
Abstract
The aquatic environment has long been a sink for diverse chemicals released as a result of human activities. In order to assess the associated risks for the aquatic life, both the chemicals’ occurrences (exposure) as well as their effects in the living organisms (hazard) need to be known. These two aspects are studied by environmental chemistry and toxicology, respectively. Mass spectrometry has been successfully applied in both fields, as it can be used not only for measuring the pollutants in different environmental compartments, but also for gaining novel insights into the molecular mechanisms of toxicity. We here discuss the application of mass spectrometry in environmental chemistry and toxicology, illustrated by several case studies drawn from research carried out at our institute.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


Keywords
- Wiley Mass Spectral Database
- Global Proteome
- Effect-directed Analysis (EDA)
- Lake Brienz
- Hydrophilic Interaction Chromatography (HILIC)
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
10.1 Introduction
Environmental sciences have evolved from being able to detect a limited set of environmental contaminants found at relatively high concentrations to where thousands of targets can be detected in a data-independent way. Examples for the first case are accidents that were catastrophic for the people involved, but raised awareness of the imminent risk of certain industrial procedures. One example is the Seveso disaster of 1976, where a runaway reaction, caused by overheated steam, led to the accidental release of the highly toxic dioxin 2,3,7,8-tetrachlorodibenzodioxin (TCDD) in kg amounts [1]. Another industrial accident, considered the world’s worst, happened in 1984 in Bhopal, India, when water leaking into a tank containing methyl isocyanate initiated an exothermic reaction that led to the release of 40 tons of methyl isocyanate, killing over 3000 people within a day [2]. Finally, in 1986 a fire in a storehouse for chemicals at Schweizerhalle in Northern Switzerland and the subsequent run-off of firefighting water contaminated the River Rhine from Basel to its estuary near Rotterdam in The Netherlands, wiping the ecosystem clean of life along long stretches [3]. All three disasters produced high concentrations of pollutants in a very short time and were caused by neglected safety features or design errors, such as a missing retention basin. The Schweizerhalle case was the only one involving a multitude of initially unknown chemicals potentially responsible for the observed toxic effects. Both, the Seveso and the Schweizerhalle incident kick-started the development of highly specific and sensitive mass spectrometers. For instance, in order to accurately quantify the dioxin congeners of differing toxicity in the presence of other polychlorinated compounds (such as polychlorinated biphenyls – PCBs), mass spectrometers providing a resolution m/Δm of better than 10,000 were required. At that time double-focusing sector field instruments were the only commercially available mass spectrometers capable of such a feat.
Another limitation was given by the fact that target analytes had to be ionized by electron ionization (EI), the most commonly used technique in the 1970s and 1980s. This limited the range of chemicals amenable for mass spectrometry (MS) analysis to those being volatile, i.e. having a mass typically below 1000 Dalton, and being thermally stable. Only in the 1990s, with the advent of electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI), the field of MS applications expanded towards polar and ionic target analytes. In addition to that, ESI extended the accessible mass range by orders of magnitude, due to the fact that it produces multiply charged species. This in turn provided the possibility to analyze biological macromolecules such as enzymes, allowing for instance to investigate stress response of organisms on the molecular level [4, 5].
Figure 10.1 illustrates the fields of application of gas chromatography (GC)-MS and liquid chromatography (LC)-MS in a biased personal representation. Of course the polar part can be partially addressed with GC-MS after derivatization.

The “Universe of Chemicals” as seen from the point of view of a mass spectrometry-practicing environmental scientist (author’s biased personal representation). The lower left part of the diagram can be analyzed using GC-MS, while the polar and ionic high molecular weight compounds have only become accessible with soft desorption ionization techniques such as electrospray, typically used in LC-MS
10.2 The Universe of Chemicals
The “Universe of Chemicals” shown schematically in Fig. 10.1 consists of millions of substances. According to the Chemical Abstracts Service (CAS – an operating division of the American Chemical Society) the 100 millionth CAS registry number was assigned on June 29, 2015 [6]. Over 300,000 of the 100 million registered chemicals are inventoried/regulated substances. Of course, not all of these will be necessarily present in the aquatic environment. However, even if it is only a fraction of these 300,000 plus regulated chemicals that pose a risk, the number is likely still beyond what can routinely be monitored simultaneously. In addition to the sheer size of the chemical universe, it also covers a wide range of physical-chemical properties, from apolar with logKow > 5, to polar and ionic, from volatile to non-volatile. Furthermore, the speciation of a polar or ionic species depends on pH and potentially other (charged) chemicals present in the matrix, thus strongly influencing sorption onto sediments, interaction with natural organic matter and uptake into aquatic organisms [7]. To simultaneously capture this multitude of chemicals, mixed-mode enrichment [8, 9] and separation [10] are required. A combination of four different separation mechanisms has been achieved by coupling two commercial mixed-mode columns, containing C18 that also acts like hydrophilic interaction chromatography (HILIC) down to 70% organic, and either weak anion (WAX) or weak cation exchanger phases (WCX – see Fig. 10.2). An environmental extract is injected onto a C18 trap, which captures hydrophobic compounds, while anionic or cationic chemicals pass through to the WAX-WCX columns.

Schematic of the multimode chromatographic setup. (a) Sample loading followed by a HILIC (hydrophilic interaction chromatography) and IEX (ion exchange) run; 3–21 min. (b) Reversed phase (RP) run after switching of the divert valve; 21–36 min. The flow rate of the isocratic delivery pump was reduced to 0 after 3 min. (c) Gradient program – Eluent A: Acetonitrile (ACN) with 3% H2O, 3 mM NH4HCO3 (pH 7.3), Eluent B: H2O with 10% ACN, 30 mM NH4HCO3 (pH 7.3) ([10], reproduced with permission)
They are then separated by a simultaneous gradient for HILIC, anion and cation exchange chromatography [10], caused by the increasing ionic strength in the eluents (see Fig. 10.2c).
After switching of the divert valve, this is then followed by a classical reversed-phase gradient. This unique setup allows the simultaneous separation of hydrophobic and ionic species (see Fig. 10.3).

Typical multimode separation chromatogram obtained from 18 standard compounds; 1 phenylalanine, 2 ascorbate, 3 galacturonic acid, 4 glutamate, 5 cystine, 6 hexanoic acid, 7 glutathione, 8 glucose-1-phosphate, 9 glutathione disulfide, 10 lysine, 11 tryptophan, 12 sucralose, 13 fluconazole, 14 arginine, 15 cysteine, 16 clotrimazole, 17 tocopherol, 18 dodecyl sulfate ([10], reproduced with permission)
For the environmental toxicologist the question however remains how best to identify the chemicals present in a very complex mixture that cause adverse effects in an ecosystem. One approach is to develop a hypothesis of what causes the effect and then target the chemicals known to be linked to it. Hence, when it was observed that brown trout catches had declined by 60% over a period of 20 years in Switzerland (see Fig. 10.4), the most popular hypothesis was that endocrine-disrupting compounds, specifically estrogens, were affecting reproductive success, thus lowering the population levels of brown trout as a consequence.

Observed decline in brown trout catches in Switzerland [12]
However, the following investigation lasting from 1998 to 2003 showed that (xeno)estrogens could be made potentially responsible for reduced fish fertility and hatching success only at hotspots, e.g. downstream of wastewater treatment plants (WWTP), where they indeed could be detected at effect-causing levels by chemical analysis. In the end, it turned out that one of the main reasons for the observed declining catches was deteriorated habitat quality [11].
10.3 Effect-Directed Analysis
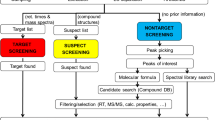
Another popular strategy, called Effect-Directed Analysis (EDA), is to prioritize for chemical analysis only the samples or fractions thereof that show an effect in an in vitro or in vivo assay (see Fig. 10.5). This allows greatly reducing the number of samples that need to be chemically analyzed. In the ideal case the measured activity can be explained by the chemicals found to be present in the sample. For example, in a Swiss survey of rivers, estradiol equivalents determined in the yeast estrogen screen (YES) were shown to correlate with estradiol equivalents calculated based on determined concentrations and relative potencies of 17ß-estrogen (E2), estrone (E1) and 17α-ethinlyestradiol (EE2), both in grab samples and extracts from passive samplers [13].

Schematic representation of the Effect-Directed Analysis (Adapted from W. Brack [14])
The same is true for tissues and body fluids as shown in the case of muscle and bile from white fish (Corregonus lavaratus) in an alpine lake in Switzerland. Here too, estrogenicity determined in the YES could be explained by the natural estrogens E2 and E1 [15]. Similarly, the main cause for estrogenicity measured in the River Thames could be attributed to 17ß-estradiol [16], and in another project, triclosan was identified as being responsible for growth inhibition observed in green algae [17].
Although this method is straightforward when established targeted analysis is used as in the case of the estrogens mentioned above, the investigation becomes much more challenging as soon as targeted analysis returns a blank and the identity of the effect-causing chemical remains unknown. In this case a differential analysis becomes necessary. The following case study will illustrate the procedure.
At the turn of the millennium, fishermen in the Bernese Oberland, Switzerland, observed an increased incidence of gonad malformations in white fish [18]. Potential causes could have been biological, e.g. genetic factors or infectious diseases, or related to environmental conditions such as temperature, habitat quality, food availability or of course water quality. In the search for causes, sediment, water and plankton samples were collected and analyzed for estrogenic activity. Figure 10.6 shows the estrogenic activity in plankton extracts, determined with two different assays, the YES, a reporter gene assay that measures estrogen receptor activation, and the E-Screen that measures estrogen-dependent cell proliferation. Both endpoints indicate estrogenic potential of a sample, which means that e.g. the plankton collected in Lake Brienz, August 2005 (BRI 08/05), is estrogenic. When comparing this sample to any sample that is not estrogenic, features found in the estrogenic but absent in non-estrogenic samples could potentially be the active compound. Identifying features of interest is done using LC-MS/MS and data-dependent analysis [19, 20]. With this technique, routinely used in global proteomics [5], everything eluting off the column is collisionally activated and the fragments generated detected.

In a first step the most intense signals, determined in a full scan spectrum, are selected and their MS/MS spectra acquired a few times. Following this, these signals are then put on an exclusion list to allow less intense features to be analyzed as well. Depending on the settings, this then also gives information related to signals of low intensity. Another technique, becoming increasingly popular in proteomics and other fields is scan-independent analysis or SWATH [23]. Instead of selecting a single precursor ion as in data-dependent acquisition mentioned above, an entire window of masses is collisionally activated and the corresponding fragments acquired. Assignment of the fragments to precursor ions is then done based on overlapping chromatographic profiles. By repeatedly shifting the window to higher masses the whole mass spectral information can be collected from a sample. This allows reanalyzing the data at a later stage, when new evidence leads to new hypotheses.
A differential analysis identifies compounds that have significantly different concentrations in an active versus an inactive sample. It provides accurate mass and fragment ion information in addition to the retention time. All this helps narrowing down the elemental composition of an unknown substance, identifying functional groups through MS/MS spectra, and the interaction with the separation phase, reflected in the retention time, informs on the physical-chemical properties of the chemical.
The seven golden rules established by the Fiehn group [24] tell us for instance that the mass 180.1020 singled out in Fig. 10.7 can only have a maximum of 15 carbon, 178 hydrogens, 11 oxygens atoms and 12 nitrogens, if it is composed of only this one element, which of course is chemically unlikely or impossible (Rule 1). The maximal number of elements can further be restricted based on the empirical information available from libraries (e.g. Wiley mass spectral database [25], or the dictionary of natural products [http://dnp.chemnetbase.com]). Another requirement for an elemental sum formula is that chemical structures can be drawn that fulfill the Lewis and Senior rules, or in other words that “molecules consisting of main group elements, especially carbon, nitrogen and oxygen, share electrons in a way that all atoms have completely filled s, p-valence shells” (Rule 2) [24]. The experimental data also contains isotopic information which helps filtering potential sum formulas even further (Rule 3). Additionally, from basic MS interpretation courses it is well known that when the monoisotopic ion M (M+· in EI or [M + H]+ in ESI) shows a signal with high intensity for the M + 2 isotope, at least one chlorine (>32%) or one bromine (>97%) is likely to be present. Similarly, the natural relative abundance of the other elements allows clearly assigning the natural isotopes to the higher isotope signals (see Fig. 10.8). For instance, when looking at the number of potential candidate sum formulas at mass 500, with a mass accuracy of 10 ppm, 266 candidates are possible. With an accuracy of 1 ppm, easily achievable with an Orbitrap, only 21 remain possible. When taking into account the isotopic distribution, the candidates are further narrowed down to 3 at 3 ppm and 2% isotope accuracy. An additional filter to reduce the number of potential sum formulas is the H/C ratio (Rule 4). As shown by Kind and Fiehn [24], the H/C ratio for most chemical compounds (99.7%) lies between 0.2 and 3.1, based on the Wiley mass spectral database (45′000 chemicals with a mass range of 30 to 1,500 Da, containing C, H, N, S, O, P, F, Cl, Br, I, Si). Similarly, other element ratios again based on the Wiley library help filter the data further (Rule 5): F/C 0–1.5, Cl/C and Br/C 0–0.8, N/C 0–1.3, O/C 0–1.2, P/C 0–0.3, S/C 0–0.8, Si/C 0–0.5; these ranges cover 99.7% of the library entries. Rule 6 performs an element probability check and Rule 7 only applies to GC/MS data, since it does a trimethylsilylation (TMS) check for derivatized compounds.

Differential analysis of an estrogenic plankton extract from Lake Brienz (BRI 08/05 in Fig. 10.6) versus an inactive sample using the software SIEVE (ThermoScientific, USA). The x-axis gives the significance of the difference, the y-axis the log ratio of the areas (estrogenic/inactive). The shaded area corresponds to a p-value < 0.02 and a ratio >10

Isotopic fine structure of the M + 2 isotope of ciprofloxacin ([M + H]+ = C17H19O3N3F), a fluoroquinolone antibiotic. The relative abundance of the various isotopic signals of an experimental spectrum needs to match the simulated distribution (m/Δm 10,000,000)
Not all of these rules are needed to find a sum formula that fits the ion at m/z 180.1020, since its mass is quite low (see Table 10.1). Table 10.2 lists possible candidate sum formulas for the protonated molecular ion [M + H]+ of 180.1020 Da. With a mass accuracy of 5 ppm, routinely achieved on time-of-flight instruments and Orbitraps, only seven candidates are possible, using the elements C, H, N, S, O, P, F, Cl, Br, I, and Si. C2H14ON7Si and C3H14N7S (marked with an X in Fig. 10.9) can be ruled out because of their high H/C and N/C ratios. Figure 10.9 shows the intensities of the M + 1 and M + 2 isotope peaks relative to the monoisotopic ion, of the ten candidates for ion m/z 180.1020 listed in Table 10.2. If the unknown were carrying a chlorine, the isotope pattern would be telling (see Fig. 10.9, the two points at M + 2 > 30%). What Fig. 10.9 shows is that the ten candidates can easily be distinguished based on their isotopic pattern (Rule 3), if the relative intensities of the isotopes are reproducible, which is a given with current instrumentations. But candidate 1 in Table 10.2 is not only the prime candidate based on isotopic distribution, but also because this investigation targeted estrogenic compounds. Most estrogens have a steroid structure with ring A being phenolic. This means that the double bond equivalents (DBE) must be four or higher (three for the double bonds of the aromatic system, and one for the ring).

Relative intensities of the M + 1 and M + 2 isotopes of the ten candidate sum formulas. The top candidate is circled. The crossed-out candidates did not comply with Rule 4 and 5. The two candidates containing one chlorine are clearly separated with M + 2 > 30%
This then identifies candidate 1 from Table 10.2 as the only possible candidate. A potential structure, which fits the sum formula and MS/MS data could be proposed and synthesized, but turned out to be non-estrogenic in the YES.
Even though the sum formula of the compound responsible for the estrogenic activity found in the plankton extract from Lake Brienz could unequivocally be determined, the structure remains unknown. Nonetheless, the case study presented here nicely shows the power of the EDA approach.
10.4 Mass Spectrometry for Characterizing Interactions of Chemicals with Organisms
EDA described above depends on the availability of established bioassays measuring biological responses that can be mapped onto a particular toxicity pathway leading to an apical outcome of interest. However, cellular and physiological changes occurring in response to environmentally relevant chronic exposure to low levels of contaminant mixtures can be rather subtle and not easy to pinpoint with ‘traditional’ bioassays, as these may suffer from inferior sensitivity. To develop novel bioassays for EDA that cover additional toxicity pathways, in particular those leading to sublethal effects as a result of chronic exposure, a solid understanding of how a chemical or a chemical mixture affects an organism is needed. However, for many chemicals present in the environment the understanding of their effects and mechanisms of toxicity, both individually and in a mixture, is currently incomplete.
Interactions between chemicals and living organisms are typically characterized by two aspects – toxicokinetics and toxicodynamics [27, 28]. Toxicokinetics covers everything that an organism does to and with a chemical, i.e. uptake, biotransformation, and excretion, but also internal distribution to the target sites where the chemical or its metabolite will actually exert its toxic action(s). These are addressed by toxicodynamics, i.e. what a chemical does to an organism. This includes for example, causing DNA adduct formation [29], oxidizing membrane lipids [5], or interacting with a nuclear receptor and by that triggering a molecular cascade resulting in gene expression changes and metabolite alterations [30]. MS-based techniques can be applied to obtain both toxicokinetic and toxicodynamic data.
Internal chemical concentrations of chemicals and their transformation products can be determined for biological specimens with the same approaches used for chemical measurements in environmental compartments [8, 27]. Taking samples at different time points, performing depuration experiments, or carrying out non-targeted or targeted metabolite screening allows constructing time-resolved profiles of chemical uptake, biotransformation, and excretion [31, 32]. Furthermore, MS-imaging techniques such as matrix-assisted laser desorption-ionization (MALDI) can be used to study the internal distribution of chemicals. This is done by measuring the analyte of interest on histological sections from different body parts and organs. MALDI MS imaging (MALDI MSI) has for instance been successfully applied to study the internal distribution of cocaine in zebrafish larvae. This study showed that, following waterborne exposure, the chemical distributes not only to the expected site of action, the brain, but also to other body sites, notably melanin-containing cells along the body and in the eyes [32]. These data were critical to the interpretation of behavioral changes occurring in this model organism in response to waterborne cocaine, in particular for understanding the differences in responses observed in zebrafish larvae and mammals.
When investigating the toxicodynamics of chemical interaction with the organism, in particular when looking at molecular mechanisms of toxicity, MS can provide data on gene expression and cellular signaling cascades. MS-based approaches are especially useful when looking at proteins (proteomics) and metabolites (metabolomics) [33].
On the protein level, both global and targeted MS-based techniques can be used to characterize the protein abundance as well as post-translational modifications of proteins. For example, Multidimensional Protein Identification Technology (MudPIT) allows the simultaneous characterization of several thousands of protein species as well as their alterations in response to toxicants. Thanks to the advancement of sequencing techniques and the availability of sequencing information, global proteomics analysis can now be carried out not only in the relatively well-characterized species often used in the lab, such as zebrafish (Danio rerio) [34] or green alga (Chlamydomonas reinhardtii) [5, 35], but also in field-relevant species such as mussels (Mytilus galloprovincialis) [4]. Recently, global proteomics was also used to investigate the identity of proteins forming a corona of silver nanoparticles (AgNP) in a gill cell line from rainbow trout (Oncorhynchus mykiss) [36]. MS can also be used to study the epigenome and epigenetic alterations, which are often governed by specific interactions between nucleic acids and proteins [37]. One such example are the histone proteins whose post-translational modifications play a crucial role in determining the chromatin state and hence activation or silencing of transcription in particular chromatin regions [38]. Diverse MS-based techniques are being increasingly applied to study these epigenetic marks [39]. Overall, global proteomics is well suited for obtaining initial proteome information needed to investigate diverse ecotoxicological questions in various species.
One important drawback of global proteomics is its general bias towards higher abundance proteins, as this limits its ability to characterize all relevant proteins that could play a role in a particular toxicity mechanism. Meta-analyses performed with mammalian [40] and zebrafish [41] global proteomics data have shown that many proteins commonly reported to be altered in global differential proteomics studies may in fact represent non-specific general stress responses, while more toxicant-specific responses, commonly occurring among the lower-abundance proteins, such as transcription factors or xenobiotic-metabolizing enzymes, cannot be characterized by global proteomics due to insufficient sensitivity.
Indeed, many aspects of the general stress response are shared among different species and stressors, since they represent a reaction to any form of macromolecular damage, with common features being manifested once the insult exceeds a certain threshold, regardless of the causative stressor [43]. These features may include gross adjustments in energy metabolism, degradation of cellular components, but also upregulation of defense and damage repair mechanisms. However, in parallel or often even preceding the general stress response developing with an ensuing damage, many other, potentially more stressor-specific responses do occur in the cell. These include rapid gene expression changes, activation or inhibition of specific enzymes or transporters, recruitment of transcription factors, or alterations in the levels of signaling molecules. These multiple toxicity pathways specifically activated in response to different stressors, have a tendency to converge on a few common general stress responses, which will be manifested once the damage becomes too high.
When approached from the gene expression point of view (see Fig. 10.10), the responses that can be grouped under the general stress response umbrella may be considered as markers of effect (e.g. consequences of damaging influences occurring on the cellular level, such as oxidative stress or inhibition of ATP synthesis), while those responses that are stressor-specific may be denoted as markers of exposure. It is this group of responses that may allow gaining detailed insights into specific mechanisms of action for each toxicant, allowing to distinguish between different toxicant groups, but also to predict their toxicity and potency [42]. Meta-analysis of global proteomics studies performed in zebrafish has shown that these datasets tend to over-represent the proteins belonging to the general stress response group, and under-represent those that can be considered to be involved in more specific responses [41]. This bias is a consequence of the overall bias of global proteomics towards better detection of higher abundance proteins. Thus, compared to transcriptomics and sequencing technologies that have a superior sensitivity and can thus deliver information on both markers of exposure and markers of effect, the most commonly applied MS-based proteomics techniques (i.e. global proteomics) appear capable of providing comprehensive information only for the latter group.

Applicability of transcriptomics and proteomics for studying chemical effects on gene expression (Schematic representation of gene expression trajectories adapted from Van Straalen and Feder [42])
However, information on mRNA expression is not a reliable proxy for protein expression [5] due to multiple additional levels of control coming into play after the mRNA synthesis [44]. Therefore, the improvement of sensitivity and versatility of high-throughput proteomics approaches is a crucial prerequisite for improving our ability to study and understand molecular mechanisms of toxicity. Fortunately, sensitivity for protein detection can be significantly improved by using targeted proteomics. With this technique, specific MS/MS transitions of peptides representative of target proteins of interest are monitored, allowing to focus on a few peptides instead of attempting to simultaneously measure all analytes present. Due to significantly shorter MS run durations compared to global approaches, targeted proteomics can also be used to measure panels of select proteins across multiple samples and conditions, thus providing detailed comparative data for proteins of interest. For example, targeted proteomics was used to characterize expression of multiple proteins potentially involved in sex determination and differentiation in zebrafish, at multiple time points during gonad differentiation [45]. Targeted proteomics has also been used to monitor a panel of proteins expected to respond to a particular chemical class, for example glucocorticoids [46], in search of biomarkers of exposure or effects of these chemicals. Furthermore, targeted proteomics methods have been developed for glutathione-S-transferases to study their involvement and significance for phase II biotransformation of xenobiotics in zebrafish [47]. Despite these advances, the sensitivity of MS-based proteomics applied in environmental toxicology currently remains suboptimal and may require further technical advancements, such as the employment of laser microdissection or cellular sorting techniques to allow collecting and analyzing cells of one type, or incorporation of dedicated enrichment workflows to allow increasing the concentration of analytes of interest.
It is commonly accepted that any physiological change in the organism in response to a stressor is accompanied by certain molecular and biochemical alterations, and in fact it can be viewed as an apical adverse outcome of such alterations. Hence, obtaining a better understanding of molecular responses may allow linking to adverse outcomes at higher levels of organization, which may even provide the means for predicting such outcomes. This assumption forms the basis of the adverse outcome pathway (AOP) concept [48] which deals with organizing the knowledge on the progression of toxicity through multiple levels of biological organization, starting with a molecular initiating event and culminating in an adverse outcome of regulatory relevance, occurring on the organism or population level [49]. Data generated by various ‘-omics’ techniques can be used to hypothesize on the mechanisms of action, helping to construct putative AOPs potentially triggered by certain toxicants [50]. However, it is important to realize that a mere demonstration of certain molecular changes is not enough for an AOP, and experimental studies should be designed in a way allowing to understand not only the molecular alterations but ideally also the apical outcomes of relevance perceived to affect the fitness and survival chances of the whole organism [48].
10.5 Conclusions
In conclusion, MS-based techniques have become essential in environmental toxicology and chemistry, (i) for determining chemicals in environmental and biological compartments, (ii) for identifying unknown chemical stressors using effect-directed analysis, scan-dependent and –independent techniques and (iii) for helping to fill the knowledge gaps regarding the molecular mechanisms of toxicity and apical effects caused by a certain chemical. Thus, MS plays an important role in both fundamental and applied research in environmental toxicology, supporting environmental risk assessment and management.
References
Bertazzi PA, Bernucci I, Brambilla G, Consonni D, Pesatori AC (1998) The Seveso studies on early and long-term effects of dioxin exposure: a review. Environ Health Perspect 106:625–633
Eckermann I (2005) The Bhopal saga – causes and consequences of the world's largest industrial disaster. Universities Press, Hyderabad
Giger W (2009) The Rhine red, the fish dead – the 1986 Schweizerhalle disaster, a retrospect and longterm impact assessment. Environ Sci Pollut Res 16:S98–S111
Oliveira IB, Groh KJ, Stadnicka-Michalak J, Schonenberger R, Beiras R, Barroso CM, Langford KH, Thomas KV, Suter MJ-F (2016) Tralopyril bioconcentration and effects on the gill proteome of the Mediterranean mussel Mytilus galloprovincialis. Aquat Toxicol 177:198–210
Pillai S, Behra R, Nestler H, Suter MJ-F, Sigg L, Schirmer K (2014) Linking toxicity and adaptive responses across the transcriptome, proteome, and phenotype of Chlamydomonas reinhardtii exposed to silver. Proc Natl Acad Sci 111:3490–3495
C.A. Service, CAS assigns the 100 millionth CAS registry number® to a substance designed to treat acute myeloid leukemia, in 2015
Schwarzenbach RP, Escher BI, Fenner K, Hofstetter TB, Johnson CA, von Gunten U, Wehrli B (2006) The challenge of micropollutants in aquatic systems. Science 313:1072–1077
Ammann AA, Macikova P, Groh KJ, Schirmer K, Suter MJ-F (2014) LC-MS/MS determination of potential endocrine disruptors of cortico signalling in river and waste water. Anal Bioanal Chem 406:7653–7665
Kern S, Baumgartner R, Helbling DE, Hollender J, Singer H, Loos MJ, Schwarzenbach RP, Fenner K (2010) A tiered procedure for assessing the formation of biotransformation products of pharmaceuticals and biocides during activated sludge treatment. J Environ Monit 12:2100–2111
Ammann AA, Suter MJ-F (2016) Multimode gradient high performance liquid chromatography mass spectrometry method applicable to metabolomics and environmental monitorin. J Chromatogr A 1456:145–151
Burkhardt-Holm P, Giger W, Güttinger H, Ochsenbein U, Peter A, Scheurer K, Segner H, Staub E, Suter MJ-F (2005) Where have all the fish gone? The reasons why fish catches in Swiss rivers are declining. Environ Sci Technol 35:441A–447A
Friedl C (1999) Fischfangrückgang in schweizerischen Fliessgewässern. Swiss Agency Environ For Landscape (SAFEL) 32
Vermeirssen ELM, Korner O, Schoenenberger R, Suter MJ-F, Burkhardt-Holm P (2005) Characterization of environmental estrogens in river water using a three pronged approach: active and passive water sampling and the analysis of accumulated estrogens in the bile of caged fish. Environ Sci Technol 39:8191–8198
Brack W (2003) Effect-directed analysis: a promising tool for the identification of organic toxicants in complex mixtures? Anal Bioanal Chem 337:397–407
Liedtke A, Schoenenberger R, Eggen RIL, Suter MJ-F (2009) Internal exposure of whitefish (Coregonus lavaretus) to estrogens. Aquat Toxicol 93:158–165
Thomas KV, Hurst MR, Matthiessen P, Waldock MJ (2001) Characterization of estrogenic compounds in water samples collected from United Kingdom estuaries. Environ Toxicol Chem 20:2165–2170
Bandow N, Altenburger R, Streck G, Brack W (2009) Effect-directed analysis of contaminated sediments with partition-based dosing using green algae cell multiplication inhibition. Environ Sci Technol 43:7343–7349
Bernet D, Wahli T, Kueng C, Segner H (2004) Frequent and unexplained gonadal abnormalities in whitefish (central alpine coregonus sp.) from an alpine oligotrophic lake in Switzerland. Dis Aquat Org 61:137–148
Schymanski EL, Singer HP, Longrée P, Loos M, Ruff M, Stravs MA, Ripollés Vidal C, Hollender J (2014) Strategies to characterize polar organic contamination in wastewater: exploring the capability of high resolution mass spectrometry. Environ Sci Technol 48:1811–1818
Singer HP, Wössner A, McArdell CS, Fenner K (2016) Rapid screening for exposure to “non-target” pharmaceuticals from wastewater effluents by combining HRMS-based suspect screening and exposure modeling. Environ Sci Technol 50:6698–6707
Liedtke A, Schoenenberger R, Eggen RIL, Suter MJ-F (2009) unpublished results
Soto AM, Sonnenschein C, Chung KL, Fernandez MF, Olea N, Serrano FO (1995) The e-screen assay as a tool to identify estrogens – an update on estrogenic environmental-pollutants. Environ Health Perspect 103:113–122
Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11: PMID 22261725
Kind T, Fiehn O (2007) Seven golden rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics 8:1–20
Wiley, Wiley Registry of Mass Spectral Data, 2016
Kind T, Fiehn O (2006) Metabolomic database annotations via query of elemental compositions: mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics 7:234
Hidasi AO, Groh KJ, Suter MJ-F, Schirmer K (2017) Clobetasol propionate causes immunosuppression in zebrafish (Danio rerio) at environmentally relevant concentrations, ecotox. Ecotox Environ Safe 138:16–24
Ashauer R, Agatz A, Albert C, Ducrot V, Galic N, Hendriks J, Jager T, Kretschmann A, O'Connor I, Rubach MN, Nyman AM, Schmitt W, Stadnicka J, van den Brink PJ, Preuss TJ (2011) Toxicokinetic-toxicodynamic modeling of quantal and graded sublethal endpoints: a brief discussion of concepts. Environ Toxicol Chem 30:2519–2524
Madureira DJ, Weiss FT, Van Midwoud P, Helbling DE, Sturla SJ, Schirmer K (2014) Systems toxicology approach to understand the kinetics of benzo(a)pyrene uptake, biotransformation, and DNA adduct formation in a liver cell model. Chem Res Toxicol 27:443–453
Mottaz H, Schoenenberger R, Fischer S, Eggen RIL, Schirmer K, Groh KJ (2017) Dose-dependent effects of morphine on lipopolysaccharide (LPS)-induced inflammation, and involvement of multixenobiotic resistance (MXR) transporters in LPS efflux in teleost fish. Environ Pollut 221:105–115
Di Paolo C, Groh KJ, Zenneg M, Vermeirssen E, Murk A, Eggen RIL, Hollert H, Werner I, Schirmer K (2015) Early life exposure to PCB126 results in delayed mortality and growth impairment in the zebrafish larvae. Aquat Toxicol 169:168–178
Kirla KT, Groh KJ, Steuer AE, Poetzsch M, Banote RK, Stadnicka-Michalak J, Eggen RI, Schirmer K, Kraemer T (2016) Zebrafish larvae are insensitive to stimulation by cocaine: importance of exposure route and toxicokinetics. Toxicol Sci 154:183–193
Groh KJ, Suter MJ-F (2014) Mass spectrometry in environmental toxicology. Chimia (Aarau) 68:140–145
Groh KJ, Nesatyy VJ, Segner H, Eggen RI, Suter MJ-F (2011) Global proteomics analysis of testis and ovary in adult zebrafish (Danio rerio). Fish Physiol Biochem 37:619–647
Nestler H, Groh KJ, Schoenenberger R, Eggen RI, Suter MJ-F (2012) Linking proteome responses with physiological and biochemical effects in herbicide-exposed Chlamydomonas reinhardtii. J Proteome 75:5370–5385
Yue Y, Behra R, Sigg L, Suter MJ-F, Pillai S, Schirmer K (2016) Silver nanoparticle-protein interactions in intact rainbow trout gill cells. Environ Sci Nano 3:1174–1185
Vandegehuchte MB, Janssen CR (2011) Epigenetics and its implications for ecotoxicolog. Ecotoxicology 20:607–624
Bannister AJ, T. Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21: 381-395
Brunner AM, Tweedie-Cullen RY, Mansuy IM (2012) Epigenetic modifications of the neuroproteome. Proteomics 12:2404–2420
Petrak J, Ivanek R, Toman O, Cmejla R, Cmejlova J, Vyoral D, Zivny J, Vulpe CD (2008) Deja vu in proteomics: a hit parade of repeatedly identified differentially expressed proteins. Proteomics 8:1744–1749
Groh KJ, Suter MJ-F (2015) Stressor-induced proteome alterations in zebrafish: a meta-analysis of response patterns. Aquat Toxicol 159:1–12
van Straalen NM, Feder ME (2012) Ecological and evolutionary functional genomics–how can it contribute to the risk assessment of chemicals? Environ Sci Technol 46: 3-9
Kultz D (2005) Molecular and evolutionary basis of the cellular stress response. Annu Rev Physiol 67:225–257
Schwanhaeusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2012) Global quantification of mammalian gene expression control. Nature 473:337–342
Groh KJ, Schoenenberger R, Eggen RI, Segner H, Suter MJ-F (2013) Analysis of protein expression in zebrafish during gonad differentiation by targeted proteomics. Gen Comp Endocrinol 193:210–220
Hidasi AO (2016) Personal communication, Eawag
Tierbach A (2016) Personal communication, Eawag
Groh KJ, Carvalho RN, Chipman JK, Denslow ND, Halder M, Murphy CA, Roelofs D, Rolaki A, Schirmer K, Watanabe KH (2015) Development and application of the adverse outcome pathway framework for understanding and predicting chronic toxicity: I. Challenges and research needs in ecotoxicology. Chemosphere 120:764–777
Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, Mount DR, Nichols JW, Russom CL, Schmieder PK, Serrano PK, Tietge JE, Villeneuve DL (2010) Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem 29:730–741
Sturla SJ, Boobis AR, FitzGerald RE, Hoeng J, Kavlock RJ, Schirmer K, Whelan M, Wilks MF, Peitsch MC (2014) Systems toxicology: from basic research to risk assessment. Chem Res Toxicol 27:314–329
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media B.V.
About this paper
Cite this paper
Groh, K.J., Suter, M.J.F. (2017). Mass Spectrometry in Environmental Chemistry and Toxicology. In: Banoub, J., Caprioli, R. (eds) Molecular Technologies for Detection of Chemical and Biological Agents. NATO Science for Peace and Security Series A: Chemistry and Biology. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-1113-3_10
Download citation
DOI: https://doi.org/10.1007/978-94-024-1113-3_10
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-1112-6
Online ISBN: 978-94-024-1113-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)