
Abstract
The identification of pathways that regulate longevity in an evolutionarily conserved manner is a major focus of modern biogerontology. Interest in the sirtuin family of deacetylases/deacylases/ADP-ribosyltransferases began with the observation that increased expression of Sir2 extends replicative lifespan in budding yeast. The seven mammalian sirtuin homologs have been the focus of intense scrutiny for their potential impacts on health- and lifespan. Here we review studies of the mammalian sirtuin SIRT6. SIRT6 plays multiple roles in metabolic homeostasis and genome integrity through modification of histones and other protein targets; consequently SIRT6 suppresses many age-associated pathologies such as neoplasia, cardiac hypertrophy, and glucose intolerance. SIRT6 overexpression results in extended lifespan in male mice, suggesting that SIRT6 may represent a true functional ortholog of yeast Sir2, and supporting an evolutionarily conserved role for sirtuins in longevity.
*Author contributed equally with all other contributors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
7.1 Introduction
Aging is a conserved but poorly understood biological phenomenon. In diverse invertebrate and mammalian model organisms , advancing age is associated with functional decline and impaired stress resistance. In humans, most common afflictions – type 2 diabetes (T2D) , cardiovascular disease , cancer , and neurodegeneration , among many others – are strongly associated with advancing age . Conversely, genetic and environmental interventions that promote increased longevity typically also delay or even prevent many age-associated pathologies . Therefore, an understanding of molecular mechanisms of aging offers the possibility of improved treatments for many common diseases. This realization has led to a hunt for pathways that regulate health- and lifespan, at least some of which function in a conserved manner across different phyla.
Sirtuins are a conserved family of NAD+-dependent deacetylases/deacylases/ADP-ribosyltransferases that promote longevity in budding yeast , mammalian healthspan, and, in the case of at least two sirtuins (SIRT1 and SIRT6 ), extended lifespan in mice. The founding member of this family is Silent Information Regulator 2 (Sir2) , a budding yeast protein that promotes chromatin silencing to regulate genomic stability and increase replicative lifespan (Longo et al. 2012). Mammals possess seven sirtuins, termed SIRT1-SIRT7 (Frye 2000). Mammalian sirtuins share a conserved central catalytic domain, but differ at their N- and C-termini, domains that help to confer upon these proteins divergent biological properties. In this chapter, we focus on SIRT6 , a mammalian sirtuin with a wide spectrum of biological functions, and its roles in suppressing diseases of aging (summarized in Fig. 7.1).

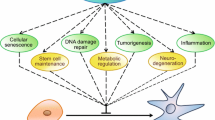
Suppression of disease and promotion of healthspan by SIRT6 . SIRT6 improves overall healthspan by impacting diverse physiological processes. SIRT6 crystal structure was obtained from RCSB Protein Data Bank (Pan et al. 2011) (Figure was produced using images from Servier Medical Art (www.servier.com))
SIRT6 is a predominately nuclear protein that associates with heterochromatin (Liszt et al. 2005; Michishita et al. 2005; Mostoslavsky et al. 2006; Xiao et al. 2010). However, SIRT6 and the invertebrate homologue SIR-2.4 has been observed in the cytoplasm , where they are critical for formation of cytoplasmic stress granules (Jedrusik-Bode et al. 2013; Michishita et al. 2005; Simeoni et al. 2013). Biochemically, SIRT6 functions as a lysine deacetylase , a mono-ADP-ribosyltransferase , and a long-chain fatty acid deacylase (Jiang et al. 2013; Liszt et al. 2005; Mao et al. 2011; Michishita et al. 2008). In vitro studies suggest that SIRT6 possesses very modest deacetylase activity (Pan et al. 2011). However, in vivo SIRT6 functionally deacetylates at least four targets: two histone sites (H3K9 and H3K56) (Michishita et al. 2008; Michishita et al. 2009; Yang et al. 2009), and two non-histone proteins (CtIP and GCN5) (Dominy et al. 2012; Kaidi et al. 2010). SIRT6 mono-ADP ribosylates PARP1 (poly (ADP-ribose) polymerase 1) to promote DNA repair (Mao et al. 2011), and the nuclear corepressor protein, KAP1 (KRAB-associated protein 1), facilitating the interaction of KAP1 with HP1α to maintain the silent heterochromatic state of repetitive DNA (Van Meter et al. 2014). SIRT6 promotes secretion of the pro-inflammatory cytokine TNFα, via removal of long-chain fatty acyl groups in this protein (Jiang et al. 2013).
Initial insights into SIRT6 function derived from studies of SIRT6-deficient cells and mice (Mostoslavsky et al. 2006). SIRT6-deficient fibroblasts and ES cells show poor growth and genomic instability . Sirt6 knockout (KO) mice appear normal at birth but soon manifest growth retardation, perhaps in part due to very low serum IGF1 levels. These animals display a premature aging-like phenotype and show severe pleiotropic defects , indicating that SIRT6 impacts multiple processes important for organismal health (summarized in Fig. 7.2). They are frail, with a hunched posture (lordokyphosis ) and lose most of their white adipose tissue (WAT) , including subcutaneous fat. At approximately two weeks of age, blood glucose levels begin to decline precipitously in Sirt6 KO animals. These mice also show rapid depletion of lymphocytes via a massive wave of apoptosis. The latter phenotype represents a systemic rather than a cell-autonomous defect , as SIRT6-deficient bone marrow cells are able to efficiently reconstitute the lymphocyte compartment of irradiated wild-type recipient mice (Mostoslavsky et al. 2006). Clearly, SIRT6 plays crucial roles in cellular and organismal homeostasis.

Schematic overview of SIRT6 functions . Through its deacetylase, mono-ADP-ribosyltransferase, and deacylase activities, SIRT6 affects activities of transcription factors and other proteins, causing alterations in key cellular processes (black boxes). Through these activities, SIRT6 suppresses multiple metabolic and age-associated pathologies (ovals)
As described below, there has been significant recent progress in elucidating molecular functions of SIRT6. A common theme that has emerged is that SIRT6 negatively regulates the transcriptional output of key cellular signaling pathways by deacetylating histones at their target promoters. This is in contrast to SIRT1 , the most well-studied sirtuin family member. Though SIRT1 can also deacetylate histones , it exerts many of its functions by directly deacetylating transcription factors and other non-histone targets themselves (Guarente 2011). Much of the more recent functional analysis of SIRT6 has been carried out using tissue-specific Sirt6 KO animals , avoiding the early postnatal lethality associated with global SIRT6 deficiency , and permitting a finer dissection of SIRT6’s roles. In this chapter we review various functions of SIRT6, and potential impacts of this protein on human disease.
7.2 SIRT6 Is a Key Metabolic Regulator
As noted above, SIRT6 is required for maintenance of glucose homeostasis , a role critical for organismal survival. Two different Sirt6 germline KO mouse strains have been described (Mostoslavsky et al. 2006; Xiao et al. 2010). Both show severe hypoglycemia and greatly reduced serum IGF1 levels. On a pure 129SvJ strain background, under standard husbandry conditions, SIRT6 deficiency results in completely penetrant postnatal lethality by one month of age (Mostoslavsky et al. 2006). However, in an outbred background , a minority of SIRT6-deficient mice survives this hypoglycemia and lives into adulthood (Xiao et al. 2010). Glucose supplementation of these SIRT6-deficient mice further improves their survival, proving that hypoglycemia is a major cause of death in the absence of SIRT6 during postnatal development.
Recent work has elucidated multiple roles for SIRT6 in glucose homeostasis . SIRT6 controls blood glucose levels by regulating at least three distinct pathways: HIF-1α signaling , insulin/IGF-like signaling (IIS) , and gluconeogenesis (Dominy et al. 2012; Xiao et al. 2010; Xiong et al. 2013; Zhong et al. 2010). HIF-1α is a major metabolic regulator ; under low oxygen or glucose conditions, HIF-1α promotes a shift from oxidative metabolism to glycolysis (Koh and Powis 2012). HIF-1α promotes expression of multiple genes encoding proteins in the glycolytic cascade; conversely, HIF-1α drives increased expression of PDK1, in turn inhibiting carbon flow into mitochondria. The glucose transporter GLUT1 is another key HIF-1α target. SIRT6 functions as a repressor of HIF-1α transcriptional output by deacetylating H3K9 at the promoters of HIF-1α target genes, and also by reducing overall HIF-1α levels (the latter through mechanisms that remain obscure). Hence, in the absence of SIRT6, elevated HIF-1α activity results in increased glycolysis and glucose uptake, most evident in skeletal muscle and brown adipose tissue, culminating in hypoglycemia (Zhong et al. 2010). Increased glycolytic gene expression has also been identified in the livers of hepatic-specific Sirt6 KOs (Kim et al. 2010).
In addition to GLUT1, SIRT6-deficient mice show increased abundance of the glucose transporter GLUT4 at the cell membrane (Xiao et al. 2010). Unlike GLUT1, GLUT4 translocation to the plasma membrane is promoted by IIS. Despite lower serum insulin and IGF1 levels in Sirt6 mutant mice, IIS is actually much more active in these animals (Sundaresan et al. 2012; Xiao et al. 2010). This may reflect the role for SIRT6 as a co-repressor of c-JUN, since c-JUN promotes expression of many genes involved in IIS (Sundaresan et al. 2012). c-JUN is a component of the activator protein 1 (AP-1) transcription factor , which is involved in diverse processes such as apoptosis, cell proliferation and development (Dunn et al. 2002).
SIRT6 also suppresses hepatic gluconeogenesis by deacetylating the acetyltransferase GCN5, activating it to acetylate PGC-1α (Dominy et al. 2012). PGC-1α is a transcriptional co-activator that functions as a regulator of mitochondrial biogenesis and other metabolic processes (Puigserver and Spiegelman 2003). In the liver, PGC-1α promotes gluconeogenesis by co-activation of FoxO1 and Hnf4 (Puigserver et al. 2003; Rhee et al. 2003). Acetylation of PGC-1α inhibits PGC-1α transcriptional activity (Rodgers et al. 2005). Hence, via activation of GCN5 (Lerin et al. 2006), SIRT6 inhibits hepatic glucose production (Dominy et al. 2012). The tumor suppressor p53 may also serve as an important regulator of gluconeogenesis, via regulation of a SIRT6/FOXO1 axis. P53 induces hepatic SIRT6 expression. SIRT6, in turn, promotes nuclear exclusion of the FOXO1 transcription factor, and subsequent downregulation of genes involved in gluconeogenesis. Knockdown of hepatic Sirt6 expression renders mice resistant to the hypoglycemic effects of p53 (Zhang et al. 2014). The twin roles of SIRT6 in suppressing glycolysis while simultaneously inhibiting hepatic glucose output might superficially seem at odds with one another. These functions might be rationalized as a means to avoid futile cycling of glucose production and breakdown (Dominy et al. 2012).
The relationship between SIRT1 and SIRT6 in the context of PGC-1α acetylation is also intriguing. SIRT1 deacetylates PGC-1α to activate its transcriptional function, thus opposing the activity of SIRT6/GCN5 (Rodgers et al. 2005). Consistent with antagonistic functions of these sirtuins, protein kinase A (PKA) suppresses SIRT6 activity while stimulating SIRT1 function (Dominy et al. 2010; Gerhart-Hines et al. 2007; Nin et al. 2012). However, these findings seem inconsistent with the published role of SIRT1 in stimulating SIRT6 expression (Kim et al. 2010). Further studies are required to elucidate the precise interplay between SIRT1 and SIRT6 in response to varied dietary conditions.
In addition to this role in glucose homeostasis , SIRT6 also controls hepatic fatty acid metabolism by regulating expression of genes involved in this process, via H3K9 deacetylation at target promoters. SIRT6 suppresses accumulation of triglycerides in hepatocytes by inhibiting fatty acid uptake and synthesis, while promoting their breakdown via β-oxidation (Kim et al. 2010). A number of genes involved in these processes are regulated by the nuclear receptor PPARγ . PPARγ induces expression of genes that regulate lipid metabolism and adipocyte differentiation (Zhang et al. 2013a), including angiopoietin-like protein 4 (ANGPTL4) and adipocyte fatty acid binding protein (A-FABP) . ANGPTL4 negatively regulates lipoprotein lipase , which hydrolyzes serum triglycerides into free fatty acids, and thus mediates triglyceride clearance from the blood (Kim et al. 2010). A-FABP is a chaperone for cytosolic fatty acids , elevated levels of which are associated with obesity and metabolic syndrome (Xu et al. 2006). Furthermore, SIRT6 binds to the promoter of DGAT1, a key enzyme in triglyceride synthesis, repressing its expression (Kim et al. 2010). Through this mechanism, SIRT6 protects against fatty liver formation in response to a high fat diet (HFD) . SIRT6 also inhibits pancreatic inflammation under these dietary conditions (Kanfi et al. 2010). Human fatty livers exhibit lower levels of SIRT6, and liver specific Sirt6 knockout mice develop fatty liver and hypercholesterolemia, in particular LDL cholesterol (Tao et al. 2013a; Wu et al. 2014). Conversely, overexpression of SIRT6 protects the liver from excessive lipid accumulation (Kanfi et al. 2010) and lowers serum LDL cholesterol in response to a HFD by recruiting FOXO3 and repressing transcription of Pcks9 and Srebf1/2, major regulators of cholesterol homeostasis (Elhanati et al. 2013; Tao et al. 2013b). The SREBPs (sterol regulatory element binding proteins) are transcription factors that bind the sterol regulatory element DNA sequence, and are required for cholesterol and fatty acid biosynthesis (Tao et al. 2013b). In addition to suppressing their expression, SIRT6 indirectly suppresses SREBP1/2 cleavage into their active forms , and SIRT6 inactivates SREBP1 through enhancing its phosphorylation (Elhanati et al. 2013). SIRT6 directly interacts with the circadian proteins CLOCK (circadian locomotor output cycles kaput) and ARNTL (aryl hydrocarbon receptor nuclear translocator-like, or BMAL ) to modulate their recruitment to chromatin, thereby controlling circadian-dependent metabolism , including fatty acid synthesis and β-oxidation through inhibition of SREBP-1-mediated transcription of target genes (Masri et al. 2014).
Given the roles of SIRT6 in glucose and lipid homeostasis, it is perhaps not surprising that it plays protective roles against obesity and T2D, both common age-associated pathologies (Dominy et al. 2012; Schwer et al. 2010). Brain-specific Sirt6 KO mice become obese in adulthood, associated with reduced levels of pituitary growth hormone and the hypothalamic factors proopiomelanocortin (POMC) , single-minded homolog 1 (SIM1) and brain-derived neurotrophic factor (BDNF) . These factors have all been linked to obesity in humans. SIRT6 deficiency in the brain causes hyperacetylation of H3K9 and H3K56, possibly leading to dysregulation of these and likely many other genes (Schwer et al. 2010). Furthermore, ectopic expression of SIRT6 in a mouse model of diabetes reduces hepatic glucose output and normalizes serum glucose levels (Dominy et al. 2012). Thus, roles of SIRT6 in regulating obesity-associated gene expression and glucose and lipid metabolism might conceivably be exploited therapeutically. It will be of great interest to assess roles for SIRT6 in other metabolically active tissues such as skeletal muscle, adipose tissue, and pancreatic β-cells. Similarly, it remains an outstanding question whether the depletion of WAT observed in global Sirt6 KOs indicates a primary role for SIRT6 in maintaining WAT, or a secondary consequence of hypoglycemia and overall disordered metabolism in these animals.
7.3 Regulation of SIRT6
Despite its central role in maintenance of metabolic homeostasis , relatively little is known regarding regulation of SIRT6 expression and enzymatic activity. Like other sirtuins, SIRT6 requires the metabolic cofactor NAD+ for activity. In response to fasting or long term calorie restriction, SIRT6 protein levels are elevated in brain, heart and WAT (Kanfi et al. 2008; Kim et al. 2010), promoting a metabolic switch from glycolysis to oxidative phosphorylation (Dominy et al. 2012; Kim et al. 2010; Xiao et al. 2010; Zhong et al. 2010). However, there is conflicting evidence regarding the underlying mechanism of altered Sirt6 expression. SIRT6 levels are decreased in livers of obese and diabetic mice (Dominy et al. 2012). Conversely, Sirt6 mRNA levels rise in liver and subcutaneous fat in response to severe weight loss; possibly due to decreased inflammation as TNFα can suppress Sirt6 expression (Moschen et al. 2013). Likewise, SIRT6 protein levels increase in response to caloric restriction. Kanfi and colleagues reported that SIRT6 protein levels, but not mRNA levels, rise in response to fasting, due to stabilization of the SIRT6 protein (Kanfi et al. 2008). However, Kim and coworkers found that induction of Sirt6 during fasting occurs transcriptionally and requires SIRT1 (Kim et al. 2010). They found that SIRT1 deacetylates FOXO3A to allow FOXO3A to form a complex with NRF1 and induce Sirt6 gene expression (Kim et al. 2010). As noted above, PKA also inhibits SIRT6 expression, while simultaneously increasing SIRT1 levels (Dominy et al. 2010; Gerhart-Hines et al. 2007; Nin et al. 2012). The ubiquitin ligase CHIP (carboxyl terminus of Hsp70-interacting protein) ubiquitinates and consequently stabilizes SIRT6, by preventing SIRT6’s interaction with other ubiquitin ligases (Ronnebaum et al. 2013). Likewise, SIRT6 is protected from proteasomal degradation by the ubiquitin-specific peptidase USP10 (Lin et al. 2013).
SIRT1 is regulated by a complex network of interactors and post-translational modifications (Revollo and Li 2013); analogously SIRT6 activity is governed by means other than regulation of expression levels. Physiological concentrations of free fatty acids increase SIRT6’s catalytic activity in vitro (Feldman et al. 2013). SIRT6 is post-translationally modified in response to both oxidative and nitrosative stress, and the catalytic activity of recombinant SIRT6 is stimulated upon nitration of tyrosine 257 (Hu et al. 2015). A recent study suggested that SIRT6 is more active when interacting with nucleosomes than on isolated histones (Gil et al. 2013).
Other studies report alterations of SIRT6 levels and/or activity under various pathological conditions; however it remains unclear for the most part how this occurs mechanistically. In one case, recent data (discussed below) suggest that altered c-JUN and c-FOS signaling inhibit Sirt6 expression during induction of hepatocellular carcinoma (Min et al. 2012). SIRT6 associates with nuclear chromatin and upon stress induction, e.g. TNFα treatment, SIRT6 relocalizes dynamically to different promoters (Kawahara et al. 2011). Similarly, SIRT6 relocalizes to sites of DNA damage, perhaps via interaction with DNA repair machinery (McCord et al. 2009). The interaction of SIRT6 with chromatin appears to be mediated at least in part by transcription factors. Kawahara and coworkers demonstrated that SIRT6 and RELA bind to a large panel of common promoter sites of genes involved in processes such as cell cycle progression, immune system development, suppression of apoptosis, and glycolysis. At a large fraction of these promoter sites, binding of SIRT6 was dependent on RELA (Kawahara et al. 2011). Also, SIRT6-bound promoters were enriched for several transcription factor motifs, notably SP1, STAT1/3 and FOXO1/4 (Kawahara et al. 2011) and thus it is reasonable to hypothesize that those transcription factors may also be necessary to recruit SIRT6 to these specific promoters in response to stimuli. Conversely, numerous post-translational modifications present on SIRT6 that have been identified by mass spectrometry (cf. www.phosphosite.org), whose functions have not yet been elucidated, could regulate the interaction of SIRT6 with specific transcriptional activators and/or repressors.
7.4 SIRT6 Regulates Inflammation
Increased sterile inflammation is a common feature of aging in many mammalian tissue types (Agrawal et al. 2009; Agrawal et al. 2010). Evidence from global and tissue-specific Sirt6 KOs suggests that SIRT6 has important roles in limiting the inflammatory response. As part of their overall degenerative syndrome, SIRT6-deficient mice develop severe colitis with erosion of the intestinal mucosa (Mostoslavsky et al. 2006). In outbred SIRT6-deficient mice that survive hypoglycemic crisis, inflammation develops in the liver, where it eventually leads to fibrosis, as well as, to a lesser extent, in the kidneys, pancreas and lung (Xiao et al. 2012). In the context of hepatic inflammation, using tissue-specific knockouts, it was shown that SIRT6 in lymphocytes and macrophages, but not in hepatocytes, is required to suppress this phenotype. Liver inflammation coincides with increased expression of numerous pro-inflammatory genes , including Mcp- 1 and IL-6, in Kuppfer cells and T-cells . Mechanistically, SIRT6 binds the transcription factor c-JUN at the promoters of these pro-inflammatory genes, where it deacetylates acH3K9 and inhibits c-JUN transcriptional output (Xiao et al. 2012).
In addition to c-JUN, SIRT6 also inhibits the transcriptional output of NF-kB signaling resulting in decreased expression of genes involved in aging (Kawahara et al. 2009). NF-kB is a family of transcription factors implicated in multiple processes such as inflammation, cell death, proliferation, and development. The NF-kB protein family consists of five members: RELA (p65), RELB, c-REL, p50 and p52, among which RELA interacts with SIRT6 . Under basal conditions, they are retained in the cytoplasm by an inhibitor of NF-kB (IkB) family member. In response to diverse stimuli, such as the inflammatory cytokine TNFα, NF-kB is released and consequently translocates to the nucleus where it can activate expression of its target genes . The transcriptional output of NF-kB is dependent on various co-regulators and chromatin modulators, including SIRT6 (Wan and Lenardo 2010). SIRT6 attenuates NF-kB mediated gene expression by deacetylating histone H3 lysine K9 (H3K9) at the promoters of NF-kB target genes (Kawahara et al. 2009). Reduced NF-kB signaling can partially rescue the lethality of SIRT6 deficiency (Kawahara et al. 2009). Therefore, factors other then hypoglycemia may contribute to the lethality of SIRT6 deficiency.
Overexpression of SIRT6 can reduce arthritis in a collagen-induced arthritis mouse model by blocking NF-kB transcriptional output and consequently diminishes secretion of pro-inflammatory cytokines (Lee et al. 2013). SIRT6 may also mediate the inflammatory response through the pro-inflammatory cytokine TNFα, however their interplay is somewhat complex. Xiao and coworkers showed that TNFα protein levels are elevated in SIRT6-deficient macrophages under both basal and lipopolysaccharide stimulated conditions (Xiao et al. 2012). Conversely, Van Gool and colleagues reported that SIRT6 increases TNFα protein levels at a post-transcriptional level, indicating that under some conditions, SIRT6 may actually promote secretion of pro-inflammatory cytokines (Van Gool et al. 2009; Xiao et al. 2012). A recent study revealed that SIRT6 stimulates TNFα secretion by removing long-chain fatty acyl groups of lysine 19 and 20 in this protein (Jiang et al. 2013). Treatment of HeLa cells with TNFα increases SIRT6 translocation to NF-kB/RELA target promoters (Kawahara et al. 2009). Further studies are needed to clarify interactions between TNFα and SIRT6; it is possible that a negative feedback loop exists in which SIRT6 inhibits TNFα from stimulating NF-kB mediated transcription of pro-inflammatory genes.
In addition to roles for SIRT6 in modulating c-JUN and NF-kB function, the function of SIRT6 in metabolism may be relevant in its suppression of inflammation. Both lymphocytes and macrophages shift their metabolism from respiration to aerobic glycolysis upon activation (Ardawi and Newsholme 1982; Garedew and Moncada 2008). Conversely, cells that limit inflammation, such as regulatory T-cells , show relatively low levels of glycolysis (O’Neill and Hardie 2013). Therefore, it is possible that increased glycolysis occurring in the absence of SIRT6 preferentially drives activation of pro-inflammatory cells.
Overall, most studies have identified roles for SIRT6 in suppressing inflammation. Based on known functions of SIRT6, it is possible that SIRT6 activators might be useful in treating age-associated chronic inflammatory diseases such as diabetes, and cardiovascular and autoimmune diseases.
7.5 SIRT6 Promotes Genomic Stability via Diverse Mechanisms
Initial studies of SIRT6-deficient cells revealed that SIRT6 plays a major role in genome integrity (Mostoslavsky et al. 2006). Sirt6 KO cells show reduced proliferation, an elevated incidence of chromosomal abnormalities, and increased sensitivity to DNA damaging agents. Originally it was hypothesized that SIRT6 might play a role in base excision repair (BER) , pathways that repair small DNA lesions , including those induced by oxidative insult (Parsons and Dianov 2013). This hypothesis was based on the spectrum of sensitivities of SIRT6-deficient cells, as well as the ability of the catalytic domain of Polβ , the major polymerase involved in BER, to rescue the genotoxin sensitivity associated with SIRT6 deficiency (Mostoslavsky et al. 2006). In support of an involvement of SIRT6 in BER, overexpression of SIRT6 can suppress oxidative DNA damage in porcine fetal fibroblasts , possibly by enhancing BER (Xie et al. 2012). SIRT6 protein levels dramatically decrease in human fibroblasts from older donors. SIRT6 is able to rescue the acquired BER defect in human fibroblasts from aged donors in a plasmid-reporter assay of BER efficiency. Rescue of the BER defect is dependent upon PARP1, and requires both the deacetylase and the ADP-ribosyltransferase activities of SIRT6 (Xu et al. 2015).
In contrast to the limited mechanistic insight into how SIRT6 facilitates BER, there has been significant progress made in understanding how SIRT6 promotes DNA double strand break (DSB) repair . DNA DSBs represent a severe threat to cell viability. They are repaired via three major pathways: classical non-homologous end-joining (C-NHEJ) , homologous recombination (HR) , and alternative end-joining (A-EJ) (Boboila et al. 2012). Overexpression of SIRT6 increases clearance of γH2AX foci and accelerates overall DSB repair , via multiple mechanisms (Mao et al. 2011). A recent study showed that, in response to DNA insult, SIRT6 is recruited to the breakage site where it interacts with the helicase SNF2H. This complex remodels chromatin around the DSB and recruits various DNA repair factors such as BRCA1 and 53BP1 (Toiber et al. 2013). SIRT6 stimulates both C-NHEJ and HR by activating PARP1 (Mao et al. 2011). PARP1 binds and stabilizes broken DNA ends, and mediates the recruitment of other DNA repair factors. In this context, ectopically-expressed SIRT6 can rescue the decline of HR capacity associated with replicative exhaustion (Mao et al. 2012). SIRT6 is required for optimal recruitment of the C-NHEJ factor DNA-PKcs to DNA DSB breaks, an effect potentially occurring via modulation of local chromatin structure by SIRT6 (McCord et al. 2009). SIRT6 promotes HR by deacetylating and activating CtIP, a factor required for DNA end resection to generate ssDNA for initiation of HR (Kaidi et al. 2010).
SIRT6 also deacetylates acH3K56 (Michishita et al. 2009; Yang et al. 2009). H3K56 acetylation levels are normally very low in mammalian cells , but increase dramatically in the setting of SIRT6 deficiency (Yang et al. 2009). Improper regulation of H3K56 acetylation in mammalian cells leads to impaired cell cycle progression, sensitivity to genotoxins and spontaneous DNA damage (Michishita et al. 2009; Yang et al. 2009; Yuan et al. 2009), phenotypes reminiscent of SIRT6 deficiency. The exact mechanism(s) through which deacetylation of acH3K56 by SIRT6 might repress DNA damage accumulation are not fully understood. In wild-type cells, SIRT6 is among the initial factors recruited to the damage site, where it rapidly deacetylates H3K56 (Toiber et al. 2013). It has been hypothesized that H3K56 acetylation is asymmetric around the replication fork during S-phase: acetylated behind the fork on newly synthesized DNA, and non-acetylated ahead of the replication fork. Thus, H3K56 acetylation may allow newly-replicated DNA to be distinguished from template DNA for proper targeting of HR during S-phase (Munoz-Galvan et al. 2013). H3K56 acetylation is also required for chromatin reassembly following DSB repair (Chen et al. 2008) and for recovery from post-repair checkpoint arrest following UV irradiation (Battu et al. 2011). In SIRT6-deficient cells , which show dramatically elevated levels of H3K56 acetylation, it is possible that any or all of these processes may be perturbed. It should be noted however that SIRT6-deficient cells are not hypersensitive to UV, despite their dramatic H3K56 hyperacetylation (Mostoslavsky et al. 2006).
In addition to these roles for SIRT6 in promoting global genome stability, SIRT6 in human cells plays a role in stabilizing telomeres specifically. In normal human cells , SIRT6 deacetylates H3K9 and H3K56 at telomeres to promote telomeric heterochromatinization and association of telomeric binding proteins (Michishita et al. 2009; Tennen et al. 2011). Hyperacetylation of telomeric chromatin in the absence of SIRT6 disrupts the interaction of telomeric regions with WRN, a protein involved in telomere maintenance, which is mutated in the premature aging-like disorder Werner Syndrome . Telomeric attrition is a major cause of replicative senescence in human cells; indeed SIRT6 depletion in human fibroblasts causes premature cellular senescence (Michishita et al. 2009; Tennen et al. 2011). Furthermore, SIRT6 is essential for maintaining telomere position effect (TPE) (Tennen et al. 2011), a phenomenon by which telomere-proximal genes are silenced. TPE is lost with replicative aging in yeast. This role of SIRT6 is reminiscent of the function of yeast Sir2 in promoting heterochromatinization of the rDNA array and telomeric regions to suppress recombination and promote increased replicative lifespan (Dang et al. 2009; Longo and Kennedy 2006). In contrast, despite the fact that SIRT6 also deacetylates H3K9 at telomeres in mouse cells, telomeres are much longer in laboratory mice than humans, and thus do not apparently display dysfunction upon SIRT6 deletion (Michishita et al. 2008). It will be of interest to determine whether SIRT6 has roles in stabilizing other non-telomeric heterochromatic loci in mammalian cells. Overall, SIRT6 plays many roles through which it can maintain the genomic integrity of the cell .
7.6 SIRT6 Extends Mammalian Lifespan
Interest in the sirtuin protein family in the context of the biology of aging began with the observation that Sir2 overexpression in budding yeast extends longevity in this organism (Kaeberlein et al. 1999). Therefore, the finding that SIRT6 overexpression increases median and maximal lifespan in male (but not female) mice on an C57BL6/J-BALB/cOlaHsd mixed background represents an extremely significant milestone in sirtuin biology (Kanfi et al. 2012). The mechanisms underlying this effect are not entirely clear. Sahin and colleagues observed an age-dependent increase in methylation of the human SIRT6 promoter, suggesting that SIRT6 expression may decrease with age (a hypothesis that has not yet been directly tested) (Sahin et al. 2014). In this regard, an extra Sirt6 gene copy might conceivably compensate for a normal age-associated reduction in SIRT6 levels. Reduced IGF1 levels are observed in male SIRT6 overexpressors; reductions in IIS are associated with increased lifespan in mice as well as invertebrates (Kenyon 2010; Lombard and Miller 2014). However, this effect is typically more pronounced in female animals, whereas the impact of SIRT6 overexpression on lifespan is seen in male mice only. It is likely that other functions of SIRT6 may be relevant for its pro-longevity role. A higher incidence of spontaneous tumors is observed in male mice compared to females (Kanfi et al. 2012); thus, a tumor suppressor role of SIRT6 might explain why lifespan extension is only observed in male SIRT6-overexpressors (Lombard and Miller 2012).
Other roles of SIRT6 may also be relevant for its pro-longevity effects. The functions of SIRT6 in DNA repair , maintenance of genomic integrity and epigenetic silencing could contribute to increased longevity of SIRT6-overexpressing male mice. As described above, through its histone deacetylase activity , SIRT6 attenuates activities of HIF-1α and NF-kB. Both of these factors have been implicated in regulating aging. Data on HIF-1α and longevity are somewhat controversial. Deletion of HIF-1α can extend lifespan in C. elegans by inhibiting IIS (Zhang et al. 2009). However, others have reported that overexpression of HIF-1α causes lifespan extension, possibly by reducing mitochondrial respiration and thus ROS production, and/or by acting as a stress response factor (Mehta et al. 2009). Hence, both deletion and overexpression of HIF-1α may have beneficial, context-dependent effects, and it is possible that increased levels of SIRT6 could cause lifespan extension by inhibiting HIF-1α. Moreover, pharmacological inhibition of NF-kB signaling can extend lifespan in both male and female Drosophila (Moskalev and Shaposhnikov 2011). NF-kB activity increases with age in mammals, promoting increased tissue inflammation (Baker et al. 2011). Blocking the age-associated increase in NF-kB levels in the skin of aged mice reverts the gene expression profile to that observed in young animals (Adler et al. 2007). Inhibition of NF-kB signaling in the mammalian hypothalamus increases mouse lifespan (Zhang et al. 2013b). Therefore it is possible that SIRT6 overexpression might attenuate age-associated NF-kB-mediated inflammation, helping to preserve tissue function and/or centrally-regulated organismal metabolic homeostasis.
7.7 SIRT6 Suppresses Cardiac Hypertrophy and Promotes Cardiac Stress Resistance
We now turn to a discussion of SIRT6’s roles in modulating age-associated disease. Cardiac hypertrophy is a condition characterized by cardiac enlargement, occurring either physiologically in response to normal stimuli such as pregnancy or exercise, or as a consequence of disease states (the latter referred to as pathological hypertrophy ) . Even in the absence of overt stress stimuli , thickening of the ventricular wall occurs with age. In humans, hypertension is a frequent cause of pathological cardiac hypertrophy . Age-associated cardiac hypertrophy is characterized by loss of cardiomyocytes, interstitial fibrosis, and hypertrophy of the remaining cells. Pathological cardiac hypertrophy and consequent ventricular dysfunction is thought to be a mostly irreversible process, which can eventually result in cardiac failure (Dai et al. 2012; Olivetti et al. 2000).
Mice induced to develop cardiac hypertrophy have elevated NF-kB activity in cardiomyocytes , and inhibition of RELA in these mice can prevent this phenotype (Gupta et al. 2008). Likewise, overexpression of SIRT6 ameliorates hypertrophy in vitro and inhibits the increase in hypertrophic marker genes in cardiomyocytes by repressing NF-kB gene expression (Yu et al. 2013). SIRT6 expression in rat cardiac fibroblasts is upregulated in response to pro-fibrotic stimuli: pressure overload or angiotensin II (Ang II) treatment . Cells treated with Sirt6 siRNA display hallmarks of myofibroblast differentiation, such as increased cell proliferation, extracellular matrix deposition and higher expression α-smooth muscle actin (α-SMA) . SIRT6 overexpression reverses α-SMA expression in Sirt6 KD or Ang II–treated cardiac fibroblasts. NF-kB DNA binding and transcriptional activities are enhanced upon SIRT6 depletion. Pharmaceutical or siRNA-mediated inhibition of NF-kB reversed the expression of myofibroblast markers of differentiation, indicating that SIRT6 inhibits NF-kB signaling to block differentiation of cardiac fibroblasts (Tian et al. 2014). Sundaresan and colleagues showed that a large reduction in SIRT6 levels occurs in cardiac hypertrophy in both human and mouse hearts (Sundaresan et al. 2012). In contrast, Yu and coworkers reported that SIRT6 levels are elevated in cardiac hypertrophy in rats, but that this coincides with decreased SIRT6 activity due to a concomitant reduction in NAD+ levels (Yu et al. 2013). Whole-body Sirt6 KO and cardiomyocyte-specific Sirt6 KO mice spontaneously develop cardiac hypertrophy as early as two months post-partum, characterized by increased cardiomyocyte size, degenerative cellular changes, and increased expression of aging-associated cytoskeletal proteins, as well as fibrotic and apoptotic markers. Conversely, SIRT6 overexpression protects animals against induction of cardiac hypertrophy (Sundaresan et al. 2012). In the absence of SIRT6, both c-JUN and NF-kB are hyperactive, and silencing either of these transcription factors can prevent hypertrophy in vitro (Gupta et al. 2008; Sundaresan et al. 2012; Yu et al. 2013).
The underlying mechanism(s) through which SIRT6 promotes cardiac health are not fully understood. Sundaresan and colleagues reported that SIRT6 protects against cardiac hypertrophy by inhibiting IIS, and Sirt6 KO hearts showed elevated expression of proteins involved in this pathway. Furthermore, pharmacological inhibition of IGF1 signaling was able to protect Sirt6 KO mice from development of cardiac hypertrophy (Sundaresan et al. 2012). Conversely, SIRT6 overexpression was able to decrease the expression of these proteins in vivo. Likewise, both mouse and human hypertrophic hearts showed increased levels of phosphorylated AKT and IGFR in comparison to controls, indicative of hyperactivity of the IIS pathway. Indeed, suppression of IIS in Drosophila can prevent the age-associated decline in cardiac performance. However, the role of IIS in cardiac health may be at odds with this mechanism, as previous studies have reported that age-dependent heart failure is associated with low serum IGF1 levels in elderly with no history of heart disease (Dai et al. 2012), and treatment of cardiomyocytes with a locally-produced IGF1 isoform can protect these cells from hypertrophy in a SIRT1 dependent manner (Vinciguerra et al. 2010). Therefore, the hypertrophy observed in Sirt6 KO mice might not mimic age-related cardiac hypertrophy, and possibly other age-associated changes in elderly hearts could contribute to the opposing effects of IIS on cardiac health.
7.8 SIRT6 Is an Intestinal Tumor Suppressor
An increased incidence of neoplasia is a major feature of aging in mammals. SIRT6’s functions in regulating glucose homeostasis, genomic stability and cellular senescence have prompted multiple groups to assess roles for SIRT6 in cancer. Recent work has revealed that SIRT6 functions as a tumor suppressor, at least in part by modulating cellular metabolism and cellular proliferation (Sebastian et al. 2012). SIRT6-deficient mouse embryonic fibroblast cell lines, immortalized by serial passage or p53 knockdown, show increased proliferation relative to controls. In contrast to controls, these cells are able to form colonies in vitro and tumors in vivo. These results are consistent with a cell-autonomous tumor suppressor role for SIRT6. Furthermore, conditional deletion of Sirt6 in intestinal epithelial cells in the Apc min adenomatosis model results in a 3-fold increase in the number of tumors, which are larger and of higher grade than lesions in littermate controls. SIRT6 mRNA levels are reduced in human colorectal cancers (CRCs) relative to normal tissue, and the SIRT6 gene is deleted in a substantial proportion of human cancer cell lines. These data support a tumor suppressor role for SIRT6 in mice, and potentially in humans as well (Sebastian et al. 2012).
Molecularly, SIRT6 deficiency leads to increased levels of glycolytic gene expression. Ribosomal biogenesis and glutaminase expression are also elevated in the absence of SIRT6 ; these are regulated by the proto-oncogene c-MYC . Mechanistically, SIRT6 interacts with c-MYC and deacetylates H3K56 at the promoters of c-MYC target genes, attenuating their expression. Inhibition of c-MYC in the absence of SIRT6 reduces cellular proliferation and inhibits tumor growth, indicating that SIRT6 acts as a tumor suppressor at least in part by inhibiting c-MYC activity (Sebastian et al. 2012).
In addition to targeting c-MYC, SIRT6 inhibits HIF-1α transcriptional activity resulting in decreased glycolysis (Sebastian et al. 2012; Zhong et al. 2010). Cancer cells , and other rapidly dividing cell types, shift their energy production from mitochondrial respiration to glycolysis and lactate production (Warburg effect) (Warburg 1956). Upon SIRT6 ablation, increased HIF-1α activity causes reprogramming of cellular metabolism by enhancing glucose uptake and glycolysis, conferring tumorigenic potential upon SIRT6-deficient cells (Sebastian et al. 2012). Conversion of pyruvate to lactate is the rate-limiting step in glycolysis, and blocking this step enhances mitochondrial respiration, reduces proliferation and inhibits colony formation of SIRT6-deficient cells. Treatment of adenoma-prone SIRT6-deficient mice with dichloroacetate (DCA) , a small molecule that promotes mitochondrial respiration , reverts increased tumorigenesis of these mice in vivo (Sebastian et al. 2012). Finally, lower SIRT6 protein expression in locally metastatic CRCs is associated with increased propensity for relapse. Consistent with this finding, reduced SIRT6 expression has been reported in hepatocellular carcinomas (HCC) , and low levels of SIRT6 are associated with more rapid recurrence in patients with this disease. Furthermore, liver-specific Sirt6 KO mice show elevated expression of HCC biomarkers (e.g. AFP, IGF2 and H19) and re-expression of SIRT6 in HCC cell lines sensitizes these cells to chemotherapeutic drugs resulting in increased apoptosis (Marquardt et al. 2013). This indicates that decreased SIRT6 expression in some tumors is correlated with more aggressive clinical behavior.
SIRT6 plays tumor suppressor functions in other contexts as well. Overexpression of SIRT6 has been reported to induce apoptosis specifically in cancer cells via activation of p53 or p73 (Van Meter et al. 2011). The tumor suppressor p53 is mutated or inactivated in most human cancers. Both p53 and its homolog p73 are involved in cell cycle regulation and induction of apoptosis (Murray-Zmijewski et al. 2006). Additionally, SIRT6 can prevent tumor formation in liver cells by blocking RELA-mediated expression of survivin to promote cell survival (Min et al. 2012). Survivin is a member of the inhibitor of apoptosis (IAP) family and inhibits cell death. It is mainly expressed during embryogenesis and, with the exception of a few cell types , is not normally expressed in adult tissue (Church and Talbot 2012). In human dysplastic liver nodules, c-JUN interferes with c-FOS transcriptional output. As Sirt6 is a target of c-FOS, c-JUN thereby inhibits Sirt6 expression. Decreased SIRT6 levels correlate with increased acH3K9 levels at the survivin promoter, allowing for increased NF-kB-driven expression of this gene (Min et al. 2012). Survivin is only upregulated during the initiation phase of liver cancer, as survivin levels were not altered in normal livers or in advanced hepatic carcinomas (Min et al. 2012). Furthermore, a recent study showed that SIRT6 mRNA levels are decreased in human non-small cell lung cancer (NSCLC) , and that overexpression of SIRT6 in lung cancer cell lines decreased cellular proliferation by inhibiting the expression of the oncogenic transcription factor TWIST1 (Han et al. 2014). Finally, in breast cancer cell lines, SIRT6 is phosphorylated by AKT and consequently degraded by MDM2. Inhibition of MDM2-mediated degradation of SIRT6 suppresses cellular proliferation, and low levels of phosphorylated SIRT6 (Ser 338) are positively correlated with breast cancer patient survival (Thirumurthi et al. 2014). These findings indicate that SIRT6 functions as a tumor suppressor in multiple tissues, and suggest that SIRT6 activators could be useful therapeutic tools for cancer treatment, potentially in both early-stage and advanced lesions.
In contrast to the tumor suppressor function of SIRT6 in colorectal and hepatic carcinomas (Khongkow et al. 2013; Marquardt et al. 2013; Sebastian et al. 2012) elevated SIRT6 levels are associated with chemotherapeutic drug resistance in MCF-7 breast cancer cells (Khongkow et al. 2013). While SIRT6 expression is elevated in several human tumor types (Bauer et al. 2012; Khongkow et al. 2013; Liu et al. 2013), SIRT6 knockdown specifically in prostate cancer cells sensitizes them to chemotherapeutic agents and decreases cellular survival (Liu et al. 2013). Deacetylation of the tumor suppressor proteins FOXO3A and p53 by SIRT6, resulting in reduced cell cycle arrest and apoptosis in response to chemotherapeutic treatment, has been proposed to explain this effect. However, importantly there currently is no direct evidence in vitro that SIRT6 can deacetylate these proteins. Khongkow and colleagues reported that high nuclear levels of SIRT6 correlated with a poor prognosis in breast cancer patients (Khongkow et al. 2013). This group also identified a fraction of SIRT6 in the cytoplasm; in contrast to nuclear SIRT6 , cytoplasmic SIRT6 levels correlate with a better clinical outcome in breast cancer. It will be of interest to determine the identities of the cytoplasmic proteins that SIRT6 targets in breast carcinoma cells – for example, whether SIRT6 regulates stress granule formation in this context – or alternatively whether SIRT6 localization in this compartment represents simply a means to attenuate nuclear SIRT6 function.
SIRT6 also appears to play an oncogenic role in the skin. In squamous cell carcinoma (SCC) , SIRT6 expression is negatively regulated by mIR-34a; mIR-34a promotes differentiation of benign and malignant keratinocytes , whereas SIRT6 is highly expressed in SCC and contributes to maintenance of the undifferentiated state (Lefort et al. 2013). Similarly, another study found that SIRT6 suppresses AMPK signaling and enhances COX-2 expression in keratinocytes in response to UV irradiation, thereby promoting inflammation, survival, and oncogenesis (Ming et al. 2014). Overall, the tumor suppressor and oncogenic functions of SIRT6 likely reflect the diverse substrates and roles of this protein. Given the underlying complexity of SIRT6 function, it is perhaps not surprising that the roles of SIRT6 in neoplasia are context- and tissue-specific, a phenomenon which has also been described for the sirtuin proteins SIRT1 and SIRT3 (Morris 2013).
7.9 Conclusion
SIRT6 is a multi-faceted protein that maintains organismal healthspan via diverse molecular roles in mammals. Current data point to mammalian SIRT6 as a functional ortholog of the yeast Sir2 protein; like yeast Sir2, SIRT6 promotes heterochromatin stability and increased longevity. SIRT6 attenuates the transcriptional output of key transcription factors such as HIF-1α, c-JUN, and c-MYC to regulate cellular processes such as glucose and adipose tissue metabolism, cellular senescence, and inflammation. SIRT6 also promotes genome integrity through multiple mechanisms, potentially contributing to lifespan extension. Loss of SIRT6 exerts severe pathological consequences: cardiac hypertrophy, diabetes, liver steatosis, chronic inflammation and cancer.
Since sirtuins require NAD+ for their enzymatic activity, an age-associated decline in NAD+ levels would be predicted to impair SIRT6 function and potentially recapitulate the pathological effects of SIRT6 deficiency or knockdown. Measurements of NAD+ levels have been reported in several studies; NAD+ decreases with age in multiple tissues in worms, rodents, and humans (Braidy et al. 2011; Braidy et al. 2014; Massudi et al. 2012; Mouchiroud et al. 2013; North et al. 2014; Ramsey et al. 2008). An emerging literature suggests that SIRT6 overexpression or restoration of NAD+ may be beneficial in mouse models of metabolic disease and other pathological states (Canto et al. 2012; Gomes et al. 2013; North et al. 2014; Stein and Imai 2014). Current research is focused on the discovery and characterization of putative SIRT6 activators with the goal of mitigating age-associated disease processes and perhaps promote longevity. Small molecules that supplement cellular NAD+ levels extend lifespan in C. elegans, and could play beneficial roles in mammals via activation of SIRT6 and other sirtuins.
References
Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY (2007) Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev 21:3244–3257
Agrawal A, Tay J, Ton S, Agrawal S, Gupta S (2009) Increased reactivity of dendritic cells from aged subjects to self-antigen, the human DNA. J Immunol 182:1138–1145
Agrawal A, Tay J, Yang GE, Agrawal S, Gupta S (2010) Age-associated epigenetic modifications in human DNA increase its immunogenicity. Aging (Albany NY) 2:93–100
Ardawi MS, Newsholme EA (1982) Maximum activities of some enzymes of glycolysis, the tricarboxylic acid cycle and ketone-body and glutamine utilization pathways in lymphocytes of the rat. Biochem J 208:743–748
Baker RG, Hayden MS, Ghosh S (2011) NF-kappaB, inflammation, and metabolic disease. Cell Metab 13:11–22
Battu A, Ray A, Wani AA (2011) ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res 39:7931–7945
Bauer I, Grozio A, Lasiglie D, Basile G, Sturla L, Magnone M, Sociali G, Soncini D, Caffa I, Poggi A et al (2012) The NAD+-dependent histone deacetylase SIRT6 promotes cytokine production and migration in pancreatic cancer cells by regulating Ca2+ responses. J Biol Chem 287:40924–40937
Boboila C, Alt FW, Schwer B (2012) Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol 116:1–49
Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R (2011) Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS ONE 6:e19194
Braidy N, Poljak A, Grant R, Jayasena T, Mansour H, Chan-Ling T, Guillemin GJ, Smythe G, Sachdev P (2014) Mapping NAD(+) metabolism in the brain of ageing Wistar rats: potential targets for influencing brain senescence. Biogerontology 15:177–198
Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P et al (2012) The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 15:838–847
Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK (2008) Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134:231–243
Church DN, Talbot DC (2012) Survivin in solid tumors: rationale for development of inhibitors. Curr Oncol Rep 14:120–128
Dai DF, Chen T, Johnson SC, Szeto H, Rabinovitch PS (2012) Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal 16:1492–1526
Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL (2009) Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459:802–807
Dominy JE Jr, Lee Y, Gerhart-Hines Z, Puigserver P (2010) Nutrient-dependent regulation of PGC-1alpha’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta 1804:1676–1683
Dominy JE Jr, Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, Ruan HB, Feldman J, Pierce K, Mostoslavsky R et al (2012) The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell 48:900–913
Dunn C, Wiltshire C, MacLaren A, Gillespie DA (2002) Molecular mechanism and biological functions of c-Jun N-terminal kinase signalling via the c-Jun transcription factor. Cell Signal 14:585–593
Elhanati S, Kanfi Y, Varvak A, Roichman A, Carmel-Gross I, Barth S, Gibor G, Cohen HY (2013) Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep 4:905–912
Feldman JL, Baeza J, Denu JM (2013) Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem 288:31350–31356
Frye RA (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273:793–798
Garedew A, Moncada S (2008) Mitochondrial dysfunction and HIF1alpha stabilization in inflammation. J Cell Sci 121:3468–3475
Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P (2007) Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. Embo J 26:1913–1923
Gil R, Barth S, Kanfi Y, Cohen HY (2013) SIRT6 exhibits nucleosome-dependent deacetylase activity. Nucleic Acids Res 41:8537–8545
Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP et al (2013) Declining NAD(+) Induces a Pseudohypoxic State Disrupting Nuclear-Mitochondrial Communication during Aging. Cell 155:1624–1638
Guarente L (2011) Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med 364:2235–2244
Gupta S, Young D, Maitra RK, Gupta A, Popovic ZB, Yong SL, Mahajan A, Wang Q, Sen S (2008) Prevention of cardiac hypertrophy and heart failure by silencing of NF-kappaB. J Mol Biol 375:637–649
Han Z, Liu L, Liu Y, Li S (2014) Sirtuin SIRT6 suppresses cell proliferation through inhibition of Twist1 expression in non-small cell lung cancer. Int J Clin Exp Pathol 7:4774–4781
Hu S, Liu H, Ha Y, Luo X, Motamedi M, Gupta MP, Ma JX, Tilton RG, Zhang W (2015) Posttranslational modification of Sirt6 activity by peroxynitrite. Free Radic Biol Med 79:176–185
Jedrusik-Bode M, Studencka M, Smolka C, Baumann T, Schmidt H, Kampf J, Paap F, Martin S, Tazi J, Muller KM et al (2013) The sirtuin SIRT6 regulates stress granule formation in C. elegans and mammals. J Cell Sci 126:5166–5177
Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R et al (2013) SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496:110–113
Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13:2570–2580
Kaidi A, Weinert BT, Choudhary C, Jackson SP (2010) Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 329:1348–1353
Kanfi Y, Shalman R, Peshti V, Pilosof SN, Gozlan YM, Pearson KJ, Lerrer B, Moazed D, Marine JC, de Cabo R et al (2008) Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett 582:543–548
Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, Kronfeld-Schor N, Cohen HY (2010) SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 9:162–173
Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY (2012) The sirtuin SIRT6 regulates lifespan in male mice. Nature 483:218–221
Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY et al (2009) SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136:62–74
Kawahara TL, Rapicavoli NA, Wu AR, Qu K, Quake SR, Chang HY (2011) Dynamic chromatin localization of Sirt6 shapes stress- and aging-related transcriptional networks. PLoS Genet 7:e1002153
Kenyon CJ (2010) The genetics of ageing. Nature 464:504–512
Khongkow M, Olmos Y, Gong C, Gomes AR, Monteiro LJ, Yague E, Cavaco TB, Khongkow P, Man EP, Laohasinnarong S et al (2013) SIRT6 modulates paclitaxel and epirubicin resistance and survival in breast cancer. Carcinogenesis 34:1476–1486
Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH et al (2010) Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab 12:224–236
Koh MY, Powis G (2012) Passing the baton: the HIF switch. Trends Biochem Sci 37:364–372
Lee HS, Ka SO, Lee SM, Lee SI, Park JW, Park BH (2013) Overexpression of SIRT6 suppresses inflammatory responses and bone destruction in collagen-induced arthritic mice. Arthritis Rheum 65(7):1776–17785
Lefort K, Brooks Y, Ostano P, Cario-Andre M, Calpini V, Guinea-Viniegra J, Albinger-Hegyi A, Hoetzenecker W, Kolfschoten I, Wagner EF et al (2013) A miR-34a-SIRT6 axis in the squamous cell differentiation network. EMBO J 32:2248–2263
Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P (2006) GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab 3:429–438
Lin Z, Yang H, Tan C, Li J, Liu Z, Quan Q, Kong S, Ye J, Gao B, Fang D (2013) USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep 5:1639–1649
Liszt G, Ford E, Kurtev M, Guarente L (2005) Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem 280:21313–21320
Liu Y, Xie QR, Wang B, Shao J, Zhang T, Liu T, Huang G, Xia W (2013) Inhibition of SIRT6 in prostate cancer reduces cell viability and increases sensitivity to chemotherapeutics. Protein & cell 4:702–710
Lombard DB, Miller RA (2012) Ageing: sorting out the sirtuins. Nature 483:166–167
Lombard DB, Miller RA (2014) Aging, disease, and longevity in mice. Annu Rev Gerontol Geriatr 34:93–138
Longo VD, Kennedy BK (2006) Sirtuins in aging and age-related disease. Cell 126:257–268
Longo VD, Shadel GS, Kaeberlein M, Kennedy B (2012) Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab 16:18–31
Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science 332:1443–1446
Mao Z, Tian X, Van Meter M, Ke Z, Gorbunova V, Seluanov A (2012) Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci U S A 109:11800–11805
Marquardt JU, Fischer K, Baus K, Kashyap A, Ma S, Krupp M, Linke M, Teufel A, Zechner U, Strand D et al (2013) SIRT6 dependent genetic and epigenetic alterations are associated with poor clinical outcome in HCC patients. Hepatology 58(3):1054–1064
Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, Xiao C, Roqueta-Rivera M, Deng C, Osborne TF, Mostoslavsky R et al (2014) Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell 158:659–672
Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ (2012) Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7:e42357
McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL et al (2009) SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 1:109–121
Mehta R, Steinkraus KA, Sutphin GL, Ramos FJ, Shamieh LS, Huh A, Davis C, Chandler-Brown D, Kaeberlein M (2009) Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 324:1196–1198
Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 16:4623–4635
Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC et al (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452:492–496
Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, Chua KF (2009) Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8:2664–2666
Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, Chen L, Scheuch H, Zheng H, Qin L et al (2012) Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat Cell Biol 14:1203–1211
Ming M, Han W, Zhao B, Sundaresan NR, Deng CX, Gupta MP, He YY (2014) SIRT6 promotes COX-2 expression and acts as an oncogene in skin cancer. Cancer Res 74:5925–5933
Morris BJ (2013) Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med 56:133–171
Moschen AR, Wieser V, Gerner RR, Bichler A, Enrich B, Moser P, Ebenbichler CF, Kaser S, Tilg H (2013) Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J Hepatol 59:1315–1322
Moskalev A, Shaposhnikov M (2011) Pharmacological inhibition of NF-kappaB prolongs lifespan of Drosophila melanogaster. Aging (Albany NY) 3:391–394
Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM et al (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124:315–329
Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K et al (2013) The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154:430–441
Munoz-Galvan S, Jimeno S, Rothstein R, Aguilera A (2013) Histone H3K56 acetylation, Rad52, and non-DNA repair factors control double-strand break repair choice with the sister chromatid. PLoS Genet 9:e1003237
Murray-Zmijewski F, Lane DP, Bourdon JC (2006) p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 13:962–972
Nin V, Escande C, Chini CC, Giri S, Camacho-Pereira J, Matalonga J, Lou Z, Chini EN (2012) Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J Biol Chem 287:23489–23501
North BJ, Rosenberg MA, Jeganathan KB, Hafner AV, Michan S, Dai J, Baker DJ, Cen Y, Wu LE, Sauve AA et al (2014) SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J 33:1438–1453
O’Neill LA, Hardie DG (2013) Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493:346–355
Olivetti G, Cigola E, Maestri R, Lagrasta C, Corradi D, Quaini F (2000) Recent advances in cardiac hypertrophy. Cardiovasc Res 45:68–75
Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM (2011) Structure and biochemical functions of SIRT6. J Biol Chem 286:14575–14587
Parsons JL, Dianov GL (2013) Co-ordination of base excision repair and genome stability. DNA Repair (Amst) 12:326–333
Puigserver P, Spiegelman BM (2003) Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 24:78–90
Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D et al (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 423:550–555
Ramsey KM, Mills KF, Satoh A, Imai S (2008) Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 7:78–88
Revollo JR, Li X (2013) The ways and means that fine tune Sirt1 activity. Trends Biochem Sci 38:160–167
Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, Spiegelman BM (2003) Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad Sci U S A 100:4012–4017
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118
Ronnebaum SM, Wu Y, McDonough H, Patterson C (2013) The ubiquitin ligase CHIP prevents SirT6 degradation through noncanonical ubiquitination. Mol Cell Biol 33:4461–4472
Sahin K, Yilmaz S, Gozukirmizi N (2014) Changes in human sirtuin 6 gene promoter methylation during aging. Biomed Rep 2:574–578
Schwer B, Schumacher B, Lombard DB, Xiao C, Kurtev MV, Gao J, Schneider JI, Chai H, Bronson RT, Tsai LH et al (2010) Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc Natl Acad Sci U S A 107:21790–21794
Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D et al (2012) The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151:1185–1199
Simeoni F, Tasselli L, Tanaka S, Villanova L, Hayashi M, Kubota K, Isono F, Garcia BA, Michishita-Kioi E, Chua KF (2013) Proteomic analysis of the SIRT6 interactome: novel links to genome maintenance and cellular stress signaling. Sci Rep 3:3085
Stein LR, Imai S (2014) Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J 33:1321–1340
Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V et al (2012) The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med 18:1643–1650
Tao R, Xiong X, DePinho RA, Deng CX, Dong XC (2013a) FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J Biol Chem 288:29252–29259
Tao R, Xiong X, DePinho RA, Deng CX, Dong XC (2013b) Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J Lipid Res 54:2745–2753
Tennen RI, Bua DJ, Wright WE, Chua KF (2011) SIRT6 is required for maintenance of telomere position effect in human cells. Nat Commun 2:433
Thirumurthi U, Shen J, Xia W, LaBaff AM, Wei Y, Li CW, Chang WC, Chen CH, Lin HK, Yu D et al (2014) MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Sci Signal 7:ra71 71-10
Tian K, Liu Z, Wang J, Xu S, You T, Liu P (2014) Sirtuin-6 inhibits cardiac fibroblasts differentiation into myofibroblasts via inactivation of nuclear factor kappaB signaling. Transl Res 13:00409–00405
Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, Sebastian C, Cosentino C, Martinez-Pastor B, Giacosa S et al (2013) SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell 51:454–468
Van Gool F, Galli M, Gueydan C, Kruys V, Prevot PP, Bedalov A, Mostoslavsky R, Alt FW, De Smedt T, Leo O (2009) Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat Med 15:206–210
Van Meter M, Mao Z, Gorbunova V, Seluanov A (2011) SIRT6 overexpression induces massive apoptosis in cancer cells but not in normal cells. Cell Cycle 10:3153–3158
Van Meter M, Kashyap M, Rezazadeh S, Geneva AJ, Morello TD, Seluanov A, Gorbunova V (2014) SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat Commun 5:5011
Vinciguerra M, Santini MP, Claycomb WC, Ladurner AG, Rosenthal N (2010) Local IGF-1 isoform protects cardiomyocytes from hypertrophic and oxidative stresses via SirT1 activity. Aging (Albany NY) 2:43–62
Wan F, Lenardo MJ (2010) The nuclear signaling of NF-kappaB: current knowledge, new insights, and future perspectives. Cell Res 20:24–33
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Wu T, Liu YH, Fu YC, Liu XM, Zhou XH (2014) Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann Clin Lab Sci 44:410–418
Xiao C, Kim HS, Lahusen T, Wang RH, Xu X, Gavrilova O, Jou W, Gius D, Deng CX (2010) SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J Biol Chem 285:36776–36784
Xiao C, Wang RH, Lahusen TJ, Park O, Bertola A, Maruyama T, Reynolds D, Chen Q, Xu X, Young HA et al (2012) Progression of chronic liver inflammation and fibrosis driven by activation of c-JUN signaling in Sirt6 mutant mice. J Biol Chem 287:41903–41913
Xie X, Zhang H, Gao P, Wang L, Zhang A, Xie S, Li J (2012) Overexpression of SIRT6 in porcine fetal fibroblasts attenuates cytotoxicity and premature senescence caused by D-galactose and tert-butylhydroperoxide. DNA Cell Biol 31:745–752
Xiong X, Tao R, DePinho RA, Dong XC (2013) Deletion of hepatic FoxO1/3/4 genes in mice significantly impacts on glucose metabolism through downregulation of gluconeogenesis and upregulation of glycolysis. PLoS One 8:e74340
Xu A, Wang Y, Xu JY, Stejskal D, Tam S, Zhang J, Wat NM, Wong WK, Lam KS (2006) Adipocyte fatty acid-binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clin Chem 52:405–413
Xu Z, Zhang L, Zhang W, Meng D, Zhang H, Jiang Y, Xu X, Van Meter M, Seluanov A, Gorbunova V et al (2015) SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 14:269–276. doi:210.4161/15384101.15382014.15980641
Yang B, Zwaans BM, Eckersdorff M, Lombard DB (2009) The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle 8:2662–2663
Yu SS, Cai Y, Ye JT, Pi RB, Chen SR, Liu PQ, Shen XY, Ji Y (2013) Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF-kappaB-dependent transcriptional activity. Br J Pharmacol 168:117–128
Yuan J, Pu M, Zhang Z, Lou Z (2009) Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle 8:1747–1753
Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA (2009) The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS One 4:e6348
Zhang F, Kong D, Lu Y, Zheng S (2013a) Peroxisome proliferator-activated receptor-gamma as a therapeutic target for hepatic fibrosis: from bench to bedside. Cell Mol Life Sci 70:259–276
Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, Li B, Liu G, Cai D (2013b) Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 497:211–216
Zhang P, Tu B, Wang H, Cao Z, Tang M, Zhang C, Gu B, Li Z, Wang L, Yang Y et al (2014) Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc Natl Acad Sci U S A 111:10684–10689
Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T et al (2010) The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140:280–293
Acknowledgements
Work in our laboratory is supported by NIH awards R01GM101171 and R21CA177925 (to D.L.), and by the Glenn Foundation for Medical Research. B.M.M.Z. was supported by a Rackham Fellowship. W.G. is supported by NIH Training Grant T32-AG000114.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Zwaans, B.M.M., Giblin, W., Lombard, D.B. (2016). Diverse Roles for SIRT6 in Mammalian Healthspan and Longevity. In: Houtkooper, R. (eds) Sirtuins. Proteins and Cell Regulation, vol 10. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-0962-8_7
Download citation
DOI: https://doi.org/10.1007/978-94-024-0962-8_7
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-0961-1
Online ISBN: 978-94-024-0962-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)