
Abstract
The survival of cellular life depends on the accurate and coordinated maintenance of biological processes at the single-cell level such as cell-cycle progression, differentiation, metabolism, development, and programmed cell death (Rudel and Sommer 2003; Hanahan and Weinberg 2011; DeBerardinis and Thompson 2012). Consequently, simultaneous regulation of complex intracellular programs is heavily reliant on the precision of gene expression at the transcriptional level. Eukaryotic gene expression begins typically with the assembly of transcription-related protein complexes and cofactors on DNA before genetic information is transcribed into messenger RNA molecules, through the recruitment of RNA polymerase and cofactors, allowing for downstream protein translation (Lee and Young 2000). Sequence-specific DNA-binding transcription factors (TFs) are an integral part of the transcriptional machinery that regulate gene expression rates through the recognition and binding to precise DNA motifs (enhancer regions or response elements) resulting in either transcriptional activation or repression (Robertson et al. 2006) through further interaction with co-regulators and histone modifiers (HATs, HDACs) (Schaefer et al. 2011). Whole-genome studies have predicted 2000–3000 TFs in the human genome (Babu et al. 2004; Kummerfeld and Teichmann 2006; Venter et al. 2001), and bioinformatics, transcriptome analysis estimates that TFs account for ~8–10% of human genes expressed (Messina et al. 2004; Kummerfeld and Teichmann 2006).
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


2.1 Introduction to Transcription Factors and Diseases
The survival of cellular life depends on the accurate and coordinated maintenance of biological processes at the single-cell level such as cell-cycle progression, differentiation, metabolism, development, and programmed cell death (Rudel and Sommer 2003; Hanahan and Weinberg 2011; DeBerardinis and Thompson 2012). Consequently, simultaneous regulation of complex intracellular programs is heavily reliant on the precision of gene expression at the transcriptional level. Eukaryotic gene expression begins typically with the assembly of transcription-related protein complexes and cofactors on DNA before genetic information is transcribed into messenger RNA molecules, through the recruitment of RNA polymerase and cofactors, allowing for downstream protein translation (Lee and Young 2000). Sequence-specific DNA-binding transcription factors (TFs) are an integral part of the transcriptional machinery that regulate gene expression rates through the recognition and binding to precise DNA motifs (enhancer regions or response elements) resulting in either transcriptional activation or repression (Robertson et al. 2006) through further interaction with co-regulators and histone modifiers (HATs, HDACs) (Schaefer et al. 2011). Whole-genome studies have predicted 2000–3000 TFs in the human genome (Babu et al. 2004; Kummerfeld and Teichmann 2006; Venter et al. 2001), and bioinformatics, transcriptome analysis estimates that TFs account for ~8–10% of human genes expressed (Messina et al. 2004; Kummerfeld and Teichmann 2006).
DNA-binding transcription factors are typically modular and generally contain a DNA-binding domain (DBD) which controls DNA binding and gene specificity, and a transactivation domain (TAD) to regulate transcription through interaction with protein factors of the transcriptional machinery. The basis of DNA selectivity lies within the DBD, which can be classified based on structure and function. Three classes are most prolifically expressed in humans: the C2H2 zinc finger, homeodomain, and helix-loop-helix families (Vaquerizas et al. 2009). TADs are generally disordered and less structured than DBDs within TF families, allowing for promiscuity in protein interaction and cofactor recruitment. Distinct categories of TADs are observed in different classes of TFs and are grouped based on the amino acid composition: acidic, isoleucine-rich, proline-rich, and glutamine-rich domains (Mermod et al. 1989; Okuda et al. 2016; Mognol et al. 2016; Hibino et al. 2016). In addition, the nine amino acid transactivation domain (9aaTAD) is a class common to eukaryotic transcription factors (Piskacek et al. 2007). Table 2.1 shows seven of the most cited TFs in the literature to date (Vaquerizas et al. 2009).
As expected, the deregulation of proper transcriptional activity has been associated with many human diseases. For example, mutations in the transcription factors HNF1beta, HNF1alpha, and HNF4alpha have been linked to maturity-onset diabetes of the young (MODY) by respectively affecting differentiation processes in the pancreas and decreasing glucose-dependent insulin secretion in beta-cells (Maestro et al. 2007). The autoimmune regulator AIRE, a transcription factor expressed in the thymus, is responsible for the identification and negative selection of self-reactive T-cells, and its inactivation causes type-I autoimmune polyendocrinopathy syndrome (APS-1) (Kyewski and Klein 2006). Aberrant gene expression from deregulated transcription factors can occur at the genetic level as a result of increased TF expression (increased copy number from gene duplication, epigenetic modifications, or chromosomal translocations), or at the protein level (post-translational modifications or a derailment in biochemical pathways like protein turnover rates). Examples include translocation of the AML1 (or RUNX1) transcription factor (resulting in oncogenic fusion proteins like AML1-ETO) commonly associated with several forms of leukemia (Licht 2001; Lukasik et al. 2002), and the HPV (human papillomavirus)-related viral protein E6 which, when present in cells during viral infections, facilitates the degradation of the tumor-suppressor p53 and promotes cervical carcinogenesis (Mantovani and Banks 2001). Additionally, mutations in cis-acting regulatory DNA elements as well as inactivating mutations within the reading frame of a gene can affect mRNA splicing, protein translation, or protein structure, and have all been described and linked to disease phenotypes (Lee and Young 2013).
2.2 Transcription Factors in Cancer
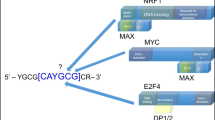
Cellular transformation and the development of cancer have been acutely linked to numerous transcription factors responsible for distinct cellular processes. The tumor-promoting c-Myc transcription factor is the most frequently amplified onco-protein in human cancers (Lin et al. 2012). c-Myc recognizes and binds DNA enhancer motifs (E-boxes) through heterodimerization with another TF, Max (myc-associated factor x) (Blackwood and Eisenman 1991), to elevate expression of genes involved in cell proliferation and metabolism, promoting cell growth (Ji et al. 2011). Furthermore, tumorigenic cellular programs have also been ascribed to simultaneous activation of transcriptional networks. Elevated transcriptional activity of TAL1 is observed in 40% of all T-ALL (T-cell acute lymphoblastic leukemia) cases and has been reported as a master regulator of transcriptional circuitries involving other TFs including RUNX, GATA3, HEB, and E2A (Sanda et al. 2012).
The rest of this chapter focuses on three transcription factors: the p53 tumor suppressor, estrogen receptor, and NF-κB; their roles in cancer; as well as past and current technologies designed at targeting them for diagnostic purposes.
2.2.1 The p53 Tumor Suppressor
The p53 tumor suppressor (also known as the guardian of the genome) is a master regulator that sits at a central node within a sophisticated network of cellular programs (Lane 1992). It functions primarily as a transcription factor that acts to safeguard the genomic integrity of an organism by inducing biochemical pathways that ultimately determine cell fate (including cell cycle arrest, apoptosis, senescence)p53 responds to upstream stress signals and prevents cellular transformation caused by genetic aberrations (Vogelstein et al. 2000). Stress signals that activate p53 take many forms but typically result in DNA mutations or chromosomal damage when left unchecked. Examples include DNA or chromosomal breakages, ionizing radiation, hypoxia, dNTP depletion, and glucose starvation (Bieging et al. 2014).
p53 shares significant homology with its family members, transcription factors p63 and p73, and they are each organized to carry several critical domains including an N-terminal transactivation domain (TAD), a proline-rich domain (PD), a well-ordered DNA-binding domain (DBD), an oligomerization domain (OD), and an unstructured carboxy-terminal domain (CTD) (Vousden and Lane 2007). In addition, p63 and p73 possess a sterile α-motif (SAM) domain that participates in protein-protein interactions (Thanos and Bowie 1999). The physiological functions of p53’s unstructured CTD have been highly controversial and both early reports and in vitro experiments have suggested an auto-inhibitory role possibly through interacting with the DBD (Hupp et al. 1992; Goh et al. 2010). However, recent animal and biochemical studies reveal more evidence of the CTD’s involvement in p53-DNA interaction, particularly in the selectivity and coordinated binding of p53 to DNA response elements and also in the precise induction of p53 response in cells (Laptenko et al. 2015, 2016). There are also postulations that the CTD can help mediate sequence-specific p53-DNA binding through weak interactions between the positively charged lysine-rich regions and the negatively charged phosphate DNA backbone (Friedler et al. 2005). This interaction may also facilitate the sensing of DNA damage and expedite DNA repair (Reed et al. 1995). The DNA-binding core of p53 is responsible for interacting with DNA in a sequence-specific manner through a highly ordered domain that is well conserved within the protein family (Belyi et al. 2010). The DBD core structure consists of a β-sandwich scaffold consisting of two antiparallel β-sheets projecting a loop-sheet-helix motif and two additional large loops that make DNA contacts (Cho et al. 1994). p53 functions as a TF by recognizing and binding cognate DNA elements known as p53 response elements (p53-RE) which contain two palindromic half-site decamers, each carrying the consensus sequence 5′-RRRC(A/T)(T/A)GYYY-3′ (where Y = pyrimidine and R = purine) separated by a spacer ranging from 0 to 13 base pairs (el-Deiry et al. 1992). A stable complex (dimer of dimers) is formed with each monomer contacting a 5-bp quarter site when p53 tetramerizes on DNA, resulting in a close to 100-fold increase in binding affinity over monomeric units alone (Balagurumoorthy et al. 1995). The DNA core motif C(A/T)(T/A)G within each decamer half-site, in particular, has been shown to have a profound influence on p53 DBD binding (Wang et al. 2009a). Wild-type p53 is known to regulate hundreds of gene targets, through transcriptional activation or repression, by interacting with DNA REs located across the entire genome. More than 200 RE sites have been established as empirically verifiable p53 response elements (Menendez et al. 2009; Riley et al. 2008; Zeron-Medina et al. 2013), with thousands more possible p53-binding sites identified through predictive algorithms and whole-genome studies (Tebaldi et al. 2015; Smeenk et al. 2008; Chang et al. 2014; Sammons et al. 2015). Furthermore, the low intrinsic thermodynamic stability of the p53 core (9-min half-life at body temperature), a likely result of evolutionary adaptations, has been linked to structural plasticity, allowing for interaction with diverse protein partners and DNA sequences (Joerger and Fersht 2010). Indeed, gene expression regulating p53-REs have shown considerable degeneracy in sequence and size, seen in noncanonical motifs like half- and three-quarter sites (Jordan et al. 2008; Tebaldi et al. 2015).
In the classical p53 response, cellular stress stimuli result in the activation of p53 modifiers like ATM (ataxia telangiectasia mutated), ATR (ataxia telangiectasia and Rad3-related protein), and CHK1/2 (checkpoint kinase 1/2) serine/threonine protein-kinase which phosphorylates p53 at key residues leading to the stabilization of intracellular protein levels, nuclear accumulation, and increased transcriptional activity on target genes (Cheng and Chen 2010). Acetylation of lysine residues found in the DBD and CTD, through the recruitment of histone or lysine acetyltransferases, can further contribute to this process (Dornan and Hupp 2001; Lambert et al. 1998). The precision of p53 gene target selection is regulated at many levels including p53 post-translational modifications (such as phosphorylation, acetylation, and ubiquitination of precise residues) (Meek and Anderson 2009), histone remodeling factors (HATs, HDACs), as well as p53-protein interaction (protein cofactors and p53 isoforms) (Khoury and Bourdon 2011; Beckerman and Prives 2010). The result is a downstream augmentation of canonical p53 cellular responses through the upregulation of classic gene targets including p21, GADD45, and 14–3-3σ which mediate growth arrest and DNA repair (Hermeking et al. 1997; Chin et al. 1997; el-Deiry et al. 1993), as well as Puma, Bax, and Noxa that induces apoptosis (Nakano and Vousden 2001; Miyashita and Reed 1995), all by virtue of high-affinity p53-REs (Weinberg et al. 2005).
Transcriptional upregulation is also achieved through the ablation of an auto-regulated negative feedback mechanism mediated by Mdm2, a p53 target gene product. p53 activity is kept low under normal cellular conditions by Mdm2, a ubiquitin E3-ligase capable of inactivating p53 through TAD binding and sequestration, followed by cytosolic translocation and ubiquitin-dependent proteasome degradation through the modification of lysine residues with poly-ubiquitin chains (Kussie et al. 1996; Lohrum et al. 2001). Overexpression of Mdm2 leads to the attenuation of the p53 response and promotes cancer development. High levels of Mdm2 are found in several types of human malignancies and hence it constitutes a promising therapeutic and prognostic target in cancer (Rayburn et al. 2005; Andre et al. 2014).
The crucial role of p53 in cancer development is obvious when examining Li-Fraumeni patients who carry germline mutations in the p53 encoding TP53 gene, resulting in cancer predisposition at a young age (particularly sarcomas and cancers of the breast, brain, and adrenal glands) (Malkin 2011). The derailment of p53 pathways leading to constitutive proliferative, or pro-survival, cellular signals can be seen in almost all human cancers, particularly in above 50% of cases where p53 exists in TP53 mutations which results in mutation-inactivated forms with compromised transcriptional functions. In human breast cancer, TP53 is one of the most frequently mutated genes (up to 80% in certain subtypes), and demonstrates a correlation between mutation type (e.g. insertational/deletion or missense mutations) and molecular subtype (Powell et al. 2014; Ciriello et al. 2015). In 75% of TP53 mutations which results in fully translated proteins carrying a single-amino acid mutation, 95% reside in the DNA-binding core domain causing varying extents of structural perturbations, abating wild-type DNA binding (Bullock and Fersht 2001). Several particular mutations, known as the “hotspot” mutations, occur most frequently in human cancers (G245S, R273H, R248Q, R175H, R282W, and R249S). In addition to losing the ability to bind canonical p53 DNA REs and transcribing p53 target genes (loss of function), these mutants are also known to possess tumorigenic functions (oncogenic gain-of-function) and provide poor disease prognosis (Joerger and Fersht 2007; Powell et al. 2014). In particular, mutants R273H and R175H have been shown to associate with other DNA-binding transcription factors to increase tumor aggressiveness and metastasis through numerous mechanisms such as exerting a dominant-negative effect over tumor-suppressing TFs (p63, p73) or associating with oncogenic TFs (eg. ETS2) to induce pro-survival programs and promote chromosomal instability (Lu et al. 2013; Martinez et al. 2016; Solomon et al. 2012; Song et al. 2007). Indeed, the requirement for TP53 mutation as an early initiation event in pathogenesis shows almost complete penetrance in high-grade serous ovarian carcinoma (Vang et al. 2016), where mutant p53 contributes to anoikis resistance (Cai et al. 2015) and tissue invasion (Iwanicki et al. 2016).
2.2.2 Estrogen Receptor (ER)
The intracellular estrogen receptors (ERα and ERβ) belong to the class of nuclear receptor (NR) superfamily of ligand-regulated transcription factors and respond to the sex steroid hormone estrogen. It functions primarily in the maintenance of the female reproductive system, but also in physiological processes including skeletal, neuroendocrine, cardiovascular, and immune systems (Swedenborg et al. 2009).
ER, like other members of the NR family, show highly conserved functional domains which comprise an N-terminus transactivation domain (AF-1), a DBD, a hinge domain containing the nuclear localization signal, and a C-terminus ligand-binding and transactivation domain (LBD and AF-2) (Le Romancer et al. 2011). ERα and ERβ have high homology within their DBD (~96%) and differ functionally by their N-termini transcriptional activity (AF-1 domain) which regulates hormone-independent transcription (Kuiper et al. 1996). Both forms of ER are expressed widely in many tissues and their relative expression in cells determines tissue-specific responsiveness to the presence of estrogen (Thomas and Gustafsson 2011). Additionally, numerous amino acid residues on ER are susceptible to post-translational modifications which influences transcriptional function, DNA selectivity, and interaction with ER-coregulators (Le Romancer et al. 2011).
Estrogen-dependent tumorigenesis has been linked to the development of many cancer types including breast, ovary, colon, and prostate (Shang 2007), and is largely ascribed to the transcriptional activity of ERα which is responsible for mediating pro-survival and proliferative signals (Liang and Shang 2013). In contrast ERβ has been reported to inhibit estrogen-dependent cell growth and also displays ERα antagonism when ectopically expressed in ERα-positive breast cancer cells, reducing cell proliferation (Strom et al. 2004). Estrogen exists predominantly as 17β-estradiol (E2) in cells, but can also take other forms like estrone (E1) and estriol (E3). Long-term exposure to estrogenic compounds, such as in hormone replacement therapy (HRT), constitutes a major risk factor in developing breast cancer (Narod 2011). Upon ligand binding, estrogen receptors are activated through homodimerization, further allowing binding to estrogen response elements (ERE) in the nucleus. Receptor dimerization has also been reported to result in interaction with other transcription factors (such as p53, NF-κB, RUNX1), allowing ER to regulate gene expression in the absence of estrogen REs (Jerry et al. 2010; Stender et al. 2010).
2.2.3 Nuclear Factor Kappa B (NF-κB)
The nuclear factor NF-κB, originally discovered as a transcriptional regulator of immunoglobulin kappa-light-chain-activated B cells, is now known to be widely expressed in many cell types and mediates the inflammatory response, as part of the innate immune system, as well as cellular growth and death (Wan and Lenardo 2009).
The family of NF-κB transcription factors contains five members (RelA, RelB, c-Rel, NF-κB1, and NF-κB2) and regulates transcription in a modular way by forming homo- and heterodimers with each other in virtually every possible permutation. All members are evolutionarily conserved and carry the approximately 300-residue Rel homology domain (RHD) responsible for dimerization, nuclear localization, and DNA interaction (Wan and Lenardo 2009). RelA, RelB, and c-Rel each contain one or more C-terminal transactivation domains (TA) (May and Ghosh 1997) but are usually inhibited in quiescent cells by interacting with members of the IκB family (inhibitors of NF-κB). In addition to obscuring the nuclear localization signals on NF-κB proteins and preventing nuclear translocation when bound (Jacobs and Harrison 1998), IκB family proteins (IκBα, IκBβ, IκBε, IκBγ, IκBζ, BCL-3, and precursors p105 and p100) (Wan and Lenardo 2009) all carry characteristic ankyrin repeats responsible for associating with RHD DNA-binding domains and render NF-κB proteins transcriptionally inactive (Verma et al. 1995). Release from IκB inhibition is required for NF-κB transcriptional activity and occurs in part through proteasome-dependent degradation of the inhibitors. The process is catalyzed through the poly-ubiquitination of lysine residues on IκB molecules by unique E3-ligases (SCFβTrCP, beta-transducin repeat-containing protein) through the recognition of a specific double-phosphorylated substrate catalyzed by IKK complexes (IκB kinase) (Karin and Ben-Neriah 2000). The remaining two NF-κB family proteins, NF-κB1 (p105) and NF-κB2 (p100), lack a transactivation domain and are synthesized as precursors that remain inactive through the negative self-regulating ankyrin repeats they carry. These inhibitory domains are cleaved during the maturation process resulting in the active forms, p50 and p52 (Hayden and Ghosh 2012). Homodimers of p50 and p52 act as transcriptional repressors as they bind κB DNA elements but lack a transactivation domain. NF-κB TFs recognize and bind DNA motifs containing the κB consensus sequence 5′GGGRNYYYCC3′ (where R = purine, Y = pyrimidine, and N = any nucleotide) (Chen et al. 1998). Additionally, precise transcriptional response and DNA-binding selectivity are also achieved through post-translational modifications of NF-κB complexes and from different combinations of heterodimerization, resulting in cellular context-dependent activity. For instance, phosphorylation of serine 536 on RelA can result in IκBα dissociation, nuclear accumulation, and enhanced transcriptional activity (Sasaki et al. 2005), but also enhances RelB association, leading to decreased κB DNA sites binding in the nucleus (Jacque et al. 2005).
In the classical pathway of NF-κB activation, upstream receptor-mediated signals (for example Toll-like receptor (TLR) stimulation from bacterial cell wall lipopolysaccharides or tumor necrosis factor receptor (TNFR) activation) lead to activation of IKK complexes which phosphorylate and target IκB family members to the proteasome, hence liberating NF-κB for nuclear localization and activation of inflammatory responses. While acute inflammation can lead to activation of cytotoxic immunity against transformed cells, chronic inflammation has been associated with pro-tumorigenic outcomes. Stimulation of NF-κB pathways can result in pro-survival signals in cells as a response to withstand physiological causes of the inflammation (Hoesel and Schmid 2013). Furthermore, release of ROS (reactive oxygen species) by neutrophils can also cause DNA damage and propagate cancer-driving mutations (Liou and Storz 2010). RANK (receptor activator of NF-κB) belongs to the TNF receptor family and is etiologically linked to some forms of metastatic bone tumors and mammary carcinoma through the activation of NF-κB signaling (Hanada et al. 2011). Recent studies using mouse models have presented RANK receptor and its ligand RANKL as potential diagnostic and therapeutic targets of breast cancer in carriers of BRCA1 mutations (Nolan et al. 2016). Furthermore, NF-κB signaling is also reported to drive cancer aggression by regulating EMT (epithelial mesenchymal transition) and can contribute to metastasis as well as angiogenesis by upregulating VEGF and increasing tumor vascularization (vascular endothelial growth factor) (Huber et al. 2004; Xie et al. 2010).
2.3 Detecting Transcription Factors in Cancer Diagnostics
As illustrated in the sections above, transcription factors in general (and in particular p53, ER, and NF-κB) are acutely linked to cellular transformation and cancer development by eliciting erroneous cellular programs or transcriptional functions through TF-DNA interaction. The ability to qualitatively and quantitatively assess DNA-binding functions of transcription factors using clinical samples will undoubtedly provide valuable information on disease prognosis or opportunities for early disease detection. For example, stabilization of ERα proteins associated with certain unique DNA-binding properties may result in the early detection of pro-survival cells (Fan et al. 2015). Oncogenic point mutations in NF-κB and RelA have also been detected in Hodgkin lymphomas, likely carrying altered but specific DNA-binding signatures (Hoesel and Schmid 2013).
Conventional and early methods for detecting TF-DNA binding are crude and often only semiquantitative. They are also labor intensive, have low throughput and sensitivity, and frequently require the use of radioactive materials to increase signal-to-noise detection. Examples include electrophoretic mobility shift assay (EMSA) which involves the electrophoretic separation of protein-DNA complexes using non-denaturing agarose or polyacrylamide gels followed by visualization. TF-DNA complexes can be further “super-shifted” when the overall molecular weight of the complex is increased by the addition of a TF-specific antibody. DNA fragments are often radioactively labeled to increase the detection limit and sensitivity of the assay (Fried 1989). In DNase footprinting assay, DNA fragments mixed with a protein of interest (a transcription factor) are later subjected to restriction endonuclease digestion. Binding of a protein to a specific region on the DNA provides protection from endonuclease activity resulting in different fragmentation patterns and allows the identification of DNA sequence involved in binding (Brenowitz et al. 1986). As before, 32P-labeled DNA can be used (through PCR amplification using radioactively labeled primers) for increased signal detection. Enzyme-linked immunosorbent assay (ELISA) is a common serological diagnostic technique which involves the use of protein-specific antibodies. In the classic approach, antigens (or targets of interest) are first immobilized on a solid matrix (typically a polystyrene microtiter plate) through either adsorption or a “capture” antibody (sandwich ELISA). Next, an antigen-targeting “detection” antibody is added followed by an enzyme-linked (e.g., horseradish peroxidase, HRP) secondary antibody which produces a chromogenic or fluorogenic signal when mixed with the appropriate substrate solution, giving an indication of the amount of antigen present in the sample (Lequin 2005). In ELISA, the “capture” and “detection” moieties can be replaced by many protein-protein or protein-chemical interacting modules, including streptavidin, biotin, peptides, protein affinity tags, and nucleic acids, making this technique modular and flexible. Jagelska and colleagues reconfigured the classic ELISA format to measure p53-DNA binding by immobilizing biotin-conjugated p53 DNA response element onto streptavidin-treated plates. p53-containing samples were then added and detected using a p53-specific antibody (Jagelska et al. 2002). Although ELISA is amenable to high-throughput applications and can be highly specific, the success is heavily reliant on the availability of good antibodies and faces caveats like moderate sensitivity and low signal-to-noise. In the following sections, we examine new technologies developed more recently to interrogate transcription factors functionally and their ability to bind DNA sequence—specifically through the unique integration of materials, reagents, and techniques.
2.4 Optical Biosensor for Detecting Transcription Factors
Optical biosensors are powerful tools for the functional study of transcription factors due to their high specificity, sensitivity, and cost-effectiveness as compared to conventional bioassays like EMSA and DNase footprinting (Garner and Revzin 1981; Galas and Schmitz 1978). Optical biosensors typically comprise optically labeled probes and optical transducers to facilitate the detection of protein functions. In the last decade, technological development of optical biosensors has experienced significant growth in studying sequence-specific TF-DNA interactions due to the increase demands for direct, real-time, and label-free sensing. Three different types of optical sensing techniques including colorimetric, fluorescence, and surface plasmon resonance (SPR) have been employed extensively for this purpose and are discussed here.
2.4.1 Fluorescence Assays
Fluorescence assays are one of the most widely applied optical techniques to study protein-DNA interactions. Noureddine and coworkers developed a fluorescent microsphere-based technique termed MAPD (microsphere assay for protein-DNA binding) that can measure p53-DNA binding in a multiplexed platform. Microspheres carrying individual fluorescent signatures are annealed with different p53 response elements of varying binding affinities (p21, PUMA, consensus sequence A and C, GADD45, and non-binding control DNA) and exposed to p53-activating drug (doxorubicin)-treated whole-cell lysates containing endogenous p53 in a multiplexed reaction. Using fluorescently tagged antibody to detect bead-bound p53 molecules, a profile of relative fluorescence intensity detailing p53 binding levels (from DNA binding) for each respective microsphere-RE is generated, showing the degree of sequence-specific DNA interactions (Noureddine et al. 2009). MAPD assay was highly sensitive and could accurately discern the binding affinities of wild-type and mutant p53 (R175H and S121F) towards different REs as well as sequences carrying single-nucleotide mutations (SNPs). Additionally, MAPD binding data also correlated to results from luciferase transactivation reporter assay in cells, demonstrating biological relevance (Noureddine et al. 2009). In another study that targets the p53 pathway, Goh and colleagues developed a biosensor using a conditionally fluorescing molecular rotor conjugate. Molecular rotors are a unique class of fluorescent chemicals that can undergo twisted intramolecular charge transfer (TICT), according to an optical property of conditional fluorescence when excited in a sterically restrictive molecular environment (due to a red-shifted fluorescence emission instead of non-radiative torsional relaxation (Grabowski et al. 2003)). Julolidine rotor, when conjugated to the 12.1 peptide sequence (p53 N-terminal analogue that binds Mdm2 protein), behaved as a switchable molecular sensor for the presence of Mdm2 proteins. Experimental and computational data shows that the binding-induced alpha helix of the peptide-rotor conjugate can be subtly altered through single-amino acid substitution to suit the modality of protein-protein interaction and fluorescence turn-on sensitivity (Goh et al. 2014). Through the use of a cell-penetrating fluorophore with the aggregation-induced emission (AIE) property, the application was further developed for detecting p53 transcriptional activity in live cells through microscopy imaging by visualizing the increase in Mdm2 production following p53 induction (Geng et al. 2015). In a separate microscopy technique, streptavidin-coated magnetic beads conjugated to different DNA-REs displayed preferential binding when exposed to different variants of the p53 transcription factor (binding-competent wild-type p53 or inactivated mutant p53) visualized through the use of a fluorescently labeled anti-p53 antibody. The authors further demonstrate a multiplexing function by attaching different fluorescent dyes with unique DNA sequences (Ong et al. 2012). In a similar concept from a seperate technolgy with higher throughput capabilities, biotinylated single-nucleotide polyphormic p53 proteins are microarrayed on neutravidin-dextran coated glass slides functionally assessed through binding to fluorescently, or radioactively labelled GADD45 DNA-RE (Boutell et al. 2004).
The basic concept of fluorescence resonance energy transfer (FRET) describes energy transfer between two chromophores where a donor chromophore in the ground state initially transfers energy to an acceptor chromophore through non-radioactive dipole-dipole coupling (Jares-Erijman and Jovin 2003). Here, the proximity between acceptor and donor chromophore plays an essential role in producing an effective energy transfer, typically in the range of 10–100 Å. Apart from distance, the spectral overlap integral (the effective overlap between acceptor chromophore’s absorption/excitation spectrum and emission spectrum of the donor chromophore) is another key determinant of FRET efficacy. The efficiency of energy transfer (E) decreases very rapidly with increasing distance (r) between the donor and acceptor, according to the relationship E α [1 + (r/R 0)6]−1, where R 0 is the distance at which E is 50%.
Ambra and coworkers developed a FRET-based protein-DNA binding assay for the successful detection of an active form of NF-κB, p50 (Giannetti et al. 2006). FRET was harnessed to study TF-DNA binding interaction between p50 proteins and double-strand DNA (dsDNA) immobilized in a glass capillary. The complementary sequence of the single-stranded DNA (ssDNA) is labeled with a Cy5 dye, and the p50 protein with a black hole quencher (BHQ-3), constituting an effective FRET pair. A change in fluorescence intensity occurs when p50 interacts with the DNA duplex. Accordingly, the optimal emission wavelength of Cy5-labeled DNA (670 nm) overlaps effectively with the excitation wavelength of BHQ-3 quencher (636 nm) when p50 binds DNA, resulting in a 90% drop in fluorescence intensity relative to pure Cy5 alone. Despite the effectiveness of this assay, fluorescence labeling of proteins may limit its practical application, making label-free methods more attractive. Xingfen et al. developed a label-free FRET-based assay to study interactions between NF-κB and its target DNA (Liu et al. 2013). In this study, binding of the NF-κB protein to its DNA response elements shields it from digestion by exonuclease III (Fig. 2.1). The fluorescent cationic conjugated polymer (CCP) then interacts with the DNA duplex through strong electrostatic interactions with the DNA phosphate backbone, resulting in highly efficient FRET activity due to the presence of intercalated SYBR green by dsDNA that remains intact from p50 protection. In the absence of a sequence-specific binding protein, the enzyme digests the DNA duplex into single-stranded DNA fragments, preventing FRET activity. Furthermore, by using label-free hairpin DNA molecules containing two protein-binding site (PBS) as detection probes, an even lower detection limit of 1 pg/mL (with low error rates) has been achieved to detect NF-κB in HeLa cell nuclear extract. In another application, graphene oxide (GO) was used as the fluorochrome quencher. In this study, a FAM-labeled ssDNA carrying an NF-κB recognition site at the stem region of the hairpin conformation associates strongly onto the surface of the GO matrix (due to π-stacking interacting forces between the GO sheet and nucleotide bases) leading to fluorescence quenching. However, addition of NF-κB which binds to the κB consensus site on the hairpin leads to DNA desorption from the GO surface and FAM emission (Liu et al. 2012).

Schematic illustration for the detection of DNA-binding protein (NF-κB) using hairpin DNA (hpDNA) and cationic conjugated polymer (CCP) (reproduced with permission from Liu et al. (2013), Copyright Elsevier, 2012)
Certain fluorescence applications for sensing protein-DNA interactions can be labor intensive and unsuitable for complex biological samples. Molecular beacons are a class of facile, yet sensitive autonomous molecular sensors. In an early study, Heyduk and Heyduk developed a FRET-based molecular beacon for the detection of CAP proteins (a bacterial TF). The technique involves the use of a pair of DNA fragments each carrying a CAP-binding half-site conjugated to either an acceptor or a donor fluorochrome. In addition, each pair of half-sites carry a short complementary overhang which will only anneal when brought into proximity of each other during TF binding resulting in FRET and fluorescence activity. The background signal from spontaneous annealing of half-sites is kept low by making adjustments to probe concentration and complementary sequence (Heyduk and Heyduk 2002). Fang and colleagues developed a molecular beacon variant consisting of a hairpin-shaped single-stranded oligonucleotide labeled with a fluorophore/quencher pair at opposite ends. The oligonucleotide probe is designed to adopt a stem-and-loop structure in solution, bringing the fluorophore and quencher in close proximity which results in fluorescence quenching (Wang et al. 2009b; Fang et al. 2000). Additionally, the loop portion contains a sequence that is complementary to a target sequence which upon hybridization to target nucleic acids changes from a hairpin shape to the more rigid rodlike double helix. This conformational change forces the two arms of the hairpin to straighten, hence separating fluorophore and quencher and resulting in fluorescent activation (Vallée-Bélisle and Plaxco 2010; Tyagi 2009). The early utility of molecular beacons confined to ssDNA or ssDNA-binding proteins detection soon expanded to include more targets. Alexis and team developed a TF beacon strategy (Wang et al. 2009b; Stojanovic and Kolpashchikov 2004) based on the concept of structure-switching oligonucleotide probes. Accordingly, DNA probes conjugated to a fluorophore and quencher probe at two specific residues are designed with sequences that allow switching between two states of stem-loop structures in constant equilibrium. In the “non-binding” state, the formation of two smaller stem-loops results in the adjacent placement of dye and quencher leading to fluorescence quenching. In a second “binding-competent” conformation, the oligonucleotide probe takes a larger single stem-loop structure where the quencher is positioned distally from the fluorophore and also displays a TF-binding site at the stem region (Fig. 2.2). The addition of appropriate transcription factors (TATA-binding protein, NF-κB, and Myc-Max heterodimers) stabilizes and shifts the equilibrium towards the ‘binding-competent’ form, leading to an increase in fluorescence activity. Additionally, the authors show that either conformation within the equilibrium can be stabilized by altering the probe’s DNA sequence at the stem region (Vallee-Belisle et al. 2011).

Transcription factor (TF) beacons for the quantitative detection of DNA binding activity. DNA sequences containing the recognition site for a specific DNA-binding protein (here shown as red stem for TATA-binding protein (TBP)) are engineered into switches alternating between “non-binding” (left) and “binding-competent” (right) conformations. Binding of the protein to response element site present only in the “binding-competent” form shifts the switch’s conformational equilibrium and stabilizes it in this form, which is associated with an increase in fluorescence due to the separation of dye and quencher (reproduced with permission from Vallée-Bélisle et al. (2011), Copyright American Chemical Society, 2011)
In an even more sophisticated application of molecular beacon sensors, Zhang and coworkers describe a procedural method which generates a self-perpetuating signal amplification. The method begins with the protection of a specific DNA site from exonuclease III digestion through NF-κB (p50) binding. This leads to the liberation of a single-stranded ‘reporter DNA’ fragment which hybridizes with a stem-loop beacon probe (carrying a quencher/fluorophore pair) resulting in a dsDNA fragment containing a restriction endonuclease site not present before. Cleavage of this restriction site releases the fluorophore into the solution (hence increasing the fluorescence signal) and simultaneously releases the reporter DNA fragment, allowing it to target another stem-loop DNA probe, creating a self-perpetuating signal cycle (Fig. 2.3) (Zhang et al. 2016).

Schematic representation of the TMDA fluorescence assay. Exonuclease III protection of DNA1/DNA2 duplex from NF-κB binding results in the release of reporter DNA fragments which hybridizes with stem-loop DNA (and fluorophore/quencher pair), constituting a Nb.BbvCI restriction site. Cleavage of this site releases free fluorophore into solution and returns reporter DNA fragment for hybridization process again (reproduced with permission from Zhang et al. (2016), Copyright Elsevier, 2016)
In another study, fluorescent readout from real-time qPCR cycling was used to sensitively and exponentially amplify detection signals from p53-DNA binding experiments. Double-stranded DNA probes each consisting of a different p53 response element (p21, PUMA, RGC, P2XM) placed adjacent to a qPCR quantifiable tag were used to detect sequence-specific DNA binding by immuno-capturing p53-DNA complexes before bound DNA are eluted and analyzed. Specific binding towards each RE was quantified by normalizing RE-binding signals against background signals from binding non-consensus DNA (qPCR tag alone), conveying absolute sequence-specific DNA binding values, and correlated well with published affinity constants. Furthermore, binding to different REs can be multiplexed in a single reaction by “barcoding” each RE with a unique qPCR tag that can be subsequently addressed with different primer sets (Goh et al. 2010). More recently, Sha et al. designed an elaborate sensor based on hairpin DNA cascade amplifier (HDCA). A dsDNA containing NF-kB p50 response element is first mixed with a specially designed ssDNA trigger in the presence of Ag+ to form a triplex. In the presence of p50, the triplex is destabilized leading to the release of the ssDNA trigger. The released trigger is then able to activate the HDCA, leading to the hybridization of specific hairpin probes, which in turn acts as an effective template for the formation of fluorescent CuNPs (Sha et al. 2016). This fluorescence-based biosensing strategy is ultrasensitive, achieving a detection limit of 0.096 pM with very high reproducibility.
2.4.2 Surface Plasmon Resonance
Surface plasmon resonance (SPR) relies on changes in refractive index at the surface/solution interface upon the binding of analyte for real-time measurements. SPR has been frequently applied for real-time monitoring of TF-DNA binding. To study the conformational effects of ligand binding on estrogen receptor alpha (ERα) and the induced selectivity towards different DNA elements (ERE or nonspecific DNA), SPR was combined with quartz crystal microbalance with dissipation monitoring (QCM-D) (Peh et al. 2007; Su et al. 2006). Here, it was observed that specific ERα-ERE complexes adopted a more compact conformation as compared to nonspecific complexes. QCM-D thus allowed for the study of conformational changes arising from ER-DNA interactions. Further evaluation of the binding capacity of ERα to ERE revealed that ligand binding affected viscoelasticity and structural conformations of protein-DNA complexes. SPR was used in this study to complement QCM as a tool for direct quantitative analysis of protein-DNA binding, as well as to elucidate ligand-dependent ERα binding capacity.
Apart from a direct detection of TF-DNA binding, additional surface modifications can allow for multiple detection modes and additional utility. As demonstrated by Wang et al., a sandwich assay format was adopted to achieve low detection limits and simultaneous measurement of total proteins using cancer cell lysates. In addition, wild-type p53 and mutant p53 were interrogated simultaneously by a dual-channel SPR technique. The surface of the SPR chip was co-immobilized with both the consensus dsDNA, to which wild-type p53 has high affinity, and monoclonal antibodies allowing the capture and quantitation of both wild-type and mutant p53 proteins (Wang et al. 2009c). This technique offers several advantages such as low detection limits for p53 proteins (10.6 pM for wild-type p53 and 1.06 pM for total p53 proteins), high specificity, and the feature of quantifying mutant p53 levels through signal differences between wild-type and total p53 proteins. Moreover, the dynamic range of the assay is impressive, allowing accurate measurement of p53 over a wide concentration range (Fig. 2.4).

Schematic diagram showing SPR-based detection of wild-type p53 through consensus DNA response elements (left), and total p53 proteins using monoclonal antibodies (right) in separate fluidic channels over a gold sensor chip functionalized with dextran (reproduced with permission from Wang et al. (2009c), Copyright American Chemical Society 2009)
2.4.3 Colorimetric Assay
Colorimetric assays are highly applicable as point-of-care diagnostics due to their instrument-free nature. Generally, noble metals such as gold or silver nanoparticles are suitable as colorimetric indicators due to their excellent extinction coefficients and strong distance-dependent optical properties (Wang et al. 2009c; Liu et al. 2009; Thaxton et al. 2006). Numerous colorimetric techniques have been developed for the sensitive and visually enabled analysis of metal ions, small molecules, proteins, as well as transcription factor-DNA binding. For example, colorimetric assays have been designed to sense estrogen receptor (ER) and specificity protein 1 (SP1) using metal nanoparticle probes (Tan et al. 2010a, b, 2011, 2013, 2014; Seow et al. 2015). One example is the measurement of ERα binding to its response elements (ERE), which for the purpose of this scheme, involved half-sites of the full response element conjugated on metal nanoparticles. Interaction of ER with nanoparticle-ERE probes leads to a decrease in aggregation (red spheres) of DNA-metal nanoparticles from the introduction of steric protection forces between the nanoparticles in the presence of salt (Fig. 2.5).

Schematic diagram of AuNP colorimetric sensing of ER-DNA binding principle. Gold nanoparticles (AuNPs) are modified to carry either half of an ERE sequence (v1 and v2) with a 3-base complementary overhang and will aggregate spontaneously when mixed (middle). Addition of KCl salt reduces charge repulsion between DNA-AuNPs and promotes rapid particle aggregation and a consequent solution color change from red to purple (left). Addition of ERα results in DNA binding to full ERE sequence and exerts steric force to stabilize AuNPs resulting in solution color to remain red (right) (reproduced with permission from Tan et al. (2010b), Copyright American Chemical Society, 2010)
Yan and group reported a user-friendly and sensitive colorimetric method to detect NF-kB p50 with an isothermal exponential amplification reaction (EXPAR) approach. Sequence-specific binding of p50 to a specially designed dsDNA results in the blocking of exonuclease III activity at a position which preserves and releases a ssDNA “DNA trigger” molecule to initiate the EXPAR cycle. DNA triggers anneal with an EXPAR ssDNA template allowing the synthesis of the antisense strand (in the presence of DNA polymerases) and the introduction of a nicking endonuclease site in between two copies of DNA triggers (Fig. 2.6). Endonuclease activity at this site leads to the release of a DNA trigger molecule which participates in another EXPAR cycle, creating an exponential increase in ssDNA trigger molecules which eventually serves as reporter oligonucleotides by aggregating AuNP probes through sequence complementarity and producing a color change (Fig. 2.6) (Zhang et al. 2012). However, in the absence of an appropriate DNA-binding protein, exonuclease III quickly degrades the DNA duplex preventing EXPAR amplification and resulting in no AuNP aggregation.

Schematic diagram showing EXPAR-based colorimetric assay. Binding of p50 leads to the protection of dsDNA and subsequent release of trigger DNA molecule cycling and signal amplification in further EXPAR reactions. Trigger ssDNA also acts as reporter oligonucleotides that promote AuNP aggregation through DNA hybridization producing a colorimetric readout (reproduced with permission from Zhang et al. (2012), Copyright American Chemical Society, 2012)
2.5 Electrical Biosensors for Transcription Factor Detection
Electrical biosensors which include electrochemical sensors and electronic sensors are usually accurate, fast, and sensitive methods for molecular sensing. In addition, they provide operating simplicity, the option for miniaturization, cost-effectiveness, and have attracted much attention in the area of point-of-care diagnostics.
Electrochemical biosensors based on DNA-mediated charge transport offer an interesting approach to study transcription factor-DNA binding. Gorodetsky et al. demonstrated the use of DNA-modified microelectrodes to rapidly detect nanomolar concentrations of TATA-binding proteins (TBP), a ubiquitous transcription factor (Gorodetsky et al. 2008). The double-stranded TBP-specific REs were immobilized on microelectrodes via Au-S chemistry and the distal ends of REs were modified with redox-active Nile Blue to give electrochemical signals. The binding of TBP bends the duplex RE and decreases the DNA-mediated reduction of Nile Blue, thus lowering the electrochemical signal. This electrochemical sensor can also be easily modified and applied to other TFs. It is also worth highlighting that the use of a microelectrode array can further allow the multiplexed detection of a panel of TFs on a single chip.
Besides electrochemical biosensors, metal oxide semiconductor field-effect transistor (MOSFET) is another popular electrical biosensor consisting of four terminals including the source, gate, drain, and substrate. Operation of the MOSFET depends on the electric field to control the size and shape of a channel from the source to the drain. Upon exposure to an analyte, a gate modulates the flow of electrons through the channel, thereby inducing changes in the drain current. Based on this principle, Han et al. pioneered the design of a field effect transistor (FET)-based biosensor to evaluate the DNA binding activity of wild-type and mutant p53 proteins. The MOSFET was immobilized with p53-specific GADD45 REs. As shown in Fig. 2.7, a significant increase in drain current was observed upon the addition of 100 nM wild-type p53, whereas addition of mutant p53 protein (R248W) gives no response (Han et al. 2010). This label-free method also allows real-time monitoring of p53-DNA binding. However, no detailed calibration has yet been performed to determine the limit of detection for this MOSFET; thus more studies are necessary to further apply this technology.

Real-time monitoring of mutant p53 using MOSFET. Plot of drain current versus time in response to mutant p53 (R248W) and wild-type p53 on a single MOSFET device at an applied voltage of 2.0 V (reproduced with permission from Han et al. (2010), Copyright Elsevier, 2010)
More recently, transcription factor biosensing at the level of single molecules has been successfully achieved by Squires et al. using a solid-state nanopore platform. Nanopores are label-free and ultrasensitive biosensors that are usually used to characterize biopolymers such as DNA, RNA, or proteins at the single-molecule level. An electrical field is applied to the nanopore to guide movement of the biopolymer into the nanopore, thus allowing the study of individual molecules. The ability to rapidly measure hundreds of samples and to resolve fine structural features alludes to the potential of nanopores in TF sensing. In this study, the DNA-binding domain of the early growth response protein 1 (EGR1), also known as zinc finger protein zif268, was used as the model TF (Squires et al. 2015). zif268-DNA binding was detected according to current blockage sublevels and duration of translocation through the nanopore. It was also demonstrated that different binding modes of zif268 will give rise to distinct current blockage patterns, demonstrating the feature of characterizing TF protein conformation. This unique nanopore technique provides a novel way to study transcription factor-DNA binding at the single-molecule level and will undeniably unveil new information about detailed molecular interaction.
2.6 Other Sensing Technology
In this section, we briefly visit TF sensing techniques using alternative detection methods as well as powerful protein sensing methods that could be repurposed for the functional sensing of transcription factors.
As an alternative to ELISA, Oberlander et al. developed a scintillation proximity assay (SPA) to measure total p53 protein in cell extracts. SPA beads are embedded scintillants, which give out light when they come close to radioactive compounds. The SPA beads are first immobilized with capture antibodies, and in the presence of p53 proteins, associate with biotinylated anti-53 antibodies. Addition of 35S-labeled streptavidin triggers the SPA, allowing photometric detection. This assay is sensitive enough to detect very low levels of p53 (50–300 pg) in small volumes of biological extracts, but requires the use of harmful radioactive labels (Oberlander et al. 2010). In a more recent EXPAR-based TF sensing technique, Ma and coworkers describe the detection of NF-κB p50 activity with remarkable sensitivity (10 fM) through the use of a dual-EXPAR scheme and G-quadruplex DNAzyme as reporter molecules. Binding of NF-κB p50 to a unique dsDNA provides protection from the nuclease activity of exonuclease I and III, sequentially added to disintegrate unbound DNA molecules. Intact DNA copies remaining serve as a template for RNA polymerase, producing RNA trigger molecules. RNA triggers then prime EXPAR ssDNA templates for DNA polymerization, producing a DNA duplex containing two trigger copies and an HRP-mimicking DNAzyme separated by nicking endonuclease sites. Cleavage of these sites releases more DNA trigger molecules (to initiate additional EXPAR cycles), a DNAzyme reporter molecule and the initial RNA trigger-EXPAR DNA template-bound fragment for elongation by DNA polymerase again; hence constituting a self-perpetuating signal amplification cycle. DNAzyme reporter molecules produced eventually catalyze a luminol-dependent chemiluminescence signal in the presence of hemin (Ma et al. 2014).
A powerful technique that is highly amenable for detecting TF-DNA interaction was developed by Langer et al. and involves a biochip with electrically actuated DNA levers able to sensitively detect hydrodynamic conformational changes. This biochip consists of four individually addressable flow channels on a glass substrate (Langer et al. 2013). Within each channel, six gold microelectrodes are immobilized with Cy3-labeled dsDNA. When positive potential is applied, the Au electrode attracts the negatively charged DNA molecules, leading to fluorescence quenching. When the potential is reversed, the DNA reverts back to an upright state, leading to fluorescence recovery. An epifluorescence setup is used to measure the change in fluorescence intensity during the DNA switching process. By applying designated capture sequences to the DNA fragment’s distal ends, the DNA levers can specifically bind target proteins from solution. Protein binding slows down the DNA switching motion which is correlated to its hydrodynamic size. In addition, this method led to the development of an analytical model that predicts the hydrodynamic diameter of the bound protein from the kinetics of DNA-protein motion. This approach has also been successfully applied to detect post-translational modifications such as phosphorylation and glycosylation of proteins. Other notable advantages include microelectrode arrays for multiplexing and low sample consumption through the use of microfluidics.
2.7 Concluding Remarks
The last decade has witnessed a rapid advancement in the integration of biology, chemistry, and physics to yield novel, highly sensitive hybrid biosensors. This parallels the trend seen for latest-generation DNA-sequencing technologies that continue to push the limits of throughput and accuracy. The different detection methods reviewed here offer unique advantages each but present their own caveats. The popularity of fluorescence assays can be attributed to their high sensitivity, specificity, and multiplexing feasibility, but often require special labels. SPR techniques are label-free and provide real-time monitoring of binding kinetics but are less sensitive, and require extensive optimization and instrumentation. While colorimetric assays are more amenable as point-of-care detection tools due to their instrument-free and visually permissive detection modes, they often face the limitations of sample solution color. Electrical sensors can be highly sensitive down to the single-molecule level, but are prone to environmental interferences and may require expensive setups or complicated fabrication of sensor chips. Further integration, coupled with exciting advances in site-specific protein labeling (Proft 2010; Ravikumar et al. 2015), will advance the development of next-generation biosensors. These will find important use in multiplexed liquid biopsies to detect the ever-increasing number of clinically significant biomarkers.
References
Andre F, Bachelot T, Commo F, Campone M, Arnedos M, Dieras V, Lacroix-Triki M, Lacroix L, Cohen P, Gentien D, Adelaide J, Dalenc F, Goncalves A, Levy C, Ferrero JM, Bonneterre J, Lefeuvre C, Jimenez M, Filleron T, Bonnefoi H (2014) Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol 15:267–274
Ascenzi P, Bocedi A, Marino M (2006) Structure-function relationship of estrogen receptor alpha and beta: impact on human health. Mol Asp Med 27:299–402
Babu MM, Luscombe NM, Aravind L, Gerstein M, Teichmann SA (2004) Structure and evolution of transcriptional regulatory networks. Curr Opin Struct Biol 14:283–291
Balagurumoorthy P, Sakamoto H, Lewis MS, Zambrano N, Clore GM, Gronenborn AM, Appella E, Harrington RE (1995) Four p53 DNA-binding domain peptides bind natural p53-response elements and bend the DNA. Proc Natl Acad Sci U S A 92:8591–8595
Beckerman R, Prives C (2010) Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2:a000935
Beishline K, Azizkhan-Clifford J (2015) Sp1 and the ‘hallmarks of cancer’. FEBS J 282:224–258
Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, Levine AJ (2010) The origins and evolution of the p53 family of genes. Cold Spring Harb Perspect Biol 2:a001198
Bieging KT, Mello SS, Attardi LD (2014) Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 14:359–370
Blackwood EM, Eisenman RN (1991) Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251:1211–1217
Boutell et al (2004) Proteomics 4(7):1950–1958
Brenowitz M, Senear DF, Shea MA, Ackers GK (1986) Quantitative DNase footprint titration: a method for studying protein-DNA interactions. Methods Enzymol 130:132–181
Bullock AN, Fersht AR (2001) Rescuing the function of mutant p53. Nat Rev Cancer 1:68–76
Cai Q, Yan L, Xu Y (2015) Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene 34:3315–3324
Chang GS, Chen XA, Park B, Rhee HS, Li P, Han KH, Mishra T, Chan-Salis KY, Li Y, Hardison RC, Wang Y, Pugh BF (2014) A comprehensive and high-resolution genome-wide response of p53 to stress. Cell Rep 8:514–527
Chen FE, Huang DB, Chen YQ, Ghosh G (1998) Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature 391:410–413
Cheng Q, Chen J (2010) Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 9:472–478
Chin PL, Momand J, Pfeifer GP (1997) In vivo evidence for binding of p53 to consensus binding sites in the p21 and GADD45 genes in response to ionizing radiation. Oncogene 15:87–99
Cho Y, Gorina S, Jeffrey PD, Pavletich NP (1994) Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 265:346–355
Ciriello et al (2015) Cell 8;163(2):506–519. doi:10.1016/j.cell.2015.09.033
DeBerardinis RJ, Thompson CB (2012) Cellular metabolism and disease: what do metabolic outliers teach us? Cell 148:1132–1144
Dornan D, Hupp TR (2001) Inhibition of p53-dependent transcription by BOX-I phospho-peptide mimetics that bind to p300. EMBO Rep 2:139–144
Eferl R, Wagner EF (2003) AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 3:859–868
el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B (1992) Definition of a consensus binding site for p53. Nat Genet 1:45–49
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75:817–825
Fan D, Liu SY, van Hasselt CA, Vlantis AC, Ng EK, Zhang H, Dong Y, Ng SK, Chu R, Chan AB, Du J, Wei W, Liu X, Liu Z, Xing M, Chen GG (2015) Estrogen receptor alpha induces prosurvival autophagy in papillary thyroid cancer via stimulating reactive oxygen species and extracellular signal regulated kinases. J Clin Endocrinol Metab 100:E561–E571
Fang X, Li JJ, Tan W (2000) Using molecular beacons to probe molecular interactions between lactate dehydrogenase and single-stranded DNA. Anal Chem 72:3280–3285
Fried MG (1989) Measurement of protein-DNA interaction parameters by electrophoresis mobility shift assay. Electrophoresis 10:366–376
Friedler A, Veprintsev DB, Freund SM, von Glos KI, Fersht AR (2005) Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure 13:629–636
Galas DJ, Schmitz A (1978) DNAase footprinting a simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res 5:3157–3170
Garner MM, Revzin A (1981) A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res 9:3047–3060
Geng J, Goh WLP, Zhang C, Lane D, Liu B, Ghadessy FJ, Tan YN (2015) A highly sensitive fluoresce light-up probe for real-time detection of endogenous protein target and its antagonism in live cells. J Mater Chem B 3:5933–5937
Giannetti A, Citti L, Domenici C, Tedeschi L, Baldini F, Wabuyele MB, Vo-Dinh T (2006) FRET-based protein–DNA binding assay for detection of active NF-κB. Sensors Actuators B Chem 113:649–654
Gilmore TD (2006) Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25:6680–6684
Goh W, Lane D, Ghadessy F (2010) Development of a novel multiplex in vitro binding assay to profile p53-DNA interactions. Cell Cycle 9:3030–3038
Goh WL, Lee MY, Joseph TL, Quah ST, Brown CJ, Verma C, Brenner S, Ghadessy FJ, Teo YN (2014) Molecular rotors as conditionally fluorescent labels for rapid detection of biomolecular interactions. J Am Chem Soc 136:6159–6162
Gorodetsky AA, Ebrahim A, Barton JK (2008) Electrical detection of TATA binding protein at DNA-modified microelectrodes. J Am Chem Soc 130:2924–2925
Grabowski ZR, Rotkiewicz K, Rettig W (2003) Structural changes accompanying intramolecular electron transfer: focus on twisted intramolecular charge-transfer states and structures. Chem Rev 103:3899–4032
Han SH, Kim SK, Park K, Yi SY, Park H-J, Lyu H-K, Kim M, Chung BH (2010) Detection of mutant p53 using field-effect transistor biosensor. Anal Chim Acta 665:79–83
Hanada R, Hanada T, Sigl V, Schramek D, Penninger JM (2011) RANKL/RANK-beyond bones. J Mol Med (Berl) 89:647–656
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hayden MS, Ghosh S (2012) NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26:203–234
Heinlein CA, Chang C (2004) Androgen receptor in prostate cancer. Endocr Rev 25:276–308
Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B (1997) 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1:3–11
Heyduk T, Heyduk E (2002) Molecular beacons for detecting DNA binding proteins. Nat Biotechnol 20:171–176
Hibino E, Inoue R, Sugiyama M, Kuwahara J, Matsuzaki K, Hoshino M (2016) Interaction between intrinsically disordered regions in transcription factors Sp1 and TAF4. Protein Sci 25:2006–2017
Hoesel B, Schmid JA (2013) The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer 12:86
Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T (2004) NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest 114:569–581
Hupp TR, Meek DW, Midgley CA, Lane DP (1992) Regulation of the specific DNA binding function of p53. Cell 71:875–886
Iwanicki MP, Chen HY, Iavarone C, Zervantonakis IK, Muranen T, Novak M, Ince TA, Drapkin R, Brugge JS (2016) Mutant p53 regulates ovarian cancer transformed phenotypes through autocrine matrix deposition. JCI Insight 1:e86829
Jacobs MD, Harrison SC (1998) Structure of an IkappaBalpha/NF-kappaB complex. Cell 95:749–758
Jacque E, Tchenio T, Piton G, Romeo PH, Baud V (2005) RelA repression of RelB activity induces selective gene activation downstream of TNF receptors. Proc Natl Acad Sci U S A 102:14635–14640
Jagelska E, Brazda V, Pospisilova S, Vojtesek B, Palecek E (2002) New ELISA technique for analysis of p53 protein/DNA binding properties. J Immunol Methods 267:227–235
Jares-Erijman EA, Jovin TM (2003) FRET imaging. Nat Biotechnol 21:1387–1395
Jerry DJ, Dunphy KA, Hagen MJ (2010) Estrogens, regulation of p53 and breast cancer risk: a balancing act. Cell Mol Life Sci 67:1017–1023
Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, Doan HM, Fan J, Cheadle C, Fallahi M, Cleveland JL, Dang CV, Zeller KI (2011) Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One 6:e26057
Joerger AC, Fersht AR (2007) Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 26:2226–2242
Joerger AC, Fersht AR (2010) The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb Perspect Biol 2:a000919
Jordan JJ, Menendez D, Inga A, Noureddine M, Bell DA, Resnick MA (2008) Noncanonical DNA motifs as transactivation targets by wild type and mutant p53. PLoS Genet 4:e1000104
Karin M, Ben-Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 18:621–663
Khoury MP, Bourdon JC (2011) p53 isoforms: an intracellular microprocessor? Genes Cancer 2:453–465
Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA (1996) Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A 93:5925–5930
Kummerfeld SK, Teichmann SA (2006) DBD: a transcription factor prediction database. Nucleic Acids Res 34:D74–D81
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274:948–953
Kyewski B, Klein L (2006) A central role for central tolerance. Annu Rev Immunol 24:571–606
Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R, Brady JN (1998) Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem 273:33048–33053
Lane DP (1992) Cancer. p53, guardian of the genome. Nature 358:15–16
Langer A, Hampel PA, Kaiser W, Knezevic J, Welte T, Villa V, Maruyama M, Svejda M, Jähner S, Fischer F, Strasser R, Rant U (2013) Protein analysis by time-resolved measurements with an electro-switchable DNA chip. Nat Commun 4:2099
Laptenko O, Shiff I, Freed-Pastor W, Zupnick A, Mattia M, Freulich E, Shamir I, Kadouri N, Kahan T, Manfredi J, Simon I, Prives C (2015) The p53 C terminus controls site-specific DNA binding and promotes structural changes within the central DNA binding domain. Mol Cell 57:1034–1046
Laptenko O, Tong DR, Manfredi J, Prives C (2016) The tail that wags the dog: how the disordered C-terminal domain controls the transcriptional activities of the p53 tumor-suppressor protein. Trends Biochem Sci 41:1022–1034
Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L (2011) Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr Rev 32:597–622
Lee TI, Young RA (2000) Transcription of eukaryotic protein-coding genes. Annu Rev Genet 34:77–137
Lee TI, Young RA (2013) Transcriptional regulation and its misregulation in disease. Cell 152:1237–1251
Lequin RM (2005) Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin Chem 51:2415–2418
Liang J, Shang Y (2013) Estrogen and cancer. Annu Rev Physiol 75:225–240
Licht JD (2001) AML1 and the AML1-ETO fusion protein in the pathogenesis of t(8;21) AML. Oncogene 20:5660–5679
Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA (2012) Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151:56–67
Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44:479–496
Liu J, Cao Z, Lu Y (2009) Functional nucleic acid sensors. Chem Rev 109:1948–1998
Liu JJ, Song XR, Wang YW, Chen GN, Yang HH (2012) A graphene oxide (GO)-based molecular beacon for DNA-binding transcription factor detection. Nanoscale 4:3655–3659
Liu X, Ouyang L, Cai X, Huang Y, Feng X, Fan Q, Huang W (2013) An ultrasensitive label-free biosensor for assaying of sequence-specific DNA-binding protein based on amplifying fluorescent conjugated polymer. Biosens Bioelectron 41:218–224
Lohrum MA, Woods DB, Ludwig RL, Balint E, Vousden KH (2001) C-terminal ubiquitination of p53 contributes to nuclear export. Mol Cell Biol 21:8521–8532
Lu X, Liu DP, Xu Y (2013) The gain of function of p53 cancer mutant in promoting mammary tumorigenesis. Oncogene 32:2900–2906
Lukasik SM, Zhang L, Corpora T, Tomanicek S, Li Y, Kundu M, Hartman K, Liu PP, Laue TM, Biltonen RL, Speck NA, Bushweller JH (2002) Altered affinity of CBF beta-SMMHC for Runx1 explains its role in leukemogenesis. Nat Struct Biol 9(9):674
Ma F, Yang Y, Zhang CY (2014) Ultrasensitive detection of transcription factors using transcription-mediated isothermally exponential amplification-induced chemiluminescence. Anal Chem 86:6006–6011
Maestro MA, Cardalda C, Boj SF, Luco RF, Servitja JM, Ferrer J (2007) Distinct roles of HNF1beta, HNF1alpha, and HNF4alpha in regulating pancreas development, beta-cell function and growth. Endocr Dev 12:33–45
Malkin D (2011) Li-fraumeni syndrome. Genes Cancer 2:475–484
Mantovani F, Banks L (2001) The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene 20:7874–7887
Martinez, LA. Mutant p53 and ETS2, a Tale of Reciprocity. Front Oncol 6, 35 (2016).
May MJ, Ghosh S (1997) Rel/NF-kappa B and I kappa B proteins: an overview. Semin Cancer Biol 8:63–73
Meek DW, Anderson CW (2009) Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol 1:a000950
Menendez D, Inga A, Resnick MA (2009) The expanding universe of p53 targets. Nat Rev Cancer 9:724–737
Mermod N, O'Neill EA, Kelly TJ, Tjian R (1989) The proline-rich transcriptional activator of CTF/NF-I is distinct from the replication and DNA binding domain. Cell 58:741–753
Messina DN, Glasscock J, Gish W, Lovett M (2004) An ORFeome-based analysis of human transcription factor genes and the construction of a microarray to interrogate their expression. Genome Res 14:2041–2047
Meyer N, Penn LZ (2008) Reflecting on 25 years with MYC. Nat Rev Cancer 8:976–990
Milde-Langosch K (2005) The Fos family of transcription factors and their role in tumourigenesis. Eur J Cancer 41:2449–2461
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293–299
Mognol GP, Carneiro FR, Robbs BK, Faget DV, Viola JP (2016) Cell cycle and apoptosis regulation by NFAT transcription factors: new roles for an old player. Cell Death Dis 7:e2199
Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7:683–694
Narod (2011) Nat Rev Clin Oncol 8(11):669–676
Nolan E, Vaillant F, Branstetter D, Pal B, Giner G, Whitehead L, Lok SW, Mann GB, Rohrbach K, Huang LY, Soriano R, Smyth GK, Dougall WC, Visvader JE, Lindeman GJ (2016) RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med 22:933–939
Noureddine MA, Menendez D, Campbell MR, Bandele OJ, Horvath MM, Wang X, Pittman GS, Chorley BN, Resnick MA, Bell DA (2009) Probing the functional impact of sequence variation on p53-DNA interactions using a novel microsphere assay for protein-DNA binding with human cell extracts. PLoS Genet 5:e1000462
Oberlander S, Xie T, Chandrachud U, Gal S (2010) Scintillation proximity assay for total p53 protein as an alternative to ELISA. J Immunol Methods 360:173–177
Okuda M, Araki K, Ohtani K, Nishimura Y (2016) The interaction mode of the acidic region of the cell cycle transcription factor DP1 with TFIIH. J Mol Biol 428:4993–5006
Ong HJ, Siau JW, Zhang JB, Hong M, Flotow H, Ghadessy F (2012) Analysis of p53 binding to DNA by fluorescence imaging microscopy. Micron 43:996–1000
Peh WY, Reimhult E, Teh HF, Thomsen JS, Su X (2007) Understanding ligand binding effects on the conformation of estrogen receptor α-DNA complexes: a combinational quartz crystal microbalance with dissipation and surface plasmon resonance study. Biophys J 92:4415–4423
Piskacek S, Gregor M, Nemethova M, Grabner M, Kovarik P, Piskacek M (2007) Nine-amino-acid transactivation domain: establishment and prediction utilities. Genomics 89:756–768
Powell et al (2014) Cancer Discov 4(4):405–414
Proft T (2010) Sortase-mediated protein ligation: an emerging biotechnology tool for protein modification and immobilisation. Biotechnol Lett 32:1–10
Ravikumar Y, Nadarajan SP, Yoo TH, Lee CS, Yun H (2015) Unnatural amino acid mutagenesis-based enzyme engineering. Trends Biotechnol 33:462–470
Rayburn E, Zhang R, He J, Wang H (2005) MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets 5:27–41
Reed M, Woelker B, Wang P, Wang Y, Anderson ME, Tegtmeyer P (1995) The C-terminal domain of p53 recognizes DNA damaged by ionizing radiation. Proc Natl Acad Sci U S A 92:9455–9459
Riley T, Sontag E, Chen P, Levine A (2008) Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9:402–412
Robertson G, Bilenky M, Lin K, He A, Yuen W, Dagpinar M, Varhol R, Teague K, Griffith OL, Zhang X, Pan Y, Hassel M, Sleumer MC, Pan W, Pleasance ED, Chuang M, Hao H, Li YY, Robertson N, Fjell C, Li B, Montgomery SB, Astakhova T, Zhou J, Sander J, Siddiqui AS, Jones SJ (2006) cisRED: a database system for genome-scale computational discovery of regulatory elements. Nucleic Acids Res 34:D68–D73
Rudel D, Sommer RJ (2003) The evolution of developmental mechanisms. Dev Biol 264:15–37
Sammons MA, Zhu J, Drake AM, Berger SL (2015) TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res 25:179–188
Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, Ma W, Tatarek J, Ahn Y, Kelliher MA, Jamieson CH, Staudt LM, Young RA, Look AT (2012) Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell 22:209–221
Sasaki CY, Barberi TJ, Ghosh P, Longo DL (2005) Phosphorylation of RelA/p65 on serine 536 defines an I{kappa}B{alpha}-independent NF-{kappa}B pathway. J Biol Chem 280:34538–34547
Schaefer U, Schmeier S, Bajic VB (2011) TcoF-DB: dragon database for human transcription co-factors and transcription factor interacting proteins. Nucleic Acids Res 39:D106–D110
Seow N, Tan YN, Yung L-YL, Su X (2015) DNA-directed assembly of Nanogold dimers: a unique dynamic light scattering sensing probe for transcription factor detection. Sci Rep 5:18293
Sha L, Zhang X, Wang G (2016) A label-free and enzyme-free ultra-sensitive transcription factors biosensor using DNA-templated copper nanoparticles as fluorescent indicator and hairpin DNA cascade reaction as signal amplifier. Biosens Bioelectron 82:85–92
Shang Y (2007) Hormones and cancer. Cell Res 17:277–279
Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M (2008) Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res 36:3639–3654
Solomon H, Buganim Y, Kogan-Sakin I, Pomeraniec L, Assia Y, Madar S, Goldstein I, Brosh R, Kalo E, Beatus T, Goldfinger N, Rotter V (2012) Various p53 mutant proteins differently regulate the Ras circuit to induce a cancer-related gene signature. J Cell Sci 125:3144–3152
Song H, Hollstein M, Xu Y (2007) p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol 9:573–580
Squires A, Atas E, Meller A (2015) Nanopore sensing of individual transcription factors bound to DNA. Sci Rep 5:11643
Stender JD, Kim K, Charn TH, Komm B, Chang KC, Kraus WL, Benner C, Glass CK, Katzenellenbogen BS (2010) Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol 30:3943–3955
Stojanovic MN, Kolpashchikov DM (2004) Modular aptameric sensors. J Am Chem Soc 126:9266–9270
Strom A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA (2004) Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci U S A 101:1566–1571
Su X, Lin C-Y, O'Shea SJ, Teh HF, Peh WY, Thomsen JS (2006) Combinational application of surface plasmon resonance spectroscopy and quartz crystal microbalance for studying nuclear hormone receptor-response element interactions. Anal Chem 78:5552–5558
Swedenborg E, Power KA, Cai W, Pongratz I, Ruegg J (2009) Regulation of estrogen receptor beta activity and implications in health and disease. Cell Mol Life Sci 66:3873–3894
Tan YN, Lai A, Su X (2014) Interrogating cooperative interactions of transcription factors with composite DNA elements using gold nanoparticles. Sci Adv Mater 6:1460–1466
Tan YN, Lee KH, Su X (2011) Study of single-stranded DNA binding protein–nucleic acids interactions using unmodified gold nanoparticles and its application for detection of single nucleotide polymorphisms. Anal Chem 83:4251–4257
Tan YN, Lee KH, Su X (2013) A study of DNA design dependency of segmented DNA-induced gold nanoparticle aggregation towards versatile bioassay development. RSC Adv 3:21604–21612
Tan YN, Su X, Liu ET, Thomsen JS (2010a) Gold-nanoparticle-based assay for instantaneous detection of nuclear hormone receptor− response elements interactions. Anal Chem 82:2759–2765
Tan YN, Su X, Zhu Y, Lee JY (2010b) Sensing of transcription factor through controlled-assembly of metal nanoparticles modified with segmented DNA elements. ACS Nano 4:5101–5110
Tebaldi T, Zaccara S, Alessandrini F, Bisio A, Ciribilli Y, Inga A (2015) Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genomics 16:464
Thanos CD, Bowie JU (1999) p53 family members p63 and p73 are SAM domain-containing proteins. Protein Sci 8:1708–1710
Thaxton CS, Georganopoulou DG, Mirkin CA (2006) Gold nanoparticle probes for the detection of nucleic acid targets. Clin Chim Acta 363:120–126
Thomas C, Gustafsson JA (2011) The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer 11:597–608
Tyagi S (2009) Imaging intracellular RNA distribution and dynamics in living cells. Nat Methods 6:331–338
Vallee-Belisle A, Bonham AJ, Reich NO, Ricci F, Plaxco KW (2011) Transcription factor beacons for the quantitative detection of DNA binding activity. J Am Chem Soc 133:13836–13839
Vallée-Bélisle A, Plaxco KW (2010) Structure-switching biosensors: inspired by nature. Curr Opin Struct Biol 20:518–526
Vang R, Levine DA, Soslow RA, Zaloudek C, Shih Ie M, Kurman RJ (2016) Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: a Rereview of cases lacking TP53 mutations in the cancer genome atlas ovarian study. Int J Gynecol Pathol 35:48–55
Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM (2009) A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10:252–263
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C et al (2001) The sequence of the human genome. Science 291:1304–1351
Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S (1995) Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev 9:2723–2735
Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408:307–310
Vousden KH, Lane DP (2007) p53 in health and disease. Nat Rev Mol Cell Biol 8:275–283
Wan F, Lenardo MJ (2009) Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb Perspect Biol 1:a000067
Wang B, Xiao Z, Ren EC (2009a) Redefining the p53 response element. Proc Natl Acad Sci U S A 106:14373–14378
Wang K, Tang Z, Yang CJ, Kim Y, Fang X, Li W, Wu Y, Medley CD, Cao Z, Li J (2009b) Molecular engineering of DNA: molecular beacons. Angew Chem Int Ed 48:856–870
Wang Y, Zhu X, Wu M, Xia N, Wang J, Zhou F (2009c) Simultaneous and label-free determination of wild-type and mutant p53 at a single surface plasmon resonance chip preimmobilized with consensus DNA and monoclonal antibody. Anal Chem 81:8441–8446
Weinberg RL, Veprintsev DB, Bycroft M, Fersht AR (2005) Comparative binding of p53 to its promoter and DNA recognition elements. J Mol Biol 348:589–596
Xie TX, Xia Z, Zhang N, Gong W, Huang S (2010) Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol Rep 23:725–732
Zeron-Medina J, Wang X, Repapi E, Campbell MR, Su D, Castro-Giner F, Davies B, Peterse EF, Sacilotto N, Walker GJ, Terzian T, Tomlinson IP, Box NF, Meinshausen N, De Val S, Bell DA, Bond GL (2013) A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell 155:410–422
Zhang K, Wang K, Zhu X, Xie M (2016) Sensitive detection of transcription factors in cell nuclear extracts by using a molecular beacons based amplification strategy. Biosens Bioelectron 77:264–269
Zhang Y, Hu J, Zhang C-y (2012) Sensitive detection of transcription factors by isothermal exponential amplification-based colorimetric assay. Anal Chem 84:9544–9549
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Goh, W.L., Assah, E., Zheng, X.T., Lane, D.P., Ghadessy, F.J., Tan, Y.N. (2017). Transcription Factors as Detection and Diagnostic Biomarkers in Cancer. In: Chandra, P., Tan, Y., Singh, S. (eds) Next Generation Point-of-care Biomedical Sensors Technologies for Cancer Diagnosis. Springer, Singapore. https://doi.org/10.1007/978-981-10-4726-8_2
Download citation
DOI: https://doi.org/10.1007/978-981-10-4726-8_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4725-1
Online ISBN: 978-981-10-4726-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)