
Abstract
Atherosclerosis underlies the majority of cardiovascular diseases and is accepted as a primary cause of mortality worldwide. Matrix metalloproteinases (MMPs) and their endogenous tissue inhibitors (TIMPs) perform complex roles during the progression and development of atherosclerosis and subsequent plaque instability. Proposed actions of MMPs include extracellular matrix remodeling alongside regulation of vascular cell proliferation, migration and apoptosis including cell types such as monocytes, macrophages and vascular smooth muscle cells. As such, a large body of evidence from both in vitro and in vivo studies has shown that individual MMPs and TIMPs are utilized by distinct cell types to regulate their behavior. Consequently, it is now accepted that some MMPs promote the growth and development of advanced atherosclerotic plaques in experimental models whilst others do not . Similarly, human genetic and pathological findings reveal some MMPs correlate with vulnerable atherosclerotic plaque phenotypes, whereas others associate with stable lesions. Furthermore, broad-spectrum MMP inhibition in both mouse and man has proved ineffective at protecting from atherosclerotic plaque progression and instability. Considering the divergent effects MMPs exert on atherosclerotic lesions, selectively targeting individual deleterious MMPs may serve as a more efficacious therapeutic strategy. For example, our recent data demonstrate that a selective MMP-12 inhibitor retards atherosclerotic plaque progression in the apolipoprotein E (Apoe) mouse atherosclerosis model, whilst also promoting plaque stabilization through reducing monocyte recruitment into plaques whilst augmenting fibrosis. Similar studies have been conducted assessing MMP-13 inhibition. Accordingly, as our knowledge of the complex roles MMPs play during the development, progression and rupture of atherosclerotic plaques expands, new impetus is required for clinical trials evaluating the therapeutic potential of selective MMP inhibition, especially in the context of atherosclerosis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Atherosclerotic plaque development and progression is the principal underlying cause of cardiovascular disease, now reported as the primary cause of mortality and morbidity in developed countries [1]. Atherosclerosis is characterized by the accumulation of lipids (atheroma) and fibrous elements (sclerosis) within major arteries sustaining the heart (coronary arteries) and the brain (carotid arteries) [1]. It has been defined as a chronic, autoimmune-like disease, which develops in the presence of elevated circulating lipid levels [2]. Atherosclerotic plaque formation and progression is usually clinically silent. However, plaque rupture followed by thrombus formation and subsequent vessel occlusion can precipitate several clinical events including myocardial infarction, stroke, and peripheral vascular disease. The main underlying trigger for plaque rupture is ascribed to the loss of extracellular matrix (ECM) proteins, such as elastin and collagen, alongside decreased smooth muscle cells content within the plaques, which commonly corresponds to areas of marked inflammation [3]. These areas are characterized by the presence of foam cell macrophages, B- and T-cells, mast cells and smaller amounts of other white bloods cells [4]. Over the last quarter of a century, a large number of pathological and experimental studies have been conducted in this field to elucidate the pathophysiology of atherosclerotic lesion development, progression, and rupture. One of the principal goals in cardiovascular research is to find suitable targets to allow the development of new therapies, aimed at specific cell types or select molecules, attributed deleterious roles in atherosclerotic disease onset and progression. Matrix metalloproteinases (MMPs) have been implicated in all the stages of atherosclerosis, from plaque development to plaque rupture, through a large body of published work [5, 6]. Elevated expression levels of MMPs including MMP-1, -2, -7, -8, -9, -11, -12, -13, and -14 have been identified in human atherosclerotic plaques (see Table 1) [7,8,9,10,11,12,13,14,15]. Moreover, the majority of increased MMP expression within atherosclerotic lesions is specifically located to macrophages-rich areas (shoulder regions and around the lipid core) suggesting that macrophage-derived MMPs may play a key role in atherosclerotic plaque progression. Furthermore, considering MMPs have been proposed to induce plaque rupture in a dual manner: by direct degradation of ECM proteins (such as elastin and collagen) and by promoting the death of vascular smooth muscle cells (VSMCs), the main cell type responsible for ECM synthesis within the plaque [16]; many studies have focused their attention on inhibitors of MMPs as a therapeutic strategy to stabilize and perhaps induce regression of atherosclerosis [17].
2 Matrix Metalloproteinases (MMPs)
Matrix metalloproteinases (MMPs), also named matrixins, are a large family of at least 24 proteolytic enzymes having a role is several physiological and pathological processes such as morphogenesis, angiogenesis, tissue repair, wound healing and remodeling. For these reasons, MMPs are involved in several pathologies including cancer progression and atherosclerosis, highlighting them as key therapeutic targets for medical research. The MMPs are multi-domain enzymes capable to degrade both ECM components and several non-ECM molecules. MMPs share similarity with two other proteinase families: ADAMs (a disintegrin and metalloproteinase family) and ADAMTSs (ADAM with thrombospondin motifs), as they all contain a zinc atom and a conserved methionine in the catalytic domain, and collectively consist the Metzincin family [18]. Due to its destructive capabilities, MMP activity is tightly regulated by a family of endogenous inhibitors named the tissue inhibitors of metalloproteinases (TIMPs) that, together with MMPs, are responsible the maintenance and balance of ECM homeostasis during physiological and pathological conditions. This equilibrium contributes to multiple other processes such as differentiation, growth, inflammation, migration, and apoptosis due in part to the capacity of MMPs to target non-ECM substrates. MMP and TIMP expression, which exhibit distinct tissue/cell, temporal and spatial differences, are tightly regulated by numerous molecules including inflammatory cytokines, hormones, growth factors, and physical cell–cell and cell–matrix interactions (as reviewed by [19]).
3 MMP Classification and Structure
MMPs share some structural homology. Usually they present:
-
(i)
Signal peptide at the N-Terminus: a hydrophobic sequence of 18–30 residues responsible for intracellular trafficking from the Golgi apparatus to the cell membrane which is cleaved during secretion [19].
-
(ii)
Pro - peptide: a highly conserved motif responsible for pro-MMP latency [19].
-
(iii)
Catalytic Domain: which contains a zinc-binding site responsible for the endopeptidase activity of MMPs.
-
(iv)
Hinge Domain: known also as a linker peptide, it is situated between the catalytic domain and the hemopexin-like domain. It stabilizes the collagenolytic activity due to the presence of several proline residues.
-
(v)
Hemopexin - like Domain: positioned at the C-terminus, it has strong sequence similarity to the serum protein hemopexin and an extensive range of roles amongst diverse MMPs [18].
Nevertheless, there are notable structural differences between MMPs that confer diverse biological properties. Based on their domain organization, MMPs can be divided into six groups [20] (see Fig. 1).

Domain structure for the major classes of MMPs. Diagram illustrating the differing domain structures of the major MMP classes, including the pro-domain, catalytic domain with the active site zinc (Zn) bound to cysteine residues within this domain and “cysteine switch-residue” in the pro-domain, the hinge domain, the hemopexin-domain, the fibronectin-like type II repeats, and in some cases for MT-MMPs, either a transmembrane domain or a glycophosphatidylinositol (GPI)-anchor domain
-
(i)
MMPs presenting the pro-domain and the catalytic domain. This group includes MMP-7 and MMP-26, also known as Matrilysins.
-
(ii)
MMPs containing the pro-domain, the catalytic domain, the hinge domain, and the hemopexin-like domain. This group contains several MMPs with diverse substrate specificities; MMP-1, -8, -13 (Collagenases), MMP-3, -10, -11 (Stromelysins), MMP-12 (Metalloelastase), MMP-20 (Enamelysin), MMP-19, MMP-22, and MMP-28.
-
(iii)
MMPs comprising the pro-domain, a catalytic domain containing fibronectin-like repeats, the hinge domain and the hemopexin-like domain. In this group there are MMP-2 and -9, also named Gelatinases for their affinity to degrade gelatin.
-
(iv)
The transmembrane type I MMPs are a group of MMPs that present, together with the pro-domain, the catalytic domain, hinge domain and the hemopexin-like domain, a transmembrane domain at the N-terminus. This domain allows this group of MMPs to localize on to the cell membrane, projecting toward the extracellular space. This characteristic facilitates pericellular matrix degradation, and hence plays a prominent role in directing cell migration [21]. This group includes MMP-14, -15, -16, and -24.
-
(v)
The transmembrane type II MMPs include one single MMP, MMP-23. Differently from the type I, type II MMP has the transmembrane domain at the C-terminus and additionally present an IgG-like domain.
-
(vi)
There is a third group of membrane-type MMPs localized to the cell membrane via glycosylphosphatidyl inositol (GPI) anchor on the N-terminus. This group includes MMP-17 and -25.
4 MMP Activation and Inhibition
MMPs are produced as zymogens; the interaction between the pro-domain and the catalytic domain keep the MMP in an inactive conformation. In order to achieve full activation of these enzymes the pro-domain has to be cleaved, an essential regulatory step toward MMP activation [19]. Activation of the biologically inactive MMP (pro-form) follows a multi-step sequence of events also known as ‘stepwise activation’. First, the cleavage of a ‘proteinase susceptible bait region’ through the action of plasma or bacterial proteinases, destabilise the cysteine–Zn2+ negative interaction within the pro-domain, resulting in a MMP intermediate form. To achieve full activation, the in-trans activity of other intermediary or active MMPs is required in order to fully remove the inhibitory pro-domain [19]. Some MMPs are completely activated intracellularly by furin or other pro-protein convertases and then either translocated to the cell membrane or secreted, as active enzymes. MMP activity is closely regulated by endogenous inhibitors (such as TIMPs and α2-macroglobulin), proteolysis, or internalization and recycling [19]. In addition, other proteins with the ability to inhibit MMPs have been described, including the reversion-inducing cysteine-rich protein with Kazal motifs (RECK), tissue factor pathway inhibitor-2 (TFPI-2), and the pro-collagen C-terminal proteinase enhancer (PCPE). Nevertheless, TIMPs are the most potent endogenous inhibitors of MMPs and therefore considered key regulators in the physiological regulation of MMP activity. Four TIMPs have been identified within vertebrates, TIMP-1, -2, -3, and -4, which exhibit diverse inhibitory actions toward different MMP family members [22]. For example, TIMP-1 has a poor inhibitory effect on MMP-9, -14, -15, -16, and -24. TIMPs also harbor the ability to inhibit members of both the ADAM and ADAMTS family of proteinases [22]. TIMP expression is tissue specific and, similarly to MMPs, is finely regulated during development and remodeling. Most of their inhibitory capacity has been ascribed to the N-Terminal domain since it is able to form, when isolated, a stable native molecule with an inhibitory effect on MMPs [23]. TIMPs are normally secreted proteins, however they can localize to the cell membrane associated with membrane protein, including several MT-MMPs. Regulation of the equilibrium between MMPs and TIMPs is essential in homeostasis. Alterations in this balance can trigger patho-physiological conditions associated with atypical ECM turnover of the matrix and/or dysregulation of processes involved in wound healing, remodeling and inflammation. Cardiovascular disease, cancer, arthritis, and neurological disorders are all examples of pathologies where an imbalance between MMPs and TIMPs is apparent [23].
5 MMPs and Atherogenesis
5.1 Early Stage: Pathological Intimal Thickening
Atherogenesis is a multi-step sequence of events that leads to atherosclerotic plaque formation on the luminal side of major arteries. In humans, it is a process that develops and evolves over several decades, beginning with early lesions that can occur during childhood. The development of atherosclerotic lesions is dependent on multiple risk factors which can be genetic or modifiable in nature, including hypercholesterolemia, smoking, high blood pressure, sedentary lifestyle, and diabetes [24]. In man, the first event that generally occurs branch points within major arteries, is the formation of an early lesion, commonly termed pathological intimal thickening (although sometimes referred to as fatty streaks) [25]. These early lesions are characterized by the accumulation of lipid-laden macrophages (also called foam-cell macrophages due to their appearance under the microscope), within a preexisting smooth muscle and ECM-rich intima. Raised levels of low density lipoprotein (LDL) within the blood stream, alongside alterations in shear stress, the presence of free radicals such as reactive oxygen species (ROS), or exposure to infection-related pathogens, can result endothelial damage. A damaged endothelium is subject to inflammatory activation that triggers expression of adhesion molecules (including vascular cell adhesion molecule-1; VCAM-1) that mediate leukocyte recruitment. Such adhesion molecules facilitate a transient contact, allowing leukocyte rolling at the luminal surface of the vessel wall. After firm adhesion to the endothelium, monocytes and lymphocytes transmigrate, penetrating into the tunica intima (the innermost layer of the artery) driven by a chemoattractant gradient, through molecules including monocyte chemoattractant protein-1 (MCP-1). Monocyte recruitment is considered a fundamental process during early lesion formation and atherosclerosis onset. In order to invade the arterial wall, monocytes are required to degrade the physical barrier represented by the ECM, therefore it is essential that they possess potent protease activity. Human monocytes constitutively express several MMPs and TIMPs including MMP-8, MMP-12, MMP-19, TIMP-1, and TIMP-2. Whereas upon adhesion and in response to inflammatory stimuli they can be activated and subsequently upregulate the expression of MMP-1, MMP-3, MMP-10, and MMP-14 via the stimulation of MAP kinase and NF-κB transcription factors [26]. Specifically, MMP-14 expression and activity is necessary for monocyte endothelial transmigration and invasion this process can be blocked by MMP-14 inhibition either by a neutralizing antibody, recombinant TIMP-2 or gene silencing in vitro [27,28,29,30]. Moreover, MMP14 inhibition of activated circulating monocytes by a neutralizing antibody, retards monocyte recruitment into existing atherosclerotic lesions in mouse model of atherosclerosis [30]. Once within the intima monocytes differentiate into macrophages in response to several stimuli, in particular the Colony Stimulating Factors (CSFs), which concomitantly drives the expression of scavenger receptors, growth factors, cytokines giving rise to a survival impulse [31]. Accordingly, recently recruited monocytes at sites within the artery where lipoproteins have accumulated and after their differentiation into macrophages, begin to internalize the modified lipoproteins from the surrounding areas, through their cell-surface scavenger receptors. This process results in transformation of macrophages into foam-cells macrophages (FCMs) [32]. Macrophages also interact with T-cells which are also recruited to developing plaques, inducing an array of immune and inflammatory responses including the expression of adhesion molecules, MMPs, cytokines, apoptotic mediators, and pro-thrombotic activities, which collectively drive an inflammatory amplification loop and therefore promoting atherosclerotic plaque progression [33]. Intra-plaque macrophages and foam-cells express a diverse range of MMPs and TIMPs [34]. In particular, it has been observed that MMP-7, MMP-9, and TIMP-3 expression is induced during macrophage differentiation in vitro [26], whereas MMP-1, MMP-3, and MMP-12 expression can be induced in macrophages in response to inflammatory mediators and cytokines [34]. However, within atherosclerotic plaques, most macrophages are lipid-laden and therefore characterized as foam cell macrophages, therefore the accumulation of lipid within macrophages may exert the most dominant role on MMP and TIMP regulation. Indeed, immunohistochemistry (a valuable method for studying atherosclerotic plaque composition alongside macrophages and foam-cell macrophages in situ), has revealed the expression of MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, MMP-11, MMP-12, MMP-13, MMP-14, and MMP-16 in foam-cell macrophages within lesions (Table 1). Moreover, the detection of matrix proteolysis (as assessed by in situ zymography) [35, 36] alongside the presence of cleaved collagen fibres at the corresponding sites [13], suggest that at least some of the MMPs expressed within these regions are in an active form. Recent direct evidence has demonstrated that MMP-1, MMP-3, and MMP-14 are over-expressed in foam-cell macrophages isolated from human plaques [37, 38] and that in vivo generated foam-cell macrophages from cholesterol-fed rabbits display heightened expression of MMP-1, MMP-3, MMP-12, and MMP-14, when compared to nonlipid-laden macrophages [39]. Additionally, increased expression and activity of MMP-14 in a sub-population of rabbit foam cell macrophages was associated with a concomitant loss of TIMP-3 expression, resulting in their increased invasiveness, proteolytic activity, and susceptibility to undergo apoptosis [40]. Therefore, the presence of foam-cell macrophage-derived MMPs within the atherosclerotic lesions may direct disease progression and predict future clinical outcome.
5.2 MMPs and Atherosclerotic Plaque Progression
One of the principal processes that determines the progression of a pathological intimal thickening toward the development of a mature atherosclerotic plaque is the formation of a fibrous cap which overlies a recently formed lipid core. The fibrous cap originates following the organized migration of vascular smooth muscle cells (VSMCs) from the tunica media (the middle layer of the artery that lies between the tunica intima on the inside and the tunica externa on the outside) toward the arterial lumen, alongside the continual growth of VSMCs already resident within the intimal thickening. The VSMCs overlying the lipid core proliferate and produce fibrous ECM components, such as collagen and fibronectin, providing a structural barrier that separates the thrombogenic lipid core from the blood stream, providing strength and hemodynamic stability to the developing lesion. In addition to the production of MMPs and TIMPs, macrophages within the plaque secrete numerous cytokines and mediators such as platelet-derived growth factor (PDGF), heparin-binding epidermal growth factor (EGF), and insulin-like growth factor (IGF) that facilitate the mobilization and recruitment of VSMCs [41]. In order to expedite their migration, VSMCs need to release themselves from their cell–cell and cell–matrix interactions which act as physical barriers—dysregulated MMP activity directs this process. Studies investigating the role of MMP activity on VSMC migration have focused their attention principally on MMP-2, MMP-9, and MMP-14, presumably due to these MMPs harboring the ability to degrade the basement membrane protein collagen type IV [16]. MMP-2 has been shown to augment VSMC migration across basement membrane proteins in vitro [42, 43], and MMP-9 overexpression can also promote the migratory capacity of isolated VSMCs [44]. A comprehensive study revealed MMP-14 is critical during VSMC migration, facilitating VSMCs to first degrade and then infiltrate 3-D collagen barriers, including the arterial wall [45]. These findings have been substantiated through subsequent in vivo studies utilizing genetically modified mice lacking either MMP-2, MMP-9, or MMP-14 which all reported attenuated VSMC migration. MMP-3 has also been shown to promote VSMC migration, predominantly through the activation of MMP-9 [46]. A role for the collagenase MMP-13 in VSMC migration has been documented, induced through an Akt-ERK dependent pathway [47]. Additionally, MMPs can contribute to VSMC migration by cleavage of nonmatrix substrates. For instance, MMP-14 can cleave and shed from the cell membrane CD44 (a cell surface hyaluronan receptor), promoting increased cell motility [48]. Conversely, intact CD44 can serve as a docking station for secreted MMP-7 and MMP-9 on the VSMC membrane, localizing their proteolytic activity to the cell surface and potentially facilitating cell migration [49, 50]. MMP activity has also been linked to VSMC proliferation. Similarly to migration, proliferation requires the removal of cell–cell and cell–matrix interactions, which otherwise exert an inhibitory effect on cell division. Cadherins are a family of adhesion proteins involved in cell–cell contact regulation of proliferation, and have recently been identified as new substrates of MMP activity [51]. Cadherins also serve as membrane receptors for cell signaling transduction and their cleavage by MMPs can modulate β-catenin nuclear translocation (a member of Wnt/wingless signaling pathway), known to activate the transcription of several pro-proliferative genes [51]. Indeed, MMP-7 and MMP-12 can induce N-cadherin cleavage/shedding, and through β-catenin signaling, promote VSMC proliferation [52]. Taken together, MMP-directed VSMC growth and migration participates in fibrous cap formation and therefore plays a prominent role in atherosclerotic plaque formation—but is considered beneficial as it protects the developing plaque from instability.
5.3 MMPs and Unstable Plaque Development and Rupture
During atherosclerotic plaque progression, foam-cell macrophages undergo cell death via apoptosis or necrosis. Macrophage and foam-cell death promotes the establishment and expansion of an extracellular lipid-rich core, which is highly thrombogenic and harbors the potential to destabilise advanced atherosclerotic plaques. Unsurprisingly, a role for MMPs has been suggested in macrophage and foam-cell apoptosis [53]. For example, macrophage and foam-cell susceptibility to undergo apoptosis can be retarded by inhibition of MMP-12 or MMP-14 activity, through use of a selective inhibitor or a neutralizing antibody, respectively [40, 54]. Accordingly, TIMP-2 and TIMP-3 can both reduce foam-cell macrophage apoptosis, in part through inhibition of MMP-14-dependent N-Cadherin cleavage [30, 40, 55]. Furthermore, loss of TIMP-2 in vivo increases the number of apoptotic macrophages within atherosclerotic plaques of hypercholesteroleamic mice, whilst TIMP-1 depletion had no effect [30]. Therefore, uncontrolled cell death and subsequent lipid core enlargement contributes to plaque progression and is associated with plaque instability and propensity to rupture [56]. As can know be appreciated, the stability of atherosclerotic plaques is determined by its composition, specifically the VSMC and fibrous ECM content (which reflects the thickness and strength of the fibrous cap), together with the macrophage and lipid content (which reveals the size and possible rate of expansion of the lipid core) [57]. The vast majority of acute coronary events originate from atherosclerotic plaque instability, notably the rupture of the fibrous cap and ensuing leakage of the thrombogenic lipid core into the arterial lumen, triggering thrombosis [58]. As such, clinical symptoms, including myocardial infarction or stroke, are often a result of plaque rupture and subsequent thrombus formation, resulting in distal impairment of blood flow or embolization and consequent ischemia. Indeed, fibrous cap disruption leads to the exposure of highly thrombogenic plaque constituents such as tissue factor (TF), lipids or modified collagen fragments. The interaction of these factors with the flowing blood results in thrombus formation by triggering activation of the coagulation cascade [59]. As earlier discussed, mature atherosclerotic plaques are characterized by a soft and highly thrombogenic lipid-rich core and associated macrophages infiltration, which is encapsulated by a VSMC and ECM-rich fibrous cap that provides structural integrity [56]. However, atherosclerotic lesions are heterogeneous in nature and can vary in fibrous cap thickness and lipid-core size; different combinations of these two variables results in different plaque phenotypes and susceptibility to rupture, with diverse clinical outcome. Pathological studies of human coronary artery atherosclerotic plaques permit histological discrimination between stable and unstable (also defined as vulnerable or rupture-prone) atherosclerotic plaques [25, 56]. Characteristically, stable plaques constitute of a thick fibrous cap, particularly enriched with VSMCs and collagen, and a small lipid core with reduced macrophage accumulation. Plaques with thick caps (and nonstenotic) are generally clinically silent. However, unstable plaques typically present with a large lipid-core and a thin fibrous cap [5] and are characterized by a high number of macrophages plus other inflammatory cell types; and are commonly referred to as thin-cap fibro-atheromas (TCFAs). Histological and in vivo animal studies have demonstrated that inflammation (T-cells and macrophages) not only promotes atherosclerotic plaque formation, but also contributes to plaque destabilization [5]. Foam-cell macrophages produce several pro-inflammatory cytokines such as IFNγ, which in addition to mediating inflammatory responses, can also inhibit VSMC collagen synthesis [60]. As discussed earlier, within atherosclerotic plaques, macrophages are a major source of proteolytic enzymes, especially MMPs, alongside a plethora of inflammatory mediators in plaques, and are therefore considered to play a fundamental in ECM degradation (i.e., collagen and elastin) and subsequent fibrous cap weakening [5]. There is also evidence that macrophage-dependent MMP activity can promote fibrous cap thinning through potentiating VSMC death. For example, MMP activity may detrimentally affect VSMC survival by disrupting cell–matrix interactions and therefore attenuating matrix-dependent survival signals [16]. The cleavage of death signal molecules and their receptors from the cell surface can trigger apoptosis through autocrine and paracrine processes. A number of MMPs including MMP-7 are able to generate the pro-apoptotic factor TNFα through proteolytic cleavage of pro-TNFα [16]. In addition, MMP-7 can cleave Fas ligand (FasL) to its pro-apoptotic soluble form (sFasL) [61]. Interestingly, MMP-7, TNFα, and FasL all co-localize in human atherosclerotic plaques, suggesting this apoptotic triptych may contribute to formation and expansion of the lipid-rich core [62]. The lateral aspects of an atherosclerotic plaque (commonly termed the shoulder regions) are the sites considered most prone to rupture, and reside between the lipid-rich core and the thinnest part of the fibrous cap. These areas are characterized by accumulations of macrophages and particularly foam-cell macrophages, alongside notable neovascularization [63]. Pathological studies of human atherosclerotic plaques have revealed that macrophages, VSMCs, lymphocytes, and endothelial cells within the rupture-prone shoulder regions express MMP-1, MMP-3, and MMP-9 [64]. MMP-2, MMP-7, MMP-11, MMP-12, MMP-13, MMP-14, and MMP-16 levels are also elevated at the shoulder regions of unstable plaques [8, 10, 12,13,14,15], where increased MMP activity and substrate cleavage has also been documented [7, 11, 13, 36]. These findings suggest that MMP expression and activity is strongly associated atherosclerotic plaque progression, highlighting them as therapeutic targets and predictors of clinical outcome in patients with advanced atherosclerotic disease.
6 MMPs as Therapeutic Target for Atherosclerosis
Animal models of atherosclerosis have been widely utilized to investigate the pathogenesis of plaque formation, progression, and instability with the objective of identifying novel therapeutic targets to prevent the clinical manifestations associated with atherosclerosis. Rabbits have been used in multiple studies as several strains spontaneously develop atherosclerotic plaques when fed a high-fat diet. However, most atherosclerosis in vivo studies are conducted in mouse models, despite the fact that wild-type mice atherosclerosis-resistant even after prolonged periods of high fat feeding. The two most commonly used mouse models of atherosclerosis are genetically modified where a key gene of the cholesterol transport pathway has been deleted; these genes are Apolipoprotein E (Apoe) or LDL receptor (Ldlr), thus rendering them hypercholesteroleamic [65,66,67]. These mice develop atherosclerotic lesions throughout the arterial tree including similar sites to plaque formation in man, even when fed on a normal diet [68]. However, on consumption of a high-fat diet, atherogenesis is significantly accelerated in either Apoe or Ldlr deficient mice, although the hypercholesterolemia is more marked in the Apoe deficient animals. There are also striking similarities in lesion development and progression between both models and humans, as early lesions closely resemble fatty streaks whilst longer periods of high-fat feeding produce complex advanced lesions [68, 69]. A multitude of studies have been conducted in Apoe deficient mice (and to a lesser degree Ldlr knockout mice) to investigate the roles of MMPs in atherosclerosis. Such studies have utilized genetically modified mice which have global or cell specific knockout or over-expression of a single MMP/TIMP, or treated with a potential therapeutic agent that targets select or all MMPs. These studies have aided the elucidation of potential pathogenic roles of multiple MMPs and TIMPs in atherosclerosis plaque progression and stability (summarized in Table 2).
6.1 Overexpression Studies
Dissimilar to humans, mice do not constitutively express MMP-1. However, when human MMP-1 was over-expressed exclusively in macrophages of Apoe deficient mice, an unexpected reduction in plaque size, and collagen content was observed [70]. In contrast, macrophage-specific over-expression of pro-MMP-9 did not affect atherosclerotic plaques [71]. However, in a collar-induced carotid artery model of atherosclerosis in Apoe deficient mice, local over-expression of pro-MMP-9 promoted intra-plaque hemorrhage [72]. Furthermore, the transplantation of transduced stem cells permitting the over-expression of an active form of MMP-9, increased plaque progression [71]. Similarly, macrophage-specific over-expression of active of MMP-12 in transgenic rabbits, augmented plaque size and markers of inflammation [73], suggesting that MMP-9 and MMP-12 activation may promote atherosclerosis progression (summarized in Table 2).
6.2 Knockout Studies
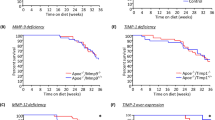
A number of studies have been conducted in Apoe knockout mice which are also deficiency for a single MMP or TIMP to elucidate the roles for a selected MMP/TIMP in atherosclerotic plaque formation. Interestingly, these studies have revealed that MMPs exert protective and detrimental effects on atherosclerosis. For instance, Mmp2 knockout mice exhibits a reduction in plaque size, attributed in part to a reduction in VSMC content and implying that plaque stability is compromised in the absence of MMP-2 [74], as MMP-2 is necessary for VSMC migration and intimal formation in vivo [75]. Equally, although Mmp3 deletion resulted in larger aortic and brachiocephalic plaques, a reduction in VSMC number was observed, associated with an increased number of buried fibrous layers (a surrogate marker of plaque instability), suggesting that MMP-3 may promotes plaque stability through promoting VSMC accumulation [76, 77]. Indeed, after carotid ligation, Mmp3 knockout mice shows decreased VSMC migration and associated neo-intimal formation [46]. Likewise Mmp9 deficient mice develop larger plaques with an increased number of buried fibrous layers, and a concomitant reduction in VSMC content [77]. Taken together with findings from an arterial injury model demonstrating MMP-9 promotes VSMC migration and concomitant neo-intimal formation [46], these studies support a beneficial role for MMP-9 in promoting plaque stability through favouring VSMC accumulation. However in another study assessing aortic plaques in mmp9 KO studies, revealed no change in plaque area and a reduced number of lesions, although they also suggested that plaque VSMC number was lowered in Mmp9 deficient animals [78].
In contrast, an increase in VSMC content was reported within the brachiocephalic plaques of mmp7 knockout mice [77], in agreement with a pro-apoptotic role attributed to MMP-7 on VSMCs [79], and indicating a deleterious role for this MMP in atherosclerosis. Mmp8 deficient mice show reduced plaque size and macrophage number but increased collagen content, suggesting MMP-8 promotes plaque progression [80]. Several lines of evidence have strongly indicated a detrimental role for MMP-12 in plaque progression and instability. Mmp12 deficiency results in smaller brachiocephalic artery plaques, with a reduced number of macrophages and buried fibrous layers [77] and diminished indicators of elastin degradation [78]. Moreover, the ratio between macrophages and VSMCs within the plaques of Mmp12 knockout mice are favourably increased toward VSMCs, in part due to reduced monocyte/macrophage invasion and apoptosis [54], suggesting MMP-12 promotes plaque instability. Further studies in a rabbit model of atherosclerosis have confirmed a detrimental role for MMP-12 in atherosclerosis [73]. Collectively these findings strongly imply that MMP-12 promotes plaque progression and instability. Whilst exerting moderate effects on plaque size, macrophage and VSMC content, mice with either global deletion of MMP-13 or macrophage-specific loss of MMP-14 exhibit a marked increase in plaque fibrillar collagen content, indicating significant roles for these two MMPs in collagen degradation and consequently plaque destabilization [81, 82].
Consequently, these studies imply that some MMPs, such as MMP-2, -3, and -9, exert a protective effect on atherosclerotic plaque progression by promoting VSMC growth and consequent fibrous cap formation. Contrastingly, other MMPs including MMP-7, -8, -12, -13, and -14, may promote plaque instability via increased inflammation, matrix degradation and apoptosis, therefore increasing the propensity of plaque rupture (summarized in Table 2).
6.3 Inhibitor Studies
Although Timp1 deficient mice had larger aortic atherosclerotic lesions with enhanced MMP activity, accompanied with heightened macrophage and lipid content [83], Timp2 knockout mice display a more unstable plaque phenotype than their Timp1 deficient counterparts [30]. These plaques were characterized by increased necrotic core size, buried fibrous layers, macrophage number, and macrophages undergoing apoptosis and proliferation; they also presented reduced collagen and VSMC content, indicative of reduced stability [84]. Equally, Timp3 deficiency in Apoe knockout mice increased lesion size within the aorta and at the aortic root, associated with heightened macrophage accumulation [85]. As therefore expected, systemic over-expression of TIMP-1 or TIMP-2, via adenovirus-mediated gene transfer, reduced lesion development and plaque progression in Apoe knockout mice [55, 86]. Additionally, gene transfer long term over-expression of TIMP-2, but not TIMP-1, arrested progression of established plaques at least in part by constraining monocyte/macrophage invasion and their susceptibility to apoptosis [55]. These findings lend robust support for MMP inhibition as a therapeutic strategy to prevent plaque progression and destabilization. Accordingly, there have been numerous endeavours by academia and industry to develop and deploy synthetic inhibitors of MMPs. Nevertheless, broad spectrum inhibitors containing zinc-chelating groups (such as thiol or hydroxamate groups, or tetracycline derivates) have given inconsistent results. Administration of hydroxamic acid-based, nonselective MMPs inhibitors to either Lldr knockout or Apoe KO deficient mice revealed no beneficial effects on plaque development or progression [87, 88]. Likewise, doxycycline (a commonly used antibiotic with known nonspecific MMP inhibitory ability) failed to prevent atherosclerosis development in Apoe deficient mice [89]. Furthermore, two independent, randomized, double-blind, and placebo controlled clinical trials involving treatment with of patients with symptomatic coronary and carotid artery disease with doxycycline, did not favourably influence plaque composition or clinical outcome [90, 91]. In contrast, use of a highly selective MMP-12 inhibitor, RXP470.1, arrested plaque progression and improved stability in Apoe deficient mice with preexisting atherosclerosis [54]. In response to MMP-12 inhibition, lesions exhibited reduced lipid core expansion and macrophage apoptosis, increased VSMC to macrophage ratio, decreased plaque calcification, and attenuated elastin degradation [54]. These results, together with a reduction of buried fibrous layers, reflected those observed previously in Mmp12/Apoe double knockout mice [77]. Similarly, a second study where a highly specific MMP-13 inhibitor was deployed, revealed intra-plaque collagenolytic activity was reduced and associated with preservation of fibrillar collagen content within plaques [92], mirroring the effects also witnessed in Mmp13 deficient mice [81]. Taken together, considering broad spectrum MMP inhibition failed to exert any striking benefits on atherosclerosis in either clinical or animal studies, whilst selective MMP inhibition was beneficial in mice, support the tenet that individual MMPs (and therefore possibly TIMPs) play divergent roles in disease development and progression. Consequently, these proof-of-principle studies in mice provide an incentive to translate selective MMP inhibitor treatment into human atherosclerotic patients (summarized in Table 2).
6.4 microRNA Regulation of MMPs
microRNAs (miRs) are small noncoding RNA molecules of approximately 22 nucleotides in length which have the ability to post-transcriptionally regulate gene expression. They are transcribed by polymerase II in the nucleus and are initially produced as primary miRs (pri-miRs). These pri-miRs are processed to miR precursors (pre-miRs) by RNAse III Drosha before they can be exported to the cytoplasm where they are eventually processed into mature and biologically functional miRs through the action of another RNAse III named Dicer. Mature miRs are able to target and bind the 3′ untranslated regions (3′-UTR) of messenger RNA (mRNA) and modulate their expression. It has been predicted that miRs may modulate up to 90% of mammalian genes and therefore play fundamental roles in regulating cellular function [93]. Numerous studies have recently investigated that ability of miRs to regulate MMP expression. For instance, the 3′UTR region of MMP-1 is targeted and regulated by miR-526 [94], which could have potential implications for collagenolysis in plaques. MMP-2 is a direct target of miR-29b, and consequently miR-29b over-expression can inhibit VSMC migration and proliferation and subsequent neo-intimal formation [95]. MMP-3 has been identified and validated as a putative target of miR-93, as such miR-93 over-expression in human nucleus pulposus cells promoted collagen accumulation [96]. In osteocarcinoma, miR-539 plays a key role in inhibiting osteosarcoma cell invasion and migration through regulating MMP-8 expression in osteosarcoma cells [97]. Direct targeting of MMP-9 by miR-204 can suppress trophoblast-like cell invasion, contributing to the development of pre-eclampsia [98]. Furthermore, MMP-9 expression may also be indirectly regulated by miR-497 via direct targeting of MEK1 in endothelial cells, in response to the anti-hyperlipidaemia drug probucol [99]. Another study conducted in chondrocytes revealed miR-320 was able to directly target and down-regulate MMP-13 expression during chondrogenesis, and vice versa during inflammatory osteoarthritis [100]. Numerous microRNAs have been identified and predicted to target and regulate the expression of MMP-14. miR181a-5p has been shown to downregulate MMP-14 expression by direct targeting of its 3′UTR, reducing cancer cell invasion, and angiogenesis [101]. Similarly, miR-9 can inhibit neuroblastoma cell invasion, metastasis, and angiogenesis by targeting of MMP-14 mRNA [102]. With regard to atherosclerosis, MMP-14 protein expression can be directly modulated by miR-24 in macrophages in response to GM-CSF, influencing the invasive capacity of macrophages [84]. Consequently, administration of a locked nucleic acid (LNA)-miR-24 inhibitor significantly exacerbated preexisting atherosclerosis in Apoe deficient mice, through increasing lesion size, macrophage content, and MMP-14 expression [84]. Moreover, miR-24 expression correlate with more stable coronary plaques in humans, suggesting a protective role of miR-24 in atherosclerosis, presumably through decreased MMP-14 activity [84]. Finally, miR-712 is induced in response to shear stress in endothelial cells of Apoe deficient mice, and through targeting of TIMP-3, exerts a detrimental effect on atherosclerosis via promotion of endothelial inflammation [103]. Collectively, these findings suggest modulation of microRNA may serve as a valuable tool for regulating MMP and TIMP expression in atherosclerosis, highlighting these important and powerful molecules as significant targets for medical intervention.
7 Conclusions
Through studies conducted in isolated cells and animal models, alongside human pathological and clinical findings, MMPs have been established to play a fundamental role in cardiovascular diseases, especially the development, progression, and rupture of atherosclerotic plaques. Seminal studies utilizing animal models that permit genetic modulation of individual MMPs or TIMPs has allowed the identification of specific roles select MMPs exert on all vascular cell types, and the ensuing significance to atherosclerosis. Collectively, this large body of work has demonstrated that modulation of MMP expression/activity can halt and even reverse atherosclerosis, whilst disappointingly broad-spectrum MMP inhibition does not replicate these effects, presumably due perturbation of both beneficial and detrimental MMPs (summarized in Fig. 2). Therefore, it is acknowledged and necessary to generate and deploy inhibitors which harbor restricted specificity towards selected MMPs, including MMP-12 and MMP-13, to facilitate transition to man—particularly in the context of atherosclerotic plaque stabilization.

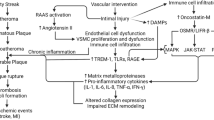
Divergent roles of MMPs in atherosclerotic plaque progression and stability. Hypothetical model of the potential beneficial and deleterious roles of MMPs and TIMPs during atherosclerotic plaque progression and rupture. Matrix metalloproteinase (MMP)-2, -3, and -9 can facilitate vascular smooth muscle cell (VSMC) migration from the media into the developing atherosclerotic plaque where they participate in fibrous cap formation and maintenance, thus promoting plaque stability. In opposition, MMP-1, MMP-8, MMP-12, MMP-13, and MMP-14 can degrade extracellular matrix proteins present in the fibrous cap whilst also encouraging the recruitment and accumulation of monocytes and macrophages, and their subsequent susceptibility to apoptosis as foam cells—which collectively enhance lipid core expansion, thrombogenicity of the plaque, and thinning of the fibrous cap. Consequently, the stability of the plaque is compromised and vulnerable to plaque rupture and ensuing thrombus formation. More recently, microRNA (miR) have been identified which can regulate MMP and TIMP expression/activity, exerting direct effects on plaque progression
References
Virmani R, Burke AP, Farb A et al (2006) Pathology of the vulnerable plaque. J Am Coll Cardiol 47(8):C13–C18
Libby P (2012) Inflammation in Atherosclerosis. Arterioscler Thromb Vasc Biol 32(9):2045–2051
Davies MJ (1996) Stability and instability: two faces of coronary atherosclerosis—the Paul Dudley White lecture 1995. Circulation 94(8):2013–2020
Hansson GK (2005) Mechanisms of disease—inflammation, atherosclerosis, and coronary artery disease. New Engl J Med 352(16):1685–1695
Newby AC (2007) Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med 17(8):253–258
Dollery CM, Libby P (2006) Atherosclerosis and proteinase activation. Cardiovasc Res 69(3):625–635
Galis ZS, Sukhova GK, Lark MW et al (1994) Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest 94:2493–2503
Li Z, Li L, Zielke HR et al (1996) Increased expression of 72-kd type IV collagenase (MMP-2) in human aortic atherosclerotic lesions. Am J Pathol 148:121–128
Henney AM, Wakeley PR, Davies MJ et al (1991) Localization of stromelysin gene expression in atherosclerotic plaques by in situ hybridization. Proc Natl Acad Sci 88:8154–8158
Halpert I, Sires UI, Roby JD et al (1996) Matrilysin is expressed by lipid-laden macrophages at sites of potential rupture in atherosclerotic lesions and localizes to areas of versican deposition, a proteoglycan substrate for the enzyme. Proc Natl Acad Sci USA 93(18):9748–9753
Herman MP, Sukhova GK, Libby P et al (2001) Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation 104:1899–1904
Schönbeck U, Mach F, Sukhova GK et al (1999) Expression of stromelysin-3 in atherosclerotic lesions: regulation via CD40-CD40 ligand signaling in vitro and in vivo. J Exp Med 189:843–853
Sukhova GK, Schonbeck U, Rabkin E et al (1999) Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation 99:2503–2509
Rajavashisth TB, Xu X-P, Jovinge S et al (1999) Membrane type 1 matrix metalloproteinase expression in human atherosclerotic plaques. Circ Res 99:3101–3109
Uzui H, Harpf A, Liu M et al (2002) Increased expression of membrane type 3-matrix metalloproteinase in human atherosclerotic plaque. Role of activated macrophages and inflammatory cytokines. Circ Res 106:3024–3030
Johnson JL (2007) Matrix metalloproteinases: influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev Cardiovasc Ther 5(2):265–282
Newby AC, Johnson JL (2005) Genetic strategies to elucidate the roles of matrix metalloproteinases in atherosclerotic plaque growth and stability. Circ Res 97(10):958–960
Murphy G (2010) Fell-Muir lecture: metalloproteinases: from demolition squad to master regulators. Int J Exp Path 91(4):303–313
Nagase H, Visse R, Murphy G (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69(3):562–573
Visse R, Nagase H (2003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases. Circ Res 93:827–839
Itoh Y, Seiki M (2006) MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol 206(1):1–8
Baker AH, Edwards DR, Murphy G (2002) Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci 115:3719–3727
Brew K, Nagase H (2010) The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. BBA-Mol Cell Res 1803(1):55–71
Golledge J, Norman PE (2010) Atherosclerosis and abdominal aortic aneurysm: cause, response, or common risk factors? Arterioscler Thromb Vasc Biol 30(6):1075–1077
Virmani R, Kolodgie FD, Burke AP et al (2000) Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol 20(5):1262–1275
Reel B, Sala-Newby GB, Huang WC et al (2011) Diverse patterns of cyclooxygenase-independent metalloproteinase gene regulation in human monocytes. Br J Pharmacol 163(8):1679–1690
Matias-Roman S, Galvez BG, Genis L et al (2005) Membrane type 1-matrix metalloproteinase is involved in migration of human monocytes and is regulated through their interaction with fibronectin or endothelium. Blood 105(10):3956–3964
May AE, Schmidt R, Bulbul O et al (2005) Plasminogen and matrix metalloproteinase activation by enzymatically modified low density lipoproteins in monocytes and smooth muscle cells. Thromb Haemostasis 93(4):710–715
Sithu SD, English WR, Olson P et al (2007) Membrane-type 1-matrix metalloproteinase regulates intracellular adhesion molecule-1 (ICAM-1)-mediated monocyte transmigration. J Biol Chem 282(34):25010–25019
Di Gregoli K, George SJ, Jackson CL et al (2016) Differential effects of tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2 on atherosclerosis and monocyte/macrophage invasion. Cardiovasc Res 109(2):318–330
Di Gregoli K, Johnson JL (2012) Role of colony-stimulating factors in atherosclerosis. Curr Opin Lipidol 23(5):412–421
Libby P, Ridker PM, Hansson GK (2009) Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol 54(23):2129–2138
Schonbeck U, Libby P (2001) CD40 signaling and plaque instability. Circ Res 89:1092–1103
Newby AC (2012) Matrix metalloproteinase inhibition therapy for vascular diseases. Vasc Pharmacol 56(5–6):232–244
Galis ZS, Sukhova GK, Libby P (1995) Microscopic localisation of active proteases by in situ zymography: detection of matrix metalloproteinase activity in vascular tissue. Faseb J 9:974–980
Johnson JL, Jackson CL, Angelini GD et al (1998) Activation of matrix-degrading metalloproteinases by mast cell proteases in atherosclerotic plaques. Arterioscler Thromb Vasc Biol 18:1707–1715
Monaco C, Gregan SM, Navin TJ et al (2009) Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation 120(24):2462–2469
Johnson JL, Jenkins NP, Huang W-C et al (2014) Relationship of MMP-14 and TIMP-3 expression with macrophage activation and human atherosclerotic plaque vulnerability. Mediat Inflamm 2014(ID276457):17
Thomas AC, Sala-Newby GB, Ismail Y et al (2007) Genomics of foam cells and nonfoamy macrophages from rabbits identifies arginase-1 as a differential regulator of nitric oxide production. Arterioscler Thromb Vasc Biol 27:571–577
Johnson JL, Sala-Newby GB, Ismail Y et al (2008) Low tissue inhibitor of metalloproteinases 3 and high matrix metalloproteinase 14 levels defines a subpopulation of highly invasive foam-cell macrophages. Arterioscler Thromb Vasc Biol 28(9):1647–1653
Libby P, Ridker PM, Maseri A (2002) Inflammation and atherosclerosis. Circulation 105:1135–1143
Pauly RR, Passaniti A, Bilato C et al (1994) Migration of cultured vascular smooth muscle cells through a basement membrane barrier requires type IV collagenase activity and is inhibited by cellular differentiation. Circ Res 75:41–54
Haque NS, Fallon JT, Pan JJ et al (2004) Chemokine receptor-8 (CCR8) mediates human vascular smooth muscle cell chemotaxis and metalloproteinase-2 secretion. Blood 103(4):1296–1304
Mason DP, Kenagy RD, Hasenstab D et al (1999) Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle migration and alters remodeling in the injured rat carotid artery. Circ Res 85:1179–1185
Filippov S, Koenig GC, Chun T-H et al (2005) MT1-matrix metalloproteinase directs arterial wall invasion and neointima formation by vascular smooth muscle cells. J Exp Med 5:663–671
Johnson JL, Dwivedi A, Somerville M et al (2011) Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler Thromb Vasc Biol 31(9):e35–e44
Yang SW, Lim L, Ju S et al (2015) Effects of matrix metalloproteinase 13 on vascular smooth muscle cells migration via Akt–ERK dependent pathway. Tissue Cell 47(1):115–121
Kajita M, Itoh Y, Chiba T et al (2001) Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J Cell Biol 153(5):893–904
Yu WH, Woessner JF, McNeish JD et al (2002) CD44 anchors the assembly of matrilysin/MMP-7 with heparin-binding epidermal growth factor precursor and ErbB4 and regulates female reproductive organ remodeling. Gene Dev 16(3):307–323
Johnson C, Galis ZS (2004) Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler Thromb Vasc Biol 24(1):54–60
George SJ, Beeching CA (2006) Cadherin: catenin complex: a novel regulator of vascular smooth muscle cell behaviour. Atherosclerosis 188(1):1–11
Dwivedi A, Slater SC, George SJ (2009) MMP-9 and -12 cause N-cadherin shedding and thereby {beta}-catenin signalling and vascular smooth muscle cell proliferation. Cardiovasc Res 81(1):178–186
Haider N, Hartung D, Fujimoto S et al (2009) Dual molecular imaging for targeting metalloproteinase activity and apoptosis in atherosclerosis: molecular imaging facilitates understanding of pathogenesis. J Nucl Cardiol 16(5):753–762
Johnson JL, Devel L, Czarny B et al (2011) A selective matrix metalloproteinase-12 inhibitor retards atherosclerotic plaque development in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol 31(3):528–535
Johnson JL, Baker AH, Oka K et al (2006) Suppression of atherosclerotic plaque progression and instability by tissue inhibitor of metalloproteinase-2: involvement of macrophage migration and apoptosis. Circulation 113(20):2435–2444
Davies MJ (2000) The pathophysiology of acute coronary syndromes. Heart 83(3):361–366
Davies MJ (2001) Going from immutable to mutable atherosclerotic plaques. Am J Cardiol 88(4A):2F–9F
Falk E, Nakano M, Bentzon JF et al (2013) Update on acute coronary syndromes: the pathologists’ view. Eur Heart J 34(10):719–728
Davì G, Patrono C (2007) Platelet activation and atherothrombosis. New Engl J Med 357(24):2482–2494
Amento EP, Ehsani N, Palmer H et al (1991) Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 11(5):1223–1230
Powell WC, Fingleton B, Wilson CL et al (1999) The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr Biol 9(24):1441–1447
Geng Y-J, Libby P (2002) Progression of atheroma: a struggle between death and procreation. Arterioscler Thromb Vasc Biol 22:1370–1380
de Boer OJ, van der Wal AC, Becker AE (2000) Atherosclerosis, inflammation, and infection. J Pathol 190:237–243
Newby AC (2005) Dual role of matrix metalloproteinases (matrixins) in neointima formation and atherosclerotic plaque rupture. Physiol Rev 85(1):1–31
Plump AS, Smith JD, Hayek T et al (1992) Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 71:343–353
Nakashima Y, Plump AS, Raines EW et al (1994) ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb Vasc Biol 14(1):133–140
Ishibashi S, Brown MS, Goldstein JL et al (1993) Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest 92(2):883–893
Jackson CL, Bennett MR, Biessen EAB et al (2007) Assessment of unstable atherosclerosis in mice. Arterioscler Thromb Vasc Biol 27:714–720
Van der Laan PA, Reardon CA, Getz GS (2004) Site specificity of atherosclerosis. Arterioscler Thromb Vasc Biol 24:12–22
Lemaître V, O’Byrne TK, Borczuk AC et al (2001) ApoE knockout mice expressing human matrix metalloproteinase-1 in macrophages have less advanced atherosclerosis. J Clin Invest 107(10):1227–1234
Gough PJ, Gomez IG, Wille PT et al (2006) Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest 116(1):59–69
de Nooijer R, Verkleij CJ, von der Thuesen JH et al (2006) Lesional overexpression of matrix metalloproteinase-9 promotes intraplaque hemorrhage in advanced lesions, but not at earlier stages of atherogenesis. Arterioscler Thromb Vasc Biol 26:340–346
Liang J, Liu E, Yu Y et al (2006) Macrophage metalloelastase accelerates the progression of atherosclerosis in transgenic rabbits. Circulation 113:1993–2001
Kuzuya M, Nakamura K, Sasaki T et al (2006) Effect of MMP-2 deficiency on atherosclerotic lesion formation in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 26(5):1120–1125
Kuzuya M, Kanda S, Sasaki T et al (2003) Deficiency of gelatinase a suppresses smooth muscle cell invasion and development of experimental intimal hyperplasia. Circulation 108(11):1375–1381
Silence J, Lupu F, Collen D et al (2001) Persistence of atherosclerotic plaque but reduced aneurysm formation in mice with stromelysin-1 (MMP-3) gene inactivation. Arterioscler Thromb Vasc Biol 21:1440–1445
Johnson JL, George SJ, Newby AC et al (2005) Divergent effects of matrix metalloproteinases -3, -7, -9 and -12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci USA 102(43):15575–15580
Luttun A, Lutgens E, Manderveld A et al (2004) Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation 109(11):1408–1414
Williams H, Johnson JL, Jackson CL et al (2010) MMP-7 mediates cleavage of N-cadherin and promotes smooth muscle cell apoptosis. Cardiovasc Res 87(1):137–146
Laxton RC, Hu Y, Duchene J et al (2009) A role of matrix metalloproteinase-8 in atherosclerosis. Circ Res 105(9):921–929
Deguchi JO, Aikawa E, Libby P et al (2005) Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation 112(17):2708–2715
Schneider F, Sukhova GK, Aikawa M et al (2008) Matrix metalloproteinase-14 deficiency in bone marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation 117(7):931–939
Silence J, Collen D, Lijnen HR (2002) Reduced atherosclerotic plaque but enhanced aneurysm formation in mice with inactivation of the tissue inhibitor of metalloproteinase-1 (TIMP-1) gene. Circ Res 90:897–903
Di Gregoli K, Jenkins N, Salter R et al (2014) MicroRNA-24 regulates macrophage behavior and retards atherosclerosis. Arterioscler Thromb Vasc Biol 34(9):1990–2000
Stöhr R, Cavalera M, Menini S et al (2014) Loss of TIMP3 exacerbates atherosclerosis in ApoE null mice. Atherosclerosis 235(2):438–443
Rouis M, Adamy C, Duverger N et al (1999) Adenovirus-mediated overexpression of tissue inhibitor of metalloproteinase-1 reduces atherosclerotic lesions in apolipoprotein E-deficient mice. Circulation 100:533–540
Prescott MF, Sawyer WK, Von Linden-Reed J et al (1999) Effect of matrix metalloproteinase inhibition on progression of atherosclerosis and aneurysm in LDL receptor-deficient mice overexpressing MMP-3, MMP-12, and MMP-13 and on restenosis in rats after balloon injury. Ann NY Acad Sci 878:179–190
Johnson JL, Fritsche-Danielson R, Behrendt M et al (2006) Effect of broad-spectrum matrix metalloproteinase inhibition on atherosclerotic plaque stability. Cardiovasc Res 71(3):586–595
Manning MW, Cassis LA, Daugherty A (2003) Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 23:483–488
Brown DL, Desai KK, Vakili BA et al (2004) Clinical and biochemical results of the metalloproteinase inhibition with subantimicrobial doses of doxycycline to prevent acute coronary syndromes (MIDAS) pilot trial. Arterioscler Thromb Vasc Biol 24:733–738
Axisa B, Loftus IM, Naylor AR et al (2002) Prospective, randomized, double-blind trial investigating the effect of doxycycline on matrix metalloproteinase expression within atherosclerotic carotid plaques. Stroke 33:2858–2864
Quillard T, Tesmenitsky Y, Croce K et al (2011) Selective inhibition of matrix metalloproteinase-13 increases collagen content of established mouse atherosclerosis. Arterioscler Thromb Vasc Biol 31(11):2464–2472
Ha M, Kim VN (2014) Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15(8):509–524
Kim K-H, Jung J-Y, Son ED et al (2015) miR-526b targets 3[prime] UTR of MMP1 mRNA. Exp Mol Med 47:e178
Lee J, Lim S, Song B-W et al (2015) MicroRNA-29b inhibits migration and proliferation of vascular smooth muscle cells in neointimal formation. J Cell Biochem 116(4):598–608
Jing W, Jiang W (2015) MicroRNA-93 regulates collagen loss by targeting MMP3 in human nucleus pulposus cells. Cell Prolif 48(3):284–292
Jin H, Wang W (2015) MicroRNA-539 suppresses osteosarcoma cell invasion and migration in vitro and targeting matrix metallopeptidase-8. Int J Clin Exp Pathol 8(7):8075–8082
Yu Y, Wang L, Liu T et al (2015) MicroRNA-204 suppresses trophoblast-like cell invasion by targeting matrix metalloproteinase-9. Biochem Biophys Res Commun 463(3):285–291
Wang Y-Y, Li H, Wang X-H et al (2016) Probucol inhibits MMP-9 expression through regulating miR-497 in HUVECs and apoE knockout mice. Thromb Res 140:51–58
Meng F, Zhang Z, Chen W et al (2016) MicroRNA-320 regulates matrix metalloproteinase-13 expression in chondrogenesis and interleukin-1β-induced chondrocyte responses. OARS 24(5):932–941
Li Y, Kuscu C, Banach A et al (2015) miR-181a-5p inhibits cancer cell migration and angiogenesis via downregulation of matrix metalloproteinase-14. Cancer Res 75(13):2674–2685
Zhang H, Qi M, Li S et al (2012) microRNA-9 targets matrix metalloproteinase 14 to inhibit invasion, metastasis, and angiogenesis of neuroblastoma cells. Mol Cancer Ther 11(7):1454–1466
Son DJ, Kumar S, Takabe W et al (2013) The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat Commun 4
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Di Gregoli, K., Johnson, J.L. (2017). Role of Matrix Metalloproteinases in the Development and Progression of Atherosclerosis. In: Chakraborti, S., Chakraborti, T., Dhalla, N. (eds) Proteases in Human Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-10-3162-5_20
Download citation
DOI: https://doi.org/10.1007/978-981-10-3162-5_20
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3161-8
Online ISBN: 978-981-10-3162-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)