
Abstract
Primary liver cancer is the second most common cause of death due to cancer worldwide, and a vast majority of primary liver cancer is the hepatocellular carcinoma (HCC) subtype. Although a number of etiologies are recognized to cause HCC, HBV infection accounts for over 50% of diagnosed HCC cases. HBV infection leads to the development of HCC through a variety of mechanisms. Indirectly, viral infection leads to repetitive liver injury through oxidative stress, the immune response, and telomere shortening, eventually causing cellular transformation. Integration of viral DNA into the host genome can indirectly lead to carcinogenesis by inducing genomic instability, as well as directly causing oncogenesis by preferentially integrating into genomic loci of known oncogenes. The activity of the HBV X protein (HBx) has been demonstrated to directly promote transformation and hepatocarcinogenesis in a number of ways, including activating cancer-related pathways and altering cellular epigenetic and noncoding RNA expression. In this chapter, we will summarize the current scientific knowledge of the ways in which HBV infection can lead to HCC.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Globally, primary liver cancer represents the fifth most commonly diagnosed cancer and the second most common cause of death due to cancer (Theise 2014). Hepatocellular carcinoma (HCC) accounts for a majority (85–90%) of primary liver cancers and leads to an estimated 600,000 deaths per year (Jemal et al. 2011). Geographically, the incidence of HCC is not distributed equally, with greater than 80% of HCC cases occurring in East Asia or sub-Saharan Africa. China alone accounts for over 50% of global HCC cases (El-Serag 2012).
There are a number of recognized etiologies leading to the development of HCC including liver cirrhosis, alcoholic liver disease, exposure to aflatoxin B1, diabetes, and obesity. However, the main etiology of HCC is hepatitis B virus (HBV) or hepatitis C virus (HCV) infection (Sanyal et al. 2010). The association between HCC development and chronic HBV infection was first reported in 1975 (Blumberg et al. 1975). Chronic HBV infection is now recognized to account for over 50% of HCC cases worldwide. Over 350 million people are chronic HBV carriers, with 75% of these cases occurring in Asian countries. Prospective cohort studies have shown that people who are chronically infected with HBV have a 5- to 100-fold increased risk of developing HCC compared to non-infected persons. (El-Serag 2012).
Most cases of HCC caused by HBV occur in patients with cirrhosis (70–90%), which is a significant risk factor for HCC (Yang et al. 2011). Other factors also contribute to an increased risk of developing HBV-related HCC. For example, demographic factors, including male sex and advanced age, are associated with an increased risk of HCC. Exposure to aflatoxin B1 or alcohol consumption also raises the risk of developing HBV-related HCC. Patients infected with HBV have a 200-fold greater risk of HCC if they are obese (body mass index ≥30) and have diabetes mellitus, indicating an importance of metabolic factors. HCC risk is also dependent on viral factors. High levels of HBV DNA and HBV replication are associated with increased risk. The genotype of HBV also plays a role. At least eight genotypes of HBV have been identified, and increased risk of developing HCC is associated with HBV genotype C in Asian populations, genotype A in Africans, and genotype F in Alaskan natives (Kew 2010). In addition, seropositivity for hepatitis B surface antigen (HBsAg) remains one of the greatest risk factors for HCC, varying from a sevenfold risk increase in Japan to 60- to 98-fold risk increase in Taiwan. Seroclearance of HBsAg reduces the risk of HBV-related HCC (Burns and Thompson 2014).
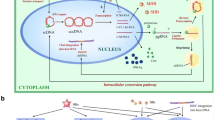
Understanding the epidemiological characteristics of HBV-related HCC remains an important field of study, but it is also important to understand the cellular mechanisms by which HBV infection leads to the development of HCC. Molecular carcinogenesis of HBV-related HCC is caused by indirect mechanisms such as oxidative stress, the immune response, and telomere shortening, the “mixed” mechanism of DNA insertion leading to genomic instability and insertional mutagenesis, and direct oncogenesis caused by the HBV X protein (HBx) (Fig. 8.1). In this chapter, we will review the oncogenic mechanisms by which HBV viral infection contributes to the development and progression of HCC.

Indirect and direct mechanisms contribute to HBV-related HCC carcinogenesis. Indirect mechanisms of carcinogenesis due to liver injury include oxidative stress, the immune response, and telomere shortening. Direct mechanisms of carcinogenesis are due to the oncogenic activities of HBx protein. Viral DNA integration into the genome promotes carcinogenesis indirectly by inducing genomic instability and directly via insertional mutagenesis
2 Indirect Oncogenic Roles of HBV: Inflammation and Liver Injury
Numerous studies have shown that HBV infection leads to inflammation and liver injury . A mouse model study that examined the progression of disease following HBV infection demonstrated that overproduction of the hepatitis B virus large envelope protein and high concentrations of HBsAg led to phenotypic changes within the liver, beginning with chronic hepatitis, followed by the development of regenerative nodules and oval cell hyperplasia, liver cell adenomas, and finally HCC (Dunsford et al. 1990). No histopathological changes were seen in age- and sex-matched controls. This study showed that HBV infection could directly lead to inflammation, regenerative hyperplasia, and injury to hepatocytes, with the potential to induce transforming mutations (Dunsford et al. 1990). There are several mechanisms by which HBV infection may lead to liver injury and HCC, including inducing oxidative stress, the immune response, and telomere length (Fig. 8.1).
2.1 Oxidative Stress
Oxidative stress represents an important mechanism by which HBV infection may lead to HCC. An overabundance of reactive oxygen species (ROS) can cause damage to cellular lipids, proteins, or DNA, which could potentially lead to chromosomal mutagenesis and carcinogenesis. ROS may also activate signaling pathways involved in cellular proliferation (Ha et al. 2010). DNA damage induced by oxidative stress and ROS has been observed within the livers of mice with chronic active hepatitis. The accumulation of 8-oxo-2′-deoxyguanosine, an adduct of deoxyguanosine that is modified by oxidation, increased as liver disease progressed. Livers exhibiting advanced disease including nodular hyperplasia, adenomas, and HCC showed the greatest increase in 8-oxo-2′-deoxyguanosine, indicating the highest amount of oxidative DNA damage (Hagen et al. 1994). DNA damage induced by ROS has also been seen in HBV-infected patients, which was measured by the accumulation of 8-hydroxydeoxyguanosine, a promutagenic DNA lesion induced by hydroxyl radicals (Fujita et al. 2008).
Several patient studies have examined changes in the liver due to HBV-related disease, with an increase in oxidative stress observed with increasing viral replication status and disease severity (Bolukbas et al. 2005). A study in Taiwan followed patients with HBV-associated HCC for over 20 years and found that these patients exhibited extensive oxidative damage during clinical disease progression (Tsai et al. 2009). Another study compared patients with inactive HBsAg carrier state (IHBCS) infection, patients with chronic HBV infection, and healthy control patients. The study found that patients with chronic HBV infection had significantly higher measured levels of total oxidative stress and lipid peroxidation compared to IHBCS patients and healthy controls. Antioxidant status was decreased significantly in chronic HBV patients (Duygu et al. 2012).
2.2 Immune Response
The association between the immune cell response and cancer is well established (Balkwill and Mantovani 2001). A decrease in the immune response can facilitate viral replication and tumor formation, whereas overactive immune response and inflammation can lead to changes in tissue architecture and tissue remodeling, apoptosis, and DNA alterations, which may also promote carcinogenesis (Budhu and Wang 2006). HBV infection is controlled by antiviral cytotoxic T cells. In patients with active hepatitis who ultimately clear the virus, the T cell response is vigorous. In patients with chronic hepatitis, the T cell response is weaker, destroying a portion of infected cells, leading to a continuous cycle of cellular destruction and regeneration, resulting in liver injury and HCC development. A murine model demonstrated that hepatocellular injury can be induced by overexpression of the HBV large envelope protein, leading to cellular regeneration, DNA damage, clonal expansion, and ultimately HCC (Nakamoto et al. 1998). Liver cell injury initiated and mediated by HBV infection has the potential to sustain carcinogenesis in the absence of viral integration, HBx expression, or genotoxic agents. This study suggested that an ineffective T cell response may be a principal oncogenic factor in the development of HCC during chronic hepatitis infection (Nakamoto et al. 1998). Indeed, an impaired immune response has been detected in patients with chronic HBV infection. For example, circulating and liver-residing regulatory T cells (Tregs) were increased in HCC patients with HBV infection, which suppressed the immune response induced by the HBV antigen and the HCC tumor antigen, thus inhibiting tumor immunosurveillance and promoting tumor progression (Zhang et al. 2010).
2.3 Telomere Length
In normal cells, the length of telomeres, segments of short nucleotide repeats that cap the ends of chromosomes, become progressively shorter with each cell division. Too short telomeres can cause chromosomal instability; thus, healthy cells will enter cellular senescence when telomeres reach a certain length. Maintaining telomere length, thus avoiding cellular senescence, is a key process in the life cycle of tumor cells, leading to immortalization. A study examining patient samples with normal livers, chronic active hepatitis, liver cirrhosis, and HCC found a progressive shortening of telomeres with each stage. However, telomerase, an enzyme that stabilizes and lengthens telomeres by adding nucleotides onto the chromosomal ends, was also detected in high levels in all HCC patient samples examined. This study suggested that rapid cellular proliferation and turnover due to viral-induced liver injury causes a multistep process of HCC carcinogenesis, beginning with progressive telomere shortening and eventually leading to telomerase activation (Miura et al. 1997).
3 “Mixed” Mechanism of HBV-Related Oncogenesis: DNA Integration
After entering the nucleus, HBV DNA can integrate into host cell chromosomes, allowing the virus to continually replicate in the cell. HBV DNA integration likely occurs prior to HCC tumor development, as integration is found in patients during the acute infection stage (Murakami et al. 2004). Integrated HBV DNA is found in the hepatocyte genome in a vast majority (85–90%) of HBV-related HCC cases and is considered to be one of the most important ways in which HBV causes a pro-oncogenic effect (Minami et al. 2005). There are several mechanisms by which HBV DNA integration can cause a carcinogenic effect. Indirectly, HBV can lead to oncogenesis by inducing genomic instability, whereas causing alterations in oncogenic gene expression via insertional mutagenesis is considered to be a direct mechanism of HBV-related oncogenesis (Fig. 8.1).
3.1 Genomic Instability
Genomic instability is a hallmark of cancer. Chromosomal instability, in which chromosome structure progressively changes in cancer cells compared to normal cells, is common in many types of cancer (Negrini et al. 2010). The integration of HBV DNA into the genome has been shown to cause structural alternations in the chromosomal DNA of hepatocytes. Microdeletions in host DNA at HBV DNA integration sites are thought to be integral to the mechanism of integration. Macrodeletions , as well as amplification of chromosomal DNA at HBV DNA integration sites, have been reported. Chromosomal translocations have also been observed, in which the DNA from two chromosomes is joined to each end of an integrated viral DNA sequence (Idilman et al. 1998). HCC that is associated with HBV infection has been shown to contain more genomic instability via allelic deletions and amplifications, caused by HBV DNA integration, than HCC that is caused by other etiologies (Lee et al. 2008). These alterations could lead to alterations in expression of genes that are related to cancer such as tumor-suppressor genes or proto-oncogenes.
3.2 Insertional Mutagenesis
Initially, HBV DNA integration into the host genome was considered to occur at random, thus constituting an indirect mechanism of oncogenesis. However, with the implementation of next-generation sequencing, recent studies have shown that there are recurrent sites into which HBV DNA preferentially integrates. A number of these integration sites include genomic loci that code for genes that are highly well-known to regulate proliferation and cell survival in cancer, and the integration of viral DNA causes changes in gene expression at these loci. For example, multiple studies have shown that HBV DNA preferentially integrates into the gene encoding TERT, telomerase reverse transcriptase, the catalytic subunit of telomerase, the enzyme responsible for maintaining telomere length for inhibiting cellular senescence (see Sect. 2.3), allowing a cell to become immortal. Other common recurrent sites of integration include genes encoding MLL4, a histone methyltransferase known to critically regulate epigenetics and gene expression in cancer, as well as CCNE1, cyclin E1, which regulates the cell-cycle transition from G1 to S phase and is known to be overexpressed in cancer (Matsubara and Tokino 1990). It is possible that viral integration indeed starts as a random process but that certain integration sites with oncogenic potential can be selectively enriched during hepatocarcinogenesis.
It should also be noted that HBV DNA does not completely integrate into the host cell genome unaltered. The viral DNA itself is subject to rearrangements and genomic alterations such as deletions. Thus, the integration of viral DNA into the genome may also lead to functional or structural alterations of proteins via protein fusions containing protein coded from viral DNA and host cell DNA. These so-called chimeric proteins may have novel functions that promote carcinogenesis. The cyclin A gene has been identified as a site of HBV viral DNA integration in which an HBV-cyclin A fusion protein was coded. This protein contained a substitution of 152 amino acids of the N-terminal portion of the cyclin A gene by 156 amino acids of the HBV middle surface protein. The resulting fusion protein demonstrated tumorigenic properties (Pollicino et al. 2011). In another case, HBV DNA integrated into the gene that encoded the retinoic acid receptor β, causing overexpression of the protein because its expression was under the control of the integrated HBV preS1 viral promotor. This fusion protein also demonstrated carcinogenic properties (Garcia et al. 1993).
4 Direct Oncogenic Roles of HBV: HBx Protein
The HBx gene of HBV encodes the 154 amino acid, 17 kDa HBx protein. This protein does not directly bind to DNA but acts as a transactivator of viral gene expression via protein-protein interactions and modulating signaling pathways. HBx protein is required for viral replication and is critical for protecting infected hepatocytes from immune destruction. HBx also enhances HCC carcinogenesis in a number of ways (Geng et al. 2015). Several studies have shown that HBx RNA and protein expression is found in HCC tumor cells without active viral replication (Su et al. 1998; Peng et al. 2005). HBx protein itself can also be mutated, and the C-terminal region of HBx, produced by truncation, has also been shown to contribute to HCC carcinogenesis.
HBx protein interacts with, and oftentimes enhances, many key proteins and pathways that are involved in HCC carcinogenesis. For example, transforming growth factor β (TGF-β) can act as a tumor suppressor by inhibiting epithelial cell growth. However, the TGF-β signaling pathway can also shift to promote oncogenesis, and it has been demonstrated that HBx can stimulate this change in the early stages of HCC carcinogenesis (Murata et al. 2009). In this section, we will discuss the various mechanisms by which HBx has been implicated in the maintenance and progression of HCC tumor cells (Fig. 8.1, Table 8.1).
4.1 HBx Affects Cancer-Related Pathways
It has been speculated that HBx can act as a growth factor for HCC tumor cells, by activating several pathways that have been implicated in the initiation and maintenance of cancer cells such as the Jak/STAT signaling pathway (Lee and Yun 1998) and the RAS/RAF/MAPK pathway (Benn and Schneider 1994). HBx also directly binds to the C-terminus of the tumor-suppressor p53. Inactivation mutations or deletion of p53 is one of the most common pan-cancer alterations. HBx inhibits the activity of p53, including p53-mediated apoptosis, by sequestering p53 in the cytoplasm and preventing it from entering the nucleus (Wang et al. 1994). HBx mutants, caused by the HBx gene overlapping with the HBV core promoter region, also indirectly modulate p53 by upregulating the S phase kinase-associated protein 2 (SKP2) . SKP2 contributes to the downregulation of p53 via ubiquitination and degradation, thus contributing to tumorigenesis (Yan et al. 2015).
The nuclear factor (NF)-κB protein complex is a key transcription factor in cancer biology, regulating inflammation and cell death. The activation of the NF-κB pathway plays a known role in hepatocarcinogenesis (Luedde and Schwabe 2011). HBx stimulates the expression of metastasis-associated protein 1 (MTA1) , via interactions with NF-κB. MTA1 is a chromatin modulator that is involved in tumor initiation and the inflammatory response (Bui-Nguyen et al. 2010). The pleotropic cytokine interleukin 6 (IL6), which is involved in immune and inflammatory response, is also upregulated due to the enhancement of two NF-κB subunits to target gene DNA (Lee et al. 1998). HBx increases the expression of known NF-κB target genes, including several matrix metalloproteinases (MMPs), which are involved in metastasis, and vascular endothelial growth factor (VEGF), a key regulator of angiogenesis (Liu et al. 2010a). HBx may also play a role in tumorigenesis by upregulating and stabilizing hypoxia-inducible factor 1α (HIF-1α), a key regulator of hypoxia, which stimulates angiogenesis (Moon et al. 2004). HBx also stimulates the upregulation of angiopoietin-2 (Ang2), a pro-angiogenic growth factor that is involved in vascular remodeling and development during carcinogenesis (Sanz-Cameno et al. 2006).
The Wnt/β-catenin pathway plays an important role in embryonic development and also contributes to the homeostasis of stem cells. The transcription of downstream target genes by β-catenin is regulated by the β-catenin destruction complex, which modulates the amount of stabilized β-catenin that can enter the nucleus. However, aberrant β-catenin signaling can lead to the improper transcription of target genes such as VEGF, oncoprotein c-Myc, and cyclin D1, an important cell-cycle regulator, leading to tumorigenesis. HBx competitively binds to the adenomatous polyposis coli (APC) protein, which is part of the β-catenin destruction complex, causing the release, stabilization, and nuclear localization of β-catenin, leading to the transcription of downstream Wnt pathway targets (Hsieh et al. 2011). β-catenin levels are also directly regulated by glycogen synthase kinase 3 (GSK3) activity, which directly phosphorylates β-catenin , priming β-catenin for ubiquitination and degradation. HBx can inhibit the activity of GSK3, leading the Wnt/β-catenin pathway activation (Cha et al. 2004).
4.2 HBx Promotes “Stemness” of HCC Cells
High tumor heterogeneity represents a hallmark of HCC. Tumor-initiating cancer stem cells (CSCs) are found within HCC tumors and are associated with aggressive disease and poor prognosis. It has been demonstrated that HBx protein promotes “stemness” characteristics in HCC cells. HBx activates the expression of classic CSC markers Oct-4, Nanog, and Klf-4, which are associated with self-renewal. HBx expression is also associated with decreased amounts of E-cadherin. Loss of E-cadherin leads to epithelial to mesenchymal transition and thus increased metastasis and invasion. In addition, the expression of epithelial cell adhesion molecule (EpCAM), a marker of HCC tumor-initiating stem cells, has been detected in the nuclei of patients who are infected with HBV-related HCC (Arzumanyan et al. 2011). EpCAM is also a direct transcriptional target of Wnt/β-catenin signaling (Yamashita et al. 2009), which is known to be activated by HBx, as discussed in Sect. 4.1.
4.3 HBx Induces Epigenetic Changes
Epigenetics, in which gene expression is controlled by modifications to genetic DNA such as DNA methylation , are also associated with cancer. Tumors exhibit a global hypomethylation of DNA, yet silencing of tumor suppressors by hypermethylation (Esteller 2008). In addition to directly modifying cancer-related signaling pathways through signal activation and protein-protein interaction, HBx also causes changes in gene expression through epigenetic modifications of the promoter region of genes, causing the downregulation of gene expression. Aberrant hypermethylation of the retinoic acid receptor-β(2) (RAR-β(2)) gene has been detected in HBV-induced HCC. HBx has been shown to upregulate the expression of DNA methyltransferases 1 and 3A (DNMT1, DNMT3A), which induces promoter hypermethylation of RAR-β(2). Downregulation of RAR-β(2) expression leads to the subsequent downregulation of G1-checkpoint regulators p16, p21, and p27, which desensitizes cells to growth inhibition signaled by retinoic acid (Jung et al. 2010). HBx can also induce the direct hypermethylation and downregulation of p16ink4a expression via DNMT1 and DNMT3A (Zhu et al. 2010). Activation of DNMT1 by HBx has also been associated with the downregulated expression of E-cadherin, a tumor suppressor that is associated with HCC CSCs (see Sect. 4.2) (Lee et al. 2005). Indirectly, HBx expression is associated with hypermethylation of the promoters for the genes encoding ankyrin-repeat-containing, SH3-domain-containing, and proline-rich-region-containing proteins (ASPP). The ASPP family of proteins is known to regulate cellular apoptosis via interaction with p53. ASPP downregulation was associated with increased tumor growth and decreased sensitivity to apoptotic stimuli (Zhao et al. 2010).
4.4 HBx Deregulates Centrosome Duplication and the Cell Cycle
The cell cycle is a highly regulated process in which DNA is replicated and cells divide to create daughter cells that are genetically identical to the parent cell. Centrosomes are key in regulating cell division and serve to organize and orient microtubules during interphase and direct the formation of bipolar spindles during mitosis. Centrosome duplication begins during the G1/S phase of the cell cycle and is completed at S phase, occurring only once per cell cycle. However, centrosome amplification in cancer cells suggests that this process is deregulated and is not cell-cycle dependent (Budhu and Wang 2005). The nuclear export receptor Crm1 and Ran GTPase form a complex that is involved in the maintenance of centrosome duplication and assembly of the mitotic spindle. HBx binds and sequesters Crm1 in the cytoplasm, causing centrosome duplication, mitotic defects, and chromosome transmission errors. The genomic instability that results from centrosome amplification and loss of faithful cell division may contribute to carcinogenesis (Forgues et al. 2003).
The fidelity of cell-cycle progression is controlled by cyclins and the subsequent activation of cyclin-dependent kinases (CDK) , which are responsible for regulating key transitions. Several studies have shown that HBx protein is capable of deregulating the cell cycle. One study demonstrated that HBx activated progression through the cell cycle, shortening the emergence from quiescence (G0) and entry into S phase, accelerating through growth checkpoint controls at G0/G1 and G2/M. The cyclin-dependent kinases CDK1 and CDK2, and their associations with cyclin A, cyclin B, and cyclin E, were activated at a quicker and increased rate by HBx (Benn and Schneider 1995). In another study, HBx induced growth arrest at the G1 phase by activating the cyclin-dependent kinase inhibitor p21waf1/cip1, inhibiting CDK2, and allowing HCC cells to evade apoptosis (Park et al. 2000). HBx induces quiescent hepatocytes to exit the G0 phase by decreasing levels of p15 and p16 and increasing levels of active G1 phase proteins cyclin D1, cyclin E, p21, p 27, and CDK4. The activation of CDK2, the CDK controlling late G1/S phase, was also inhibited, causing cells to stall in the G1 phase of the cell cycle. This study suggested that active HBV viral replication requires cells to maintain the G1 phase of growth for the most efficient replication (Gearhart and Bouchard 2010).
HBx protein can also promote HCC tumor cell immortality by overcoming senescence, growth arrest, and apoptosis. HBx upregulates the transcriptional activation of TERT, increasing telomerase activity and leading to chromosomal telomere lengthening and preventing cells from becoming senescent (Liu et al. 2010b). HBx can also cause tumor cells to overcome senescence induced by all-trans retinoic acid by promoting the downregulation of p16 and p21 proteins via DNA methylation (Park et al. 2011).
4.5 HBx Affects Mitochondrial Function
HBx may induce cellular dysfunction in HCC cells through interactions with mitochondria. Mitochondrial structures aggregate abnormally in HBV-infected HCC cells, and HBx protein is associated with these structures, leading to a potential increase in cell death (Takada et al. 1999). HBx can alter the transmembrane potential of mitochondria by physically co-localizing with the voltage-dependent anion channel HVDAC3 (Rahmani et al. 2000). HBx has also been shown to downregulate key mitochondrial enzymes that are involved in electron transport for oxidative phosphorylation and increases oxidative injury via mitochondrial reactive oxygen species and the production of lipid peroxide. These changes sensitize cells to apoptosis signals (Lee et al. 2004). It has been found that the nuclear chaperone protein heat shock protein 60 (Hsp60) physically co-localizes with HBx in the mitochondria of HCC cells and enhances apoptosis (Tanaka et al. 2004). HBx protein also activates a kinase pathway that is essential for HBV replication. This pathway is calcium-dependent and can decrease mitochondrial calcium signaling, causing mitochondrial permeability transition (Tan et al. 2009).
4.6 HBx Affects DNA Repair
The ability of cancer cells to detect and repair DNA damage is another key hallmark of cancer. The accumulation of DNA damage leads to mutagenesis and genomic instability. Multiple studies have shown that HBx interferes with nucleotide excision repair (NER). One study demonstrated that HBx can interact with X-associated protein 1(XAP-1), a human protein homologous to simian repair protein UV-damaged DNA-binding protein (UVDDB), which is thought to be involved with the first step of NER by binding to damaged DNA, thus prohibiting effective NER (Becker et al. 1998). HBx also affects NER by complexing with p53 protein and inhibiting the association of p53 with the NER transcription factor ERCC3 (Wang et al. 1994). HBx can also directly bind to NER factors XPB and XPD DNA helicases and inhibit NER (Jia et al. 1999). Another study indicated that HBx affects the expression of NER DNA repair enzyme DNA glycosylase α (hMYHα), leading to an increase in the level of 8-hydroxy-2 deoxyguanosine (8-OHdG), a mutagenic DNA adduct that serves as a cellular indicator of oxidative stress (Cheng et al. 2010).
4.7 HBx Causes Changes in Noncoding RNA
A majority of the DNA in the human genome is transcribed into RNA transcripts that are not translated into protein products. However, these noncoded transcripts still have an important function within the cell, controlling cellular pathways by modulating the expression of other noncoding RNA and mRNA from genes encoding proteins or directly interacting with proteins. There are two general classes of noncoding RNA based on size: short noncoding RNAs, including microRNAs (miRNA) , with a length of under 200 nucleotides, and long noncoding RNAs (lncRNA) that are greater than 200 nucleotides in length (Ghidini and Braconi 2015). Expression of HBx in HCC cells deregulates the expression of numerous miRNAs and lncRNAs. The HBx transcript directly downregulates the expression of two well-known tumor-suppressor microRNAs, miR-15a and miR-16-1, via viral RNA sequences that target the microRNA (Wang et al. 2013). A number of other studies have demonstrated that HBx protein indirectly affects microRNA expression. For example, one study showed that in tumor samples from patients infected with HBV, deregulated miRNAs targeted genes in pathways associated with signal transduction, cell death, and DNA damage and recombination (Ura et al. 2009). NF-κB, upregulated by HBx protein, transcribes an increased amount of miR-143, causing the repression of fibronectin type III domain-containing 3B (FNDC3B), leading to an increase in cellular invasion and migration, promoting tumor metastasis (Zhang et al. 2009). HBx expression downregulated members of the let-7 family of miRNAs, leading to an upregulation of signal transducer and activator of transcription 3 (STAT3) and an increase in cellular proliferation (Wang et al. 2010). Another study found that HBx enhances tumorigenesis by downregulating the p53-mediated activation of miR-148a, which reduces growth, invasion, and metastasis of HCC cells. A decrease in miR-148a led to the increased expression of hematopoietic pre-B-cell leukemia transcription factor-interacting protein (HPIP), which regulates cancer cell growth through the Akt/extracellular-related kinase (ERK) and mammalian target of rapamycin (mTOR) signaling pathway (Xu et al. 2013). The microRNA miR-21 has been shown in several studies to promote HCC carcinogenesis. A C-terminal-truncated HBx protein upregulated miR-21 through IL6 and STAT3 activation. This study demonstrated that miR-21 overexpression plays a critical role in hepatocyte carcinogenesis following HBV infection (Li et al. 2014). HBx downregulated the expression of programed cell death 4 (PDCD4) through miR-21. PDCD4 may induce apoptosis in cells; thus, a decrease in PCDC4 may lead to decreased apoptosis in HCC cells (Qiu et al. 2013).
Epigenetics and microRNAs are interrelated in the progression of HCC. For example, HBx can lead to epigenetic aberration via downregulation of tumor-suppressing miR-152, which causes an increase in DNMT1, leading to hypermethylation and silencing of target genes (Huang et al. 2010). Another microRNA, miR-122, is the most highly expressed liver microRNA, and reduced expression of miR-122 is associated with the progression of HCC. Peroxisome proliferator-activated receptor-gamma (PPARγ) is part of a nuclear complex that modifies gene transcription, via epigenetic changes through the recruitment of histone deacetylases or histone methyltransferases, and promotes the expression of miR-122. PPARγ is directly bound by HBx in HBV-infected hepatocytes, leading to decreased miR-122 expression and cancer progression (Song et al. 2013). MiR-132, a tumor-suppressive microRNA, represses tumor growth by inhibiting the Akt-signaling pathway. HBx epigenetically represses the expression of MiR-132 by inducing DNA methylation of the MiR-132 promoter (Wei et al. 2013).
LncRNAs have been implicated in cancer progression as well. The lncRNA highly upregulated in liver cancer (HULC) is positively associated with HCC. HBx promotes the expression of HULC by associating with the cAMP-responsive element-binding protein and activating the HULC promoter. Increased expression of HULC was also found to downregulate the expression of the tumor-suppressor protein p18, leading to increased hepatocarcinogenesis (Du et al. 2012). Another lncRNA, downregulated expression by HBx (termed lncRNA-dreh), acts as a tumor suppressor by interacting with and reducing the expression of the intermediate filament protein vimentin, preventing tumor metastasis. A decrease in lncRNA-dreh is associated with poor prognosis in HCC patients (Huang et al. 2013). The expression of a novel lncRNA, DBH-AS1, was discovered to be positively associated with HBsAg expression, as well as HCC tumor size. DBH-AS1 promotes tumorigenesis by inducing cell-cycle progression, inhibiting apoptosis, and activating the MAPK pathway. The expression of DBH-AS1 is significantly increased by HBx protein (Huang et al. 2015). HBx protein also affects lncRNA expression through the transcription regulator ecotropic viral integration site 1 (Evi1). HBx expression causes an increase in Evi1, which deregulates a cluster of lncRNAs involved in HCC progression (Huang et al. 2016). HBx expression also induces the upregulation of the lncRNA UCA1, which promotes cell growth by enhancing cell-cycle progression via the downregulation of p27 genes (Hu et al. 2016).
As discussed in Sect. 3, HBV viral DNA inserts into host DNA, causing insertional mutagenesis. HBV insertion often occurs near or within DNA that is noncoding, such as long interspersed nuclear elements (LINEs), leading to the creation of chimeric HBx-LINE fusions. One study demonstrated that the HBx-LINE1 chimeric fusion is found in 23.3% of HCC tumors that are associated with HBV and is correlated with poor survival. The authors of the study also demonstrated that HBx-LINE1 expression could affect the activity of β-catenin and activate Wnt pathway signaling, thus acting like a lncRNA with HCC promoting properties (Lau et al. 2014). The HBx-LINE1 fusion transcript also contains six binding site for the tumor-suppressive miR-122, thus sequestering and depleting miR-122 in HCC cells. The reduction of miR-122 promotes tumorigenesis by activating epithelial-mesenchymal transition (EMT), leading to changes such as activation of the Wnt/β-catenin pathway, downregulation of E-cadherin, and increased cell migration (Liang et al. 2016).
5 Concluding Remarks
HBV infection constitutes a major etiological factor for the development of primary liver cancer and remains a public health issue worldwide. HBV contributes to the carcinogenesis of HCC by various indirect and direct mechanisms (Fig. 8.1). HBV infection triggers cellular damage through oxidative stress, reactive oxygen species, the inflammatory response, and telomere shortening, leading to potential mutation and cellular transformation following repeated cycles of liver cell injury. HBV integration into the genome contributes to oncogenesis indirectly by inducing genomic instability and directly through insertional mutagenesis in or near genomic loci that code for genes that regulate cellular proliferation and survival. The integration of viral DNA also allows for the expression of the HBx protein, which directly contributes to the molecular carcinogenesis of HCC by a number of mechanisms including affecting cancer-related pathways and altering cellular epigenetics and noncoding RNA expression (Table 8.1), which promotes cellular transformation.
To date, the most effective treatment for HBV-related HCC is prevention through vaccination programs. In Taiwan, a universal nationwide childhood vaccination program was implemented in the 1980s. A 20-year follow-up revealed a significant reduction of both the number of HBV carriers, as well as the incidence of childhood HCC, with benefits being maintained into adulthood (Chang et al. 2009). For people who are already carriers of HBV, continued surveillance of disease progression is an important preventative strategy, and early interventional antiviral therapy is key for reducing the incidence of HCC. Antiviral therapy, in addition to reducing serum viral load, controlling viral replication, and promoting the serum conversion of hepatitis B e antigen, may also block the carcinogenic properties of HBx. The immunomodulator interferon alpha (INF-α) may provide a benefit to patients with cirrhosis, whereas treatment with nucleoside analogs has been shown to reduce the risk of HCC development and delay disease progression in patients with HBV. However, the development of resistance continues to be a challenge (Sanyal et al. 2010; Lai and Yuen 2013).
Despite our current understanding of the molecular mechanisms of HCC carcinogenesis that are promoted by HBV infection, there are still a number of important questions that remain to be answered. For example, there is a disconnect between the vast amount of information we have gathered regarding the various mechanisms of HBV-induced HCC carcinogenesis and the true contribution that each plays in cellular transformation. It is unknown whether the amount of HBV replication or viral integration in each patient is directly correlated to increased clonal expansion, cellular transformation, and advanced tumor progression. Additionally, it remains to be answered whether there are distinct subtypes of HBV-related HCC or how HBV may interact with other etiological factors in HCC, which may lead to unique molecular pathogenesis. Thus, the discovery of biomarkers and new therapeutic targets for the treatment of HBV-related HCC will continue to be an important field of study, which will be necessary for the development of prevention strategies and personalized treatment modalities for patients with HBV infection (Levrero and Zucman-Rossi 2016).
References
Arzumanyan A, Friedman T, Ng IO, Clayton MM, Lian Z, Feitelson MA. Does the hepatitis B antigen HBx promote the appearance of liver cancer stem cells? Cancer Res. 2011;71(10):3701–8. https://doi.org/10.1158/0008-5472.CAN-10-3951.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45.
Becker SA, Lee TH, Butel JS, Slagle BL. Hepatitis B virus X protein interferes with cellular DNA repair. J Virol. 1998;72(1):266–72.
Benn J, Schneider RJ. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci U S A. 1994;91(22):10350–4.
Benn J, Schneider RJ. Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc Natl Acad Sci U S A. 1995;92(24):11215–9.
Blumberg BS, Larouze B, London WT, Werner B, Hesser JE, Millman I, Saimot G, Payet M. The relation of infection with the hepatitis B agent to primary hepatic carcinoma. Am J Pathol. 1975;81(3):669–82.
Bolukbas C, Bolukbas FF, Horoz M, Aslan M, Celik H, Erel O. Increased oxidative stress associated with the severity of the liver disease in various forms of hepatitis B virus infection. BMC Infect Dis. 2005;5:95. https://doi.org/10.1186/1471-2334-5-95.
Budhu A, Wang XW. The role of cytokines in hepatocellular carcinoma. J Leukoc Biol. 2006;80(6):1197–213.
Budhu AS, Wang XW. Loading and unloading: orchestrating centrosome duplication and spindle assembly by Ran/Crm1. Cell Cycle. 2005;4(11):1510–4.
Bui-Nguyen TM, Pakala SB, Sirigiri RD, Xia W, Hung MC, Sarin SK, Kumar V, Slagle BL, Kumar R. NF-kappaB signaling mediates the induction of MTA1 by hepatitis B virus transactivator protein HBx. Oncogene. 2010;29(8):1179–89. https://doi.org/10.1038/onc.2009.404.
Burns GS, Thompson AJ. Viral hepatitis B: clinical and epidemiological characteristics. Cold Spring Harb Perspect Med. 2014;4(12):a024935. https://doi.org/10.1101/cshperspect.a024935.
Cha MY, Kim CM, Park YM, Ryu WS. Hepatitis B virus X protein is essential for the activation of Wnt/beta-catenin signaling in hepatoma cells. Hepatology. 2004;39(6):1683–93. https://doi.org/10.1002/hep.20245.
Chang MH, You SL, Chen CJ, Liu CJ, Lee CM, Lin SM, Chu HC, Wu TC, Yang SS, Kuo HS, Chen DS. Decreased incidence of hepatocellular carcinoma in hepatitis B vaccinees: a 20-year follow-up study. J Natl Cancer Inst. 2009;101(19):1348–55.
Cheng B, Zheng Y, Guo X, Wang Y, Liu C. Hepatitis B viral X protein alters the biological features and expressions of DNA repair enzymes in LO2 cells. Liver Int. 2010;30(2):319–26. https://doi.org/10.1111/j.1478-3231.2009.02167.x.
Du Y, Kong G, You X, Zhang S, Zhang T, Gao Y, Ye L, Zhang X. Elevation of highly up-regulated in liver cancer (HULC) by hepatitis B virus X protein promotes hepatoma cell proliferation via down-regulating p18. J Biol Chem. 2012;287(31):26302–11. https://doi.org/10.1074/jbc.M112.342113.
Dunsford HA, Sell S, Chisari FV. Hepatocarcinogenesis due to chronic liver cell injury in hepatitis B virus transgenic mice. Cancer Res. 1990;50(11):3400–7.
Duygu F, Karsen H, Aksoy N, Taskin A. Relationship of oxidative stress in hepatitis B infection activity with HBV DNA and fibrosis. Ann Lab Med. 2012;32(2):113–8. https://doi.org/10.3343/alm.2012.32.2.113.
El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142(6):1264–1273. e1261. https://doi.org/10.1053/j.gastro.2011.12.061.
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59.
Forgues M, Difilippantonio MJ, Linke SP, Ried T, Nagashima K, Feden J, Valerie K, Fukasawa K, Wang XW. Involvement of Crm1 in hepatitis B virus X protein-induced aberrant centriole replication and abnormal mitotic spindles. Mol Cell Biol. 2003;23(15):5282–92.
Fujita N, Sugimoto R, Ma N, Tanaka H, Iwasa M, Kobayashi Y, Kawanishi S, Watanabe S, Kaito M, Takei Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J Viral Hepat. 2008;15(7):498–507. https://doi.org/10.1111/j.1365-2893.2008.00972.x.
Garcia M, de The H, Tiollais P, Samarut J, Dejean A. A hepatitis B virus pre-S-retinoic acid receptor beta chimera transforms erythrocytic progenitor cells in vitro. Proc Natl Acad Sci U S A. 1993;90(1):89–93.
Gearhart TL, Bouchard MJ. The hepatitis B virus X protein modulates hepatocyte proliferation pathways to stimulate viral replication. J Virol. 2010;84(6):2675–86. https://doi.org/10.1128/JVI.02196-09.
Geng M, Xin X, Bi LQ, Zhou LT, Liu XH. Molecular mechanism of hepatitis B virus X protein function in hepatocarcinogenesis. World J Gastroenterol. 2015;21(38):10732–8. https://doi.org/10.3748/wjg.v21.i38.10732.
Ghidini M, Braconi C. Non-coding RNAs in primary liver cancer. Front Med (Lausanne). 2015;2:36. https://doi.org/10.3389/fmed.2015.00036.
Ha HL, Shin HJ, Feitelson MA, Yu DY. Oxidative stress and antioxidants in hepatic pathogenesis. World J Gastroenterol. 2010;16(48):6035–43.
Hagen TM, Huang S, Curnutte J, Fowler P, Martinez V, Wehr CM, Ames BN, Chisari FV. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. ProcNatlAcadSci USA. 1994;91(26):12808–12.
Hsieh A, Kim HS, Lim SO, Yu DY, Jung G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/beta-catenin signaling. Cancer Lett. 2011;300(2):162–72. https://doi.org/10.1016/j.canlet.2010.09.018.
Hu JJ, Song W, Zhang SD, Shen XH, Qiu XM, Wu HZ, Gong PH, Lu S, Zhao ZJ, He ML, Fan H. HBx-upregulated lncRNA UCA1 promotes cell growth and tumorigenesis by recruiting EZH2 and repressing p27Kip1/CDK2 signaling. Sci Rep. 2016;6:23521. https://doi.org/10.1038/srep23521.
Huang J, Wang Y, Guo Y, Sun S. Down-regulated microRNA-152 induces aberrant DNA methylation in hepatitis B virus-related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology. 2010;52(1):60–70. https://doi.org/10.1002/hep.23660.
Huang JF, Guo YJ, Zhao CX, Yuan SX, Wang Y, Tang GN, Zhou WP, Sun SH. Hepatitis B virus X protein (HBx)-related long noncoding RNA (lncRNA) down-regulated expression by HBx (Dreh) inhibits hepatocellular carcinoma metastasis by targeting the intermediate filament protein vimentin. Hepatology. 2013;57(5):1882–92. https://doi.org/10.1002/hep.26195.
Huang JF, Wang Y, Liu F, Liu Y, Zhao CX, Guo YJ, Sun SH. EVI1 promotes cell proliferation in HBx-induced hepatocarcinogenesis as a critical transcription factor regulating lncRNAs. Oncotarget. 2016;7(16):21887–99. 10.18632/oncotarget.7993.
Huang JL, Ren TY, Cao SW, Zheng SH, Hu XM, Hu YW, Lin L, Chen J, Zheng L, Wang Q. HBx-related long non-coding RNA DBH-AS1 promotes cell proliferation and survival by activating MAPK signaling in hepatocellular carcinoma. Oncotarget. 2015;6(32):33791–804. 10.18632/oncotarget.5667.
Idilman R, De Maria N, Colantoni A, Van Thiel DH. Pathogenesis of hepatitis B and C-induced hepatocellular carcinoma. J Viral Hepat. 1998;5(5):285–99.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
Jia L, Wang XW, Harris CC. Hepatitis B virus X protein inhibits nucleotide excision repair. Int J Cancer. 1999;80:875–9.
Jung JK, Park SH, Jang KL. Hepatitis B virus X protein overcomes the growth-inhibitory potential of retinoic acid by downregulating retinoic acid receptor-beta2 expression via DNA methylation. J Gen Virol. 2010;91(Pt 2):493–500. https://doi.org/10.1099/vir.0.015149-0.
Kew MC. Epidemiology of chronic hepatitis B virus infection, hepatocellular carcinoma, and hepatitis B virus-induced hepatocellular carcinoma. Pathol Biol (Paris). 2010;58(4):273–7. https://doi.org/10.1016/j.patbio.2010.01.005.
Lai CL, Yuen MF. Prevention of hepatitis B virus-related hepatocellular carcinoma with antiviral therapy. Hepatology. 2013;57(1):399–408. https://doi.org/10.1002/hep.25937.
Lau CC, Sun T, Ching AK, He M, Li JW, Wong AM, Co NN, Chan AW, Li PS, Lung RW, Tong JH, Lai PB, Chan HL, To KF, Chan TF, Wong N. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell. 2014;25(3):335–49. https://doi.org/10.1016/j.ccr.2014.01.030.
Lee JM, Wong CM, Ng IO. Hepatitis B virus-associated multistep hepatocarcinogenesis: a stepwise increase in allelic alterations. Cancer Res. 2008;68(14):5988–96. https://doi.org/10.1158/0008-5472.CAN-08-0905.
Lee JO, Kwun HJ, Jung JK, Choi KH, Min DS, Jang KL. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene. 2005;24(44):6617–25. https://doi.org/10.1038/sj.onc.1208827.
Lee Y, Park US, Choi I, Yoon SK, Park YM, Lee YI. Human interleukin 6 gene is activated by hepatitis B virus-X protein in human hepatoma cells. Clin Cancer Res. 1998;4(7):1711–7.
Lee YH, Yun Y. HBx protein of hepatitis B virus activates Jak1-STAT signaling. J Biol Chem. 1998;273(39):25510–5.
Lee YI, Hwang JM, Im JH, Lee YI, Kim NS, Kim DG, Yu DY, Moon HB, Park SK. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J Biol Chem. 2004;279(15):15460–71. https://doi.org/10.1074/jbc.M309280200.
Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64(1 Suppl):S84–S101. https://doi.org/10.1016/j.jhep.2016.02.021.
Li CH, Xu F, Chow S, Feng L, Yin D, Ng TB, Chen Y. Hepatitis B virus X protein promotes hepatocellular carcinoma transformation through interleukin-6 activation of microRNA-21 expression. Eur J Cancer. 2014;50(15):2560–9. https://doi.org/10.1016/j.ejca.2014.07.008.
Liang HW, Wang N, Wang Y, Wang F, Fu Z, Yan X, Zhu H, Diao W, Ding Y, Chen X, Zhang CY, Zen K. Hepatitis B virus-human chimeric transcript HBx-LINE1 promotes hepatic injury via sequestering cellular microRNA-122. J Hepatol. 2016;64(2):278–91. https://doi.org/10.1016/j.jhep.2015.09.013.
Liu H, Shi W, Luan F, Xu S, Yang F, Sun W, Liu J, Ma C. Hepatitis B virus X protein upregulates transcriptional activation of human telomerase reverse transcriptase. Virus Genes. 2010a;40(2):174–82. https://doi.org/10.1007/s11262-009-0441-3.
Liu LP, Liang HF, Chen XP, Zhang WG, Yang SL, Xu T, Ren L. The role of NF-kappaB in hepatitis b virus X protein-mediated upregulation of VEGF and MMPs. Cancer Invest. 2010b;28(5):443–51. https://doi.org/10.3109/07357900903405959.
Luedde T, Schwabe RF. NF-kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8(2):108–18. https://doi.org/10.1038/nrgastro.2010.213.
Matsubara K, Tokino T. Integration of hepatitis B virus DNA and its implications to hepatocarcinogenesis. Mol Biol Med. 1990;7:243–60.
Minami M, Daimon Y, Mori K, Takashima H, Nakajima T, Itoh Y, Okanoue T. Hepatitis B virus-related insertional mutagenesis in chronic hepatitis B patients as an early drastic genetic change leading to hepatocarcinogenesis. Oncogene. 2005;24(27):4340–8.
Miura N, Horikawa I, Nishimoto A, Ohmura H, Ito H, Hirohashi S, Shay JW, Oshimura M. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet Cytogenet. 1997;93(1):56–62.
Moon EJ, Jeong CH, Jeong JW, Kim KR, Yu DY, Murakami S, Kim CW, Kim KW. Hepatitis B virus X protein induces angiogenesis by stabilizing hypoxia-inducible factor-1alpha. FASEB J. 2004;18(2):382–4. https://doi.org/10.1096/fj.03-0153fje.
Murakami Y, Minami M, Daimon Y, Okanoue T. Hepatitis B virus DNA in liver, serum, and peripheral blood mononuclear cells after the clearance of serum hepatitis B virus surface antigen. J Med Virol. 2004;72(2):203–14. https://doi.org/10.1002/jmv.10547.
Murata M, Matsuzaki K, Yoshida K, Sekimoto G, Tahashi Y, Mori S, Uemura Y, Sakaida N, Fujisawa J, Seki T, Kobayashi K, Yokote K, Koike K, Okazaki K. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology. 2009;49(4):1203–17. https://doi.org/10.1002/hep.22765.
Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV. Immune pathogenesis of hepatocellular carcinoma. J Exp Med. 1998;188(2):341–50.
Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–8. https://doi.org/10.1038/nrm2858.
Park SH, Jung JK, Lim JS, Tiwari I, Jang KL. Hepatitis B virus X protein overcomes all-trans retinoic acid-induced cellular senescence by downregulating levels of p16 and p21 via DNA methylation. J Gen Virol. 2011;92(Pt 6):1309–17. https://doi.org/10.1099/vir.0.029512-0.
Park US, Park SK, Lee YI, Park JG, Lee YI. Hepatitis B virus-X protein upregulates the expression of p21waf1/cip1 and prolongs G1–>S transition via a p53-independent pathway in human hepatoma cells. Oncogene. 2000;19(30):3384–94. https://doi.org/10.1038/sj.onc.1203674.
Peng Z, Zhang Y, Gu W, Wang Z, Li D, Zhang F, Qiu G, Xie K. Integration of the hepatitis B virus X fragment in hepatocellular carcinoma and its effects on the expression of multiple molecules: a key to the cell cycle and apoptosis. Int J Oncol. 2005;26(2):467–73.
Pollicino T, Saitta C, Raimondo G. Hepatocellular carcinoma: the point of view of the hepatitis B virus. Carcinogenesis. 2011;32(8):1122–32. https://doi.org/10.1093/carcin/bgr108.
Qiu X, Dong S, Qiao F, Lu S, Song Y, Lao Y, Li Y, Zeng T, Hu J, Zhang L, Zhang L, Fan H. HBx-mediated miR-21 upregulation represses tumor-suppressor function of PDCD4 in hepatocellular carcinoma. Oncogene. 2013;32(27):3296–305. https://doi.org/10.1038/onc.2013.150.
Rahmani Z, Huh KW, Lasher R, Siddiqui A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J Virol. 2000;74(6):2840–6.
Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist. 2010;15(Suppl 4):14–22. https://doi.org/10.1634/theoncologist.2010-S4-14.
Sanz-Cameno P, Martin-Vilchez S, Lara-Pezzi E, Borque MJ, Salmeron J, Munoz de Rueda P, Solis JA, Lopez-Cabrera M, Moreno-Otero R. Hepatitis B virus promotes angiopoietin-2 expression in liver tissue: role of HBV x protein. Am J Pathol. 2006;169(4):1215–22. https://doi.org/10.2353/ajpath.2006.051246.
Song K, Han C, Zhang J, Lu D, Dash S, Feitelson M, Lim K, Wu T. Epigenetic regulation of MicroRNA-122 by peroxisome proliferator activated receptor-gamma and hepatitis b virus X protein in hepatocellular carcinoma cells. Hepatology. 2013;58(5):1681–92. https://doi.org/10.1002/hep.26514.
Su Q, Schroder CH, Hofmann WJ, Otto G, Pichlmayr R, Bannasch P. Expression of hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinomas. Hepatology. 1998;27(4):1109–20.
Takada S, Shirakata Y, Kaneniwa N, Koike K. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene. 1999;18(50):6965–73.
Tan C, Guo H, Zheng M, Chen Y, Huang W. Involvement of mitochondrial permeability transition in hepatitis B virus replication. Virus Res. 2009;145(2):307–11. https://doi.org/10.1016/j.virusres.2009.08.001.
Tanaka Y, Kanai F, Kawakami T, Tateishi K, Ijichi H, Kawabe T, Arakawa Y, Kawakami T, Nishimura T, Shirakata Y, Koike K, Omata M. Interaction of the hepatitis B virus X protein (HBx) with heat shock protein 60 enhances HBx-mediated apoptosis. Biochem Biophys Res Commun. 2004;318(2):461–9. https://doi.org/10.1016/j.bbrc.2004.04.046.
Theise ND. Liver cancer. World cancer report 2014. Lyon, France: International Agency for Research on Cancer; 2014.
Tsai SM, Lin SK, Lee KT, Hsiao JK, Huang JC, Wu SH, Ma H, Wu SH, Tsai LY. Evaluation of redox statuses in patients with hepatitis B virus-associated hepatocellular carcinoma. Ann Clin Biochem. 2009;46(Pt 5):394–400. https://doi.org/10.1258/acb.2009.009029.
Ura S, Honda M, Yamashita T, Ueda T, Takatori H, Nishino R, Sunakozaka H, Sakai Y, Horimoto K, Kaneko S. Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology. 2009;49(4):1098–112. https://doi.org/10.1002/hep.22749.
Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci U S A. 1994;91:2230–4.
Wang Y, Lu Y, Toh ST, Sung WK, Tan P, Chow P, Chung AY, Jooi LL, Lee CG. Lethal-7 is down-regulated by the hepatitis B virus x protein and targets signal transducer and activator of transcription 3. J Hepatol. 2010;53(1):57–66. https://doi.org/10.1016/j.jhep.2009.12.043.
Wang Y, Jiang L, Ji X, Yang B, Zhang Y, Fu XD. Hepatitis B viral RNA directly mediates down-regulation of the tumor suppressor microRNA miR-15a/miR-16-1 in hepatocytes. J Biol Chem. 2013;288(25):18484–93. https://doi.org/10.1074/jbc.M113.458158.
Wei X, Tan C, Tang C, Ren G, Xiang T, Qiu Z, Liu R, Wu Z. Epigenetic repression of miR-132 expression by the hepatitis B virus x protein in hepatitis B virus-related hepatocellular carcinoma. Cell Signal. 2013;25(5):1037–43. https://doi.org/10.1016/j.cellsig.2013.01.019.
Xu X, Fan Z, Kang L, Han J, Jiang C, Zheng X, Zhu Z, Jiao H, Lin J, Jiang K, Ding L, Zhang H, Cheng L, Fu H, Song Y, Jiang Y, Liu J, Wang R, Du N, Ye Q. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest. 2013;123(2):630–45. https://doi.org/10.1172/JCI64265.
Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, Jia H, Ye Q, Qin LX, Wauthier E, Reid LM, Minato H, Honda M, Kaneko S, Tang ZY, Wang XW. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136(3):1012–24.
Yan J, Yao Z, Hu K, Zhong Y, Li M, Xiong Z, Deng M. Hepatitis B virus core promoter A1762T/G1764A (TA)/T1753A/T1768A mutations contribute to hepatocarcinogenesis by deregulating Skp2 and P53. Dig Dis Sci. 2015;60(5):1315–24. https://doi.org/10.1007/s10620-014-3492-9.
Yang JD, Kim WR, Coelho R, Mettler TA, Benson JT, Sanderson SO, Therneau TM, Kim B, Roberts LR. Cirrhosis is present in most patients with hepatitis B and hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2011;9(1):64–70. https://doi.org/10.1016/j.cgh.2010.08.019.
Zhang HH, Mei MH, Fei R, Liu F, Wang JH, Liao WJ, Qin LL, Wei L, Chen HS. Regulatory T cells in chronic hepatitis B patients affect the immunopathogenesis of hepatocellular carcinoma by suppressing the anti-tumour immune responses. J Viral Hepat. 2010;17(Suppl 1):34–43. https://doi.org/10.1111/j.1365-2893.2010.01269.x.
Zhang X, Liu S, Hu T, Liu S, He Y, Sun S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology. 2009;50(2):490–9.
Zhao J, Wu G, Bu F, Lu B, Liang A, Cao L, Tong X, Lu X, Wu M, Guo Y. Epigenetic silence of ankyrin-repeat-containing, SH3-domain-containing, and proline-rich-region- containing protein 1 (ASPP1) and ASPP2 genes promotes tumor growth in hepatitis B virus-positive hepatocellular carcinoma. Hepatology. 2010;51(1):142–53.
Zhu YZ, Zhu R, Shi LG, Mao Y, Zheng GJ, Chen Q, Zhu HG. Hepatitis B virus X protein promotes hypermethylation of p16(INK4A) promoter through upregulation of DNA methyltransferases in hepatocarcinogenesis. Exp Mol Pathol. 2010;89:268–75.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Fako, V., Wang, X.W. (2018). Molecular Carcinogenesis of HBV-Related HCC. In: Kao, JH., Chen, DS. (eds) Hepatitis B Virus and Liver Disease. Springer, Singapore. https://doi.org/10.1007/978-981-10-4843-2_8
Download citation
DOI: https://doi.org/10.1007/978-981-10-4843-2_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4842-5
Online ISBN: 978-981-10-4843-2
eBook Packages: MedicineMedicine (R0)