
Abstract
The Runt-domain (RD) transcription factors (RUNX genes) are an important family of transcriptional mediators that interact with a variety of proteins including the Hippo pathway effector proteins, YAP and TAZ. In this chapter we focus on two examples of RUNX-TAZ/YAP interactions that have particular significance in human cancer. Specifically, recent evidence has found that RUNX2 cooperates with TAZ to promote epithelial to mesenchymal transition mediated by the soluble N-terminal ectodomain of E-Cadherin, sE-Cad. Contrastingly, in gastric cancer, RUNX3 acts as a tumor suppressor via inhibition of the YAP-TEAD complex and disruption of downstream YAP-mediated gene transcription and the oncogenic phenotype. The reports highlighted in this chapter add to the growing repertoire of instances of Hippo pathway crosstalk that have been identified in cancer. Elucidation of these increasingly complex interactions may help to identify novel strategies to target Hippo pathway dysregulation in human cancer.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction – RUNX Genes and Hippo Signaling
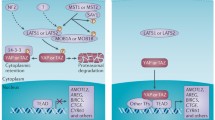
Identified initially in Drosophila melanogaster using genetic mosaic screens to identify novel tumor suppressors, the Hippo signaling pathway is a crucial regulator of organ size that is frequently dysregulated in human cancers (reviewed in Liu et al. 2012). The canonical Hippo pathway consists of the serine/threonine kinases Hippo (Hpo) and Warts, with their mammalian sterile 20-like kinases 1 and 2 (MST1/2) and large tumor suppressors 1 and 2 (LATS1/2) orthologs.
The respective adaptor proteins include Salvador (Sav; SAV1 ortholog) and Mob as tumor suppressor (Mats; Mps one binder kinase activator-like 1, MOB1 ortholog). These kinases center on Yorkie (Yki), a potent transcriptional coactivator that associates with the DNA-binding protein Scalloped (Sd) to drive transcription of genes involved in cell proliferation and survival (Justice et al. 1995; Tapon et al. 2002; Harvey et al. 2003; Pantalacci et al. 2003; Wu et al. 2003). The Yes-associated protein (YAP) and Transcriptional coactivator with PDZ-binding motif (TAZ /WWTR1) are both orthologs of Yki, and TEA domain proteins 1–4 (TEAD1–4) are the Sd mammalian orthologs. Activated by upstream signals, Hpo (MST) phosphorylates and activates Warts (LATS1/2), which in turn phosphorylates Yki (YAP/TAZ) on a specific serine residue to generate a 14-3-3 binding site, resulting in Yki (YAP/TAZ) cytoplasmic sequestration (Huang et al. 2005; Oh and Irvine 2008). For simplicity, only the mammalian nomenclature for Hippo components is referred to hereafter, unless otherwise specified.
In this chapter we explore the interplay between the Hippo signaling pathway and RUNX2 (oncogenic) and RUNX3 (tumor suppressive) proteins in carcinogenesis. We cover both the transcriptional and non-transcriptional interactions between RUNX and TAZ/YAP -TEAD , and in particular we focus on their contribution to breast and gastric cancers (Brusgard et al. 2015). The findings highlighted here may lead to the development of useful paradigms that integrate novel data with our current understanding of RUNX-Hippo crosstalk to better understand mechanisms driving cancer progression.
2 Basic Principles
2.1 Oncogenic and Tumor Suppressor Functions of the RUNX Genes
In addition to their ‘classic’ role as transcriptional regulators during development and tumorigenesis, transcription-independent functions for RUNX proteins have been identified. Indeed RUNX has been implicated in the regulation of numerous physiological processes including DNA damage and cellular stress response, mitosis, autophagy, stem cell differentiation, and chromatin remodeling (Ito et al. 2015). Several studies have identified diverse roles for RUNX proteins via their interaction with numerous oncogenic and tumor suppressor mediators such as TGFβ , p53, Wnt and YAP /TAZ . One unifying principle in RUNX crosstalk with other cellular components is the ability of RUNX proteins to antagonize or enhance tumor suppressor or oncogenic functions. Recent evidence suggests RUNX proteins compete with each other to direct specific and opposing functions in part because they share identical DNA-recognition domains on target gene promoters (Chuang et al. 2013). For example, interaction of tumor suppressors RUNX1 or RUNX3 with p53 up-regulates BAX and PUMA to drive apoptosis following DNA damage (Ozaki et al. 2013a). Conversely, RUNX2 acts as a negative regulator of p53-dependent apoptosis via formation of a RUNX2/HDAC6/p53 transcriptional complex that represses BAX and PUMA (Ozaki et al. 2013b) (for detailed depiction of RUNX-p53 interaction refer to Fig. 26.1).

Disparate regulatory functions of RUNX family members on p53 in the DNA damage response . RUNX1 and RUNX3 act as positive regulators of p53 in response to DNA damage. In contrast, the DNA damage-induced proapoptotic activity of p53 is inhibited by RUNX2 (Refer to Refs. (Ozaki et al. 2013a, b) for more details)
The oncogenic properties of RUNX2 are well established. In addition to its anti-apoptotic interaction with p53, RUNX2 attenuates the pro-apoptotic signaling of TAp73 to confer drug resistance (Ozaki et al. 2015), and negatively regulates the long non-coding RNA, MT1DP, a known tumor suppressor (Yu et al. 2014b). RUNX2 is upregulated during epithelial-mesenchymal transition (EMT) in breast and prostate cancer (Chimge et al. 2011; Baniwal et al. 2010) and increased RUNX2 abundance correlates with poor prognosis in luminal and triple-negative subtypes of breast cancer (Mcdonald et al. 2014; Brusgard et al. 2015). In animal models, RUNX2 mediates breast cancer metastasis (Barnes et al. 2004; Javed et al. 2005; Pratap et al. 2011) and was shown to promote drug resistance and escape from apoptosis (Ozaki et al. 2013b). Furthermore, RUNX2 negatively regulates mitochondrial SIRT6 and pyruvate dehydrogenase (PDH) and increases breast cancer cell glucose metabolism, which is a hallmark of cancer (Choe et al. 2015). Intriguingly, tumor suppressive functions for RUNX2 have also been described. RUNX2 promotes mammary epithelial cell differentiation (Inman and Shore 2003) and in a subset of breast cancers RUNX2 antagonizes estrogen receptor growth-stimulation (Chimge et al. 2012; Chimge and Frenkel 2013). Increased RUNX2 expression may also promote a more differentiated phenotype in osteosarcomas, providing the “brakes” against further tumor progression (Pratap et al. 2003).
2.2 The Hippo Signaling Pathway in Cancer
Increased activity of the Hippo pathway effectors YAP and TAZ has been reported in the majority of solid cancer types (Liu et al. 2012; Harvey et al. 2013; Plouffe et al. 2015; Zanconato et al. 2016). Studies in mice revealed that knockout of the upstream regulator Neurofibromin 2 (NF2), as well as LATS, MST, SAV1, and MOB1 frequently leads to cancer development (reviewed in Harvey et al. 2013). Common mechanisms of pathway dysregulation in humans include gene amplification of YAP/TAZ and epigenetic silencing of Hippo components, particularly by promoter hypermethylation. Interestingly, with the exception of NF2, somatic mutations within Hippo components are relatively rare. However numerous regulators of the core components of the Hippo pathway (MATS, SAV1, LATS and MOB1) have been identified that contribute to tumorigenesis (Liu et al. 2012). For example, hypermethylation (inhibition) of RASSF1A, a positive regulator of MST1/2, is commonly observed in breast cancer (Mehrotra et al. 2004) and may be responsible for inhibition of the Hippo pathway. Furthermore, reduced E-Cadherin expression downregulates Hippo pathway signaling and hence increases nuclear translocation and activity of TAZ/YAP (Kim et al. 2011; Harvey et al. 2013).
Increased YAP and TAZ abundance and nuclear localization is frequently observed in breast cancer (Plouffe et al. 2015). Overexpression of YAP in breast cancer cell lines promotes tumor formation in mouse xenograft models, which can be blocked by YAP knockdown (Wang et al. 2012; Chen et al. 2014). Likewise, increased TAZ abundance promotes cell transformation and EMT and correlates with a more invasive breast cancer phenotype (Lei et al. 2008; Chan et al. 2008). Mechanistically, LIFR (Leukemia inhibitory factor receptor), a suppressor of metastasis that is frequently lost in breast cancer, inactivates YAP via regulation of Hippo pathway signaling (Chen et al. 2012). Similar to RUNX2, YAP has also been reported to exhibit tumor suppressive functions. YAP knockdown in breast cancer cells increased tumor cell invasion and growth in nude mice (Yuan et al. 2008). Notably, hyperactivation of YAP alone is insufficient to give rise to tumors in normal mammary epithelial cells (Chen et al. 2014). From this study the authors hypothesize that other genetic disruptions are required to promote YAP-induced oncogenesis. Dysregulation of YAP activity was also reported to produce dysplasia (YAP overexpression) and hyperplasia (SAV1 conditional knockout) of the gastrointestinal epithelium (Harvey et al. 2013). Importantly, inactivation of the Hippo pathway does not induce gastric carcinoma, though the pathway is reported to promote development of pancreatic and colorectal cancers (Plouffe et al. 2015).
3 RUNX2 and TAZ as Oncogenes in Breast Cancer
3.1 Breast Cancer Subtypes
Breast cancer is the second leading cause of cancer-related death among women (Siegel et al. 2013). However breast cancer is a heterogeneous disease that varies significantly in terms of pathological features, metastatic potential, and response to treatment regimens (Eroles et al. 2012; Cadoo et al. 2013). Breast cancer can be divided into four broad subtypes based on their molecular signatures, namely luminal A, luminal B, triple negative basal-like, and HER2-type. Luminal subtypes are more common and generally have a better prognostic outcome compared to basal-like tumors, which tend to be more aggressive. As the name would suggest, HER2-type tumors are typically HER2 receptor-positive and thus can be treated with HER2-targeting drugs such as Herceptin or lapatinib.
3.2 RUNX2 and TAZ Expression in Breast Cancer
RUNX2 is normally expressed in developing breast epithelial cells and in the mammary stem cell population where it promotes terminal end bud differentiation (Ferrari et al. 2013; Mcdonald et al. 2014). In breast cancer cell lines however, RUNX2 promotes an osteomimetic phenotype and metastasis to bone through transcriptional activation of osteopontin, matrix metalloproteinases (MMPs), and VEGF (Barnes et al. 2004; Pratap et al. 2005, 2006). This is important since luminal breast cancers relapse predominantly to the bone microenvironment (Eroles et al. 2012; Foley et al. 2010) and account for 50 % of all metastasis-related breast cancer deaths (Ganapathy et al. 2012). As introduced above, overexpression of TAZ is observed in breast cancer patient samples (Chan et al. 2008) and cell lines (Hiemer et al. 2014), correlating with increased cell migration, tumorigenesis, invasiveness, and drug resistance (Lei et al. 2008). Notably, RUNX2 can bind YAP (Yagi et al. 1999) and TAZ (Cui et al. 2003) via interaction of the PPxY motif within its C-terminal transactivation domain with the WW domain/s of YAP/TAZ. Cooperation between RUNX and YAP /TAZ has been shown to promote cell transformation (Vitolo et al. 2007), osteoblast differentiation (Cui et al. 2003) and stem cell renewal (Varelas et al. 2008; Cordenonsi et al. 2011).
3.3 sE-Cad-Mediated EMT
EMT is typically characterized by downregulation of E-Cadherin and upregulation of vimentin (Lee et al. 2006; Thiery et al. 2009; Valastyan and Weinberg 2011). Whilst this ‘classical’ EMT is usually required for cancer progression, cells may also metastasize from the primary tumor via an alternate mechanism involving proteolytic cleavage of E-Cadherin (120 kDa) to release the soluble, N-terminal ectodomain (sE-Cad; 80 kDa) (David and Rajasekaran 2012). MMP2 and -9 and ADAM (A Disintegrin and Metalloproteinase) -15 mediate cleavage of E-Cadherin to sE-Cad (David and Rajasekaran 2012; Najy et al. 2008; Davies et al. 2001; Huguenin et al. 2008; Noe et al. 2001; Symowicz et al. 2007; Zuo et al. 2011). sE-Cad exhibits autocrine and/or paracrine activity by binding HER2 (David and Rajasekaran 2012; Inge et al. 2011; Najy et al. 2008; Brouxhon et al. 2013, 2014) and interacts with full length E-Cadherin to destabilize adherens junctions (David and Rajasekaran 2012). The effect of sE-Cad signaling is promotion of migration, invasion, and proliferation while maintaining an epithelial morphology (David and Rajasekaran 2012; Grabowska and Day 2012; Chunthapong et al. 2004; Inge et al. 2011; Kuefer et al. 2003; Najy et al. 2008). Hence sE-Cad is a useful functional metastatic biomarker for numerous cancers , including breast cancer (David and Rajasekaran 2012; Chunthapong et al. 2004; Kuefer et al. 2003; Hofmann et al. 2013; Kuefer et al. 2005).
3.4 RUNX2 Cooperates with TAZ to Promote sE-Cad-Mediated EMT
Recently, our group discovered that cooperation between RUNX2 and TAZ increases shedding of sE-Cad to promote a tumorigenic phenotype characterized by anchorage-independent growth (tumorsphere formation) in breast cancer cells (Brusgard et al. 2015). RUNX2 promotes nuclear localization of TAZ, which is a driver for tumorigenesis since TAZ knockdown reduces tumorsphere growth. Intriguingly, given that TAZ and YAP are similarly regulated, expression and localization of YAP was not affected by RUNX2 expression in these cells (Brusgard et al. 2015). MMP expression (including MMP2, which can cleave E-Cadherin) was significantly elevated in RUNX overexpressing breast cancer cells and could be inhibited by MMP inhibitors. Treatment with E-Cadherin neutralizing antibody reduced the level of sE-Cad and inhibited tumorsphere formation. Binding of sE-Cad to HER2, which is expressed in a subset of luminal breast cancers (Ithimakin et al. 2013), promotes tumorgenicity (Brouxhon et al. 2013). Treatment of RUNX2 overexpressing cells with the HER2-targeting drugs Herceptin or lapatinib inhibited tumorsphere proliferation (Brusgard et al. 2015). Taken together, these results suggest that RUNX2 and TAZ cooperate to upregulate MMP expression in breast cancer and promote an sE-Cad/HER2-mediated EMT. Our working model is therefore as such: E-Cadherin, via its influence on Hippo pathway activity (Kim et al. 2011), maintains TAZ in a cytoplasmic (inactive), tumor suppressive state. RUNX2-induced cleavage of E-Cadherin to sE-Cad inactivates the Hippo pathway, resulting in nuclear localisation of TAZ and oncogenic transformation (Fig. 26.2).

RUNX2 manifests its oncogenic activity through upregulation of a soluble form of E-Cadherin (sE-Cad) that inactivates the Hippo tumor suppressor pathway. Conversely, full-length membrane bound E-Cadherin positively regulates the Hippo signaling pathway to keep RUNX2 oncogenic function in check
3.5 Outstanding Questions and Future Directions
Correlation between RUNX2 signaling and increased TAZ nuclear localization in breast cancer cells suggests that factors which inhibit RUNX2 may restore Hippo signaling and block breast cancer progression. Mechanistically, we hypothesize that RUNX2 oncogenic activity is mediated, at least in part, by increased production of sE-Cad. However, whether sE-Cad is oncogenic, independent of RUNX2 overexpression, remains to be determined.
Furthermore, whilst RUNX2-induced TAZ nuclear localization suggests attenuation of Hippo signaling, a role for the Hippo pathway kinases MST1/2 and LATS1/2 in mediating RUNX2 oncogenic function has not been reported. To address these outstanding questions, cells could be treated directly with recombinant sE-Cad in vitro and assess the effect on Hippo signaling and tumorigenic properties.
Preliminary unpublished data from our lab indicate that treatment of breast cancer cells with recombinant sE-Cad reduces the abundance of active phosphorylated (phospho-) LATS1/2. Moreover, treatment of cells with the RUNX2 small molecule inhibitor CADD522 increased phospho-LATS1/2 as well as the total level of LATS1 protein. This is consistent with our data showing significant reduction of TAZ abundance in the nucleus upon RUNX inhibition (Brusgard et al. 2015). Though these data support a role for RUNX2 in controlling Hippo pathway activity, this does not explain why YAP is not similarly regulated by RUNX2 overexpression in these cells. This is a curious observation that should be addressed in subsequent studies. Furthermore, data from the recombinant sE-Cad experiments would suggest that TAZ activation (nuclear localization) lies downstream of RUNX2 overexpression, MMP production and increased sE-Cad shedding. Therefore the mechanism linking TAZ activation and tumorigenic transformation of breast cancer cells should be determined.
Identification of TAZ-specific oncogenic target genes may reveal novel cancer biomarkers and therapeutic targets. Since RUNX2 inactivates several tumor suppressor pathways including p53, E-Cadherin, and SIRT6/PDH metabolic regulators (Choe et al. 2015), in addition to the Hippo pathway as discussed in detail here, RUNX2 inhibition could prove very effective as a novel cancer targeting strategy. Future effort should be employed to determine whether a combination of oncogene/tumor suppressor targeting and metabolic reprogramming strategies would be effective for other tumorigenic events where RUNX2 is a driving factor.
4 RUNX3 and TEAD-YAP Regulation in Gastric Cancer
4.1 RUNX3 Is a Tumor Suppressor in Gastric Cancer
Gastric cancer is the second leading cause of cancer-related mortality worldwide and is characterized by tumor heterogeneity driven by various signaling pathways (Shah and Ajani 2010). Consistent with gastric hyperplasia observed in Runx3 knockout mice (Ito et al. 2011), loss of RUNX3 expression, typically due to hemizygous deletion or promoter hypermethylation, is observed in 60 % of human gastric cancers. Furthermore, reduced RUNX3 is causally linked to the initiation and progression of gastric cancer (Li et al. 2002; Fan et al. 2011). In gastric epithelial cells, RUNX3 cooperates with Smad/TGFβ signaling to drive expression of p21 (CIP1) (Chi et al. 2005) and BIM (Yano et al. 2006) to inhibit cell division and promote apoptosis, respectively. In the colon, RUNX3 attenuates oncogenic WNT signaling via inhibitory binding to the TCF4-β-catenin complex (Ito et al. 2008), and in the mouse lung Runx3 inhibits cellular transformation via upregulation of p19Arf and p21 in response to oncogenic K-Ras signaling (Lee et al. 2013).
4.2 The TEAD-YAP Complex Is Oncogenic in Gastric Cancer
YAP, in association with TEAD (TEAD-YAP complex), promotes cell proliferation by upregulation of target genes including connective tissue growth factor (CTGF) and Cysteine-rich angiogenic inducer 61 (CYR61) (Lai et al. 2011). YAP (Zhang et al. 2012; Lam-Himlin et al. 2006) and TEAD4 (Lim et al. 2013) have both been reported to be upregulated in gastric cancer patient samples, and expression of YAP target genes positively correlates with gastric carcinoma progression (Jiang et al. 2011; Lin et al. 2005), and patient outcome (Qiao et al. 2015). In gastric cancer cells , overexpression of a TEAD-YAP fusion protein increases anchorage-independent growth (Qiao et al. 2015), whilst YAP knockdown inhibits proliferation and metastasis (Zhang et al. 2012) and in some instances induces apoptosis (Zhou et al. 2011). Use of a YAP antagonist (Super-TDU: an inhibitor peptide mimicking the TDU region of VGLL4 that blocks YAP-TEAD binding) suppresses gastric cancer cell growth in vitro and is proposed as a therapeutic strategy to treat gastric cancer (Jiao et al. 2014).
Notably, crosstalk between RUNX and the Hippo signaling pathway, independent of direct interaction with YAP/TAZ, has been reported. Facilitated by MST2, RUNX3 and SAV1 form a complex to promote Hippo pathway-mediated cell death (Min et al. 2012). Consistent with this, expression of RUNX1 and RUNX3 inversely correlate with YAP abundance in cultured gastric cancer cells and patient samples (Qiao et al. 2015). Thus RUNX3 is a negative regulator of YAP activity and ‘low RUNX/high YAP’ expression might be a useful marker of gastric cancer progression.
4.3 RUNX3, TEAD and YAP Form a Ternary Complex
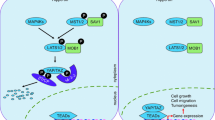
Recently, we showed that RUNX3 is a novel regulator of the TEAD-YAP complex in gastric carcinogenesis whereby RUNX3 physically interacts with TEAD, reducing its DNA-binding ability and effectively inhibiting downstream YAP signaling (Qiao et al. 2015). Mapping of the TEAD-RUNX3 interaction revealed that the Runt (DNA-binding) domain (RD) of RUNX3 is essential. We also discovered that the TEAD-RUNX3 interface overlaps with the TEAD DNA-recognition helix (Qiao et al. 2015). Even though RUNX family members share a high degree of sequence identity in their Runt domains, interaction between TEAD and RUNX2 was significantly weaker than that of RUNX1 and RUNX3, despite strong interaction between RUNX2 and YAP or TAZ (Qiao et al. 2015). Further experiments revealed that RUNX3, TEAD and YAP form a ternary complex, in which distinct domains mediate direct interaction of RUNX3 and YAP with TEAD (Qiao et al. 2015) (Fig. 26.3).

RUNX3, TEAD and YAP form a tripartite protein complex
RUNX3 binds YAP via interaction of its carboxy terminal (C) PPxY motif with the WW domain(s) of YAP. YAP’s amino terminal (N) TEAD-binding domain contacts the C-terminal region of TEAD. The Runt DNA-binding domain of RUNX3 completes the complex, associating with the N-terminus of TEAD, overlapping with TEAD’s DNA-recognition helix
4.4 RUNX3 Binding Abolishes TEAD-YAP Oncogenic Activity in Gastric Cancer
The overlap of TEAD’s DNA-recognition helix and RUNX binding interface led us to hypothesize that RUNX interaction would significantly reduce the DNA-binding ability of TEAD (see Fig. 26.4). Accordingly, RUNX3 overexpression in gastric cancer tissues correlates with downregulation of TEAD-YAP targets including CTGF , CYR61, and GLI2 (Qiao et al. 2015). Further analysis in gastric cancer cell lines revealed that RUNX3 abundance negatively correlates with YAP-induced CTGF expression, and this is associated with decreased binding of TEAD to CTGF promoters (Qiao et al. 2015). Screening of several RUNX3 mutants that are frequently observed in gastric cancer revealed that mutation of Leucine121 to Histidine (L121H) abolished its interaction with TEAD and significantly reduced the expression of CTGF (Qiao et al. 2015). In terms of biological significance, increased expression of wild-type RUNX3 (but not the L121H mutant) reduced anchorage-independent growth of gastric cancer cells in vitro, and tumor growth using nude mouse assay in vivo (Qiao et al. 2015). Moreover, overexpression of RUNX3 could block TEAD-YAP-induction of colony formation, providing evidence of a direct tumor suppressor role for RUNX3 in gastric carcinoma.

RUNX3 inhibits TEAD /YAP-mediated gene transcription
Under conditions of high RUNX3 (left panel) DNA-binding ability of TEAD is inhibited and transcription of TEAD/YAP target genes (e.g., CTGF and CYR61) is attenuated. When RUNX3 is inactivated or expressed at relatively low levels (right panel) TEAD/YAP drives transcription of oncogenic target genes
4.5 Future Perspectives and Potential for Therapeutic Application
Our recent report elucidates a novel mechanism of RUNX3 tumor suppressor activity in gastric cancer that has great potential for application in a range of human cancers driven by aberrant TEAD-YAP activity. Given the dual role of RUNX3 in regulating YAP activity via complex formation with SAV1/MST2 (Min et al. 2012) and TEAD-YAP (Qiao et al. 2015) it would be interesting to ascertain the relative contribution of these two mechanisms to RUNX3 anti-oncogenic activity in gastric cancer.
Precise control of RUNX3 and YAP expression is important during embryonic development and differentiation of the gastrointestinal tract; aberrant expression of RUNX3 (knockout) (Ito et al. 2011) or YAP (activation) (Camargo et al. 2007) promotes gastrointestinal dysplasia. The potent inhibition of TEAD-YAP by RUNX3 raises interesting questions regarding their roles and possible interaction during development. Preliminary data suggest that transcriptional activity of RUNX3 inversely correlates with TEAD4 expression (unpublished data). This mutual regulation between RUNX3 and TEAD4 might be a way to fine-tune the balance of proliferation and differentiation both during development and tumorigenesis.
Interestingly, only 35 % of TEAD-targeted genes were suppressed by RUNX3 overexpression in gastric cancer cells (Qiao et al. 2015). This suggests that inhibition of these targets might be due to adjacent RUNX3 and TEAD binding sites in their promoters, even though DNA binding does not seem to be required for RUNX3-TEAD interaction in our assays (Qiao et al. 2015). It is possible however that cognate DNA stabilizes and enhances the multicomponent RUNX3-TEAD-YAP complex, priming it for biological activity. A genome wide ChIP-seq experiment analyzing all TEAD-binding sites for proximity to RUNX motifs in gastric tissues could test this hypothesis.
In support of this, members of the Piccolo laboratory recently conducted a comprehensive ChIP-seq analysis to identify DNA-binding platforms for YAP and TAZ in breast cancer cells (Zanconato et al. 2015). Unsurprisingly, TEAD binding motifs were present in the majority of YAP/TAZ peaks. Encouragingly however, of the various DNA-binding factors proposed to cooperate with YAP/TAZ , RUNX-binding sites were the only other prominent motif identified. Moreover, for some YAP/TAZ target genes, there was a physical proximity of TEAD and RUNX binding sites in the cells analyzed (Zanconato et al. 2015).
Since RUNX3 is such a potent inhibitor of YAP in gastric cancer , these findings could lead to the development of novel RUNX3 mimicking compounds to target TEAD-YAP activity in vivo. Support for this proposal comes from studies demonstrating the efficacy of using YAP-TEAD inhibitors such as verteporfin (Liu-Chittenden et al. 2012; Yu et al. 2014a) and Super-TDU (Jiao et al. 2014) to suppress the oncogenic activity of YAP. Moreover, a publication showing forced overexpression of YAP in hematopoietic stem cells , in which RUNX1 and RUNX3 were highly expressed, did not lead to malignant cell growth (Jansson and Larsson 2012).
5 Concluding Remarks
The Hippo signaling pathway and its role in controlling the mammalian effector proteins YAP and TAZ was elucidated nearly ten years ago, yet we are still discovering novel regulators of this important signaling pathway. Recent findings from our laboratories linking RUNX2/TAZ with sE-Cad expression, and RUNX3 with TEAD -YAP in different models of cancer highlight the potential for development of effective targeting strategies for Hippo pathway dysregulation in various human pathologies. That being said, questions still remain regarding the mechanisms of YAP/TAZ regulation by the RUNX protein family.
Notably, apparent differences exist between the regulation of TAZ and YAP by RUNX2 and sE-Cad signaling in breast cancer. The Hippo pathway similarly regulates YAP and TAZ in terms of phosphorylation and nuclear localization (Hao et al. 2008; Kanai et al. 2000). Recently, we reported that YAP is a negative regulator of TAZ protein abundance in mammalian cells (Finch-Edmondson et al. 2015). This is relevant since it demonstrates that YAP and TAZ are subjected to discrete forms of regulation. Whether this direct relationship between YAP and TAZ abundance has implications for RUNX-mediated YAP/TAZ regulation remains to be determined.
Multiple isoforms of YAP harboring single (YAP1-1) or tandem (YAP1-2) WW domains are expressed in mammals (Gaffney et al. 2012). Because RUNX bind to YAP/TAZ via this key protein interaction domain, differences in the binding efficiency of RUNX to YAP1-1 or YAP1-2 isoforms may influence the signaling outcome. Especially in gastric cancer , where RUNX acts to inhibit TEAD -YAP activity, YAP isoforms that exhibit weaker binding to RUNX have the potential to be more oncogenic. Elucidation of the protein “interactome” of individual YAP isoforms may reveal striking differences in RUNX3 binding. Furthermore, since TAZ has only one WW domain, whether the number of WW domains influences RUNX interaction would be interesting to assess.
Finally, the development of CRISPR/Cas9 technology for efficient gene editing in vitro and in vivo has provided great opportunity for analyzing the effect of point mutations on protein-protein interactions. By taking advantage of clinical data signposting common mutants detected in cancer (e.g., RUNX3 mutant L121H) we can measure their effect using a biologically, and translationally relevant approach. This will enable us to better understand how mutations in critical proteins can drive cancer formation and progression, and may even pave the way for genetic engineering to combat cancer in humans.
References
Baniwal, S. K., Khalid, O., Gabet, Y., Shah, R. R., Purcell, D. J., Mav, D., et al. (2010). Runx2 transcriptome of prostate cancer cells: Insights into invasiveness and bone metastasis. Molecular Cancer, 9, 258.
Barnes, G. L., Hebert, K. E., Kamal, M., Javed, A., Einhorn, T. A., Lian, J. B., et al. (2004). Fidelity of Runx2 activity in breast cancer cells is required for the generation of metastases-associated osteolytic disease. Cancer Research, 64, 4506–4513.
Brouxhon, S. M., Kyrkanides, S., Teng, X., Raja, V., O'banion, M. K., Clarke, R., et al. (2013). Monoclonal antibody against the ectodomain of E-cadherin (DECMA-1) suppresses breast carcinogenesis: Involvement of the HER/PI3K/Akt/mTOR and IAP pathways. Clinical Cancer Research, 19, 3234–3246.
Brouxhon, S. M., Kyrkanides, S., Teng, X., O'banion, M. K., Clarke, R., Byers, S., & Ma, L. (2014). Soluble-E-cadherin activates HER and IAP family members in HER2+ and TNBC human breast cancers. Molecular Carcinogenesis, 53, 893–906.
Brusgard, J. L., Choe, M., Chumsri, S., Renoud, K., Mackerell Jr., A. D., Sudol, M., & Passaniti, A. (2015). RUNX2 and TAZ-dependent signaling pathways regulate soluble E-Cadherin levels and tumorsphere formation in breast cancer cells. Oncotarget, 6, 28132–28150.
Cadoo, K. A., Fornier, M. N., & Morris, P. G. (2013). Biological subtypes of breast cancer: Current concepts and implications for recurrence patterns. The Quarterly Journal of Nuclear Medicine and Molecular Imaging, 57, 312–321.
Camargo, F. D., Gokhale, S., Johnnidis, J. B., Fu, D., Bell, G. W., Jaenisch, R., & Brummelkamp, T. R. (2007). YAP1 increases organ size and expands undifferentiated progenitor cells. Current Biology, 17, 2054–2060.
Chan, S. W., Lim, C. J., Guo, K., Ng, C. P., Lee, I., Hunziker, W., et al. (2008). A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Research, 68, 2592–2598.
Chen, D., Sun, Y., Wei, Y., Zhang, P., Rezaeian, A. H., Teruya-Feldstein, J., et al. (2012). LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nature Medicine, 18, 1511–1517.
Chen, Q., Zhang, N., GRAY, R. S., Li, H., Ewald, A. J., Zahnow, C. A., & Pan, D. (2014). A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes & Development, 28, 432–437.
Chi, X. Z., Yang, J. O., Lee, K. Y., Ito, K., Sakakura, C., Li, Q. L., et al. (2005). RUNX3 suppresses gastric epithelial cell growth by inducing p21(WAF1/Cip1) expression in cooperation with transforming growth factor β-activated SMAD. Molecular and Cellular Biology, 25, 8097–8107.
Chimge, N. O., & Frenkel, B. (2013). The RUNX family in breast cancer: Relationships with estrogen signaling. Oncogene, 32, 2121–2130.
Chimge, N. O., Baniwal, S. K., Little, G. H., Chen, Y. B., Kahn, M., Tripathy, D., et al. (2011). Regulation of breast cancer metastasis by Runx2 and estrogen signaling: The role of SNAI2. Breast Cancer Research, 13, R127.
Chimge, N. O., Baniwal, S. K., Luo, J., Coetzee, S., Khalid, O., Berman, B. P., et al. (2012). Opposing effects of Runx2 and estradiol on breast cancer cell proliferation: In vitro identification of reciprocally regulated gene signature related to clinical letrozole responsiveness. Clinical Cancer Research, 18, 901–911.
Choe, M., Brusgard, J. L., Chumsri, S., Bhandary, L., Zhao, X. F., Lu, S., et al. (2015). The RUNX2 transcription factor negatively regulates SIRT6 expression to alter glucose metabolism in breast cancer cells. Journal of Cellular Biochemistry, 116, 2210–2226.
Chuang, L. S., Ito, K., & Ito, Y. (2013). RUNX family: Regulation and diversification of roles through interacting proteins. International Journal of Cancer, 132, 1260–1271.
Chunthapong, J., Seftor, E. A., Khalkhali-Ellis, Z., Seftor, R. E., Amir, S., Lubaroff, D. M., et al. (2004). Dual roles of E-cadherin in prostate cancer invasion. Journal of Cellular Biochemistry, 91, 649–661.
Cordenonsi, M., Zanconato, F., Azzolin, L., Forcato, M., Rosato, A., Frasson, C., et al. (2011). The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell, 147, 759–772.
Cui, C. B., Cooper, L. F., Yang, X., Karsenty, G., & Aukhil, I. (2003). Transcriptional coactivation of bone-specific transcription factor Cbfa1 by TAZ. Molecular and Cellular Biology, 23, 1004–1013.
David, J. M., & Rajasekaran, A. K. (2012). Dishonorable discharge: The oncogenic roles of cleaved E-cadherin fragments. Cancer Research, 72, 2917–2923.
Davies, G., Jiang, W. G., & Mason, M. D. (2001). Matrilysin mediates extracellular cleavage of E-cadherin from prostate cancer cells: A key mechanism in hepatocyte growth factor/scatter factor-induced cell-cell dissociation and in vitro invasion. Clinical Cancer Research, 7, 3289–3297.
Eroles, P., Bosch, A., Perez-Fidalgo, J. A., & Lluch, A. (2012). Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treatment Reviews, 38, 698–707.
Fan, X. Y., Hu, X. L., Han, T. M., Wang, N. N., Zhu, Y. M., Hu, W., et al. (2011). Association between RUNX3 promoter methylation and gastric cancer: A meta-analysis. BMC Gastroenterology, 11, 92.
Ferrari, N., Mcdonald, L., Morris, J. S., Cameron, E. R., & Blyth, K. (2013). RUNX2 in mammary gland development and breast cancer. Journal of Cellular Physiology, 228, 1137–1142.
Finch-Edmondson, M. L., Strauss, R. P., Passman, A., Sudol, M., Yeoh, G. C., & Callus, B. A. (2015). TAZ protein accumulation is negatively regulated by YAP abundance in mammalian cells. The Journal of Biological Chemistry, 290(46), 27928–27938.
Foley, J., Nickerson, N. K., Nam, S., Allen, K. T., Gilmore, J. L., Nephew, K. P., et al. (2010). EGFR signaling in breast cancer: Bad to the bone. Seminars in Cell & Developmental Biology, 21, 951–960.
Gaffney, C. J., Oka, T., Mazack, V., Hilman, D., Gat, U., Muramatsu, T., et al. (2012). Identification, basic characterization and evolutionary analysis of differentially spliced mRNA isoforms of human YAP1 gene. Gene, 509, 215–222.
Ganapathy, V., Banach-Petrosky, W., Xie, W., Kareddula, A., Nienhuis, H., Miles, G., & Reiss, M. (2012). Luminal breast cancer metastasis is dependent on estrogen signaling. Clinical & Experimental Metastasis, 29, 493–509.
Grabowska, M. M., & Day, M. L. (2012). Soluble E-cadherin: More than a symptom of disease. Frontiers in Bioscience (Landmark Edition), 17, 1948–1964.
Hao, Y., Chun, A., Cheung, K., Rashidi, B., & Yang, X. (2008). Tumor suppressor LATS1 is a negative regulator of oncogene YAP. The Journal of Biological Chemistry, 283, 5496–5509.
Harvey, K. F., Pfleger, C. M., & Hariharan, I. K. (2003). The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell, 114, 457–467.
Harvey, K. F., Zhang, X., & Thomas, D. M. (2013). The Hippo pathway and human cancer. Nature Reviews. Cancer, 13, 246–257.
Hiemer, S. E., Szymaniak, A. D., & Varelas, X. (2014). The transcriptional regulators TAZ and YAP direct transforming growth factor β-induced tumorigenic phenotypes in breast cancer cells. The Journal of Biological Chemistry, 289, 13461–13474.
Hofmann, G., Balic, M., Dandachi, N., Resel, M., Schippinger, W., Regitnig, P., et al. (2013). The predictive value of serum soluble E-cadherin levels in breast cancer patients undergoing preoperative systemic chemotherapy. Clinical Biochemistry, 46, 1585–1589.
Huang, J., Wu, S., Barrera, J., Matthews, K., & Pan, D. (2005). The hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating yorkie, the Drosophila homolog of YAP. Cell, 122, 421–434.
Huguenin, M., Muller, E. J., Trachsel-Rosmann, S., Oneda, B., Ambort, D., Sterchi, E. E., & Lottaz, D. (2008). The metalloprotease meprinβ processes E-cadherin and weakens intercellular adhesion. PloS One, 3, e2153.
Inge, L. J., Barwe, S. P., D'ambrosio, J., Gopal, J., Lu, K., Ryazantsev, S., et al. (2011). Soluble E-cadherin promotes cell survival by activating epidermal growth factor receptor. Experimental Cell Research, 317, 838–848.
Inman, C. K., & Shore, P. (2003). The osteoblast transcription factor Runx2 is expressed in mammary epithelial cells and mediates osteopontin expression. The Journal of Biological Chemistry, 278, 48684–48689.
Ithimakin, S., Day, K. C., Malik, F., Zen, Q., Dawsey, S. J., Bersano-Begey, T. F., et al. (2013). HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Research, 73, 1635–1646.
Ito, K., Lim, A. C., Salto-Tellez, M., Motoda, L., Osato, M., Chuang, L. S., et al. (2008). RUNX3 attenuates β-catenin/T cell factors in intestinal tumorigenesis. Cancer Cell, 14, 226–237.
Ito, K., Chuang, L. S., Ito, T., Chang, T. L., Fukamachi, H., Salto-Tellez, M., & Ito, Y. (2011). Loss of Runx3 is a key event in inducing precancerous state of the stomach. Gastroenterology, 140, 1536–1546.
Ito, Y., Bae, S. C., & Chuang, L. S. (2015). The RUNX family: Developmental regulators in cancer. Nature Reviews. Cancer, 15, 81–95.
Jansson, L., & Larsson, J. (2012). Normal hematopoietic stem cell function in mice with enforced expression of the Hippo signaling effector YAP1. PloS One, 7, e32013.
Javed, A., Barnes, G. L., Pratap, J., Antkowiak, T., Gerstenfeld, L. C., Van Wijnen, A. J., et al. (2005). Impaired intranuclear trafficking of Runx2 (AML3/CBFA1) transcription factors in breast cancer cells inhibits osteolysis in vivo. Proceedings of the National Academy of Sciences of the United States of America, 102, 1454–1459.
Jiang, C. G., Lv, L., Liu, F. R., Wang, Z. N., Liu, F. N., Li, Y. S., et al. (2011). Downregulation of connective tissue growth factor inhibits the growth and invasion of gastric cancer cells and attenuates peritoneal dissemination. Molecular Cancer, 10, 122.
Jiao, S., Wang, H., Shi, Z., Dong, A., Zhang, W., Song, X., et al. (2014). A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell, 25, 166–180.
Justice, R. W., Zilian, O., Woods, D. F., Noll, M., & Bryant, P. J. (1995). The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes & Development, 9, 534–546.
Kanai, F., Marignani, P. A., Sarbassova, D., Yagi, R., Hall, R. A., Donowitz, M., et al. (2000). TAZ: A novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. The EMBO Journal, 19, 6778–6791.
Kim, N. G., Koh, E., Chen, X., & Gumbiner, B. M. (2011). E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proceedings of the National Academy of Sciences of the United States of America, 108, 11930–11935.
Kuefer, R., Hofer, M. D., Gschwend, J. E., Pienta, K. J., Sanda, M. G., Chinnaiyan, A. M., et al. (2003). The role of an 80 kDa fragment of E-cadherin in the metastatic progression of prostate cancer. Clinical Cancer Research, 9, 6447–6452.
Kuefer, R., Hofer, M. D., Zorn, C. S., Engel, O., Volkmer, B. G., Juarez-Brito, M. A., et al. (2005). Assessment of a fragment of e-cadherin as a serum biomarker with predictive value for prostate cancer. British Journal of Cancer, 92, 2018–2023.
Lai, D., Ho, K. C., Hao, Y., & Yang, X. (2011). Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Research, 71, 2728–2738.
Lam-Himlin, D. M., Daniels, J. A., Gayyed, M. F., Dong, J., Maitra, A., Pan, D., et al. (2006). The hippo pathway in human upper gastrointestinal dysplasia and carcinoma: A novel oncogenic pathway. International Journal of Gastrointestinal Cancer, 37, 103–109.
Lee, J. M., Dedhar, S., Kalluri, R., & Thompson, E. W. (2006). The epithelial-mesenchymal transition: New insights in signaling, development, and disease. The Journal of Cell Biology, 172, 973–981.
Lee, Y. S., Lee, J. W., Jang, J. W., Chi, X. Z., Kim, J. H., Li, Y. H., et al. (2013). Runx3 inactivation is a crucial early event in the development of lung adenocarcinoma. Cancer Cell, 24, 603–616.
Lei, Q. Y., Zhang, H., Zhao, B., Zha, Z. Y., Bai, F., Pei, X. H., et al. (2008). TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Molecular and Cellular Biology, 28, 2426–2436.
Li, Q. L., Ito, K., Sakakura, C., Fukamachi, H., Inoue, K., Chi, X. Z., et al. (2002). Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell, 109, 113–124.
Lim, B., Park, J. L., Kim, H. J., Park, Y. K., Kim, J. H., Sohn, H. A., et al. (2013). Integrative genomics analysis reveals the multilevel dysregulation and oncogenic characteristics of TEAD4 in gastric cancer. Carcinogenesis, 35, 1020–1027.
Lin, M. T., Zuon, C. Y., Chang, C. C., Chen, S. T., Chen, C. P., Lin, B. R., et al. (2005). Cyr61 induces gastric cancer cell motility/invasion via activation of the integrin/nuclear factor-kappaB/cyclooxygenase-2 signaling pathway. Clinical Cancer Research, 11, 5809–5820.
Liu, A. M., Wong, K. F., Jiang, X., Qiao, Y., & Luk, J. M. (2012). Regulators of mammalian Hippo pathway in cancer. Biochimica et Biophysica Acta, 1826, 357–364.
Liu-Chittenden, Y., Huang, B., Shim, J. S., Chen, Q., Lee, S. J., Anders, R. A., et al. (2012). Genetic and pharmacological disruption of the TEAD–YAP complex suppresses the oncogenic activity of YAP. Genes & Development, 26, 1300–1305.
Mcdonald, L., Ferrari, N., Terry, A., BELL, M., Mohammed, Z. M., Orange, C., et al. (2014). RUNX2 correlates with subtype-specific breast cancer in a human tissue microarray, and ectopic expression of Runx2 perturbs differentiation in the mouse mammary gland. Disease Models & Mechanisms, 7, 525–534.
Mehrotra, J., Vali, M., Mcveigh, M., Kominsky, S. L., Fackler, M. J., Lahti-Domenici, J., et al. (2004). Very high frequency of hypermethylated genes in breast cancer metastasis to the bone, brain, and lung. Clinical Cancer Research, 10, 3104–3109.
Min, B., Kim, M. K., Zhang, J. W., Kim, J., Chung, K. C., Oh, B. C., et al. (2012). Identification of RUNX3 as a component of the MST/Hpo signaling pathway. Journal of Cellular Physiology, 227, 839–849.
Najy, A. J., Day, K. C., & Day, M. L. (2008). The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. The Journal of Biological Chemistry, 283, 18393–18401.
Noe, V., Fingleton, B., Jacobs, K., Crawford, H. C., Vermeulen, S., Steelant, W., et al. (2001). Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. Journal of Cell Science, 114, 111–118.
Oh, H., & Irvine, K. D. (2008). In vivo regulation of Yorkie phosphorylation and localization. Development, 135, 1081–1088.
Ozaki, T., Nakagawara, A., & Nagase, H. (2013a). RUNX Family Participates in the Regulation of p53-Dependent DNA Damage Response. International Journal of Genomics, 2013, 271347.
Ozaki, T., Wu, D., Sugimoto, H., Nagase, H., & Nakagawara, A. (2013b). Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death & Disease, 4, e610.
Ozaki, T., Sugimoto, H., Nakamura, M., Hiraoka, K., Yoda, H., Sang, M., et al. (2015). Runt-related transcription factor 2 attenuates the transcriptional activity as well as DNA damage-mediated induction of pro-apoptotic TAp73 to regulate chemosensitivity. The FEBS Journal, 282, 114–128.
Pantalacci, S., Tapon, N., & Leopold, P. (2003). The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nature Cell Biology, 5, 921–927.
Plouffe, S. W., Hong, A. W., & Guan, K. L. (2015). Disease implications of the Hippo/YAP pathway. Trends in Molecular Medicine, 21, 212–222.
Pratap, J., Galindo, M., Zaidi, S. K., Vradii, D., Bhat, B. M., Robinson, J. A., et al. (2003). Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Research, 63, 5357–5362.
Pratap, J., Javed, A., Languino, L. R., Van Wijnen, A. J., Stein, J. L., Stein, G. S., & Lian, J. B. (2005). The Runx2 osteogenic transcription factor regulates matrix metalloproteinase 9 in bone metastatic cancer cells and controls cell invasion. Molecular and Cellular Biology, 25, 8581–8591.
Pratap, J., Lian, J. B., Javed, A., Barnes, G. L., Van Wijnen, A. J., Stein, J. L., & Stein, G. S. (2006). Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Reviews, 25, 589–600.
Pratap, J., Lian, J. B., & Stein, G. S. (2011). Metastatic bone disease: Role of transcription factors and future targets. Bone, 48, 30–36.
Qiao, Y., Lin, S. J., Chen, Y., Voon, D. C., Zhu, F., Chuang, L. S., et al. (2015). RUNX3 is a novel negative regulator of oncogenic TEAD-YAP complex in gastric cancer. Oncogene, 35(20), 2664–2674.
Shah, M. A., & Ajani, J. A. (2010). Gastric cancer--an enigmatic and heterogeneous disease. Journal of the American Medical Association, 303, 1753–1754.
Siegel, R., Naishadham, D., & Jemal, A. (2013). Cancer statistics, 2013. CA: A Cancer Journal for Clinicians, 63, 11–30.
Symowicz, J., Adley, B. P., Gleason, K. J., Johnson, J. J., Ghosh, S., Fishman, D. A., et al. (2007). Engagement of collagen-binding integrins promotes matrix metalloproteinase-9-dependent E-cadherin ectodomain shedding in ovarian carcinoma cells. Cancer Research, 67, 2030–2039.
Tapon, N., Harvey, K. F., Bell, D. W., Wahrer, D. C., Schiripo, T. A., Haber, D., & Hariharan, I. K. (2002). salvador Promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell, 110, 467–478.
Thiery, J. P., Acloque, H., Huang, R. Y., & Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell, 139, 871–890.
Valastyan, S., & Weinberg, R. A. (2011). Tumor metastasis: Molecular insights and evolving paradigms. Cell, 147, 275–292.
Varelas, X., Sakuma, R., Samavarchi-Tehrani, P., Peerani, R., Rao, B. M., Dembowy, J., et al. (2008). TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nature Cell Biology, 10, 837–848.
Vitolo, M. I., Anglin, I. E., Mahoney Jr., W. M., Renoud, K. J., Gartenhaus, R. B., Bachman, K. E., & Passaniti, A. (2007). The RUNX2 transcription factor cooperates with the YES-associated protein, YAP65, to promote cell transformation. Cancer Biology & Therapy, 6, 856–863.
Wang, X., Su, L., & Ou, Q. (2012). Yes-associated protein promotes tumour development in luminal epithelial derived breast cancer. European Journal of Cancer, 48, 1227–1234.
Wu, S., Huang, J., Dong, J., & Pan, D. (2003). hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell, 114, 445–456.
Yagi, R., Chen, L. F., Shigesada, K., Murakami, Y., & Ito, Y. (1999). A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. The EMBO Journal, 18, 2551–2562.
Yano, T., Ito, K., Fukamachi, H., Chi, X. Z., Wee, H. J., Inoue, K., et al. (2006). The RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells undergoing transforming growth factor beta-induced apoptosis. Molecular and Cellular Biology, 26, 4474–4488.
Yu, F. X., Luo, J., Mo, J. S., Liu, G., Kim, Y. C., Meng, Z., et al. (2014a). Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell, 25, 822–830.
Yu, W., Qiao, Y., Tang, X., Ma, L., Wang, Y., Zhang, X., et al. (2014b). Tumor suppressor long non-coding RNA, MT1DP is negatively regulated by YAP and Runx2 to inhibit FoxA1 in liver cancer cells. Cellular Signalling, 26, 2961–2968.
Yuan, M., Tomlinson, V., Lara, R., Holliday, D., Chelala, C., Harada, T., et al. (2008). Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death and Differentiation, 15, 1752–1759.
Zanconato, F., Forcato, M., Battilana, G., Azzolin, L., Quaranta, E., Bodega, B., et al. (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nature Cell Biology, 17, 1218–1227.
Zanconato, F., Cordenonsi, M., & Piccolo, S. (2016). YAP/TAZ at the roots of cancer. Cancer Cell, 29, 783–803.
Zhang, J., Xu, Z. P., Yang, Y. C., Zhu, J. S., Zhou, Z., & Chen, W. X. (2012). Expression of Yes-associated protein in gastric adenocarcinoma and inhibitory effects of its knockdown on gastric cancer cell proliferation and metastasis. International Journal of Immunopathology and Pharmacology, 25, 583–590.
Zhou, Z., Zhu, J. S., Xu, Z. P., & Zhang, Q. (2011). Lentiviral vector-mediated siRNA knockdown of the YAP gene inhibits growth and induces apoptosis in the SGC7901 gastric cancer cell line. Molecular Medicine Reports, 4, 1075–1082.
Zuo, J. H., Zhu, W., Li, M. Y., Li, X. H., Yi, H., Zeng, G. Q., et al. (2011). Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. Journal of Cellular Biochemistry, 112, 2508–2517.
Acknowledgments
The research of MS, YQ, and MFE has been supported by generous “Seed Grants” from NUS-MBI-IMCB-A*STAR of the Republic of Singapore. AP was supported by a VA Health Services Research & Development award and a Pilot Grant project from the Marlene & Stewart Greenebaum Cancer Center. The authors would also like to acknowledge the assistance of Chun Xi Wong in preparation of the figure illustrations.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Passaniti, A., Brusgard, J.L., Qiao, Y., Sudol, M., Finch-Edmondson, M. (2017). Roles of RUNX in Hippo Pathway Signaling. In: Groner, Y., Ito, Y., Liu, P., Neil, J., Speck, N., van Wijnen, A. (eds) RUNX Proteins in Development and Cancer. Advances in Experimental Medicine and Biology, vol 962. Springer, Singapore. https://doi.org/10.1007/978-981-10-3233-2_26
Download citation
DOI: https://doi.org/10.1007/978-981-10-3233-2_26
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3231-8
Online ISBN: 978-981-10-3233-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)