
Abstract
In patients with diabetes mellitus, endothelial dysfunction is the initial step in the process of atherosclerosis and plays an important role in the development of this condition, leading to diabetic vascular complications. Oxidative stress induced by hyperglycemia and acute glucose fluctuations are associated with endothelial dysfunction through inactivating nitric oxide (NO) by excess production of reactive oxygen species (ROS). Under the condition of insulin resistance, NO production is selectively impaired, whereas endothelin-1 (ET-1) secretion is preferentially activated in endothelial cells, leading to endothelial dysfunction in obese or overweight diabetic patients. On the other hand, endothelial dysfunction might contribute to insulin resistance in skeletal muscle. Reduced NO production through oxidative stress and selective insulin resistance in endothelial cells contributes to decreased glucose uptake by skeletal muscle due to a delayed increase in insulin concentration in the interstitium of the skeletal muscle. Therefore, insulin resistance is further exacerbated through a vicious cycle of endothelial dysfunction and reduced glucose uptake by skeletal muscle. From a clinical perspective, it is important to select an appropriate intervention that is effective in improving endothelial dysfunction for treatment of patients with diabetes mellitus.
In addition to lifestyle modifications, antidiabetic agents that improve insulin sensitivity are anticipated to improve endothelial function and prevent cardiovascular events in patients with diabetes mellitus.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
4.1 Diabetic Vascular Complications
Diabetes mellitus is associated with an increased risk of microvascular and macrovascular complications. A recent study has shown that adults aged 50 years or older with diabetes mellitus die 4.6 years earlier, develop disabilities 6–7 years earlier, and spend 1–2 more years in a disabled state than do those without diabetes mellitus in the USA [1]. Microvascular complications, including retinopathy, nephropathy, and neuropathy, and macrovascular complications, including coronary artery disease, ischemic stroke, and peripheral artery disease, are important causes of morbidity and mortality in patients with diabetes mellitus. Macrovascular complications, namely, cardiovascular diseases (CVDs), are associated with increased mortality in patients with diabetes mellitus. CVD is the most common underlying cause of death, accounting for about 45% of deaths in patients with type 1 diabetes mellitus and about 50% of deaths in patients with type 2 diabetes mellitus [2]. It is therefore important to prevent the onset and progression of CVD for better prognosis in the management of patients with diabetes mellitus.
Endothelial dysfunction is the initial step in the pathogenesis of atherosclerosis and plays a key role in the development of this condition, leading to cardiovascular complications [3]. In addition, it has been demonstrated that endothelial function is an independent predictor of cardiovascular events [4]. Diabetes mellitus has been shown to be associated with endothelial dysfunction [5, 6]. For the prevention of CVD in patients with diabetes mellitus, it is therefore important to understand the causative mechanisms linking diabetes mellitus and endothelial dysfunction and to select an appropriate intervention that will effectively ameliorate endothelial function. In this section, the current understanding of the relationship between diabetes mellitus and endothelial function and treatment options are discussed.
4.2 Endothelial Function
It had been thought that the vascular endothelium was just a structural barrier separating the blood vessel wall and the inside cavity. In the 1980s, it was revealed that the vascular endothelium functions not only as a barrier but also as an endocrine organ secreting various vasoactive agents such as the vasodilators nitric oxide (NO), prostacyclin, and endothelium-derived hyperpolarizing factor (EDHF) and the vasoconstrictors endothelin-1 (ET-1), angiotensin II, and thromboxane A2 [7]. Thus, the vascular endothelium might be one of the biggest endocrine organs in the human body, with an estimated total weight equal to that of the liver and an estimated total area equal to that of six tennis courts. A healthy endothelium acts as a gatekeeper controlling vascular tone and structure by regulating the balance between vasodilation and vasoconstriction, growth inhibition and growth promotion, anti-thrombosis and pro-thrombosis, anti-inflammation and pro-inflammation, and anti-oxidation and pro-oxidation. Endothelial dysfunction refers to a condition characterized by an inability of the endothelium to maintain vascular homeostasis as a result of an imbalance between endothelium-derived relaxing and contracting factors, leading to the progression of atherosclerosis. Endothelial dysfunction is an early feature of atherosclerosis and is associated with the development of this condition in human [3]. It is expected that improvement or augmentation of endothelial function may prevent the development and progression of atherosclerosis and consequently reduce cardiovascular events. Therefore, endothelial dysfunction has emerged as a therapeutic target in patients with cardiovascular risk factors such as hypertension, dyslipidemia, and diabetes mellitus and in patients with CVD. NO, one of the various vasoactive agents released from the endothelium, has various anti-atherosclerotic effects including vasodilation, inhibition of platelet aggregation and adhesion, inhibition of leucocyte adhesion, and suppression of vascular smooth muscle cell proliferation. Considering the wide range of anti-atherosclerotic effects of NO, reduced NO bioavailability is generally referred to as endothelial dysfunction.
Diabetes mellitus is associated with endothelial dysfunction. Impaired endothelium-dependent vasodilation has been demonstrated in patients with type 1 and type 2 diabetes [8, 9]. Although the definitive pathogenesis remains unclear, several mechanisms underlying the endothelial dysfunction in patients with diabetes mellitus have been proposed.
4.3 Mechanism Underlying the Endothelial Dysfunction in Diabetes
4.3.1 Oxidative Stress
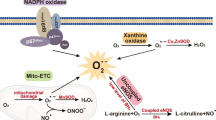
Oxidative stress has been shown to be associated with the pathology of various diseases including diabetes mellitus. Oxidative stress refers to a condition in which the balance of reactive oxygen species (ROS) and the antioxidant system is disturbed in favor of prooxidant ROS. Excessive production of ROS cannot be sufficiently counteracted by the antioxidant defense system, and the deleterious effects of ROS, such as cell proliferation, hypertrophy, apoptosis, and inflammation, become clinically evident. ROS are produced by various oxidase enzymes, including nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, uncoupled endothelial NO synthase (eNOS), mitochondrial electron transport, cyclooxygenase, glucose oxidase, and lipoxygenase. ROS include superoxide anion (O2−), hydrogen peroxide (H2O2), hydroxyl radical (OH), hypochlorous acid (HOCl), NO, and peroxynitrite (ONOO−). O2− is produced by the reduction of molecular oxygen through removal of one electron, and it serves as the precursor of other ROS such as H2O2 and OH. In addition, O2− reacts directly with NO and reduces NO bioavailability. In this context, O2− is an important source of oxidative stress associated with endothelial dysfunction. Accumulating evidence has revealed an interaction between oxidative stress and endothelial dysfunction. Excessive O2− reacts directly with NO with high affinity, resulting not only in degradation and inactivation of NO but also in formation of ONOO−, a highly potent oxidant causing lipid peroxidation, DNA damage, and cell death (Fig. 4.1). In addition, ONOO− can oxidize the essential eNOS cofactor tetrahydrobiopterin (BH4) to the biologically inactive trihydrobiopterin (BH3), leading to a deficiency of BH4. In the absence of sufficient concentrations of BH4, eNOS is converted from an NO-producing enzyme into an O2−-generating enzyme. This process is referred to as eNOS uncoupling (Fig. 4.1) [10]. Under this condition, impaired endothelial function is further exacerbated through a vicious cycle of increased oxidative stress and eNOS uncoupling, leading to a further increase in O2− production and a decrease in NO bioavailability. In diabetes mellitus, chronic hyperglycemia is known to be a major contributor to elevated oxidative stress and endothelial dysfunction. In addition, recent studies have demonstrated that acute glucose fluctuations are involved in the mechanism underlying the increased oxidative stress in diabetes mellitus.

Putative mechanism of endothelial nitric oxide synthase (eNOS) uncoupling in patients with diabetes mellitus. O 2 − indicates superoxide, BH 4 tetrahydrobiopterin, BH3 trihydrobiopterin, BH 2 dihydrobiopterin
4.3.1.1 Oxidative Stress Induced by Hyperglycemia in Diabetes
Mitochondria are the major intracellular source of O2−. Intracellular glucose oxidation starts with glycolysis, which generates pyruvate for mitochondrial catabolism to form ATP in the cytoplasm. Pyruvate transported into the mitochondria is oxidized to NADH and FADH2 by the tricarboxylic acid (TCA) cycle. NADH and FADH2 serve as donors of electrons used as energy for ATP production through oxidative phosphorylation by the electron transport chain composed of four multiprotein enzyme complexes located in the inner mitochondrial membrane. Electron transfer is coupled with the transfer of protons across the inner mitochondrial membrane. Therefore, electron transfer through the electron transport chain generates a proton gradient by pumping protons across the inner mitochondrial membrane, providing the energy to drive the ATP synthase. Under the condition of hyperglycemia, NADH and FADH2, electron donors, from the TCA cycle are increased, and the proton gradient across the inner mitochondrial membrane is increased due to the enhanced electron transfer through the electron transport chain and a concomitant increase in the proton pumping. An increase in proton gradient above a certain threshold inhibits electron transport through the electron transport chain, resulting in increased electron leak and O2− generation in mitochondria [11].
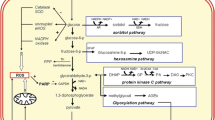
Hyperglycemia-induced overproduction of mitochondrial O2− inhibits the activity of the glycolytic enzyme glyceraldehyde-3-phosphate, the activity of which is essential for maintenance of glycolytic flux, thereby resulting in the accumulation of upstream glycolysis intermediates and increased flux of these metabolites into glucose overutilization pathways, including the polyol pathway, hexosamine pathway, protein kinase C (PKC) pathway, and advanced glycation end product (AGE) pathway (Fig. 4.2) [11]. Increased glucose flux into the polyol pathway consumes NADPH, which is required for regenerating reduced glutathione (GSH), a main intracellular antioxidant. Therefore, intracellular concentrations of reduced GSH are consequently decreased, leading to an increase in intracellular oxidative stress. Shunting of excess intracellular glucose into the hexosamine pathway increases the modification of transcriptional factor through dysregulated protein glycosylation, resulting in altered protein expression. PKC activation induced by hyperglycemia through increased diacylglycerol has various pathogenic consequences including decreased expression of eNOS, increased expression of ET-1, transforming growth factor-β and plasminogen activator inhibitor-1, and activation of NF-κB and NADPH oxidase, leading to impairment of endothelial function. AGE causes functional alterations of intracellular proteins and modifications of extracellular matrix proteins and plasma proteins. In addition, activation of RAGE, a receptor for AGEs, on endothelial cells mediates the production of ROS and activation of NF-κB. Hyperglycemia causes endothelial dysfunction through overproduction of mitochondrial O2− and diversion of glycolytic flux to alternative metabolic pathways.

Putative mechanisms of hyperglycemia-induced endothelial dysfunction in patients with diabetes mellitus. ROS indicates reactive oxygen species, GAPDH glyceraldehyde-3-phosphate, eNOS endothelial nitric oxide synthase, AGE advanced glycation end product, RAGE receptor for AGEs, AP-1 activator protein-1
4.3.1.2 Oxidative Stress Induced by Acute Glucose Fluctuations in Diabetes
Blood glucose levels are strictly regulated within a narrow range in normal subjects. However, in patients with diabetes mellitus, a rapid and large increase in blood glucose levels in the postprandial phase is observed. Recent studies have demonstrated that the postprandial acute hyperglycemia may play a significant role in the pathogenesis of diabetic vascular complications through increased oxidative stress, reduced NO bioavailability, and consequent endothelial dysfunction [12]. Glucose fluctuations induced by intermittent high glucose might be more deleterious to endothelial cells than a constant high glucose concentration. In in vitro studies, intermittent high glucose has been demonstrated to enhance apoptosis of endothelial cells via the activation of PKC and NADPH oxidase, suggesting the involvement of oxidative stress in endothelial cell injury [13, 14]. In addition, a recent clinical study has demonstrated that glucose fluctuations, obtained from continuous glucose monitoring system data by calculating the mean amplitude of glycemic excursion (MAGE), are associated with increased oxidative stress in patients with type 2 diabetes [15]. Monnier et al. reported that the mean urinary excretion rate of 8-iso PGF2α, an oxidative stress marker, strongly correlated with MAGE, whereas there were no significant correlations between the urinary excretion rates of 8-iso PGF2α and any other glucose control parameters, including HbA1c and fasting plasma glucose [15]. In addition, Torimoto et al. reported that there was a significant association between glucose fluctuations and endothelial dysfunction in patients with type 2 diabetes [16]. MAGE significantly correlated with reactive hyperemia index, a marker of endothelial function, measured by using peripheral arterial tonometry in patients with type 2 diabetes [16]. Although the precise molecular mechanisms for enhanced oxidative stress and consequent endothelial cell injury by glucose fluctuations have not been fully elucidated, a possible explanation is that constant high glucose may facilitate cellular metabolic adaptations against high glucose-induced toxic effects by consistent feedback, whereas such adaptations may be reduced during intermittent exposure to high glucose due to the absence of consistent feedback, resulting in higher glucose toxicity. HbA1c has been used as a clinical marker of glycemic exposure and as a therapeutic marker of glycemic control in treatment of patients with diabetes mellitus. However, HbA1c serves as a time-averaged measure of glycemic exposure without any information regarding glycemic variability and fluctuations. Recent large randomized trials have demonstrated the lack of significant reduction in cardiovascular events with intensive glycemic control using HbA1c as a therapeutic parameter of glucose control. In the treatment of patients with diabetes mellitus, attention should be paid to not only fasting plasma glucose and HbA1c levels but also postprandial hyperglycemia in order to protect the endothelium from oxidative injury for the prevention of vascular complications in patients with diabetes mellitus.
4.3.2 Selective Insulin Resistance in Diabetes
In addition to its essential glucose and lipid metabolic actions, insulin has several important vascular actions including NO production in endothelial cells. Insulin stimulation of endothelial cells through binding to its cognate receptor on the endothelial cell surface phosphorylates insulin receptor substrate (IRS), which stimulates phosphoinositide 3-kinase (PI 3-kinase)/Akt pathway. Akt directly phosphorylates eNOS at Ser1177, resulting in activation of eNOS and increased NO production (Fig. 4.3). Insulin also stimulates the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway and downstream release of the vasoconstrictor ET-1 in endothelial cells, which is independent of the PI 3-kinase/Akt pathway (Fig. 4.3). Under the condition of insulin resistance in endothelial cells, insulin-induced activation of the PI 3-kinase/Akt pathway and the downstream phosphorylation of eNOS are selectively impaired due to the decreased endothelial IRS function, whereas the MAPK/ERK pathway is unaffected and preferentially activated due to the compensatory hyperinsulinemia, resulting in decreased NO production and increased ET-1 secretion, a characteristic of endothelial dysfunction. It has been demonstrated that the vasodilatory effect of insulin is enhanced under the condition of ET-1 blockade in patients with type 2 diabetes but not in healthy control subjects [17]. Insulin resistance causes endothelial dysfunction through the selectively impaired PI 3-kinase/Akt pathway due to impaired IRS function and enhanced stimulation of the MAPK/ERK pathway due to compensatory hyperinsulinemia in endothelial cells.

Distinct signaling pathways mediating insulin effects on nitric oxide (NO) and endothelin. IRS indicates insulin receptor substrate, PI 3-k phosphoinositide 3-kinase, eNOS endothelial nitric oxide synthase, MEK mitogen-activated protein kinase kinase, MAPK mitogen-activated protein kinase
4.4 Insulin Resistance in Skeletal Muscle Associated with Endothelial Dysfunction
Skeletal muscle plays an important role in glucose homeostasis through insulin-induced glucose uptake. Insulin has to be delivered to the interstitium of skeletal muscle for stimulating glucose uptake by the skeletal muscle. Insulin itself acts in an NO-dependent fashion to increase interstitial insulin concentration by dilating terminal arterioles to increase the number of perfused capillaries and microvascular exchange surface area (microvascular recruitment), dilating larger resistance vessels to increase total limb blood flow, and promoting trans-endothelial transport of insulin to the interstitium of skeletal muscle [18]. Therefore, endothelial dysfunction induced by oxidative stress and selective insulin resistance in endothelial cells causes impairment of vasodilation and insulin transport across the endothelium through reduced NO bioavailability, leading to insulin resistance in skeletal muscle due to a delayed increase in insulin concentration in the interstitium and a consequent decrease in glucose uptake by skeletal muscle. Results of both animal and human studies support the association of insulin resistance with impaired vasodilator action and impaired glucose uptake in skeletal muscle [19, 20]. In clinical studies, skeletal muscle blood flow response to insulin was shown to decrease in diabetic and obese subjects compared with that in lean subjects [19]. In in vivo studies, eNOS knockout mice have been shown to exhibit insulin resistance through decreased muscle blood flow and glucose uptake by skeletal muscle [20]. Therefore, insulin resistance is further exacerbated through a vicious cycle of endothelial dysfunction induced by selective insulin resistance and reduced glucose uptake by skeletal muscle (Fig. 4.4).

A vicious cycle of endothelial dysfunction and diabetes mellitus
4.5 Current Treatment Targeting Endothelial Dysfunction in Diabetes Mellitus
From a clinical perspective, early detection of endothelial dysfunction and early intervention for maintaining endothelial function in a healthy condition are important for the prevention of future cardiovascular events in patients with cardiovascular risk factors. Therefore, in the management of patients with diabetes mellitus, it is important to select an appropriate intervention that effectively improves endothelial function for the prevention of diabetic vascular complications.
Considering the associations of endothelial dysfunction with hyperglycemia, glucose fluctuations, and insulin resistance, lifestyle modifications and pharmacological therapies aiming at lowering glucose level without hypoglycemia, flattening glucose fluctuations, and improving insulin sensitivity may be beneficial for the restoration of endothelial function and prevention of cardiovascular events.
4.5.1 Insulin Therapy
Intensive glycemic control with insulin therapy effectively reduces microvascular complications and cardiovascular events in patients with type 1 diabetes [21]. For patients with type 1 diabetes, in whom insulin resistance does not predominate and a healthy energy balance is achieved, insulin therapy may be safe and beneficial to the endothelium because of the absence of selective insulin resistance in the PI 3-kinase/Akt pathway, leading to increased production of NO in endothelial cells. However, the effect of insulin therapy on endothelial function in patients with type 2 diabetes is still controversial and may be dependent on the achieved metabolic control level [22]. It is possible that high-dose insulin therapy in obese or overweight diabetic patients with insulin resistance who are refractory to its glucose-lowering effect due to excess nutrient supply and positive energy balance may be harmful to the endothelium through an imbalance between impaired PI 3-kinase/Akt pathway and enhanced MAPK/ERK pathway activation caused by selective insulin resistance in endothelial cells, leading to endothelial dysfunction.
4.5.2 Antidiabetic Agents
An antidiabetic agent exerts its glucose-lowering effect by increasing pancreatic insulin secretion and/or ameliorating insulin sensitivity in peripheral tissues. Sulfonylureas, insulin secretagogues, could potentially have an effect similar to that of high-dose insulin therapy and could be harmful to endothelial function in obese or overweight diabetic patients with insulin resistance due to the selective insulin resistance in endothelial cells.
Considering the reciprocal relationship between insulin resistance and endothelial dysfunction, antidiabetic agents that improve insulin sensitivity are anticipated to have beneficial effects on endothelial function through restoration of PI 3-kinase/Akt signaling and downstream NO production. Thiazolidinediones, insulin sensitizers, have been shown to improve endothelium-dependent vasodilation [23, 24]. In addition, thiazolidinediones have been demonstrated to increase the expression and plasma level of adiponectin, which directly stimulates NO production from endothelial cells through activation of the PI 3-kinase/Akt pathway in patients with insulin resistance or type 2 diabetes [25, 26]. Metformin, another insulin-sensitizing agent, has also been shown to improve endothelium-dependent vasodilation with significant association with improvement of insulin resistance assessed by the homeostasis model (HOMA-IR) in patients with type 2 diabetes [27]. Therefore, pharmacological therapies targeting insulin resistance may have beneficial effects on endothelial function through improving insulin sensitivity and increasing NO production in endothelial cells in diabetic patients with insulin resistance.
Antidiabetic agents that decrease the postprandial rise in blood glucose levels are also anticipated to have beneficial effects on endothelial function through decreased oxidative stress and a consequent increase in NO bioavailability. Glinides, dipeptidyl peptidase 4 (DPP-4) inhibitors, and α-glucosidase inhibitors are antidiabetic drugs that improve the control of postprandial glucose levels. These antidiabetic agents have been shown to improve postprandial endothelial function [28,29,30,31]. However, there are conflicting reports showing that α-glucosidase inhibitors, glinides, and DPP-4 inhibitors have no significant beneficial effects or even have adverse effects on endothelial function in patients with type 2 diabetes [28, 32, 33]. Although the precise reasons for the discrepancy of the results remain unknown, some explanations, including differences in the vascular beds assessed for endothelial function, subject selection, and treatment period, have been postulated. As for glinides and DPP-4 inhibitors, there is a possibility that increased insulin secretion through their pharmacological actions could be harmful to endothelial function in obese or overweight diabetic patients due to the selective insulin resistance in endothelial cells.
4.5.3 Other Treatment
In patients with type 2 diabetes, other cardiovascular risk factors such as hypertension and dyslipidemia are highly prevalent. Therefore, a multiple risk factor intervention approach is important for the prevention of cardiovascular events in patients with type 2 diabetes. The Steno-2 study demonstrated that an intensified and targeted multifactorial intervention including behavior modifications and polypharmacologic therapy aimed at controlling several modifiable risk factors reduced the risk of cardiovascular and microvascular events by about 50% compared with a conventional strategy [34]. In the management of other risk factors in patients with type 2 diabetes, it is also important to select an appropriate intervention that will be expected to improve endothelial function, including administration of antihypertensive agents such as angiotensin-converting enzyme inhibitors and angiotensin II type I receptor blockers [35, 36], administration of statins [37], and lifestyle modifications such as aerobic exercise and body weight reduction [38, 39].
4.6 Conclusions
In patients with diabetes mellitus, endothelial dysfunction is the early feature of atherosclerosis and plays an important role in the development of this condition, leading to diabetic vascular complications. Oxidative stress induced by hyperglycemia and glucose fluctuations causes endothelial dysfunction through inactivating NO by excess production of ROS. Selective insulin resistance in the PI 3-kinase/Akt pathway and impaired downstream NO production in endothelial cells may also contribute to endothelial dysfunction, whereas endothelial dysfunction might contribute to insulin resistance. Insulin potentially regulates its own delivery to skeletal muscle in an NO-dependent fashion at multiple steps. Reduced NO production through oxidative stress and selective insulin resistance in endothelial cells causes insulin resistance in skeletal muscle due to a delayed increase in insulin concentration in the interstitium of skeletal muscle. It is clinically important to break out of the cycle of endothelial dysfunction and insulin resistance in obese or overweight diabetic patients for the prevention of cardiovascular events. In addition to lifestyle modifications such as aerobic exercise and body weight reduction, antidiabetic agents that improve insulin sensitivity are expected to ameliorate endothelial function. Although an intervention targeting the reduction of oxidative stress is theoretically attractive, clinical studies in which the effects of antioxidants on cardiovascular events in patients with diabetes mellitus were investigated have revealed disappointing outcomes. Further studies are needed to develop clinically safe and effective treatment strategies targeting oxidative stress for the prevention of cardiovascular events in patients with diabetes mellitus.
References
Bardenheier BH, Lin J, Zhuo X, Ali MK, Thompson TJ, Cheng YJ, et al. Disability-free life-years lost among adults aged >/=50 years with and without diabetes. Diabetes Care. 2016;39(7):1222–9. doi:10.2337/dc15-1095.
Morrish NJ, Wang SL, Stevens LK, Fuller JH, Keen H. Mortality and causes of death in the WHO Multinational Study of Vascular Disease in Diabetes. Diabetologia. 2001;44(Suppl 2):S14–21.
Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73(3):411–8.
Lerman A, Zeiher AM. Endothelial function: cardiac events. Circulation. 2005;111(3):363–8. doi:10.1161/01.CIR.0000153339.27064.14.
Williams SB, Cusco JA, Roddy MA, Johnstone MT, Creager MA. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1996;27(3):567–74.
Henry RM, Ferreira I, Kostense PJ, Dekker JM, Nijpels G, Heine RJ, et al. Type 2 diabetes is associated with impaired endothelium-dependent, flow-mediated dilation, but impaired glucose metabolism is not; The Hoorn Study. Atherosclerosis. 2004;174(1):49–56. doi:10.1016/j.atherosclerosis.2004.01.002.
Vane JR, Anggard EE, Botting RM. Regulatory functions of the vascular endothelium. N Engl J Med. 1990;323(1):27–36. doi:10.1056/NEJM199007053230106.
McVeigh GE, Brennan GM, Johnston GD, McDermott BJ, McGrath LT, Henry WR, et al. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1992;35(8):771–6.
Johnstone MT, Creager SJ, Scales KM, Cusco JA, Lee BK, Creager MA. Impaired endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. Circulation. 1993;88(6):2510–6.
Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33(7):829–37, 37a–37d. doi:10.1093/eurheartj/ehr304.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–20. doi:10.1038/414813a.
Ceriello A. The emerging role of post-prandial hyperglycaemic spikes in the pathogenesis of diabetic complications. Diabet Med. 1998;15(3):188–93. doi:10.1002/(SICI)1096-9136(199803)15:3<188::AID-DIA545>3.0.CO;2-V.
Risso A, Mercuri F, Quagliaro L, Damante G, Ceriello A. Intermittent high glucose enhances apoptosis in human umbilical vein endothelial cells in culture. Am J Physiol Endocrinol Metab. 2001;281(5):E924–30.
Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of protein kinase C and NAD(P)H-oxidase activation. Diabetes. 2003;52(11):2795–804.
Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol JP, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006;295(14):1681–7. doi:10.1001/jama.295.14.1681.
Torimoto K, Okada Y, Mori H, Tanaka Y. Relationship between fluctuations in glucose levels measured by continuous glucose monitoring and vascular endothelial dysfunction in type 2 diabetes mellitus. Cardiovasc Diabetol. 2013;12:1. doi:10.1186/1475-2840-12-1.
Cardillo C, Nambi SS, Kilcoyne CM, Choucair WK, Katz A, Quon MJ, et al. Insulin stimulates both endothelin and nitric oxide activity in the human forearm. Circulation. 1999;100(8):820–5.
Barrett EJ, Eggleston EM, Inyard AC, Wang H, Li G, Chai W, et al. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. Diabetologia. 2009;52(5):752–64. doi:10.1007/s00125-009-1313-z.
Laakso M, Edelman SV, Brechtel G, Baron AD. Impaired insulin-mediated skeletal muscle blood flow in patients with NIDDM. Diabetes. 1992;41(9):1076–83.
Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104(3):342–5.
Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353(25):2643–53. doi:10.1056/NEJMoa052187.
Nolan CJ, Ruderman NB, Kahn SE, Pedersen O, Prentki M. Insulin resistance as a physiological defense against metabolic stress: implications for the management of subsets of type 2 diabetes. Diabetes. 2015;64(3):673–86. doi:10.2337/db14-0694.
Fujishima S, Ohya Y, Nakamura Y, Onaka U, Abe I, Fujishima M. Troglitazone, an insulin sensitizer, increases forearm blood flow in humans. Am J Hypertens. 1998;11(9):1134–7.
Hidaka T, Nakagawa K, Goto C, Soga J, Fujii Y, Hata T, et al. Pioglitazone improves endothelium-dependent vasodilation in hypertensive patients with impaired glucose tolerance in part through a decrease in oxidative stress. Atherosclerosis. 2010;210(2):521–4. doi:10.1016/j.atherosclerosis.2009.12.011.
Yu JG, Javorschi S, Hevener AL, Kruszynska YT, Norman RA, Sinha M, et al. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes. 2002;51(10):2968–74.
Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, et al. PPARgamma ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes. 2001;50(9):2094–9.
Mather KJ, Verma S, Anderson TJ. Improved endothelial function with metformin in type 2 diabetes mellitus. J Am Coll Cardiol. 2001;37(5):1344–50.
Kato T, Inoue T, Node K. Postprandial endothelial dysfunction in subjects with new-onset type 2 diabetes: an acarbose and nateglinide comparative study. Cardiovasc Diabetol. 2010;9:12. doi:10.1186/1475-2840-9-12.
Matsubara J, Sugiyama S, Akiyama E, Iwashita S, Kurokawa H, Ohba K, et al. Dipeptidyl peptidase-4 inhibitor, sitagliptin, improves endothelial dysfunction in association with its anti-inflammatory effects in patients with coronary artery disease and uncontrolled diabetes. Circ J. 2013;77(5):1337–44.
Nakamura K, Oe H, Kihara H, Shimada K, Fukuda S, Watanabe K, et al. DPP-4 inhibitor and alpha-glucosidase inhibitor equally improve endothelial function in patients with type 2 diabetes: EDGE study. Cardiovasc Diabetol. 2014;13:110. doi:10.1186/s12933-014-0110-2.
Shimabukuro M, Higa N, Chinen I, Yamakawa K, Takasu N. Effects of a single administration of acarbose on postprandial glucose excursion and endothelial dysfunction in type 2 diabetic patients: a randomized crossover study. J Clin Endocrinol Metab. 2006;91(3):837–42. doi:10.1210/jc.2005-1566.
Ayaori M, Iwakami N, Uto-Kondo H, Sato H, Sasaki M, Komatsu T, et al. Dipeptidyl peptidase-4 inhibitors attenuate endothelial function as evaluated by flow-mediated vasodilatation in type 2 diabetic patients. J Am Heart Assoc. 2013;2(1):e003277. doi:10.1161/JAHA.112.003277.
Maruhashi T, Higashi Y, Kihara Y, Yamada H, Sata M, Ueda S, et al. Long-term effect of sitagliptin on endothelial function in type 2 diabetes: a sub-analysis of the PROLOGUE study. Cardiovasc Diabetol. 2016;15(1):134. doi:10.1186/s12933-016-0438-x.
Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med. 2003;348(5):383–93. doi:10.1056/NEJMoa021778.
Higashi Y, Sasaki S, Nakagawa K, Ueda T, Yoshimizu A, Kurisu S, et al. A comparison of angiotensin-converting enzyme inhibitors, calcium antagonists, beta-blockers and diuretic agents on reactive hyperemia in patients with essential hypertension: a multicenter study. J Am Coll Cardiol. 2000;35(2):284–91.
Ghiadoni L, Virdis A, Magagna A, Taddei S, Salvetti A. Effect of the angiotensin II type 1 receptor blocker candesartan on endothelial function in patients with essential hypertension. Hypertension. 2000;35(1 Pt 2):501–6.
Wolfrum S, Jensen KS, Liao JK. Endothelium-dependent effects of statins. Arterioscler Thromb Vasc Biol. 2003;23(5):729–36. doi:10.1161/01.ATV.0000063385.12476.A7.
Higashi Y, Sasaki S, Kurisu S, Yoshimizu A, Sasaki N, Matsuura H, et al. Regular aerobic exercise augments endothelium-dependent vascular relaxation in normotensive as well as hypertensive subjects: role of endothelium-derived nitric oxide. Circulation. 1999;100(11):1194–202.
Sasaki S, Higashi Y, Nakagawa K, Kimura M, Noma K, Sasaki S, et al. A low-calorie diet improves endothelium-dependent vasodilation in obese patients with essential hypertension. Am J Hypertens. 2002;15(4 Pt 1):302–9.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Maruhashi, T., Kihara, Y., Higashi, Y. (2018). Diabetes and Endothelial Dysfunction. In: Yamagishi, Si. (eds) Diabetes and Aging-related Complications. Springer, Singapore. https://doi.org/10.1007/978-981-10-4376-5_4
Download citation
DOI: https://doi.org/10.1007/978-981-10-4376-5_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4375-8
Online ISBN: 978-981-10-4376-5
eBook Packages: MedicineMedicine (R0)