-DNP + 1,8-DNP). The curves for Py decay are exponential nonlinear least-square fits assuming first-order reactions. The curves for nitropyrene formation are for illustrative purposes only
The nitration of PAHs is catalyzed by acids (Shiri et al. 2010). Thus, the surface acid property of mineral dust may play a role in the heterogeneous nitration of Py. The surface acid properties of solid catalysts, including clay minerals, can be examined using Fourier transform infrared spectroscopy (FT-IR) with pyridine as a probe (Parry 1963). When pyridine binds to Brønsted acid sites, pyridinium ions are produced, which have an absorption band around 1545 cm−1. In contrast, pyridine molecules coordinated to Lewis acid sites have an absorption band around 1445 cm−1. The band at 1490 cm−1 is attributed to both molecules. The spectra of pyridine adsorbed onto some substrates (CDD, ATD, montmorillonites, kaolin, and saponite) have absorption bands at (1445 and 1490) cm−1, while no absorption band is observed around 1545 cm−1, except in the cases of kaolin and montmorillonite K10 (Fig. 7.3). This suggests that CDD and ATD, as well as clay minerals, have abundant acid sites, particularly Lewis acid sites. On the contrary, the spectra of the other substrates displayed no clear peaks, indicating that they have little to no acid sites on their surfaces. The largest k obs value was obtained for the reaction on montmorillonite K10, an acid-activated clay. These results strongly suggest that substrates showing acidic surface properties have an accelerating effect on the rate of heterogeneous nitration of PAHs by NO2.

IR spectra of pyridine adsorbed on the substrates examined in the study (Reproduced from Kameda et al. 2016)
Lewis acid sites on aluminosilicates are proposed to function as electron acceptors, leading to the formation of aromatic radical cations via electron transfer (Laszlo 1987; Soma and Soma 1989). The radical cations of several kinds of PAHs, such as Py, perylene, anthracene, and benzo[a]pyrene (which form on the surface of aluminosilicates), have been identified by spectroscopic methods, such as electron spin resonance (ESR) (Garcia and Roth 2002). These cations would couple with surface NO2 to yield NPAHs (Laszlo 1987), similar to that with the nitrous acid-catalyzed (NAC) nitration mechanism (Ridd 1991). That is, the rate-determining step would be the subsequent addition of NO2 to the aromatic radical cation yielding a σ complex (Wheland intermediate), and the deprotonation of this complex would constitute the final fast step that produces the nitrocompound (Fig. 7.4). Thus, the finding that the Lewis acid property of the substrates probably plays a role in nitration suggests that the rapid formation of 1-NP on mineral dust is the result of NO2 reacting with the radical cations of Py, which form at the surface Lewis acid sites (Fig.7.4).

Proposed mechanisms for the nitration of aromatic compounds (ArH) on the acidic surface of mineral dust (Reproduced from Kameda et al. 2016). (a) Schematic illustration of the heterogeneous nitration. The gas-particle interface is divided into a gas-phase with gaseous NO2 (NO2(g)), a sorption layer with adsorbed NO2 (NO2(s)), a quasi-static surface layer with the aromatic radical cation (ArH+ •), and a particle bulk. The blue arrow indicates the adsorption and desorption fluxes of NO2. The red and purple arrows indicate chemical reactions and electron transfer, respectively. This heterogeneous chemistry is based on the Pöschl-Rudich-Ammann (PRA) framework (Shiraiwa et al. 2009). (b) Chemical reaction scheme of the nitroaromatic compound (ArNO2) formation on Lewis acid sites of the dust surface (DS+)
4 Atmospheric Formation of Hydroxynitropyrenes from a Photochemical Reaction of Particle-Associated 1-Nitropyrene
The 1-NP taken up by humans and animals is transformed into various metabolites such as hydroxynitropyrenes (OHNPs), in the presence of cytochrome P450 enzymes (Rosser et al. 1996). Several isomers of OHNP (Fig. 7.5), such as 1-hydroxy-3-nitropyrene (1-OH-3-NP), 1-hydroxy-6-nitropyrene (1-OH-6-NP), and 1-hydroxy-8-nitropyrene (1-OH-8-NP), have also been observed on airborne particles (Gibson et al. 1986; Kameda et al. 2010) and on diesel exhaust particles (DEP ) (Manabe et al. 1985; Schuetzle 1983; Schuetzle et al. 1985). Several studies have found that most OHNP isomers have lower mutagenic activity than the parent 1-NP (Ball et al. 1984; Rosser et al. 1996; Manabe et al. 1985). Recently, however, OHNPs such as 1-OH-3-NP, 1-OH-6-NP, and 1-OH-8-NP have been found to act as endocrine disruptors: they act as estrogenic, anti-estrogenic, and anti-androgenic compounds (Kameda et al. 2008, 2011a) that may cause dysfunction of human and wildlife endocrine systems, abnormal development of reproductive systems, and immunodeficiencies. In view of the influence of OHNPs on human health, we need to learn more about their environmental concentration levels , sources , and behaviors. Therefore, the formation of OHNPs including 1-OH-3-NP, 1-OH-6-NP, and 1-OH-8-NP from photochemical reactions of 1-NP was examined in laboratory experiments to clarify the occurrence of atmospheric OHNPs (Kameda et al. 2011b).

Structure of hydroxynitropyrene
Figure 7.6 shows a profile of an HPLC analysis using chemiluminescence detection (HPLC/CLD ) for the products from photoreactions of 1-NP in methanol. Five chromatographic peaks were observed in the chromatogram (symbolized as A, B, C, D, and E). The retention times of peaks B, C, D, and E were the same as those of authentic 1-OH-6-NP, 1-OH-8-NP, 1-OH-3-NP, and 1-OH-2-NP, respectively. When analyzed by LC/MS/MS, a fraction containing the photoreaction products also yielded five peaks. By comparing the retention times and the MS/MS spectra of these peaks with those of the authentic standards, four known OHNPs (1-OH-2-NP, 1-OH-3-NP, 1-OH-6-NP, and 1-OH-8-NP) were identified. For these compounds, the molecule-related ion m/z 262 ([M-H]−) together with the characteristic fragment ions m/z 232 ([M-H-NO]−) and 216 ([M-H-NO2]−) was detected in a full scan analysis. In the study, and for the first time, 1-OH-3-NP, 1-OH-6-NP, and 1-OH-8-NP were found in 1-NP photoreaction products. The unknown compound that was observed in the HPLC/CLD chromatogram was also observed in the LC/MS/MS analysis. This compound also gave a characteristic MS/MS spectrum with a molecule-related ion m/z 262 and fragment ions m/z 232 and m/z 216. The similarity between the fragmentation patterns of the unknown compound and known OHNPs indicates that the unknown compound is an isomer of OHNP. The structure of the unknown compound obtained by the preparative scale photoreaction was then determined by analysis of its1H–NMR spectrum. On the basis of chemical shifts and coupling patterns, the unknown compound contained in the photo-reaction products was identified as 1-hydroxy-5-nitropyrene (1-OH-5-NP).

Chromatograms of standard OHNPs (a) and photoreaction products of 1-NP (b) obtained by the HPLC -chemiluminescence detection system (Kameda 2011) (Reproduced with permission from Journal of Health Science Vol. 57 No. 6. Copyright 2011 The Pharmaceutical Society of Japan )
All the OHNP isomers, which were found in the 1-NP photoreaction products, were also identified in ambient airborne particles collected at a typical residential area in Osaka, Japan . In contrast, 1-OH-2-NP and 1-OH-5-NP were not found in Standard Reference Materials (SRM) 1650b and SRM 1975, which are typical DEP samples. The concentrations of the other OHNP isomers in the DEP samples were much lower than the concentration of 1-NP. On the other hand, significantly higher concentration ratios of ΣOHNP (= 1-OH-3-NP + 1-OH-6-NP + 1-OH-8-NP) to 1-NP were observed in ambient airborne particles rather than in the DEP samples. In ambient airborne particles, the mean ΣOHNP/1-NP concentration ratio of 1.4 was 35 times higher than that in SRM 1650b and 470 times higher than that in SRM 1975. The diurnal concentration of 1-NP observed at the site in Osaka increased early in the morning and late in the evening, suggesting that automotive emissions contributed to the occurrence of 1-NP. The OHNP concentrations also rose in the morning, and variations of OHNP concentrations similar to those of 1-NP were observed during the daytime. However, the concentrations of OHNPs did not increase during the evening rush hour and were low at night (i.e., in the absence of sunlight). These results support the idea that atmospheric OHNPs are predominantly formed via secondary formation processes; photochemical reactions of 1-NP are expected to have a significant effect on the occurrence of OHNPs in the atmosphere.
5 Photochemical Decomposition of Selected Nitro- and Oxy-polycyclic Aromatic Hydrocarbons on Airborne Particles Under Simulated Solar UV Irradiation
As in the case of 1-NP , photo-induced decomposition is a dominant pathway for the degradation of particle-associated PAH derivatives (Finlayson-Pitts and Pitts 2000; Kamens et al. 1988). Solar radiation in the UV spectral region can modify PAH derivatives to form new compounds that may exhibit different types of biological effects. These might include the disruption of endocrine systems and the production of reactive oxygen species (ROS) in the human body (Chung et al. 2007). Thus, it is necessary to understand the photodecomposition of PAH derivatives in order to understand their effects on humans. However, the rate constants and the quantum yields related to the photolysis of the derivatized PAHs, which are the most significant factors for photodecomposition, have not been studied well. Thus, photodecomposition experiments for selected OPAHs and NPAHs, including 3-nitrobenzanthrone (3-NBA) a nitrated aromatic ketone that is strongly mutagenic (Enya et al. 1997), were conducted on a glass surface. With this as a simple model of airborne particles, the photolysis rate constants and the quantum yields for the PAH derivatives in the system were determined (Kameda et al. 2009). Furthermore, the atmospheric lifetime of the compounds due to photodecomposition was estimated using the actinic flux on the Earth’s surface, photolysis rate constants, and quantum yields obtained in the study.
The highest photolysis rate constant was observed for 9-nitroanthracene (9-NA), while 4-nitropyrene (4-NP) and 3-NBA were found to be the most stable of the nitrated compounds under UV irradiation (Table 7.2). It is hypothesized that photoreactivity of NPAHs is governed by the orientation of the nitro group; i.e., NPAHs having nitro groups perpendicular to the aromatic ring are more easily photodecomposed than those having parallel ones (Yang et al. 1994; Warner et al. 2004). The fast photodegradations observed for 9-NA, 6-nitrobenzo[a]pyrene (6-NBaP), and 7-nitrobenz[a]anthracene (7-NBaA), which all have a perpendicular nitro group, were consistent with this hypothesis (Fig. 7.7). The photoreaction products of NPAHs were reported to include quinoid PAHs (Warner et al. 2004), as well as photoreaction products of PAHs. Benzo[c]phenanthrene-5,6-quinone (BcP-5,6-Q), which has a similar photolysis rate constant to 9-nitrophenanthrene (9-NPh), degraded the fastest of the seven OPAHs tested. Of all the substituted PAHs examined in the study, 1,2-benzanthraquinone (1,2-BAQ) was most resistant to photodecomposition. Although previous studies on the photostability of OPAHs were quite limited, Cvrčková and Ciganek (2005) reported that 9,10-phenanthraquinone (9,10-PQ) was less stable than anthraquinone (AQ) under UV irradiation. This is consistent with the results.


Structures of PAH derivatives examined in the photodecomposition experiment
The photolysis rate constants of the compounds tested under solar irradiation were estimated based on the actinic flux at the Earth’s surface (Demerjian et al. 1980) and on the quantum yields obtained in the study. Atmospheric lifetimes of the compounds due to photodecomposition were calculated to be 0.5–22 h for NPAHs and 4.5–35 h for OPAHs using the obtained photolysis rate constants. OPAHs were found to be more stable against photo-irradiation than were NPAHs . This indicates that the risk induced by OPAHs is critical for human health, because we may be continuously exposed to OPAHs due to their longer residence time in the atmosphere. Recently, it has been found that ROS , which can cause severe oxidative stress connected with inflammatory processes, is produced in larger amounts via chain reactions induced by quinoid PAHs in the human body (Chung et al. 2007). Further studies on the biological effects and on the atmospheric formation and decomposition mechanisms of OPAHs are required.
References
Atkinson R, Arey J (1994) Atmospheric chemistry of gas-phase polycyclic aromatic hydrocarbons: formation of atmospheric mutagens. Environ Health Perspect 102:117–126
Ball LM, Kohan MJ, Claxton LD, Lewtas J (1984) Mutagenicity of derivatives and metabolites of 1-nitropyrene: activation by rat liver S9 and bacterial enzymes. Mutat Res 138:113–125
Bamford HA, Bezabeh DZ, Schantz MM, Wise SA, Baker JE (2003) Determination and comparison of nitrated-polycyclic hydrocarbons measured in air and diesel particulate reference materials. Chemosphere 50:575–587
Barker CC, Emmerson RG, Periam JD (1955) The monosubstitution of triphenylene. J Chem Soc:4482–4485
Bezabeh DZ, Bamford HA, Schantz MM, Wise SA (2003) Determination of nitrated polycyclic aromatic hydrocarbons in diesel particulate-related standard reference materials by using gas chromatography/mass spectrometry with negative ion chemical ionization. Anal Bioanal Chem 375:381–388
Chung SW, Chung HY, Toriba A, Kameda T, Tang N, Kizu R, Hayakawa K (2007) An environmental quinoid polycyclic aromatic hydrocarbon, acenaphthenequinone, modulates cyclooxygenase-2 expression through reactive oxygen species generation and nuclear factor kappa B activation in A549 cells. Toxicol Sci 95:348–355
Ciccioli P, Cecinato A, Brancaleoni E, Frattoni M, Zacchei P, Miguel AH, de Castro Vasconcellos P (1996) Formation and transport of 2-nitrofluoranthene and 2-nitropyrene of photochemical origin in the troposphere. J Geophys Res 101:19567–19581
Cvrčková O, Ciganek M (2005) Photostability of polycyclic aromatic hydrocarbons (PAHs) and nitrated polycyclic aromatic hydrocarbons (NPAHs) in dichloromethane and isooctane solutions. Polycycl Aromat Compd 25:141–156
Cvrčková O, Ciganek M, Šimek Z (2006) Anthracene, chrysene, their nitro- and methyl-derivatives photostability in isooctane. Polycycl Aromat Compd 26:331–344
Cwiertny DM, Young MA, Grassian VH (2008) Chemistry and photochemistry of mineral dust aerosol. Annu Rev Phys Chem 59:27–51
Demerjian KL, Schere KL, Peterson JT (1980) Theoretical estimates of actinic (spherically integrated) flux and photolytic rate constants of atmospheric species in the lower troposphere. Adv Environ Sci Technol 10:369–459
Enya T, Suzuki H, Watanabe T, Hirayama T, Hisamatsu Y (1997) 3-nitrobenzanthrone, a powerful bacterial mutagen and suspected human carcinogen found in diesel exhaust and airborne particulates. Environ Sci Technol 31:2772–2776
Esteve W, Budzinski H, Villenave E (2004) Relative rate constants for the heterogeneous reactions of OH, NO2 and NO radicals with polycyclic aromatic hydrocarbons adsorbed on carbonaceous particles. Part 1: PAHs adsorbed on 1-2 μm calibrated graphite particles. Atmos Environ 38:6063–6072
Falkovich AH, Schkolnik G, Ganor E, Rudich Y (2004) Adsorption of organic compounds pertinent to urban environments onto mineral dust particles. J Geophys Res Atmos 109:D02208
Fernández RJ (2002) Do humans create deserts? Trends Ecol Evol 17:6–7
Finlayson-Pitts BJ, Pitts JN Jr (2000) Chemistry of the upper and lower atmosphere. Academic Press, San Diego, CA
Garcia H, Roth HD (2002) Generation and reactions of organic radical cations in zeolites. Chem Rev 102:3947–4007
Gibson TL, Korsog PE, Wolff GT (1986) Evidence for the transformation of polycyclic organic matter in the atmosphere. Atmos Environ 20:1575–1578
Inazu K, Tsutsumi N, Aika KI, Hisamatsu Y (2000) SO2-enhanced nitration of fluoranthene and pyrene adsorbed on particulate matter in the heterogeneous reaction in the presence of NO2. Polycycl Aromat Compd 20:191–203
International Agency for Research on Cancer (2013) Diesel and gasoline engine exhausts and some nitroarenes. In: IARC monographs on the evaluation of carcinogenic risks to humans, vol. 105. International Agency for Research on Cancer
Ishii S, Hisamatsu Y, Inazu K, Aika K (2000) Ambient measurement of nitrotriphenylenes and possibility of nitrotriphenylene formation by atmospheric reaction. Environ Sci Technol 34:1893–1899
Ishii S, Hisamatsu Y, Inazu K, Aika K (2001) Environmental occurrence of nitrotriphenylene observed in airborne particulate matter. Chemosphere 44:681–690
Kameda T, Inazu K, Hisamatsu Y, Takenaka N, Bandow H (2004) Determination of atmospheric nitro-polycyclic aromatic hydrocarbons and their precursors at a heavy traffic roadside and at a residential area in Osaka, Japan. Polycycl Aromat Compd 24:657–666
Kameda T, Inazu K, Hisamatsu Y, Takenaka N, Bandow H (2006) Isomer distribution of nitrotriphenylenes in airborne particles, diesel exhaust particles, and the products of gas-phase radical-initiated nitration of triphenylene. Atmos Environ 40:7742–7751
Kameda T, Akiyama A, Toriba A, Tachikawa C, Yoshita M, Tang N, Hayakawa K (2008) Evaluation of endocrine disrupting activities of monohydroxylated derivatives of 1-nitropyrene by yeast two-hybrid assay. J Health Sci 54:118–122
Kameda T, Nakayama Y, Goto T, Koyanagi T, Bandow H, Fujimori K, Toriba A, Tang N, Hayakawa K (2009) Photochemical degradation of selected nitro- and oxy-polycyclic aromatic hydrocarbons on airborne particles under simulated solar UV-irradiation. Airborne Particulates. Nova Science Publishers, New York, pp 291–307
Kameda T, Akiyama A, Toriba A, Tang N, Hayakawa K (2010) Determination of particle-associated hydroxynitropyrenes with correction for chemical degradation on a quartz fibre filter during high volume air sampling. Int J Environ Anal Chem 90:976–987
Kameda T, Akiyama A, Toriba A, Tachikawa C, Yoshita M, Tang N, Hayakawa K (2011a) Mutagenicities and endocrine-disrupting activities of 1-hydroxy-2-nitropyrene and 1-hydroxy-5-nitropyrene. J Health Sci 57:372–377
Kameda T, Akiyama A, Toriba A, Tang N, Hayakawa K (2011b) Atmospheric formation of hydroxynitropyrenes from a photochemical reaction of particle-associated 1-nitropyrene. Environ Sci Technol 45:3325–3332
Kameda T, Inazu K, Asano K, Murota M, Takenaka N, Sadanaga Y, Hisamatsu Y, Bandow H (2013) Prediction of rate constants for the gas phase reactions of triphenylene with OH and NO3 radicals using a relative rate method in CCl4 liquid phase-system. Chemosphere 90:766–771
Kameda T, Azumi E, Fukushima A, Tang N, Matsuki A, Kamiya Y, Toriba A, Hayakawa K (2016) Mineral dust aerosols promote the formation of toxic nitropolycyclic aromatic compounds. Sci Rep 6:24427
Kamens RM, Guo Z, Fulcher J, Bell D (1988) Influence of humidity, sunlight, and temperature on the daytime decay of polyaromatic hydrocarbons on atmospheric soot particles. Environ Sci Technol 22:103–108
Kawanaka Y, Sakamoto K, Wang N, Yun S (2005) Determination of nitroarenes and 3-nitrobenzanthrone in atmospheric particulate matter by gas chromatography/tandem mass spectrometry with negative ion chemical ionization. Bunseki. Kagaku 54:685–691
Laszlo P (1987) Chemical reaction on clays. Science 235:1473–1477
Ma JZ, Liu YC, He H (2011) Heterogeneous reactions between NO2 and anthracene adsorbed on SiO2 and MgO. Atmos Environ 45:917–924
Manabe Y, Kinouchi T, Ohnishi Y (1985) Identification and quantification of highly mutagenic nitroacetoxypyrenes and nitrohydoroxypyrenes in diesel-exhaust particles. Mutat Res 158:3–18
Miet K, Le Menach K, Flaud PM, Budzinski H, Villenave E (2009) Heterogeneous reactivity of pyrene and 1-nitropyrene with NO2: kinetics, product yields and mechanism. Atmos Environ 43:837–843
Nguyen ML, Bedjanian Y, Guilloteau A (2009) Kinetics of the reactions of soot surface-bound polycyclic aromatic hydrocarbons with NO2. J Atmos Chem 62:139–150
Parry EP (1963) An infrared study of pyridine adsorbed on acidic solids characterization of surface acidity. J Catal 2:371–379
Phousongphouang PT, Arey J (2003) Sources of the atmospheric contaminants, 2-nitrobenzanthrone and 3-nitrobenzanthrone. Atmos Environ 37:3189–3199
Radner F (1983) Nitration of polycyclic aromatic hydrocarbons with dinitrogen tetroxide. A simple and selective synthesis of mononitroderivatives. Acta Chem Scand B 37:65–67
Ramdahl T, Bjorseth A, Lokensgard DM, Pitts JN (1984) Nitration of polycyclic aromatic hydrocarbons adsorbed to different carriers in a fluidized-bed reactor. Chemosphere 13:527–534
Reisen F, Arey J (2005) Atmospheric reactions influence seasonal PAH and nitro-PAH concentrations in the Los Angeles basin. Environ Sci Technol 39:64–73
Ridd JH (1991) The range of radical processes in nitration by nitric acid. Chem Soc Rev 20:149–165
Rosser PF, Ramachandran P, Sangaiah R, Austin RN, Gold A, Ball LM (1996) Role of O-acetyltransferase in activation of oxidised metabolites of the genotoxic environmental pollutant 1-nitropyrene. Mutat Res 369:209–220
Salmeen IT, Pero AM, Zator R, Schuetzle D, Riley TL (1984) Ames assay chromatograms and the identification of mutagens in diesel particle extracts. Environ Sci Technol 18:375–382
Sasaki J, Aschmann SM, Kwok ESC, Atkinson R, Arey J (1997) Products of the gas-phase OH and NO3 radical-initiated reactions of naphthalene. Environ Sci Technol 31:3173–3179
Schuetzle D (1983) Sampling of vehicle emissions for chemical analysis and biological testing. Environ Health Perspect 47:65–80
Schuetzle D, Riley TL, Prater TJ, Harvey TM, Hunt DF (1982) Analysis of nitrated polycyclic aromatic-hydrocarbons in diesel particulates. Anal Chem 54:265–271
Schuetzle D, Jensen TE, Ball JC (1985) Polar polynuclear aromatic hydrocarbon derivatives in extracts of particulates: Biological characterization and techniques for chemical analysis. Environ Int 11:169–181
Shiraiwa M, Garland RM, Pöschl U (2009) Kinetic double-layer model of aerosol surface chemistry and gas-particle interactions (K2-SURF): degradation of polycyclic aromatic hydrocarbons exposed to O3, NO2, H2O, OH and NO3. Atmos Chem Phys 9:9571–9586
Shiri M, Zolfigol MA, Kruger HG, Tanbakouchian Z (2010) Advances in the application of N2O4/NO2 in organic reactions. Tetrahedron 66:9077–9106
Soma Y, Soma M (1989) Chemical reactions of organic compounds on clay surfaces. Environ Health Perspect 83:205–214
Tanaka TY, Chiba M (2006) A numerical study of the contributions of dust source regions to the global dust budget. Glob Planet Chang 52:88–104
Usher CR, Michel AE, Grassian VH (2003) Reactions on mineral dust. Chem Rev 103:4883–4939
Wang H, Hasegawa K, Kagaya S (2000) The nitration of pyrene adsorbed on silica particles by nitrogen dioxide. Chemosphere 41:1479–1484
Warner SD, Farant JP, Butler IS (2004) Photochemical degradation of selected nitropolycyclic aromatic hydrocarbons in solution and adsorbed to solid particles. Chemosphere 54:1207–1215
Yang DTC, Chou A, Chen E, Chiu LH (1994) Photodecomposition of environmental nitro-polycyclic aromatic hydrocarbons. Polycycl Aromat Compd 5:201–208
Yang XY, Igarashi K, Tang N, Lin J, Wang W, Kameda T, Toriba A, Hayakawa K (2010) Indirect- and direct-acting mutagenicity of diesel, coal and wood burning-derived particulates and contribution of polycyclic aromatic hydrocarbons and nitropolycyclic aromatic hydrocarbons. Mutat Res 695:29–34
Zielinska B, Arey J, Atkinson R, Ramdahl T, Winer AM, Pitts JN Jr (1986) Reaction of dinitrogen pentoxide with fluoranthene. J Am Chem Soc 108:4126–4132
Zielinska B, Arey J, Atkinson R, Ramdahl T, Winer AM (1989) The nitroarenes of molecular weight 247 in ambient particulate samples collected in southern California. Atmos Environ 23:223–229
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kameda, T. (2018). Atmospheric Reactions of PAH Derivatives: Formation and Degradation. In: Hayakawa, K. (eds) Polycyclic Aromatic Hydrocarbons. Springer, Singapore. https://doi.org/10.1007/978-981-10-6775-4_7
Download citation
DOI: https://doi.org/10.1007/978-981-10-6775-4_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-6774-7
Online ISBN: 978-981-10-6775-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)