
Abstract
Therapeutic drug monitoring (TDM) is a clinical science centered around the quantification of drug concentrations in body fluids. To be considered candidates for TDM drugs must possess certain characteristics, including a narrow therapeutic index; in other words, to avoid toxicity at clinical doses. The relationship between dose and systemic concentration is particularly poor and unpredictable in special populations liable to different or dynamically changing pharmacokinetics. Antimicrobial use in the critical care setting has received special attention, where adequate concentrations for efficacy are especially pertinent. This chapter reviews recent evidence for TDM of established candidate drugs, aminoglycosides and vancomycin, and emerging evidence for beta-lactams, fluoroquinolones, linezolid, colistin and daptomycin, in the critically ill patient. The use of dose adaptation software for dose adjustment is discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


9.1 Introduction
Therapeutic drug monitoring (TDM) is a clinical science centered around the quantification of drug concentrations in bodily fluids, most often serum or plasma derived from a venous blood sample. This may be for the purposes of determining lack of response (suspected poor compliance or dosing/administration errors, or of unknown cause), elevated levels following intentional or unintentional overdosing; however most often it is used for adjusting the course of therapy to achieve optimal concentrations in the systemic circulation where a “therapeutic range” or target has been defined. TDM is traditionally applied to a finite set of drugs including a limited number of antibiotics, early generation anti-epileptics, mood stabilizers and antipsychotics, immunosuppressants, specific anticancer agents and other, often older, drugs such as digoxin and theophylline. Commercially available immunoassays encompass the most widely used technique to determine drugs that are commonly monitored, principally because procedural aspects are simplified, costs are lower, and the turnaround time is faster.
Professional societies and individual authors have put forward characteristics for drugs to be considered candidates for TDM [1]:
-
There is a relationship between systemic concentrations and efficacy or toxicity, and this relationship has been evidenced and defined.
-
Knowledge of the concentration would impact clinical decision making: adjustment of dose would be difficult or impossible to perform based on clinical observation alone.
-
The relationship between the dose and circulating concentrations is poor, and large inter-patient variability exists.
-
A narrow therapeutic range; that is, the concentration required for efficacy is close to concentrations where toxicity might be observed.
While this list aims to orient a rational application of the discipline, it has driven common use to a limited list of drugs that are considered classic TDM candidates, primarily selected due to their narrow therapeutic indices; in other words, to avoid toxicity at traditionally used doses. Large inter-patient variability is an important criterion; however prior to market this is often evaluated during trials in healthy subjects or relatively homogenous target populations. Additionally, in modern drug development, strategies are employed to limit the causes of inter-patient variation observed in earlier generations of drugs, namely absorption and hepatic transformation. Care is also taken to determine toxicity at standard dosing. Thus, as concerns drug development, regulation, and health policy, TDM based on the definition and scope above has had limited clinical application, and this has continued as newer drugs have emerged.
The relationship between dose and systemic concentration is particularly poor and unpredictable in special populations liable to different and/or dynamically changing pharmacokinetics [2]. In these patients, it might be difficult to gauge if doses and administration regimens used result in appropriate systemic concentrations. In general terms, they include patients at extremes of age, complex drug regimens with likely interactions between co-administered drugs, pregnant women, and obese patients. Disease processes where pharmacokinetics may differ from an “average” patient chronically, and alter further with acute disease, include cystic fibrosis, patients with renal and hepatic disease, and hematological malignancy, amongst others. Acute and severe pathophysiological processes that can influence pharmacokinetics include sepsis, septic shock, severe burns, traumatic brain injury, major surgery, organ transplantation, and pancreatitis.
Adequate antimicrobial concentrations for efficacy are especially pertinent in the critically ill patient, in whom unique pharmacokinetic changes may result in essentially diluted concentrations resulting from standard dosing regimens. Further, illness severity and disease processes may impact access to the site of infection, and infectious organisms are often less sensitive [3]. In this context, careful dosing based on drug concentrations may improve efficacy, help to avoid resistance, or detect and control for it if it emerges during the course of treatment [4,5,6]. This theoretical clinical need would augment the list of drugs that may require TDM beyond the traditional list for which immunoassays are available, provided suitable target concentrations can be established.
While immunoassays are used in health care systems worldwide, clinical chemistry laboratories providing a specialized service based on in-house methods and chromatographic techniques are relatively few. Instrumentation involved can include High Performance Liquid Chromatography (HPLC) or LC separation, with ultraviolet (UV), mass spectrometry (MS), and more recently with tandem MS detection, which provides additional sensitivity and specificity, improved throughput and turnaround, reduced sample volumes and analysis of multiple drugs simultaneously [7]. Specific drug assays, especially for older candidate TDM drugs, might be standardized and approved by regulatory bodies, while emerging assays pose challenges to standardize between centers, and participation in proficiency testing schemes is advocated. Development of new immunoassays and improvement of existing ones by the diagnostic industry has been limited in responding to clinical need. In deed, insufficient analytical quality associated with specific immunoassays may be part of the limited acceptance of TDM in clinical practice. The clinical chemistry laboratory can thus provide an invaluable service when it is positioned to articulate with clinical teams. A dedicated TDM program may contribute to ensuring a rational approach to requests, appropriate sampling and recording of complementary information, and foster quality control and ongoing education.
9.2 Beyond “Numbers Only” TDM: Additional Considerations for Antimicrobial Drugs
Across the traditional list of TDM candidate drugs, a drug level is taken at a single time point, and related to a range representing a margin of efficacy and absence of toxicity within a given population. This is typically a trough level, taken at the end of a dose interval, but could be at any point during the dosing interval that best relates to the area under the concentration–time curve (AUC). Taking a single sample is considered convenient and cost-effective; however, timing must be precise.
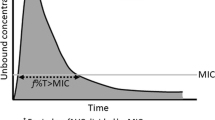
Antimicrobial activity for a given class of antimicrobial drugs is best described by one of three pharmacokinetic/pharmacodynamic (PK/PD) models: concentration-dependent, contingent on the drug’s maximal concentration above the microorganism’s minimum inhibitory concentration for the same drug (Cmax/MIC); time-dependent, dependent on the duration that the concentration is above the MIC(%T > MIC); and concentration- and time-dependent, contingent on AUC/MIC [8]. For drugs where toxicity is observed at clinical dosing schedules, such as the aminoglycosides and vancomycin, this often relates to accumulated exposure and might be best represented by AUC.
The AUC for a given dose interval can be calculated by a variety of means; however, most involve multiple samples and require specialized knowledge. Dose adaptation based on Bayesian forecasting and control, also referred to as Bayesian feedback and Target Concentration Intervention, promises several advantages. Calculations require prior information, including patient characteristics, and pharmacokinetic parameters for the drug from a similar population to the patient being treated.
Advantages of using Bayesian dose adaptation software:
-
Allows calculating an initial dose or loading dose
-
It is not necessary to wait for steady state to be achieved, and TDM can proceed from the first dosing interval
-
Allows calculating the AUC, and determining AUC-based outcome measures
-
AUC can be calculated with a minimum number of samples, often a single sample
-
Time of sampling is more flexible. An inadvertently taken sample can be useful so long as sampling time is accurately taken into account
-
If a visual representation of the concentration–time curve is provided, this is useful for educating patients (when relevant) or staff involved in the TDM process
-
For antimicrobial drugs, MIC can be included for an optimal PK/PD target
Beyond logistical challenges and the learning curve for implementation, one disadvantage of some of these software is an inability to include covariates that are not traditonal PK covariates. Biomarkers related to hepatic metabolism pose a particular challenge, as they are typically surrogates, and may vary between drugs. Examples include liver enzymes, C-reactive protein [9], and genotypes of genes of metabolic enzymes that exhibit polymorphism. Some experienced practitioners use the population modelling software NONMEM [10] for Bayesian dose adaptation in individuals, but we will foscus on specific tools here.
9.2.1 Available Dose Adaptation Tools for Clinical Use
There has been an interest in dose adaption since the 1970s; however, few centers worldwide apply the use of dedicated software to routine TDM. While there are many options, we will focus on tools that employ Bayesian methods, and have relatively large drug libraries or can accommodate additional models. Generally, these are academic initiatives, or commence as such. Most are Windows based, although some overcome system interoperability by providing web versions, also permitting use on personal smart devices. For an excellent historical review and evaluation, the reader is referred to Fuchs et al. [11].
USC*PACK was released in 1973 and represents an initiative from the Laboratory of Applied Pharmacokinetics of the University of Southern California [12]. Of the various software within the pack, MM-USC*PACK permits dose adaptation. The software has continually evolved, was briefly renamed RightDose but subsequently superseded by BestDose, which is currently being actively and commercially developed [13]. BestDose and predecessors are unique amongst the software described here in that they are based on nonparametric methods.
MWPharm was developed in 1982 at the Department of Pharmacology and Pharmacotherapy of the University of Groningen [14]. Mediware, a company originating from Charles University, Prague, continued development of the software from the late 1980s. The DOS version has been used since the 1990s in clinical pharmacology departments, including the University Medical Centre of Groningen, and national training programs for clinical pharmacists and pharmacologists in the Netherlands. Windows versions of the software have been developed by Mediware, the latest being MWPharm++ released in 2014. It is one of the few software that permits interfacing with hospital inpatient systems through Mirth TM Connect Technology. MWPharm Online is a recently released browser version.
RxKinetics is a suite of software tools for pharmacists, including Bayesian dose adaptation tools, developed by Rick Tharp, pharmacist and certified developer [15]. Antibiotic Kinetics and APK offer one-compartment models, while Kinetics offers multi-compartment models. In addition to Windows versions, Antibiotic Kinetics and APK offer versions for smart devices, and Antibiotic Kinetics offers an inexpensive iPhone application. Analyses for non-steady-state conditions are a recent initiative [16]. The website fosters an online community of users.
Two commercial solutions from the United States were released in the 1990s. Abbott Laboratories released Abbottbase Pharmacokinetic Systems [17]; however, it is no longer distributed. The original software was used widely in the United States, and is widely cited. T.D.M.S. 2000 by Healthware Incorporated was released in the 1990s and continues to be distributed [18]. A trial version permits individual calculations without the ability to save data, and is widely used.
TCIWorks, Target Concentration Intervention software, was released in 2011 and is a joint initiative of collaborators from the University of Otago, Dunedin, and the University of Queensland, Brisbane [19]. It has been widely used in Australia, primarily for challenging scenarios involving traditional TDM candidate drugs and where estimation of AUC has been advocated. TCIworks can be used on systems supporting JAVA applications, including Windows, Linux, and Mac, and is free of charge. The website is currently inaccessible and it is unclear if the software will continue to be developed; however, available versions of the software continue to be used.
DoseMe is a comprehensive software released in 2013 by an Australian proprietary company of the same name [20]. It appears to offer an extremely easy to use interface, and is supported on all platforms (Windows, Mac, Linux, Android, and iOS devices). Pricing is not disclosed on the website but appears to be in the format of an annual fee to clinical institutions. Finally, InsightRx is a recent spin-off from the University of California, San Fransisco, currently undergoing pilot studies for Busulfan and Vancomycin.
A unique initiative is the web-based service provided by the Limoges University Hospital, France [21]. While immunosuppressant dosing is the specialty (ImmunoSuppressant Bayesian dose Adjustment, ISBA platform), the more recent PK-JUST platform covers other drugs, including aminoglycoside and glycopeptide antibiotics. Clinical area is taken into account, including ICU, hematology, pediatrics, and aged care. Users enter drug levels through a form, modelling is performed and reviewed by a pharmacologist, and a report generated including dosing suggestions, a modelled pharmacokinetic curve, and historical concentration plots when relevant. The average turnaround time is 2 h. To date, the portal is free to use for international users, and a small fee is applied for national requests.
9.3 Pathophysiological Changes in the Critically Ill Patient
While other chapters of this book elaborate pathophysiological changes in the critically ill patient in greater detail, we will briefly cover some aspects here to consider the impact on circulating drug concentrations (Table 9.1). Pathophysiological changes are dynamic in these patients, and are liable to change and influence plasma concentrations over the course of therapy.
Inflammatory processes in severe infection may cause third spacing, which, in addition to supportive measures, can greatly impact the volume of distribution of hydrophilic drugs, resulting in doubled volume of distribution compared to patients who are not critically ill, and circulating concentrations might be lower than expected. Severe inflammation can also influence the metabolism of hepatically cleared drugs [9].
Hypoalbuminemia is frequently observed in the critically ill, impacting drugs that are highly protein bound. While a greater free fraction of the drug is available for the clearance of hydrophilic drugs, augmented tissue distribution can also occur, coincident with third spacing. An augmented volume of distribution is thus observed, which can be double that of patients without hypoalbuminemia. Renal clearance in these instances might be normal or augmented, leading to increased clearance of the free fraction, or impaired causing accumulation of the free fraction. Plasma/serum concentrations measured in this scenario may reflect the total rather than unbound drug, a challenge when making dosing decisions based on concentration measurements.
Infection and supportive measures may result in augmented renal clearance (creatinine > 130 mg/min) in some patients, producing lower than expected concentrations for renally cleared drugs. Progressing infection may lead to an abrupt loss of kidney function (acute kidney injury), necessitating a dialysis modality. Pre-existing renal impairment may also impact drug handling, and nephrotoxic agents may impact function over the course of treatment. Extracorporeal interventions, including renal replacement therapy, for example continuous or intermittent dialysis, sustained low-efficiency dialysis/extended daily diafiltration, and extracorporeal membrane oxygenation, impact volume of distribution and clearance, particularly for hydrophilic drugs. The outcome on circulating concentrations is difficult to predict given different modalities and large differences in procedural aspects between institutions.
9.4 Classic TDM Candidate Drugs: Aminoglycosides and Vancomycin
TDM experience with aminoglycosides and vancomycin spans several decades. They conform to the requisites as traditional TDM candidates, namely possessing narrow therapeutic indices due to nephrotoxicity and ototoxicity. Immunoassays have been available for individual drugs since the late 1970s, exhibit excellent sensitivity and are used routinely to determine plasma concentration levels.
9.4.1 Aminoglycosides
Aminoglycosides have broad-spectrum activity against gram-negative bacteria. Drugs in this class are small hydrophilic molecules, with similar pharmacokinetic properties between agents. Due to their concentration-dependent bactericidal activity, once-daily administration is the traditional dosing form in most contexts [22]. For patients in whom pharmacokinetic alterations are not expected, empirical short-term therapy with once-daily dosing will likely not require monitoring as adequate peak concentrations are expected to be achieved. For empirical treatment that extends beyond 48h, directed therapy including prolonged treatment due to resistance to other agents, combination therapy, or synergistic low-dose use, plasma concentrations should be determined to guide dosing. Higher initial dosing is suggested in severe sepsis (7 mg/kg up to 640 mg) due to altered volume of distribution [22].
In individuals with normal or augmented renal function, trough concentrations are likely to fall below the limit of detection by immunoassay platforms. It is reasonable to measure trough concentrations in individuals with impaired renal function to avoid toxicity, especially since concentration–time profiles begin to approximate continuous infusions [23]. Peak concentrations are measured 30 min following the end of the infusion. A reasonable target is 6–10 mg/L for gentamycin and tobramycin, and 12–20 mg/L for amikacin [23]. If considering local biogram data or when microbiological data are available, a Cmax/MIC ratio of 8–10 is a reasonable target, although >10 might be necessary in severely ill patients. For Bayesian calculations, some guidelines recommend that the second level after the peak be taken 6–14 h following the end of the infusion to avoid undetectable trough levels.
An excellent narrative review describes the history of nomograms and forecasting solutions for use in aminoglycoside dose adaptation [24]. Nomograms based on drug concentrations have been found to result in under-dosing in some patients [25, 26], including in the critical care setting [27]. While superior to nomograms, Sawchuk and Zaske’s computationally simple, one-compartment model for individualizing dosing requires several samples [28]. Some authors report inferiority of this method compared to Bayesian forecasting [25]. Various software based on Bayesian methods are available and several have been used in the context of aminoglycoside treatment in critically ill patients [29,30,31]. Gauthier et al. highlight the importance of using population parameters from the appropriate population in critical care patients [29]. In addition to favorable sampling conditions (single sample, flexible timing, and not having to wait for a steady-state condition), Bayesian forecasting provides individual estimates for Cmax and AUC, covering efficacy and toxicity. While minor differences in calculations between software have been observed, recommendations for the purposes of dose adaptation are similar [11, 25, 29, 32].
9.4.2 Dose Adaptation Tools for Aminoglycosides: Impact on Clinical Outcomes
Gillaizeau and colleagues performed a systematic review of clinical trials for the Cochrane collaboration evaluating computer-assisted dose adaptation in various clinical scenarios [33]. Most reports concerned anticoagulants and insulin (25/46), while five represented aminoglycosides [27, 34,35,36,37]. It is uncertain if TDM was performed in the context of once-daily dosing, although Begg, Hickling and colleagues targeted maximal concentrations in the range 6–10 mg/L [27, 34]. Interventions represented computer-supported advice from clinical pharmacists or pharmacologists, with the control arm representing dosing and adaptation based on blood levels by physicians from the treating team, either following a nomogram or a defined therapeutic range. Programs were based on the Sawchuk and Zaske linear regression model with modification [28], or Bayesian models [38, 39]. Interventions resulted in improved attainment of target concentrations, while the impact on treatment success and length of stay was significant but minor. For nephrotoxicity, despite a large cohort, reduced risk was not significant. Software-based dose adaptation was superior to targeting within a Cmax range in the two reports evaluating this outcome [27, 34].
Eleven studies involving antimicrobials were identified as within scope, but were not included in the review, concerning gentamycin, amikacin, and vancomycin [33]. Reasons cited by the authors included not randomized controlled trial, dose calculations not performed by computer, or absence of primary outcome data sought by the authors. Despite a before and after design, the work by Van Lent-Evers and colleagues represents a large, well-powered cohort and evaluation of several clinically important outcomes [40]. The TDM intervention involved pharmacy input in dosing regimens through a 24 h service, with initial dosing and adjustment calculated by MWPharm, although the population model was developed using the nonparametric NPEM2 algorithm of the USC*PACK. The model included samples from ICU patients. Prior to the intervention physicians dosed according to nomograms, and when levels were requested, pharmacy performed dose calculations using the Sawchuk and Zaske method. The intervention resulted in reduced length of hospital stay, reduced days with signs of infection, fewer individuals with nephrotoxicity, and was more cost-effective. A trend toward improved survival was observed, but was likely underpowered.
9.4.3 Vancomycin
TDM for vancomycin has a controversial history with respect to both achieving efficacy and avoiding toxicity. The 2009 consensus review for TDM represents an effort to achieve an agreement between various organizations (American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists) based on the literature available to that date [41].
Trough levels obtained at steady state are recommended for practical reasons, and assume an acceptable relationship between trough and AUC. A trough of 15 mg/L is proposed for pathogens with an MIC 1 mg/L, to achieve an AUC/MIC ratio of 400 [41]. The minimum trough level of 10 mg/L required to avoid resistance is frequently cited, and based on observations from a case with recurrent MRSA bacteremia [42] and in vitro work supported by clinical relevance [43, 44]. To interpret trough levels, samples should be taken at steady state, for example, following the fourth dose for twice-daily dosing. In patients with altered renal function, the observed half-life will change, thus impacting time to steady state. For pathogens with reduced susceptibility, for example those with MIC ≥ 2 mg/L, achievement of the requisite AUC > 800 would not be possible with conventional dosing (15 mg/kg daily based on actual body weight, ABW). A loading dose (25–30 mg/kg ABW) is advocated for severely ill patients, permitting attainment of target concentrations more rapidly; sampling in this instance can be performed following the first conventional dose. These guidelines do not recommend continuous infusion, as superiority for patient outcomes to intermittent infusion had not been evidenced. Subsequent meta-analyses did not find a significant impact on clinical success [45, 46], although nephrotoxicity appeared reduced [45,46,47]. This is not significant in the work by Hanrahan et al., although the authors provide reasoning for this. Continuous infusion may be especially useful in critically ill patients for quicker attainment of pharmacokinetic targets, together with a loading dose 35 mg/kg to rapidly achieve plasma concentrations of 20 mg/L. Sampling can occur at any point during the infusion, with this concentration likely to achieve the target AUC/MIC ratio of 400 in appropriately sensitive microorganisms.
Following the 2009 guideline, and resulting change in practice, several research groups have performed meta-analyses evaluating proposed targets; trough levels above 15 mg/L, and, as it became more frequently reported, AUC/MIC. The body of work represented was essentially observational, principally prospective and retrospective cohort studies [48,49,50,51,52]. Nephrotoxicity, typically defined by a prespecified increase in creatinine, is elevated approximately twofold in individuals with trough levels above 15 mg/L [49, 51, 52]. Van Hal et al. highlight that a dose–effect can be observed in studies reporting multiple dose strata [53,54,55,56], and that a time–effect relationship can be observed in reports noting that most nephrotoxic events occurred after 7 days of therapy [57,58,59,60]. Steinmetz et al. highlight that no cases of irreversible damage were reported amongst the reports they included. Concerning treatment failure, some authors report a modest effect after accounting for heterogeneity (OR = 0.68 (0.52–0.89), n = 611/657, high arm/low arm, respectively) [51], while others only when restricting to bacteremia (RR = 0.72 (0.59–0.88), n = 374/420) [49], or persistent bacteremia (OR = 0.3 (0.14–0.62), n = 104/129) [50].
A body of work relating AUC/MIC to outcome measures has emerged and recent meta-analyses have attempted to summarize findings [48, 50]. Authors of included reports typically calculate breakpoints determined by CART (classification and regression tree) analyses. Reports in which MICs are determined by the broth dilution method (BMD) the cutoff is around the AUC/MIC target 400 ± 15%. A twofold improvement in treatment failure is observed in patients with higher AUC/MIC breakpoints compared to lower AUC/MIC breakpoints (OR = 0.41 (0.31–0.53), n = 694/397, [50]; RR = 0.47 (0.30–0.73), n = 419/236, [48]). Of note is that the largest study included in both analyses, by the group that authored the 2009 guideline, detected a smaller effect size compared to other reports, influencing heterogeneity [54]. This group uniquely included MICs determined by both BMD and Etest methods, although Men et al. also noted relatively higher APACHE II scores relative to other reports included in their meta-analysis. In contrast to trough level based comparisons, those based on AUC/MIC thresholds demonstrate an improvement in mortality, when reported (RR = 0.47 (0.31–0.70), n = 188/132) [48].
The vast but essentially observational literature concerning monitoring and dose adjustment for vancomycin appears to favor an AUC-based approach for improving patient outcomes. The target AUC/MIC ratio of 400 seems reasonable, though the local method used to determine MICs must be taken into account. In this context, a clinical trial comparing trough- and AUC-based dose adjustment is warranted, including patients where vancomycin TDM would be rationally applied.
9.5 TDM for Other Antimicrobial Agents
TDM presently requiring chromatographic methods for quantification is limited to clinical institutions with a specialized service. Despite technical advances, turnaround times are rate limiting for dose adaptation. In the critical care setting, there is concern to ensure adequate systemic concentrations, especially important for antimicrobials that are frequently used such as broad-spectrum beta-lactams and fluoroquinolones, though in principle may apply to any antimicrobial. A case for TDM has also been made for additionally avoiding toxicity with some drugs that are last-line agents or reserved for severe or resistant infections including linezolid, colistin, and daptomycin. TDM is becoming increasingly accepted for antifungal agents, and there is emerging evidence for antiviral agents; however, these are beyond the scope of the present work and the reader is referred to a recent review [61]. Selected additional antimicrobials with some evidence for a breakpoint related to clinical outcomes are included in Table 9.2.
9.5.1 Beta-Lactams
The role of TDM for beta-lactams has gained interest for wider application, principally in the critical care setting. Various authors have reflected over the relevance, potential benefits, and challenges of beta-lactam TDM, specifically in the critically ill [3, 62,63,64]. Important distinctions between individual drugs within the class that might influence TDM-directed dosing include a significant post-antimicrobial effect for meropenem, significant protein binding with ceftriaxone and flucloxacillin, and long half-life for ceftriaxone.
Beta-lactams demonstrate a time above MIC dependent effect relationship, primarily evidenced through preclinical PK/PD models [8]. Several reports for clinically derived PK/PD indices in recent literature contrast with preclinical work and suggest a longer time above MIC may be necessary [65,66,67,68,69]. Most reports simulate plasma concentrations based on creatinine clearance, typically using population parameters from a model involving a similar population; in one report a microbiological assay was used to determine concentrations [69]. Ariano et al. report an 80% response rate for 60 individuals with fT > MIC: >75% for meropenem in bacteremia, excluding concurrent infections and renal impairment [65]. McKinnon et al. report an 82% clinical cure rate for individuals with T > MIC = 100% for ceftazidime or cefepime in 76 patients with sepsis [68]. Individual patient data for patients with AUIC < 500 are presented, thus it is possible to determine clinical cure at other breakpoints: T > MIC: >75% results in 81%, and T > MIC: >60% results in 79.4% [68]. Crandon et al. arrive at a breakpoint of fT > MIC: >60% for cefepime using CART analysis and report a microbiological success rate of 63.8%, in 56 patients with an active P. aeruginosa infection (varied sites) [66]. The Etest method was used to determine MICs. Two reports are used to promote fT > MIC × 5 as a PK/PD index. Li et al. found fCmin/MIC > 5 as the only significant predictor of clinical success in 101 adults with lower respiratory infections, the authors noting that fT > MIC 100% was achieved in most patients [67]. Tam et al. report MIC × 4.3 as an indicator for clinical success for cefepime-treated individuals with diverse gram-negative infections; 1/23 individuals manifested clinical failure [69]. DALI (Defining Antibiotic Levels in ICU patients), an international point prevalence study, explored outcomes at the cutoffs fT > MIC 50% and 100%, and fT > MIC × 4 50% and 100% [70]. Free beta-lactam concentrations in plasma were measured in a central laboratory, and related to clinical outcomes. Two hundred forty-eight patients were treated for infection, representing eight beta-lactams. MIC results were available for 34.2%, EUCAST MIC90 was used for 38.7%, and the highest possible MIC for the given beta-lactam was assumed for 27.1% of participants; the authors highlight that many centers lacked services to determine MIC. While prudent, this strategy may have influenced risk estimates related to outcomes. Positive clinical outcome was observed in individuals achieving 50% fT > MIC (OR = 1.02) and 100% fT > MIC (OR = 1.56), p < 0.03 in the multivariable model including indices for sickness severity. These data empirically suggest that 100% fT > MIC is superior to 50%; however, the optimal index may vary between agents.
These data indicate that microbial sensitivity plays an important role in achievement of PK/PD indices. Further work is required to determine an optimal parameter that is both clinically useful and practical to apply. An emerging body of work demonstrates that PK/PD indices are difficult to achieve in the critically ill, especially in the early phases of sepsis [71] and with augmented renal clearance [72,73,74,75,76]. Non-critically ill obese patients likewise present augmented renal clearance that impact target attainment of beta-lactams [77]. However, different target indices are used across reports, and some authors report a lack of relationship between augmented renal clearance and clinical success [73]. Tools based on creatinine clearance developed in critically ill populations, such as nomograms [78] or the augmented renal clearance score [79], together with optimized administration strategies (loading dose, extended/continuous infusions) may be sufficient for the purposes of dose adaptation, but further validation and wider application is warranted.
A large investigative effort has compared clinical outcomes between continuous/extended infusions and intermittent bolus dosing. Multiple meta-analyses have attempted to synthesize data from observational work and clinical trials [80,81,82,83,84,85,86,87]. Randomized controlled trials in this setting have been critiqued for including patients with lower disease severity, use of inconsistent total antibiotic doses between comparator groups, and heterogeneity between studies, including differences in pathogens and their MICs, duration of follow-up, and definitions of outcomes [83, 84, 88, 89]. A recent meta-analysis employed strict inclusion criteria, limiting inclusion to clinical trials recruiting patients with severe sepsis or septic shock [83]. Mortality at 30 days was 19.6% versus 26.3% (RR 0.74 (0.56–1.00)) and clinical cure 55.4% versus 46.3% (RR 1.20 (1.03–1.40)), for continuous infusion and intermittent bolus dosing, respectively. The authors further highlight that benefits of continuous infusions are especially pronounced in individuals treated for severe sepsis caused by non-fermenting gram-negative bacilli, and diminished in patients requiring renal replacement therapy where bolus dosing begins to approximate the kinetics of continuous infusions.
9.5.2 Fluoroquinolones
Fluoroquinolones have dose-dependent antimicrobial activity for the treatment of bacterial infections caused by gram-negative, gram-positive pathogens and mycobacteria. The best PK/PD index predicting efficacy is the AUC/MIC ratio, followed by the Cmax/MIC ratio [90, 91], and quantitatively depends on the infective pathogen. While for gram-positive microorganisms, such as Streptococcus pneumoniae, AUC/MIC has been defined to be ≥30–35, for gram-negatives it should be greater than 100 [92, 93]. A Cmax/MIC of 8–10 results in maximum antibacterial efficacy in in vivo animal models [92, 93].
Several studies have correlated an AUC/MIC of 30–60 for different fluoroquinolones (levofloxacin, ciprofloxacin, gatifloxacin, moxifloxacin, etc.) to in vitro antimicrobial activity [92,93,94] and improved clinical outcomes, such as bacterial eradication [95]. For levofloxacin, population pharmacokinetic studies have demonstrated that standard dosing of 500 mg/day results in inadequate achievement of target PK/PD indices, especially for certain patients and Gram-negative infections [96, 97]. Moxifloxacin exhibits a better PK profile, with AUC/MIC > 35 achieved in 100% of patients, where strains exhibited MICs of 1 mg/L [98]. Treatment failures with fluoroquinolones administered at standard doses (ciprofloxacin and levofloxacin) in patients with respiratory tract infections due to fluoroquinolone-resistant S. pneumoniae have been reported, especially in patients previously treated with these antimicrobials [99]. An increased dose of levofloxacin to 750 mg/day or 500 mg/12 h has been suggested. A multicenter, randomized, double-blind study demonstrated no differences in clinical success and microbiologic eradication when comparing levofloxacin dosages of 750 mg/day for 5 days with the dose of 500 mg/day for 10 days for the treatment of mild to severe community-acquired pneumonia [100]. This high-dose short course regimen maximizes its concentration-dependent bactericidal activity and may reduce resistance. A dose of 500 mg twice-daily of levofloxacin has been proposed for the treatment of early-onset ventilator-associated pneumonia in intensive care patients [101, 102]. TDM for fluoroquinolones with increased dosing might be a complementary tool to avoid toxicity.
The declining susceptibilities to fluoroquinolones of Gram-negative isolates pose an important challenge [103]. Some authors have attempted to define a PK/PD-based threshold to minimize the development of resistance. Homma et al. evaluated different clinical isolates of S. pneumoniae in vitro with various MIC and MPC (Mutant Prevention Concentration) values for levofloxacin and moxifloxacin, and propose a target AUC/MPC ≥ 13.41 or Cmax/MPC above 1.20 for complete eradication without decreased susceptibility [104].
While the PK/PD behavior of the fluoroquinolones has been widely described, few authors have evaluated the necessity or clinical benefit of TDM. Scaglione et al. report their local experience of a TDM program for ciprofloxacin, using Cmax/MIC of 10 as a target, but do not report clinical outcomes [105]. Pea et al. evaluated ciprofloxacin TDM in 89 critically ill patients and report wide and unpredictable interindividual pharmacokinetic variability. They conclude that fixed dosing of 200 or 400 mg/12 h is only useful for fully susceptible microorganisms (MIC < 0.3 mg/L), further supporting use of higher doses and potential usefulness of TDM [106]. Other centers only monitor fluoroquinolones in certain patients such as obese patients or with significant burn injuries [107]. Restricting TDM to special populations may be a rational approach [108, 109].
TDM of fluoroquinolones has been more widely applied for the treatment of tuberculosis (TB) due to the observed high pharmacokinetic drug variability [108,109,112] and the high frequency of patients with low serum concentrations [113]. Fluoroquinolones are used for the treatment of TB, including the multidrug-resistant (MDR) TB, levofloxacin and moxifloxacin being preferred due to potency and relative safety. Manika et al. report wide variability of moxifloxacin concentrations amongst patients with multidrug-resistant TB receiving the same regimen (400 mg per day), concluding that this standard dose may not be sufficient for all patients [112]. A limited-sampling strategy has been proposed [114]; however, the pharmacodynamic target for Mycobacterium tuberculosis has not been defined. In addition, the presence of low serum concentrations of anti-MDR-TB drugs might not affect the 2-month sputum conversion rate [113].
9.5.3 Linezolid
Linezolid is a member of the oxazolidinones with bacteriostatic activity against enterococci and staphylococci, and is bactericidal for most streptococci strains. Recommended dosing is 600 mg twice-daily in a fixed dose formulation, irrespective of renal or hepatic function, and pharmacokinetic parameters are claimed to be insignificantly altered by age, gender, or renal/hepatic insufficiency. Recent reports, however, evidence wide inter- and intraindividual variability [115, 116], especially amongst the critically ill or those with renal impairment [115,116,117,120]. Pea at al. suggest that TDM of linezolid may be worthwhile in 30% of individuals to avoid treatment failure or dose-dependent toxicity [121]. Patients with renal impairment, the elderly, or those with low body weight risk overexposure and toxicity, while acute illness may exacerbate linezolid-related hematological toxicity [119, 120,121,124]. On the other hand, critically ill patients are at risk of subtherapeutic levels, especially those with augmented clearance and greater volume of distribution, thus TDM might optimize dosing and prevent clinical failure [120]. Data concerning the pharmacokinetics of linezolid in patients with excessive body weight are limited and controversial [125, 126] and TDM may cover inconsistencies. Standard dose of linezolid results in suboptimal concentrations in more than 40% of pediatric patients [127]. Further, higher doses were required for pathogens with borderline susceptibility (MIC > 1 mg/L).
The main reason to perform TDM of linezolid is to avoid or prevent hematological toxicity. High linezolid trough concentrations are associated with thrombocytopenia in patients with Gram-positive bacterial infections [128]. The trough concentration limit to prevent toxicity remains to be defined. Different thresholds have been proposed including 6.5 mg/L [124], 7–10 mg/L [117, 123, 129, 130], and 22.1 mg/L [131]. It is surprising that this relationship between exposure and toxicity was not confirmed in patients receiving linezolid for the treatment of drug-resistant tuberculosis, in whom the AUC of linezolid did not associate with any drug-related adverse event [132].
Administration by continuous infusion has been proposed to optimize achievement of the PK/PD index for clinical efficacy; however, data are lacking [133]. Optimal linezolid plasma concentrations to achieve the highest clinical efficacy are unknown. Some authors have identified a trough concentration of ≥2 mg/L as a predictor of bacterial eradication [134] and a therapeutic range 2–7 mg/L has been proposed [124]. Furthermore, target concentrations should consider MIC to achieve an optimal PK/PD ratio: an AUC/MIC ratio between 80 and 120 is frequently cited. AUC calculations based on a minimal sampling strategy can be used to individualize dosing [128, 135].
9.5.4 Colistin
Colistin, or polymyxin E, is a cationic polypeptide antibiotic active against gram-negative bacteria, including multidrug-resistant strains. Its use has reemerged worldwide as rescue therapy for infections caused by multidrug-resistant bacilli, such as Pseudomonas aeruginosa, Acinetobacter baumannii, and Enterobacteriaceae species. It is administered parenterally as a prodrug, colistin methanesulfonate sodium (CMS), which is converted in vivo to the active compound, colistin. It exhibits concentration-dependent antibacterial activity. This polymyxin was developed in Japan in the 1940s–1950s but its clinical and parenteral use were abandoned in most countries due to reports of serious adverse events, such as nephrotoxicity and neurotoxicity [136]. Initial dosing regimens of CMS relied on PK/PD data from older work that lacked appropriate methods and provided unreliable findings [137]. In addition, most PK/PD studies were performed in patients with cystic fibrosis which exhibit unique PK characteristics as a population. In recent years, specific chromatographic methods for the accurate analysis of CMS and colistin have been established [138]. This has led to novel PK/PD work in animals and humans, providing updated data to optimize colistin dosing and improve its clinical efficacy, while limiting toxicities and emergence of resistance [139, 140]. This is of extreme importance in difficult-to-treat multi-resistant pathogens with no therapeutic alternatives.
A steady-state colistin trough concentration of 2–2.5 mg/L has been proposed, corresponding to a target AUC 0–24 of 60 mg h/L [138, 139]. This AUC/MIC value is based on the results of a preclinical work testing three strains each of A. baumannii and P. aeruginosa in murine thigh and lung infection models that demonstrated that an AUC 0–24/MIC of 60 h associated with an effect between stasis and 1-log kill. This target concentration of 2.5 mg/L is optimal for an MIC of 1 mg/L, and requires adjustment for other MIC values.
To date, this target concentration has not correlated with positive clinical outcomes. A randomized clinical trial assessing TDM of colistin (using Cmax/CMI) failed to demonstrate a benefit in terms of clinical cure or 30-day mortality in patients with different types of multidrug-resistant gram-negative bacterial infections [141]. Yamada et al. describe a case with bacteremia due to multidrug-resistant Pseudomonas aeruginosa who was successfully treated with colistin in conjunction with TDM [140,141,144].
One important conclusion of recent PK work is the need to administer an initial loading dose and a higher CMS maintenance dose to rapidly attain therapeutic concentrations as the manufacturer dosage recommendations are insufficient, especially in critically ill patients [139, 145, 146]. Updated dosing recommendations for intravenous colistin based on renal function vary between the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) [147]. Currently, newer dosage regimens are being widely implemented, although TDM is not routinely performed in the majority of centers; this could have implications for toxicity observed with clinical use [137]. A prospective observational cohort study demonstrated that the trough plasma level of colistin is an independent risk factor for nephrotoxicity, and that acute kidney injury is best predicted at 2.42 mg/L [148]. This value has been validated in a prospective cohort of individuals treated for multidrug-resistant gram-negative infections [149]. It is clear that the therapeutic window for colistin is narrow, with concentrations required for efficacy being quite close to those in which toxicities are observed [150].
9.5.5 Daptomycin
Daptomycin is a lipoglycopeptide with a concentration-dependent antimicrobial activity best described by AUC/MIC [151, 152]. The mean AUC/MIC value associated with a static, 1-log killing, and 2-log killing effect against S. aureus has been defined as 438, 666, and 1061, respectively [153]. In addition, an AUC/MIC ratio of <666 was associated with increased mortality in patients with gram-positive severe infections [154]. Other authors have identified that an AUC/MIC > 200 is required to prevent S. aureus resistance [155].
The approved dose of daptomycin for soft-tissue infections is 4 mg/kg daily and for bacteremia 6 mg/kg daily. For infections with a high inoculum, such as endocarditis, microbiological data suggest that doses higher than 6 mg/kg/daily are required, especially against strains with reduced daptomycin susceptibility [156]. In an in vitro PD model, a dose of 10 mg/kg was required to prevent resistance [152]; however, further clinical data are warranted [151].
Vast variability in the pharmacokinetics of daptomycin has been observed with clinical use, including high-dose regimens [154, 157]. Much of the variability could not be accounted for by clinical factors (creatinine clearance, albumin, or dose interval), suggesting the need for TDM, which may be especially useful in critical illness, severe sepsis, dynamically changing renal function, and acute kidney injury [153, 154, 158, 159]. Excessive exposure is related to musculoskeletal toxicity [154, 157, 158]. Bhavnani et al. report a Cmin breakpoint of 24.3 mg/L associated with an elevation of creatine kinase in patients treated with standard dosing (6 mg/kg/day) [160]. The clinical application of TDM for daptomycin remains limited, and the literature is represented by only a few reports [5, 157, 161, 162].
9.6 Conclusions and Future Directions
Interest in TDM for optimizing therapy is becoming rekindled, particularly for special populations. Antimicrobial use in the intensive care setting has received special attention and there is a growing body of literature to support that pathophysiological changes in critically ill patients influence circulating concentrations. Data concerning optimal PK/PD targets for emerging TDM candidates, and the clinical impact of concentration guided dose adaptation, remains limited. Clinical trials exploring the impact of monitoring on clinical outcomes are only useful when breakpoints are well established; well-conducted prospective observational studies based on measured concentrations can help to determine optimal indices. Further, wider implementation and investigation is contingent on laboratories for measurement. Sensitive and specific immunoassays for emerging candidates would aid wider implementation but also research efforts in the clinical setting. Aptamer-based technology may help to overcome the challenges of antibody-based immunoassays [163], and has been used together with a microfluidic electrochemical detector for real-time tracking of circulating drug concentrations [164]. Therapeutic drug monitoring complements innovations in other areas including microbial diagnostics and response-related biomarkers and is an important tool in achieving personalized medicine [165, 166].
References
Dasgupta A (2012) Introduction to therapeutic drug monitoring. Frequently and less frequently monitored drugs. In: Therapeutic drug monitoring, 1st edn. Elsevier, New York, p 1–29
Walson PD (1998) Therapeutic drug monitoring in special populations. Clin Chem 44:415–419
Roberts JA, Abdul-Aziz MH, Lipman J et al (2014) Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis 14:498–509. doi:10.1016/S1473-3099(14)70036-2
Fish DN, Piscitelli SC, Danziger LH (1995) Development of resistance during antimicrobial therapy: a review of antibiotic classes and patient characteristics in 173 studies. Pharmacotherapy 15:279–291
Pea F, Cojutti P, Sbrojavacca R et al (2011) TDM-guided therapy with daptomycin and meropenem in a morbidly obese, critically ill patient. Ann Pharmacother 45:e37. doi:10.1345/aph.1P745
Taccone FS, Cotton F, Roisin S et al (2012) Optimal meropenem concentrations to treat multidrug-resistant Pseudomonas aeruginosa septic shock. Antimicrob Agents Chemother 56:2129–2131. doi:10.1128/AAC.06389-11
Shipkova M, Svinarov D (2016) LC-MS/MS as a tool for TDM services: where are we? Clin Biochem 49:1009–1023. doi:10.1016/j.clinbiochem.2016.05.001
Craig WA (1998) Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26:1–10
Veringa A, Ter Avest M, Span LFR et al (2017) Voriconazole metabolism is influenced by severe inflammation: a prospective study. J Antimicrob Chemother 72:261–267. doi:10.1093/jac/dkw349
Beal S, Sheiner LB, Boeckmann A, Bauer A (2009) NONMEM user’s guides (1989–2009). Icon Development Solutions, Ellicott City
Fuchs A, Csajka C, Thoma Y et al (2013) Benchmarking therapeutic drug monitoring software: a review of available computer tools. Clin Pharmacokinet 52:9–22. doi:10.1007/s40262-012-0020-y
Jelliffe RW (1991) The USC*PACK PC programs for population pharmacokinetic modeling, modeling of large kinetic/dynamic systems, and adaptive control of drug dosage regimens. Proc Symp Comput Appl Med Care: 922–924
LAPK (2016) BestDose Software. http://www.lapk.org/bestdose.php. Accessed 28 Dec 2016
Proost JH, Meijer DK (1992) MW/Pharm, an integrated software package for drug dosage regimen calculation and therapeutic drug monitoring. Comput Biol Med 22:155–163
RxKinetics (2016) RxKinetics pharmacokinetics and nutrition software for pharmacists. In: RxKinetics Pharmacokinet. Nutr. Softw. Pharm. http://www.rxkinetics.com/. Accessed 28 Dec 2016
Tharpe R (2016) Complex non-steady-state analysis | RxRick’s Blog. http://rxkinetics.com/blog/?p=1495. Accessed 28 Dec 2016
Lacarelle B, Pisano P, Gauthier T et al (1994) Abbott PKS system: a new version for applied pharmacokinetics including Bayesian estimation. Int J Biomed Comput 36:127–130
T.D.M.S. 2000 (2016) T.D.M.S. 2000TM. http://tdms2000.com/. Accessed 28 Dec 2016
Duffull SB, Kirkpatrick CJ, Van Den Berg L (2016) TCIWorks. In: TCIWorks. tciworks.info. Accessed 2016
DoseMe (2016) DoseMe—personalised medicine: making complex simple. In: DoseMe—Pers. Med. Mak. Complex simple. https://www.doseme.com.au/. Accessed 28 Dec 2016
Limoges University Hospital (2016) Limoges University Hospital Laboratory of Pharmacology. https://pharmaco.chu-limoges.fr/. Accessed 28 Dec 2016
Antibiotic Expert Group (2010) Therapeutic guidelines: antibiotic. Therapeutic Guidelines Limited, Melbourne
Begg EJ, Barclay ML, Kirkpatrick CJ (1999) The therapeutic monitoring of antimicrobial agents. Br J Clin Pharmacol 47:23–30
Begg EJ, Barclay ML (1995) Aminoglycosides—50 years on. Br J Clin Pharmacol 39:597–603
Avent ML, Teoh J, Lees J et al (2011) Comparing 3 methods of monitoring gentamicin concentrations in patients with febrile neutropenia. Ther Drug Monit 33:592–601. doi:10.1097/FTD.0b013e31822c78e9
Martin J, Barras M, Yui NA et al (2012) Gentamicin monitoring practices in teaching hospitals—time to undertake the necessary randomised controlled trial. Clin Toxicol 2:1000146. doi:10.4172/2161-0495.1000146
Hickling K, Begg E, Moore ML (1989) A prospective randomised trial comparing individualised pharmacokinetic dosage prediction for aminoglycosides with prediction based on estimated creatinine clearance in critically ill patients. Intensive Care Med 15:233–237
Sawchuk RJ, Zaske DE (1976) Pharmacokinetics of dosing regimens which utilize multiple intravenous infusions: gentamicin in burn patients. J Pharmacokinet Biopharm 4:183–195
Gauthier T, Lacarelle B, Marre F et al (1994) Predictive performance of two software packages (USC*PACK PC and Abbott PKS system) for the individualization of amikacin dosage in intensive care unit patients. Int J Biomed Comput 36:131–134
Mar Fernández de Gatta MD, Victoria Calvo M, Ardanuy R et al (2009) Evaluation of population pharmacokinetic models for amikacin dosage individualization in critically ill patients. J Pharm Pharmacol 61:759–766. doi:10.1211/jpp/61.06.0008
Rodvold KA, Pryka RD, Kuehl PG et al (1990) Bayesian forecasting of serum gentamicin concentrations in intensive care patients. Clin Pharmacokinet 18:409–418
Duffull SB, Kirkpatrick CM, Begg EJ (1997) Comparison of two Bayesian approaches to dose-individualization for once-daily aminoglycoside regimens. Br J Clin Pharmacol 43:125–135
Gillaizeau F, Chan E, Trinquart L, et al (2013) Computerized advice on drug dosage to improve prescribing practice. Cochrane Database Syst Rev (11):CD002894. doi: 10.1002/14651858.CD002894.pub3
Begg EJ, Atkinson HC, Jeffery GM, Taylor NW (1989) Individualised aminoglycoside dosage based on pharmacokinetic analysis is superior to dosage based on physician intuition at achieving target plasma drug concentrations. Br J Clin Pharmacol 28:137–141
Burton ME, Ash CL, Hill DP et al (1991) A controlled trial of the cost benefit of computerized bayesian aminoglycoside administration. Clin Pharmacol Ther 49:685–694
Destache CJ, Meyer SK, Bittner MJ, Hermann KG (1990) Impact of a clinical pharmacokinetic service on patients treated with aminoglycosides: a cost-benefit analysis. Ther Drug Monit 12:419–426
Leehey DJ, Braun BI, Tholl DA et al (1993) Can pharmacokinetic dosing decrease nephrotoxicity associated with aminoglycoside therapy. J Am Soc Nephrol JASN 4:81–90
Burton ME, Brater DC, Chen PS et al (1985) A Bayesian feedback method of aminoglycoside dosing. Clin Pharmacol Ther 37:349–357
Sheiner LB, Rosenberg B, Melmon KL (1972) Modelling of individual pharmacokinetics for computer-aided drug dosage. Comput Biomed Res Int J 5:411–459
van Lent-Evers NA, Mathôt RA, Geus WP et al (1999) Impact of goal-oriented and model-based clinical pharmacokinetic dosing of aminoglycosides on clinical outcome: a cost-effectiveness analysis. Ther Drug Monit 21:63–73
Rybak M, Lomaestro B, Rotschafer JC et al (2009) Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health-Syst Pharm 66:82–98. doi:10.2146/ajhp080434
Sakoulas G, Gold HS, Cohen RA et al (2006) Effects of prolonged vancomycin administration on methicillin-resistant Staphylococcus aureus (MRSA) in a patient with recurrent bacteraemia. J Antimicrob Chemother 57:699–704. doi:10.1093/jac/dkl030
Tsuji BT, Rybak MJ, Cheung CM et al (2007) Community- and health care-associated methicillin-resistant Staphylococcus aureus: a comparison of molecular epidemiology and antimicrobial activities of various agents. Diagn Microbiol Infect Dis 58:41–47. doi:10.1016/j.diagmicrobio.2006.10.021
Tsuji BT, Rybak MJ, Lau KL, Sakoulas G (2007) Evaluation of accessory gene regulator (agr) group and function in the proclivity towards vancomycin intermediate resistance in Staphylococcus aureus. Antimicrob Agents Chemother 51:1089–1091. doi:10.1128/AAC.00671-06
Cataldo MA, Tacconelli E, Grilli E et al (2012) Continuous versus intermittent infusion of vancomycin for the treatment of Gram-positive infections: systematic review and meta-analysis. J Antimicrob Chemother 67:17–24. doi:10.1093/jac/dkr442
Hao J-J, Chen H, Zhou J-X (2016) Continuous versus intermittent infusion of vancomycin in adult patients: a systematic review and meta-analysis. Int J Antimicrob Agents 47:28–35. doi:10.1016/j.ijantimicag.2015.10.019
Hanrahan T, Whitehouse T, Lipman J, Roberts JA (2015) Vancomycin-associated nephrotoxicity: a meta-analysis of administration by continuous versus intermittent infusion. Int J Antimicrob Agents 46:249–253. doi:10.1016/j.ijantimicag.2015.04.013
Men P, Li H-B, Zhai S-D, Zhao R-S (2016) Association between the AUC0-24/MIC ratio of vancomycin and its clinical effectiveness: a systematic review and meta-analysis. PLoS One 11:e0146224. doi:10.1371/journal.pone.0146224
Meng L, Fang Y, Chen Y et al (2015) High versus low vancomycin serum trough regimen for Gram-positive infections: a meta-analysis. J Chemother Florence Italy 27:213–220. doi:10.1179/1973947814Y.0000000182
Prybylski JP (2015) Vancomycin trough concentration as a predictor of clinical outcomes in patients with Staphylococcus aureus bacteremia: a meta-analysis of observational studies. Pharmacotherapy 35:889–898. doi:10.1002/phar.1638
Steinmetz T, Eliakim-Raz N, Goldberg E et al (2015) Association of vancomycin serum concentrations with efficacy in patients with MRSA infections: a systematic review and meta-analysis. Clin Microbiol Infect 21:665–673. doi:10.1016/j.cmi.2015.04.003
van Hal SJ, Paterson DL, Lodise TP (2013) Systematic review and meta-analysis of vancomycin-induced nephrotoxicity associated with dosing schedules that maintain troughs between 15 and 20 milligrams per liter. Antimicrob Agents Chemother 57:734–744. doi:10.1128/AAC.01568-12
Cano EL, Haque NZ, Welch VL et al (2012) Incidence of nephrotoxicity and association with vancomycin use in intensive care unit patients with pneumonia: retrospective analysis of the IMPACT-HAP Database. Clin Ther 34:149–157. doi:10.1016/j.clinthera.2011.12.013
Kullar R, Davis SL, Levine DP, Rybak MJ (2011) Impact of vancomycin exposure on outcomes in patients with methicillin-resistant Staphylococcus aureus bacteremia: support for consensus guidelines suggested targets. Clin Infect Dis 52:975–981. doi:10.1093/cid/cir124
Lodise TP, Patel N, Lomaestro BM et al (2009) Relationship between initial vancomycin concentration-time profile and nephrotoxicity among hospitalized patients. Clin Infect Dis 49:507–514. doi:10.1086/600884
Wunderink RG, Niederman MS, Kollef MH et al (2012) Linezolid in methicillin-resistant Staphylococcus aureus nosocomial pneumonia: a randomized, controlled study. Clin Infect Dis 54:621–629. doi:10.1093/cid/cir895
Hidayat LK, Hsu DI, Quist R et al (2006) High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity. Arch Intern Med 166:2138–2144. doi:10.1001/archinte.166.19.2138
Jeffres MN, Isakow W, Doherty JA et al (2007) A retrospective analysis of possible renal toxicity associated with vancomycin in patients with health care-associated methicillin-resistant Staphylococcus aureus pneumonia. Clin Ther 29:1107–1115. doi:10.1016/j.clinthera.2007.06.014
Minejima E, Choi J, Beringer P et al (2011) Applying new diagnostic criteria for acute kidney injury to facilitate early identification of nephrotoxicity in vancomycin-treated patients. Antimicrob Agents Chemother 55:3278–3283. doi:10.1128/AAC.00173-11
Prabaker KK, Tran TP-H, Pratummas T et al (2012) Elevated vancomycin trough is not associated with nephrotoxicity among inpatient veterans. J Hosp Med 7:91–97. doi:10.1002/jhm.946
Jager NGL, van Hest RM, Lipman J et al (2016) Therapeutic drug monitoring of anti-infective agents in critically ill patients. Expert Rev Clin Pharmacol 9:961–979. doi:10.1586/17512433.2016.1172209
Huttner A, Harbarth S, Hope WW et al (2015) Therapeutic drug monitoring of the β-lactam antibiotics: what is the evidence and which patients should we be using it for? J Antimicrob Chemother 70:3178–3183. doi:10.1093/jac/dkv201
Sime FB, Roberts MS, Peake SL et al (2012) Does beta-lactam pharmacokinetic variability in critically ill patients justify therapeutic drug monitoring? A systematic review. Ann Intensive Care 2:35. doi:10.1186/2110-5820-2-35
Wong G, Sime FB, Lipman J, Roberts JA (2014) How do we use therapeutic drug monitoring to improve outcomes from severe infections in critically ill patients? BMC Infect Dis 14:288. doi:10.1186/1471-2334-14-288
Ariano RE, Nyhlén A, Donnelly JP et al (2005) Pharmacokinetics and pharmacodynamics of meropenem in febrile neutropenic patients with bacteremia. Ann Pharmacother 39:32–38. doi:10.1345/aph.1E271
Crandon JL, Bulik CC, Kuti JL, Nicolau DP (2010) Clinical pharmacodynamics of cefepime in patients infected with Pseudomonas aeruginosa. Antimicrob Agents Chemother 54:1111–1116. doi:10.1128/AAC.01183-09
Li C, Du X, Kuti JL, Nicolau DP (2007) Clinical pharmacodynamics of meropenem in patients with lower respiratory tract infections. Antimicrob Agents Chemother 51:1725–1730. doi:10.1128/AAC.00294-06
McKinnon PS, Paladino JA, Schentag JJ (2008) Evaluation of area under the inhibitory curve (AUIC) and time above the minimum inhibitory concentration (T>MIC) as predictors of outcome for cefepime and ceftazidime in serious bacterial infections. Int J Antimicrob Agents 31:345–351. doi:10.1016/j.ijantimicag.2007.12.009
Tam VH, McKinnon PS, Akins RL et al (2002) Pharmacodynamics of cefepime in patients with Gram-negative infections. J Antimicrob Chemother 50:425–428
Roberts JA, De Waele JJ, Dimopoulos G et al (2012) DALI: defining antibiotic levels in intensive care unit patients: a multi-centre point of prevalence study to determine whether contemporary antibiotic dosing for critically ill patients is therapeutic. BMC Infect Dis 12:152. doi:10.1186/1471-2334-12-152
Taccone FS, Laterre P-F, Dugernier T et al (2010) Insufficient β-lactam concentrations in the early phase of severe sepsis and septic shock. Crit Care 14:R126. doi:10.1186/cc9091
Carlier M, Carrette S, Roberts JA et al (2013) Meropenem and piperacillin/tazobactam prescribing in critically ill patients: does augmented renal clearance affect pharmacokinetic/pharmacodynamic target attainment when extended infusions are used? Crit Care 17:R84. doi:10.1186/cc12705
Huttner A, Von Dach E, Renzoni A et al (2015) Augmented renal clearance, low β-lactam concentrations and clinical outcomes in the critically ill: an observational prospective cohort study. Int J Antimicrob Agents 45:385–392. doi:10.1016/j.ijantimicag.2014.12.017
Udy AA, Lipman J, Jarrett P et al (2015) Are standard doses of piperacillin sufficient for critically ill patients with augmented creatinine clearance? Crit Care 19:28. doi:10.1186/s13054-015-0750-y
Udy AA, Putt MT, Shanmugathasan S et al (2010) Augmented renal clearance in the intensive care unit: an illustrative case series. Int J Antimicrob Agents 35:606–608. doi:10.1016/j.ijantimicag.2010.02.013
Udy AA, Varghese JM, Altukroni M et al (2012) Subtherapeutic initial β-lactam concentrations in select critically ill patients: association between augmented renal clearance and low trough drug concentrations. Chest 142:30–39. doi:10.1378/chest.11-1671
Hites M, Taccone FS, Wolff F et al (2014) Broad-spectrum β-lactams in obese non-critically ill patients. Nutr Diabetes 4:e119. doi:10.1038/nutd.2014.15
Pea F, Viale P, Cojutti P, Furlanut M (2012) Dosing nomograms for attaining optimum concentrations of meropenem by continuous infusion in critically ill patients with severe gram-negative infections: a pharmacokinetics/pharmacodynamics-based approach. Antimicrob Agents Chemother 56:6343–6348. doi:10.1128/AAC.01291-12
Akers KS, Niece KL, Chung KK et al (2014) Modified augmented renal clearance score predicts rapid piperacillin and tazobactam clearance in critically ill surgery and trauma patients. J Trauma Acute Care Surg 77:S163–S170. doi:10.1097/TA.0000000000000191
Falagas ME, Tansarli GS, Ikawa K, Vardakas KZ (2013) Clinical outcomes with extended or continuous versus short-term intravenous infusion of carbapenems and piperacillin/tazobactam: a systematic review and meta-analysis. Clin Infect Dis 56:272–282. doi:10.1093/cid/cis857
Korbila IP, Tansarli GS, Karageorgopoulos DE et al (2013) Extended or continuous versus short-term intravenous infusion of cephalosporins: a meta-analysis. Expert Rev Anti Infect Ther 11:585–595. doi:10.1586/eri.13.44
Lal A, Jaoude P, El-Solh AA (2016) Prolonged versus intermittent infusion of β-lactams for the treatment of nosocomial pneumonia: a meta-analysis. Infect Chemother 48:81–90. doi:10.3947/ic.2016.48.2.81
Roberts JA, Abdul-Aziz M-H, Davis JS et al (2016) Continuous versus intermittent β-lactam infusion in severe sepsis. A meta-analysis of individual patient data from randomized trials. Am J Respir Crit Care Med 194:681–691. doi:10.1164/rccm.201601-0024OC
Roberts JA, Webb S, Paterson D et al (2009) A systematic review on clinical benefits of continuous administration of beta-lactam antibiotics. Crit Care Med 37:2071–2078. doi:10.1097/CCM.0b013e3181a0054d
Shiu J, Wang E, Tejani AM, Wasdell M (2013) Continuous versus intermittent infusions of antibiotics for the treatment of severe acute infections. Cochrane Database Syst Rev CD008481. doi: 10.1002/14651858.CD008481.pub2
Teo J, Liew Y, Lee W, Kwa AL-H (2014) Prolonged infusion versus intermittent boluses of β-lactam antibiotics for treatment of acute infections: a meta-analysis. Int J Antimicrob Agents 43:403–411. doi:10.1016/j.ijantimicag.2014.01.027
Yang H, Zhang C, Zhou Q et al (2015) Clinical outcomes with alternative dosing strategies for piperacillin/tazobactam: a systematic review and meta-analysis. PLoS One 10:e0116769. doi:10.1371/journal.pone.0116769
Abdul-Aziz MH, Dulhunty JM, Bellomo R et al (2012) Continuous beta-lactam infusion in critically ill patients: the clinical evidence. Ann Intensive Care 2:37. doi:10.1186/2110-5820-2-37
Yusuf E, Spapen H, Piérard D (2014) Prolonged vs intermittent infusion of piperacillin/tazobactam in critically ill patients: a narrative and systematic review. J Crit Care 29:1089–1095. doi:10.1016/j.jcrc.2014.07.033
Forrest A, Nix DE, Ballow CH et al (1993) Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob Agents Chemother 37:1073–1081
Shandil RK, Jayaram R, Kaur P et al (2007) Moxifloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against Mycobacterium tuberculosis: evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob Agents Chemother 51:576–582. doi:10.1128/AAC.00414-06
Andes DR, Craig WA (1998) Pharmacodynamics of fluoroquinolones in experimental models of endocarditis. Clin Infect Dis 27:47–50
Turnidge J (1999) Pharmacokinetics and pharmacodynamics of fluoroquinolones. Drugs 58(Suppl 2):29–36
Lacy MK, Lu W, Xu X, et al (1999) Pharmacodynamic comparisons of levofloxacin, ciprofloxacin, and ampicillin against Streptococcus pneumoniae in an in vitro model of infection. Antimicrob Agents Chemother 43:672–677.
Ambrose PG, Grasela DM, Grasela TH et al (2001) Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections. Antimicrob Agents Chemother 45:2793–2797. doi:10.1128/AAC.45.10.2793-2797.2001
Kiser TH, Hoody DW, Obritsch MD et al (2006) Levofloxacin pharmacokinetics and pharmacodynamics in patients with severe burn injury. Antimicrob Agents Chemother 50:1937–1945. doi:10.1128/AAC.01466-05
Burgess DS, Hall RG (2007) Simulated comparison of the pharmacodynamics of ciprofloxacin and levofloxacin against Pseudomonas aeruginosa using pharma-cokinetic data from healthy volunteers and 2002 minimum inhibitory concentra-tion data. Clin Ther 29:1421–1427. doi:10.1016/j.clinthera.2007.07.024
Kiffer CR, Pignatari AC (2011) Pharmacodynamic evaluation of commonly prescribed oral antibiotics against respiratory bacterial pathogens. BMC Infect Dis 11:286. doi:10.1186/1471-2334-11-286
Fuller JD, Low DE (2005) A review of Streptococcus pneumoniae infection treatment failures associated with fluoroquinolone resistance. Clin Infect Dis 41:118–121. doi:10.1086/430829
Dunbar LM, Wunderink RG, Habib MP et al (2003) High-dose, short-course levofloxacin for community-acquired pneumonia: a new treatment paradigm. Clin Infect Dis 37:752–760. doi:10.1086/377539
Pea F, Di Qual E, Cusenza A et al (2003) Pharmacokinetics and pharmacodynamics of intravenous levofloxacin in patients with early-onset ventilator-associated pneumonia. Clin Pharmacokinet 42:589–598
Sánchez Navarro A, Colino Gandarillas C-I, Alvarez Lerma F et al (2005) Pharmacokinetics and pharmacodynamics of levofloxacin in intensive care patients. Clin Pharmacokinet 44:627–635
Labreche MJ, Frei CR (2012) Declining susceptibilities of gram-negative bacteria to the fluoroquinolones: effects on pharmacokinetics, pharmacodynamics, and clinical outcomes. Am J Health-Syst Pharm 69:1863–1870. doi:10.2146/ajhp110464
Homma T, Hori T, Sugimori G, Yamano Y (2007) Pharmacodynamic assessment based on mutant prevention concentrations of fluoroquinolones to prevent the emergence of resistant mutants of Streptococcus pneumoniae. Antimicrob Agents Chemother 51:3810–3815. doi:10.1128/AAC.01372-06
Scaglione F (2002) Can PK/PD be used in everyday clinical practice. Int J Antimicrob Agents 19:349–353
Pea F, Poz D, Viale P et al (2006) Which reliable pharmacodynamic breakpoint should be advised for ciprofloxacin monotherapy in the hospital setting? A TDM-based retrospective perspective. J Antimicrob Chemother 58:380–386. doi:10.1093/jac/dkl226
Roberts JA, Norris R, Paterson DL, Martin JH (2012) Therapeutic drug monitoring of antimicrobials. Br J Clin Pharmacol 73:27–36. doi:10.1111/j.1365-2125.2011.04080.x
Gao C-H, L-S Y, Zeng S et al (2014) Personalized therapeutics for levofloxacin: a focus on pharmacokinetic concerns. Ther Clin Risk Manag 10:217–227. doi:10.2147/TCRM.S59079
Montay G, Gaillot J (1990) Pharmacokinetics of fluoroquinolones in hepatic failure. J Antimicrob Chemother 26(Suppl B):61–67
Alsultan A, An G, Peloquin CA (2015) Limited sampling strategy and target attainment analysis for levofloxacin in patients with tuberculosis. Antimicrob Agents Chemother 59:3800–3807. doi:10.1128/AAC.00341-15
Long R, Barrie J, Peloquin CA (2015) Therapeutic drug monitoring and the conservative management of chronic tuberculous empyema: case report and review of the literature. BMC Infect Dis 15:327. doi:10.1186/s12879-015-1093-7
Manika K, Chatzika K, Zarogoulidis K, Kioumis I (2012) Moxifloxacin in multidrug-resistant tuberculosis: is there any indication for therapeutic drug monitoring? Eur Respir J 40:1051–1053. doi:10.1183/09031936.00202411
Lee SH, Seo K-A, Lee YM et al (2015) Low serum concentrations of moxifloxacin, prothionamide, and cycloserine on sputum conversion in multi-drug resistant TB. Yonsei Med J 56:961–967. doi:10.3349/ymj.2015.56.4.961
Pranger AD, Kosterink JGW, van Altena R et al (2011) Limited-sampling strategies for therapeutic drug monitoring of moxifloxacin in patients with tuberculosis. Ther Drug Monit 33:350–354. doi:10.1097/FTD.0b013e31821b793c
Di Paolo A, Malacarne P, Guidotti E et al (2010) Pharmacological issues of linezolid: an updated critical review. Clin Pharmacokinet 49:439–447. doi:10.2165/11319960-000000000-00000
Dryden MS (2011) Linezolid pharmacokinetics and pharmacodynamics in clinical treatment. J Antimicrob Chemother 66(Suppl 4):iv7–iv15. doi:10.1093/jac/dkr072
Matsumoto K, Takeshita A, Ikawa K et al (2010) Higher linezolid exposure and higher frequency of thrombocytopenia in patients with renal dysfunction. Int J Antimicrob Agents 36:179–181. doi:10.1016/j.ijantimicag.2010.02.019
Tsuji Y, Hiraki Y, Matsumoto K et al (2011) Thrombocytopenia and anemia caused by a persistent high linezolid concentration in patients with renal dysfunction. J Infect Chemother 17:70–75. doi:10.1007/s10156-010-0080-6
Tsuji Y, Yukawa E, Hiraki Y et al (2013) Population pharmacokinetic analysis of linezolid in low body weight patients with renal dysfunction. J Clin Pharmacol 53:967–973. doi:10.1002/jcph.133
Zoller M, Maier B, Hornuss C et al (2014) Variability of linezolid concentrations after standard dosing in critically ill patients: a prospective observational study. Crit Care 18:R148. doi:10.1186/cc13984
Pea F, Furlanut M, Cojutti P et al (2010) Therapeutic drug monitoring of linezolid: a retrospective monocentric analysis. Antimicrob Agents Chemother 54:4605–4610. doi:10.1128/AAC.00177-10
Abe S, Chiba K, Cirincione B et al (2009) Population pharmacokinetic analysis of linezolid in patients with infectious disease: application to lower body weight and elderly patients. J Clin Pharmacol 49:1071–1078. doi:10.1177/0091270009337947
Nukui Y, Hatakeyama S, Okamoto K et al (2013) High plasma linezolid concentration and impaired renal function affect development of linezolid-induced thrombocytopenia. J Antimicrob Chemother 68:2128–2133. doi:10.1093/jac/dkt133
Pea F, Viale P, Cojutti P et al (2012) Therapeutic drug monitoring may improve safety outcomes of long-term treatment with linezolid in adult patients. J Antimicrob Chemother 67:2034–2042. doi:10.1093/jac/dks153
Bhalodi AA, Papasavas PK, Tishler DS et al (2013) Pharmacokinetics of intravenous linezolid in moderately to morbidly obese adults. Antimicrob Agents Chemother 57:1144–1149. doi:10.1128/AAC.01453-12
Hamilton R, Thai XC, Ameri D, Pai MP (2013) Oral bioavailability of linezolid before and after Roux-en-Y gastric bypass surgery: is dose modification necessary in obese subjects? J Antimicrob Chemother 68:666–673. doi:10.1093/jac/dks431
Cojutti P, Maximova N, Crichiutti G et al (2015) Pharmacokinetic/pharmacodynamic evaluation of linezolid in hospitalized paediatric patients: a step toward dose optimization by means of therapeutic drug monitoring and Monte Carlo simulation. J Antimicrob Chemother 70:198–206. doi:10.1093/jac/dku337
Matsumoto K, Shigemi A, Takeshita A et al (2014) Analysis of thrombocytopenic effects and population pharmacokinetics of linezolid: a dosage strategy according to the trough concentration target and renal function in adult patients. Int J Antimicrob Agents 44:242–247. doi:10.1016/j.ijantimicag.2014.05.010
Boak LM, Rayner CR, Grayson ML et al (2014) Clinical population pharmacokinetics and toxicodynamics of linezolid. Antimicrob Agents Chemother 58:2334–2343. doi:10.1128/AAC.01885-13
Cattaneo D, Orlando G, Cozzi V et al (2013) Linezolid plasma concentrations and occurrence of drug-related haematological toxicity in patients with gram-positive infections. Int J Antimicrob Agents 41:586–589. doi:10.1016/j.ijantimicag.2013.02.020
Hiraki Y, Tsuji Y, Hiraike M et al (2012) Correlation between serum linezolid concentration and the development of thrombocytopenia. Scand J Infect Dis 44:60–64. doi:10.3109/00365548.2011.608712
Cattaneo D, Gervasoni C, Clementi E (2016) Is there still room for therapeutic drug monitoring of linezolid in patients with tuberculosis? Eur Respir J 47:1287–1288. doi:10.1183/13993003.01913-2015
De Pascale G, Fortuna S, Tumbarello M et al (2015) Linezolid plasma and intrapulmonary concentrations in critically ill obese patients with ventilator-associated pneumonia: intermittent vs continuous administration. Intensive Care Med 41:103–110. doi:10.1007/s00134-014-3550-y
Dong H, Xie J, Chen L et al (2014) Developments in the pharmacokinetic/pharmacodynamic index of linezolid: a step toward dose optimization using Monte Carlo simulation in critically ill patients. Int J Infect Dis 22:35–40. doi:10.1016/j.ijid.2014.01.016
Alffenaar J-WC, Kosterink JGW, van Altena R et al (2010) Limited sampling strategies for therapeutic drug monitoring of linezolid in patients with multidrug-resistant tuberculosis. Ther Drug Monit 32:97–101. doi:10.1097/FTD.0b013e3181cc6d6f
Falagas ME, Kasiakou SK (2005) Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin Infect Dis 40:1333–1341. doi:10.1086/429323
Zavascki AP (2014) Polymyxins for the treatment of extensively-drug-resistant Gram-negative bacteria: from pharmacokinetics to bedside. Expert Rev Anti Infect Ther 12:531–533. doi:10.1586/14787210.2014.902307
Nation RL, Li J, Cars O et al (2015) Framework for optimisation of the clinical use of colistin and polymyxin B: the Prato polymyxin consensus. Lancet Infect Dis 15:225–234. doi:10.1016/S1473-3099(14)70850-3
Garonzik SM, Li J, Thamlikitkul V et al (2011) Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 55:3284–3294. doi:10.1128/AAC.01733-10
Plachouras D, Karvanen M, Friberg LE et al (2009) Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with infections caused by gram-negative bacteria. Antimicrob Agents Chemother 53:3430–3436. doi:10.1128/AAC.01361-08
Sorlí L, Luque S, Campillo N, et al (2016) Impact of colistin therapeutic drug monitoring in a clinical setting. Amsterdam, Netherlands
Bode-Böger SM, Schopp B, Tröger U et al (2013) Intravenous colistin in a patient with serious burns and borderline syndrome: the benefits of therapeutic drug monitoring. Int J Antimicrob Agents 42:357–360. doi:10.1016/j.ijantimicag.2013.06.009
Yaita K, Sameshima I, Takeyama H et al (2013) Liver abscess caused by multidrug-resistant Pseudomonas aeruginosa treated with colistin; a case report and review of the literature. Intern Med Tokyo Jpn 52:1407–1412
Yamada T, Ishiguro N, Oku K et al (2015) Successful colistin treatment of multidrug-resistant Pseudomonas aeruginosa infection using a rapid method for determination of colistin in plasma: usefulness of therapeutic drug monitoring. Biol Pharm Bull 38:1430–1433. doi:10.1248/bpb.b15-00323
Karaiskos I, Friberg LE, Pontikis K et al (2015) Colistin population pharmacokinetics after application of a loading dose of 9 MU colistin methanesulfonate in critically ill patients. Antimicrob Agents Chemother 59:7240–7248. doi:10.1128/AAC.00554-15
Mohamed AF, Karaiskos I, Plachouras D et al (2012) Application of a loading dose of colistin methanesulfonate in critically ill patients: population pharmacokinetics, protein binding, and prediction of bacterial kill. Antimicrob Agents Chemother 56:4241–4249. doi:10.1128/AAC.06426-11
Nation RL, Garonzik SM, Li J et al (2016) Updated US and European dose recommendations for intravenous colistin: how do they perform? Clin Infect Dis 62:552–558. doi:10.1093/cid/civ964
Sorlí L, Luque S, Grau S et al (2013) Trough colistin plasma level is an independent risk factor for nephrotoxicity: a prospective observational cohort study. BMC Infect Dis 13:380. doi:10.1186/1471-2334-13-380
Horcajada JP, Sorlí L, Luque S et al (2016) Validation of a colistin plasma concentration breakpoint as a predictor of nephrotoxicity in patients treated with colistin methanesulfonate. Int J Antimicrob Agents 48:725–727. doi:10.1016/j.ijantimicag.2016.08.020
Landersdorfer CB, Nation RL (2015) Colistin: how should it be dosed for the critically ill? Semin Respir Crit Care Med 36:126–135. doi:10.1055/s-0034-1398390
Moise PA, North D, Steenbergen JN, Sakoulas G (2009) Susceptibility relationship between vancomycin and daptomycin in Staphylococcus aureus: facts and assumptions. Lancet Infect Dis 9:617–624. doi:10.1016/S1473-3099(09)70200-2
Rose WE, Leonard SN, Sakoulas G et al (2008) daptomycin activity against Staphylococcus aureus following vancomycin exposure in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother 52:831–836. doi:10.1128/AAC.00869-07
Safdar N, Andes D, Craig WA (2004) In vivo pharmacodynamic activity of daptomycin. Antimicrob Agents Chemother 48:63–68
Falcone M, Russo A, Cassetta MI et al (2013) Variability of pharmacokinetic parameters in patients receiving different dosages of daptomycin: is therapeutic drug monitoring necessary? J Infect Chemother 19:732–739. doi:10.1007/s10156-013-0559-z
Firsov AA, Smirnova MV, Lubenko IY et al (2006) Testing the mutant selection window hypothesis with Staphylococcus aureus exposed to daptomycin and vancomycin in an in vitro dynamic model. J Antimicrob Chemother 58:1185–1192. doi:10.1093/jac/dkl387
Chambers HF, Basuino L, Diep BA et al (2009) Relationship between susceptibility to daptomycin in vitro and activity in vivo in a rabbit model of aortic valve endocarditis. Antimicrob Agents Chemother 53:1463–1467. doi:10.1128/AAC.01307-08
Reiber C, Senn O, Müller D et al (2015) Therapeutic drug monitoring of daptomycin: a retrospective monocentric analysis. Ther Drug Monit 37:634–640. doi:10.1097/FTD.0000000000000196
D’Avolio A, Pensi D, Baietto L et al (2016) Daptomycin pharmacokinetics and pharmacodynamics in septic and critically ill patients. Drugs 76:1161–1174. doi:10.1007/s40265-016-0610-3
Falcone M, Russo A, Cassetta MI et al (2012) Daptomycin serum levels in critical patients undergoing continuous renal replacement. J Chemother 24:253–256. doi:10.1179/1973947812Y.0000000033
Bhavnani SM, Rubino CM, Ambrose PG, Drusano GL (2010) Daptomycin exposure and the probability of elevations in the creatine phosphokinase level: data from a randomized trial of patients with bacteremia and endocarditis. Clin Infect Dis 50:1568–1574. doi:10.1086/652767
Barreau S, Benaboud S, Kernéis S et al (2016) Staphylococcus aureus osteo-articular infection: usefulness of the determination of daptomycin serum concentration to explain a treatment failure. Int J Clin Pharmacol Ther 54:923–927. doi:10.5414/CP202538
Cohen-Wolkowiez M, Smith PB, Benjamin DK et al (2008) Daptomycin use in infants: report of two cases with peak and trough drug concentrations. J Perinatol Off J Calif Perinat Assoc 28:233–234. doi:10.1038/sj.jp.7211898
Soto D, Silva C, Andresen VM et al (2015) Monitorización terapéutica de antibióticos: Nuevas metodologías: biosensores. Rev Médica Chile 143:1050–1057. doi:10.4067/S0034-98872015000800013
Ferguson BS, Hoggarth DA, Maliniak D et al (2013) Real-time, aptamer-based tracking of circulating therapeutic agents in living animals. Sci Transl Med 5:213ra165. doi:10.1126/scitranslmed.3007095
Tsalik EL, Henao R, Nichols M et al (2016) Host gene expression classifiers diagnose acute respiratory illness etiology. Sci Transl Med 8:322ra11. doi:10.1126/scitranslmed.aad6873
Webster M, Kumar VS (2016) A blood scan for sepsis? Clin Chem 62:538–540. doi:10.1373/clinchem.2015.253682
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Stojanova, J., Luque, S. (2018). Therapeutic Drug Monitoring: More Than Avoiding Toxicity. In: Udy, A., Roberts, J., Lipman, J. (eds) Antibiotic Pharmacokinetic/Pharmacodynamic Considerations in the Critically Ill. Adis, Singapore. https://doi.org/10.1007/978-981-10-5336-8_9
Download citation
DOI: https://doi.org/10.1007/978-981-10-5336-8_9
Published:
Publisher Name: Adis, Singapore
Print ISBN: 978-981-10-5335-1
Online ISBN: 978-981-10-5336-8
eBook Packages: MedicineMedicine (R0)