
Abstract
When the activity of SIRT1 was unraveled as an NAD+-dependent deacetylase, the possibility of coupling metabolic changes to transcriptional outputs was rapidly envisioned. More than a decade later, SIRT1 has been demonstrated to be at the pinnacle of metabolic control and metabolic adaptation in diverse organisms, including mammals. The activation of SIRT1 has been shown to be protective against metabolic damage and also holds promise in the battle against neurodegenerative diseases and cancer, among others. Here, we aim to review our current knowledge on SIRT1 actions, with special emphasis on what mammalian animal models have taught us on the possibility of targeting complex diseases through SIRT1 activation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Introduction
The story of SIRT1 , and sirtuins in general, began more than three decades ago, with the identification of an enzyme, Sir2, participating in gene silencing at specific genomic regions (Ivy et al. 1986; Shore et al. 1984). However, it was not until fifteen years later that we began to truly grasp the significance of Sir2 activity. A key finding in the story of sirtuins was the identification of Sir2 as an NAD+-depedent deacetylase enzyme (Imai et al. 2000a). This meant that Sir2, and all its homologs along the evolutionary scale, used NAD+ as a cosubstrate to catalyze deacetylation of a substrate, rendering nicotinamide and 2’-O-acetyl-ADP-ribose as concomitant reaction products. The engagement of sirtuins with NAD+ as a cosubstrate is unique compared to all other deacetylase enzyme families known to date. In addition, this fundamental feature suggests that sirtuins might respond directly to changes in intracellular metabolism by “sensing” NAD+ levels (Canto and Auwerx 2012; Imai et al. 2000b). Since reversible acetylation has putative regulatory properties on the activity and function of a constellation of proteins, including histones , sirtuins were proposed to act as enzymes coupling metabolic cues to transcriptional outputs.
Among all sirtuins, SIRT1 is the most well-studied mammalian homolog of the yeast enzyme Sir2. The initial interest in this protein rapidly spread due to its possible role in eukaryote lifespan regulation (Berdichevsky et al. 2006; Canto and Auwerx 2009; Kaeberlein et al. 1999; Viswanathan and Guarente 2011; Viswanathan et al. 2005). While this is still a matter of debate (Burnett et al. 2011; Lombard et al. 2011), an overwhelming amount of evidence in animal models suggests that SIRT1 might play key role in metabolic regulation and adaptation (Canto and Auwerx 2012). This, in turn, impinges on the sensibility of organisms to develop metabolic and age-related diseases, including insulin resistance, cancer and diverse neurodegenerative pathologies. Here, we aim to review the most recent advances on SIRT1 functions, with strong emphasis on the knowledge obtained using transgenic animal models .
4.2 SIRT1 Biology
4.2.1 Basic Structure and Localization
The human SIRT1 protein spans for 747 amino acids. The structure of the protein is immaculately conserved within mammals and contains the defining conserved catalytic core of sirtuins flanked by both N- and C-terminal extensions (Michan and Sinclair 2007). These extensions span each around 240 amino acids and serve as docking and interactive platforms for regulatory proteins and substrates (Canto and Auwerx 2012; Michan and Sinclair 2007).
The expression of Sirt1 is rather ubiquitous, and can be detected at the protein level in most, if not all, mammalian tissues. However, as described later, Sirt1 expression is very plastic, and can dramatically change in response to metabolic stress in an organ, tissue and cell-autonomous fashion. In addition, the SIRT1 protein can be found in the nuclei and/or the cytosolic compartment (Michishita et al. 2005; Tanno et al. 2007). SIRT1 contains two nuclear localization signals, that drive nuclear import, and two nuclear exportation signals, that drive the export (Tanno et al. 2007). The balanced function of these different signals determines SIRT1 localization in diverse tissues. This way, SIRT1 can display a very predominant nuclear localization in some tissues and cells, such as COS-7 cells or mouse embryonic fibroblasts (McBurney et al. 2003; Sakamoto et al. 2004). In turn, SIRT1 is predominantly cytosolic in pancreatic β-cells , myotubes and cardiomyocytes (Moynihan et al. 2005; Tanno et al. 2007). Furthermore, SIRT1 can actively shuttle between these compartments in response to environmental cues. For example, inhibition of the phosphatidylinositol 3-kinase (PI3K) pathway rapidly promotes nuclear exclusion of SIRT1 (Tanno et al. 2007). These observations further support the possible interrelation between SIRT1 activity and the cellular metabolic state .
4.2.2 SIRT1 Regulation
4.2.2.1 Regulation at the Expression Level
Intracellular SIRT1 activity can be modulated at the transcriptional level. The overexpression of Sirt1 in cultured cells, tissues and organisms is enough to increase global SIRT1 activity (Banks et al. 2008; Pfluger et al. 2008; Rodgers et al. 2005; Rodgers and Puigserver 2007). At the endogenous level, the expression of Sirt1 is generally upregulated in situations of low nutrient availability and energy stress (Canto and Auwerx 2012; Nemoto et al. 2004; Noriega et al. 2011). Hence, it was not surprising to find out how many transcriptional effectors of key metabolic signals directly regulate the activity of the Sirt1 promoter.
Initial studies trying to elucidate how the expression of the Sirt1 gene increased upon glucose deprivation found out that the Sirt1 promoter is critically regulated by members of the Forkhead-O-box (FOXO) family of transcription factors (Nemoto et al. 2004). FOXOs are transcription factors whose activity is negatively modulated by the PI3K pathway via direct Akt-mediated phosphorylation (Brunet et al. 1999). Therefore, insulin and other PI3K-activating growth factors , inhibit FOXO activity (Calnan and Brunet 2008). Elegant studies from the Finkel lab demonstrated that FOXO3a is a key regulator of the activity of the Sirt1 promoter (Nemoto et al. 2004). FOXO3a modulated Sirt1 gene expression via its interaction with p53 (Nemoto et al. 2004). When nutrients are abundant, FOXO3a is exported to the cytosol and p53 is bound to the proximal region of the Sirt1 promoter, repressing its transcriptional activity. However, upon nutrient depletion , FOXO3a is no longer sequestered to the cytosol via phosphorylation, hence shuttling to the nucleus where it directly interacts with p53 at the Sirt1 promoter (Nemoto et al. 2004). The interaction between FOXO3a and p53 relieves the inhibition of Sirt1 transcription, probably by modulating the accessibility of coactivator/corepressor complexes . It was exciting to find that FOXOs can also regulate Sirt1 transcription through direct binding to the Sirt1 promoter. Indeed, a number of FOXO binding sites have been identified in more distal regions (up to 1.5 kbp) of the rat Sirt1 promoter (Xiong et al. 2011). To date, however, only FOXO1 has been shown to bind to them and positively influence SIRT1 transcription (Xiong et al. 2011), and the conservation of these binding sites has to be yet fully analyzed. While organismal confirmation of these findings needs still to be solidified, FOXOs provide an excellent link on how nutrient deprivation enhances Sirt1 transcription. Similarly, the nuclear exclusion of FOXO constitutes a beautiful mechanism to integrate hormonal signals linked to nutrient abundance and the repression of the Sirt1 promoter .
Additional mechanism by which feeding/fasting cycles might influence Sirt1 expression in diverse tissues was unraveled recently in animal models. In the fed state, the Sirt1 promoter is repressed via the direct binding of the carbohydrate response element-binding protein (ChREBP) (Noriega et al. 2011). Upon fasting, ChREBP is translocated to the cytosol and its binding region is liberated. This goes together with the activation of CREB by fasting-derived cAMP signals triggered for example, by glucagon. CREB and ChREBP can bind to similar response elements (Noriega et al. 2011). Therefore, during the fasting state, the activated CREB can bind to the regions liberated by ChREBP and activates Sirt1 transcription .
Further supporting the metabolic regulation of Sirt1 gene expression, the peroxisome proliferator-activated receptor (PPAR) family of nuclear receptors also has key roles on Sirt1 transcriptional regulation. PPARs are directly activated by different lipid species and critically control lipid anabolism and catabolism (Michalik et al. 2006). The distal region of the Sirt1 promoter contains PPAR response elements (PPREs) (Han et al. 2010; Hayashida et al. 2010), even though a thorough analysis of their mapping and conservation is still lacking. This way, it was demonstrated that PPARγ can directly bind to and repress the Sirt1 promoter (Han et al. 2010). This could explain how situations of nutrient overload, when PPARγ is activated, are generally correlated with Sirt1 downregulation, both in mice (Coste et al. 2008) and humans (Costa Cdos et al. 2010). Interestingly, the existence of PPREs also opens the door for other PPARs to regulate the Sirt1 gene. Indeed, the activation of PPARα and PPARβ can also regulate Sirt1 expression , both in a positive manner (Hayashida et al. 2010; Kim et al. 2012; Okazaki et al. 2010). However, whether this regulation occurs via the same PPREs described is far from clear. In the case of PPARβ , it has been reported that such positive regulation could rely on an alternative mechanism, where PPARβ would enhance the positive action of p21 on the Sirt1 promoter (Okazaki et al. 2010). While the possible regulation of Sirt1 expression by PPARs provides a beautiful link between lipid metabolism and SIRT1 transcriptional activity, the mechanisms remain still poorly defined and even conflictive observations have been reported on the role of PPARγ on Sirt1 gene regulation (Chiang et al. 2013). Further work in vivo will be key to fully grasp the relevance of PPARs on Sirt1 transcription.
Other transcriptional regulators have been described on the Sirt1 promoter, but with either poor understanding of their in vivo relevance or their regulatory mechanisms. For example, the poly(ADP-ribose) polymerase (PARP)-2 protein has recently been described as a powerful repressor of Sirt1 transcription by directly binding to the proximal promoter region (Bai et al. 2011a). The downregulation or genetic ablation of PARP-2 is enough to enhance basal Sirt1 expression and protein levels (Bai et al. 2011a). However, the mechanism by with PARP-2 represses the Sirt1 promoter is still nebulous, and so is the possible physiological modulation of PARP-2 inhibitory action. Another mechanism for Sirt1 transcriptional repression that deserves attention is that constituted by the hypermethylated in cancer 1 (HIC1) protein. HIC1 naturally forms a corepressor complex on the SIRT1 promoter (Chen et al. 2005). The stability of this complex is critically modulated by the presence of the C-terminal binding protein (CtBP) , which depends on NADH (Zhang et al. 2007). In situations of low NADH levels , such as in cells treated with 2-deoxyglucose – mimicking low glucose availability – the complex is destabilized and the inhibitory effect is relieved, allowing enhanced Sirt1 transcription rates, which, in turn, optimizes the adaptation to low nutrient availability (Zhang et al. 2007). In this case the molecular regulation of the HIC1:CtBP complex on the Sirt1 promoter is very well defined, but support of such findings in animal models will be required.
Another level of regulation of Sirt1 mRNA occurs through microRNAs (miRNAs) , which promote the cleavage of specific mRNAs or inhibit their translation (Neilson and Sharp 2008). To date, more than 16 miRNAs have been described to regulate Sirt1 expression and activity (Yamakuchi 2012). Among them, miR-34a has been the most widely studied. Briefly, miR-34a binds to the 3’-untranslated region of the Sirt1 mRNA in a partial complementary manner and represses its translation (Lee et al. 2010; Yamakuchi et al. 2008). Several interventions have demonstrated a strong negative correlation with Sirt1 levels in physiological situations (Lee et al. 2010; Yamakuchi 2012; Yamakuchi et al. 2008). We kindly refer the reader to other recent reviews in order to gain more insight into the fascinating new level of complexity on the regulation of Sirt1 introduced by miRNAs (Yamakuchi 2012).
4.2.2.2 Regulation by NAD+ and NAM
The fact that SIRT1 activity requires NAD+ as a mandatory cosubstrate raised the hypothesis that SIRT1 could act as an NAD+ sensor in the cell, coupling the metabolic and redox status of the cell to transcriptional outputs (Imai et al. 2000b). However, to act as a true metabolic sensor, the activity of SIRT1 should be rate-limited by NAD+ availability. But, is this really the case?
Most studies to date suggest that the Km of SIRT1 for NAD+ is around 150-200 μM (Houtkooper et al. 2010). Whether this is the true range of NAD+ availability in the cell is difficult to confirm. First, most estimates indicate that basal intracellular NAD+ levels in most cells and tissues fall in a range between 0.2 and 0.5 μM (Houtkooper et al. 2010). This originally led to think that NAD+ might not be rate-limiting for SIRT1. However, these values hardly take into account NAD+ intracellular compartmentalization. In fact, some estimates have indicated that nuclear NAD+ concentrations might be around 70 μM (Fjeld et al. 2003), which would be critically rate-limiting for SIRT1 activity. Similarly, the most commonly used techniques to measure intracellular NAD+ fail to distinguish between free (available) and protein-bound NAD+. Therefore, our current knowledge is still too preliminary to unequivocally determine NAD+ bioavailability . However, given the above considerations, it seems plausible that NAD+ could truly be rate-limiting for SIRT1 activity. Supporting this possibility, most – if not all – experimental strategies aimed to alter intracellular NAD+ levels have consistently been shown to influence SIRT1 activity (Canto and Auwerx 2012).
A number of in vivo strategies have demonstrated how increases in NAD+ levels translate into SIRT1 activation. One strategy relied in the deletion of alternative NAD+ consumers, such as PARP-1 or the cADP-ribose synthase CD38 . Both of these enzymes are avid NAD+ consumers and, therefore, their deletion should allow higher NAD+ bioavailability for SIRT1 . This way, if NAD+ was rate-limiting for SIRT1, these models should display enhanced SIRT1 activity. In line with this hypothesis, the deletion of either PARP-1 or CD38 has consistently shown a correlative increase in NAD+ levels and SIRT1 activity in most tissues examined (Aksoy et al. 2006; Bai et al. 2011b). A second strategy has relied in enhancing NAD+ biosynthesis by providing NAD+ precursors or manipulating the expression of NAD+ biosynthetic enzymes. The efforts from the Imai lab have consistently demonstrated how intraperitoneal (ip) injection of nicotinamide mononucleoside (NMN) is enough to raise NAD+ levels and robutstly increase SIRT1 activity (Yoshino et al. 2011). Parallel experiments demonstrated that ip injections of NMN prevented fructose rich diet-induced islet dysfunction, likely through SIRT1 activation (Caton et al. 2011). Similarly, the Auwerx lab has demonstrated how food supplementation with the natural NAD+ precursor nicotinamide riboside (NR) also leads to an elevation in NAD+ levels and enhanced SIRT1 activity in mouse tissues (Canto et al. 2012). The overexpression of NAD+ biosynthetic enzymes, such as the nicotinamide phosphoribosyltransferase (Nampt) or the nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) , provides an alternative way to boost NAD+ availability, which generally leads to enhanced SIRT1 activity (Araki et al. 2004; Revollo et al. 2004; Wu et al. 2011; Zhang et al. 2009). In fact, it has been reported that SIRT1 might directly interact with NMNAT1 (Zhang et al. 2009). Such a complex could potentially channel NAD+ production to fuel SIRT1 enzymatic catalysis, creating a microdomain for the regulation of SIRT1 activity . Altogether, these strategies support that boosting NAD+ availability enhances SIRT1 activity .
Many physiological challenges also promote fluctuations in NAD+ levels. In most cases, these fluctuations rarely increase NAD+ levels beyond 2-fold (Canto et al. 2009; Chen et al. 2008; Fulco et al. 2008; Rodgers et al. 2005), very much in line with the range expected to affect SIRT1 activity. In general, NAD+ levels and SIRT1 activity increase in most tissues in response to nutrient deprivation or energy stress, such as exercise (Canto et al. 2009; Costford et al. 2010), fasting (Canto et al. 2010; Rodgers et al. 2005) or calorie restriction (Chen et al. 2008). The degree of the response, however, depends on the baseline NAD+ levels and SIRT1 activity of each tissue. For example, the exercise-induced enhancement in SIRT1 activity was evident in glycolytic muscle, but not in oxidative muscle, where SIRT1 activity was already high on the basal state (Canto et al. 2009).
SIRT1 activity can also be modulated by other NAD+ metabolites. For example, it has been shown that NADH can compete with NAD+ binding to SIRT1 and inhibit SIRT1 activity (Lin et al. 2004; Schmidt et al. 2004). While this could prompt the hypothesis that SIRT1 could also act as an NADH sensor, the levels of NADH required to promote competitive inhibition are in the millimolar range, which are unlikely to be met physiologically (Schmidt et al. 2004). Therefore, NADH levels should rarely determine SIRT1 activity in most common physiological situations, even if it cannot be fully ruled out that such cases might exist.
Another prominent natural inhibitor of SIRT1 activity is nicotinamide (NAM), which is a product of the sirtuin reaction. NAM exerts end-product inhibition of SIRT1 in a non-competitive fashion with NAD+ (Anderson et al. 2003; Bitterman et al. 2002). While it is known that NAM can inhibit SIRT1 at concentrations around 200 μM or lower, the true intracellular content of NAM content is far from clear. In addition, NAM can diffuse across membranes (van Roermund et al. 1995), which further complicates compartmentalization studies. An important point is that, at low levels, NAM actually prompts activation of SIRT1, due to its property as an NAD+ precursor (Houtkooper et al. 2010; Revollo et al. 2004). Therefore, NAM at lower micromolar range might be beneficial for SIRT1 activity, while its accumulation is deleterious.
4.2.2.3 Regulation by Post-translational Modifications
The enzymatic activity of SIRT1 can also be influenced through post-translational modifications. Initial evidence for this came from a large mass spectrometry-based screening, which identified SIRT1 as a phosphorylated protein (Beausoleil et al. 2004). Two phosphoresidues , Ser27 and Ser47, on the human SIRT1 were identified, but their respective function and modulation is not yet clear. A later SIRT1-centered study revealed up to 13 phosphorylable residues , mostly located on the N- and C-terminal expansions flanking the conserved catalytic domain (Sasaki et al. 2008). Among them, the authors identified Thr530 and Ser540 as residues directly phosphorylated by cyclinB/cdk1 (Sasaki et al. 2008). The phosphorylation of these residues resulted in higher SIRT1 activity (Sasaki et al. 2008). Very much in the same line, it was reported how Ser27, Ser47 and Thr530 could be phosphorylated by JNK1, resulting in nuclear translocation and enhanced SIRT1 activation (Nasrin et al. 2009). Interestingly, the phosphorylation by JNK1 targetted SIRT1 activity to specific substrates, as it triggered the deacetylation of histone H3 but not that of p53, which is another well-established SIRT1 substrate (Nasrin et al. 2009). This is actually a key concept, as it is often overlooked that SIRT1 affects many different targets and cellular processes, sometimes with opposite effects. Therefore, SIRT1 action must be targeted to some degree. In this sense, post-translational modifications might be an attractive mechanism by which SIRT1 could be channeled to specific subsets of targets.
A constellation of additional kinases have been described to phosphorylate one or more residues of SIRT1 (see (Canto and Auwerx 2012) for review). A critical point , however, is that most of the residues identified – or their flanking sequences – are very poorly conserved, making it difficult to argue that they are key to the primordial metabolic functions described for SIRT1 orthologs across evolution. Similarly, the physiological modulation of these possible phosphorylation events is far from understood. A major change in the field was hence provided by the Puigserver lab , when they identified mouse Ser434 (human Ser442) as a phosphorylation substrate for PKA (Gerhart-Hines et al. 2011). First, because Ser442 is located in the catalytic core domain of SIRT1 and is highly conserved across species. Second, because the authors elegantly demonstrated how the phosphorylation of this residue is modulated by hormonal inputs and physiological stimuli in mice. Third, and most importantly, because the phosphorylation of this residue allowed SIRT1 to decrease the Km for NAD+ (Gerhart-Hines et al. 2011). The enhanced affinity for NAD+ via phosphorylation can explain how SIRT1 activity can change even in the absence of changes in NAD+.
SIRT1 activity can also be influenced by the addition of small ubiquitin-like modifier (SUMO) . SIRT1 is SUMOylated at Lys734 upon irradiation or treatment with toxic concentrations of hydrogen peroxide (Yang et al. 2007). Upon SUMOylation SIRT1 increases its intrinsic deacetylase activity and enhances the ability of the cell to survive to the above mentioned damaging agents (Yang et al. 2007). A key caveat of the described SUMOylation event is the poor conservation of the Lys734 residue, even within mammals. While this does not rule out the potential contribution of SUMOylation in some species, including humans, it is unlikely that SUMOylation is a major contributor of the conserved key metabolic actions of SIRT1. Moreover, further studies will have to underscore the physiological implications and regulation of SIRT1 SUMOylation.
While we are just beginning to grasp the complex interplay of post-translational modifications that occur on SIRT1, it is tempting to hypothesize that they do not act independently. Rather these modifications might interact to control substrate accessibility, modify the kinetic properties or serve as interaction platforms for SIRT1. Understanding these specific mechanisms will be essential to design future strategies to selectively channel SIRT1 into specific functions .
4.2.2.4 Regulation by Protein Interactions
Another layer of regulation is provided by the association of SIRT1 with different proteins. As described previously, SIRT1 was originally described as a transcriptional silencing enzyme. Therefore, it was not surprising that SIRT1 was found in mammalian cells and tissues forming part of corepressor complexes with the Nuclear Receptor Corepressor 1 (NCoR1) (Picard et al. 2004). In there, SIRT1 participated in silencing of the transcriptional activitiy of nuclear receptors, such as PPARγ . However, the coregulator properties of SIRT1 are not limited to silencing. For example, when associated to the PPAR coactivator 1α (PGC-1α) , it participates in the transcriptional activation of mitochondrial and fatty acid oxidation genes (Gerhart-Hines et al. 2007; Rodgers et al. 2005). Therefore, the association with different partners confers SIRT1 the ability to act both as a corepressor and as a coactivator .
A key finding in the SIRT1 field came with the simultaneous identification by two different labs of a nuclear protein, deleted in breast cancer 1 (DBC1) , as a protein forming a stable complex with SIRT1 (Kim et al. 2008; Zhao et al. 2008). DBC1 binds to the catalytic domain of SIRT1 and inhibits SIRT1 activity both in vivo and in vitro (Kim et al. 2008; Zhao et al. 2008). This way, reductions in DBC-1 prompted an increase in SIRT1 activity in cultured cells (Kim et al. 2008; Zhao et al. 2008). Mice with a germline deletion of DBC1 display a 2- to 4-fold increase in endogenous SIRT1 activity in a wide range of tissues (Escande et al. 2010). Under normal circumstances, it was estimated that at least 50 % of the total SIRT1 in liver is associated with DBC1, and that this interaction was nearly absent upon starvation, when SIRT1 activity is higher (Escande et al. 2010). In contrast, the interaction was more prominent after chronic high-fat feeding, when SIRT1 activity is decreased (Escande et al. 2010). While these evidences illustrate that the interaction of DBC1 and SIRT1 can be modulated, the nature of this plasticity has been elusive for a long time. However, it has been recently reported that this interaction can be modulated by phosphorylation events . For example, the interaction between DBC1 and SIRT1 increases following DNA damage and oxidative stress (Yuan et al. 2012). This is due to the phosphorylation of DBC1 at Thr454 by the ATM (ataxia telangiectasia-mutated) and ATR (ataxia telangiectasia and Rad3-related ) kinases, which create a second binding site for SIRT1 (Yuan et al. 2012; Zannini et al. 2012). In contrast, it has been found that the activation of the AMP-activated protein kinase (AMPK) pathway leads to the dissociation of DBC1-SIRT1 complexes (Nin et al. 2012), even though the underlying mechanism is not fully understood.
Almost simultaneous to the identification of DBC1 as a negative regulator of SIRT1, Kim and colleagues identified the active regulator of SIRT1 (AROS) as an activator of SIRT1 activity (Kim et al. 2007). The interaction of AROS with SIRT1, presumably in its catalytic domain, enhances SIRT1 activity by 2-fold (Kim et al. 2007). However, the exact nature of the action of AROS on SIRT1 and its physiological relevance has been barely explored, still constituting promising area for research .
4.2.3 SIRT1 Functions
While initially described as a transcriptional silencing enzyme , the actions of SIRT1 have unfolded as a rich universe that expands far beyond histone modifications. In fact, a very large number of non-histone protein targets have been identified. There are a number of reviews that extensively recapitulate SIRT1 targets, and we kindly refer the reader to them for further information (Canto and Auwerx 2012; Houtkooper et al. 2012).
In general, the physiological activation of SIRT1 orchestrates cellular and organismal adaptations aimed to favor survival in situations of nutrient scarcity. This way, SIRT1 activation potentiates the extraction of energy from non-carbohydrate sources , mostly through mitochondrial respiration . Given the dual localization of SIRT1, either in the cytoplasm and/or the nucleus, it is not surprising that SIRT1 targets have also been identified in both compartments (Canto and Auwerx 2012). The cytosolic targets of SIRT1 generally potentiate short-term adaptations, while the nuclear ones are transcriptional regulators by which SIRT1 controls mitochondrial and fatty acid oxidation gene expression to allow chronic adaptations (Canto and Auwerx 2012). The metabolic adaptations prompted by SIRT1 do not only impact directly on metabolic homeostasis, but also indirectly on cellular proliferation, inflammation and survival (Canto and Auwerx 2012; Houtkooper et al. 2012). Therefore, it is not surprising that SIRT1 activation has not only been linked to beneficial effects on metabolic health, but also on cancer and cognitive function, among others.
In the next sections we will analyze SIRT1 functions based on the evidences gathered on animal models, so as to favor a physiological integrative point of view (Table 4.1 and 4.2).
4.3 What Animal Models Have Taught Us
4.3.1 SIRT1 and Whole Body Metabolism
SIRT1 orthologs in lower eukaryotes have been proposed to be determinants of lifespan (Canto and Auwerx 2009). Despite the abstract notion of what lifespan determination really means and the controversial results on whether SIRT1 overexpression truly enhances longevity in lower eukaryotes (Burnett et al. 2011), it seems clear that SIRT1 activation does not enhance lifespan in mice under normal food regimes (Baur et al. 2006; Herranz et al. 2010; Pearson et al. 2008). However, SIRT1 transgenic mice are protected against the metabolic damage induced by high-fat diets (Banks et al. 2008; Pfluger et al. 2008), and SIRT1 activation prevents the curving of lifespan induced by high-fat feeding (Baur et al. 2006).
The first SIRT1 gain-of function model reported displayed several features resembling calorie restriction : they were leaner, metabolically more active, and had increased glucose tolerance (Bordone et al. 2007). Two additional SIRT1 transgenic lines were later generated, both of them concluded that mild overexpression of SIRT1 prevented against high-fat diet induced hyperglycemia, insulin resitance and fatty liver, despite no significant differences in body weight (Banks et al. 2008; Pfluger et al. 2008). Posterior efforts have also certified that lines with a higher SIRT1 overexpression have enhanced mitochondrial content and display greater mitochondrial function (Price et al. 2012). While this feature has not been deeply analyzed in the previous lines, it could provide an interesting mechanism to explain the phenotypes observed. The major caveat of this approach is that higher SIRT1 levels do not necessarily have to correlate with SIRT1 activity. This has been demonstrated recently in aging models, where reduced NAD+ availability compromises SIRT1 activity, despite higher SIRT1 content (Braidy et al. 2011).
A number of compounds have been used as SIRT1 activating compounds (STACS) , even though their specificity has been long debated (Canto and Auwerx 2012). A recent publication elegantly demonstrates that STACS directly influence the activity of SIRT1 through a specific residue, Glu230 for human SIRT1 (Hubbard et al. 2013). This mechanism for direct activation of SIRT1, however, would only affect a subset of substrates with specific structural requirements (Hubbard et al. 2013) and would not explain the effects of STACs on lower eukaryotes, where the human Glu230 residue is not conserved. While further in vivo consolidation of these observations is required, they provide an interesting conciliatory explanation on why STACs have been reported to trigger SIRT1 activation through both direct and indirect mechanism . Among all STACS, resveratrol has probably been the one receiving more attention. Indeed, mice fed with resveratrol show a number of features in common with the SIRT1 transgenic mice. Most notably, they are protected against high-fat diet induced metabolic damage and display enhanced mitochondrial function (Baur et al. 2006; Lagouge et al. 2006). The effects, however, were more marked than those observed on the transgenic, including prevention against high-fat diet induced obesity (Lagouge et al. 2006). A possible explanation for this relies on the ability of resveratrol to activate also other pathways (Canto and Auwerx 2012; Park et al. 2012). In fact, many studies suggest that the activation of SIRT1 by resveratrol, at least at the doses most commonly used in animal studies, could be an indirect consequence of AMPK activation (Canto et al. 2010; Um et al. 2010), which leads to increased NAD+ levels (Canto et al. 2009) and promotes Nampt expression (Canto et al. 2009; Fulco et al. 2008). A more recent screening for other possible small molecular SIRT1 agonists led to the identification of a second batch of compounds, among which the best characterized is SRT1720 (Milne et al. 2007). Similar to resveratrol, the treatment of mice with SRT1720 ameliorated the diabetic phenotype of obese mice (Milne et al. 2007) and prevented high-fat diet induced insulin resistance (Feige et al. 2008). Furthermore, SRT1720-fed mice displayed enhanced longevity (Minor et al. 2011). All this correlated with enhanced SIRT1 activity in the tissues of SRT1720 treated mice (Feige et al. 2008; Minor et al. 2011). However, similar to resveratrol, several questions have been raised regarding the specificity of SRT1720 on SIRT1 (Pacholec et al. 2010). In addition to that, the in vitro assays indicated that SRT1720 was a more potent activator of SIRT1 than resveratrol (Milne et al. 2007). This, however was not clearly translated in vivo, indicating either poor bioavailability or that the actions of SRT1720 in vivo on SIRT1 largely rely on an indirect activation .
There are also a number of transgenic models that indirectly affecting SIRT1 activity. For example, mice lacking DBC1 , the inhibitory endogenous interacting protein, display many features similar to SIRT1 transgenic mice, such as protection against high-fat diet-induced hepatic steatosis and inflammation (Escande et al. 2010). However, DBC1 deficient mice still developed diabetes under high-fat feeding (Escande et al. 2010), indicating that SIRT1 function is not solely controlled by DBC1. Another strategy aimed to boost SIRT1 activity has been the knockout of alternative cellular NAD+ consumers. PARP-1 is considered to be a major NAD+ consumer in the cell, and its activity can deplete intracellular NAD+ by 70 % (Bai and Canto 2012). The deletion of PARP-1 is enough to increase basal NAD+ availavility and SIRT1 activity (Bai et al. 2011b). PARP-1 deficient mice also show a number of phenotypes resembling SIRT1 transgenesis , such as enhanced energy expenditure and protection against high-fat diet-induced diabetes (Bai et al. 2011b). A similar case could be made for CD38 , another alternative cellular NAD+ consumer (Aksoy et al. 2006). Mice defective for CD38 have increased SIRT1 activity in most tissues, probably due to increased NAD+ availability, and this confers protection against many of the metabolic complications induced by high caloric diets (Barbosa et al. 2007). However, similar to the PARP-1 model, this protective phenotype is markedly more pronounced than that observed in SIRT1 transgenic. This could be mainly due to two reasons: first, that these alternative NAD+ consumers affect many other processes other than SIRT1 activity and, second, that SIRT1 transgenesis might not reach similar SIRT1 activity levels as in the PARP-1 or CD38 models, due to NAD+ availability limitations .
A final strategy to enhance NAD+ availability consists in dosing with NAD+ precursors. Intraperitoneal injections of NMN for 7 days were enough to ameliorate age and high-fat diet-induced glucose intolerance, coupled to higher SIRT1 activity (Yoshino et al. 2011). Similarly, dietary supplementation of mice with NR was enough to increase SIRT1 activity in diverse tissues (Canto et al. 2012). This was coupled to a marked enhancement of insulin sensitivity, in both chow and high-fat fed animals, as well as increased oxidative capacity and global energy expenditure (Canto et al. 2012). Altogether, these strategies illustrate how a number of strategies aimed to enhance SIRT1 activity converge into higher glucose tolerance and increased capacity for oxidative metabolism .
All the above observations raise an obvious interest in understanding how the deletion of SIRT1 could impact global metabolic homeostasis . This has proven not to be an easy task. The whole-body deletion of SIRT1 leads to elevated prenatal death rates in inbred mice (Cheng et al. 2003; McBurney et al. 2003). The very few pups that were born displayed marked cardiac and neurological problems, leading to death very early in the postnatal period (Cheng et al. 2003; McBurney et al. 2003). In order to bypass this situation, SIRT1 deletion was performed in outbred mice. These mice were viable and displayed a marked metabolic inefficiency, which impaired their ability to metabolically adapt to calorie restriction (Boily et al. 2008). Outbred mouse stocks , however, are not ideal for metabolic studies due to their heterogeneity. Recently, an inducible model has been developed in order to genetically ablate SIRT1 exclusively in adulthood (Price et al. 2012). The induction of SIRT1 knockout in adult mice did not result in any overt phenotype . Similarly, there were no obvious differences between SIRT1 KO and WT mice on most metabolic parameters, although weight gain was slightly lower in the knockouts when placed on a high-caloric diet (Price et al. 2012). Another model worth discussing is the SIRT1 heterozygous mice. The heterozygous SIRT1-KO (SIRT1+/−) mice were normal in body weight, fat content, and lean body mass relative to their WT littermates (Purushotham et al. 2012a). Similarly, they did not display any remarkable difference in a series of histologic and gene expression analyses. However, when placed in high fat diets these mice were more prone to develop hepatic steatosis and metabolic damage (Purushotham et al. 2012a; Xu et al. 2010). Overall, these models provide conclusive evidence that SIRT1 deletion leads to inefficient metabolism. While this does not dramatically manifest into an overt phenotype when fed regular diets, it renders them more prone to metabolic complications upon dietary challenges.
In order to gain knowledge on the role of SIRT1 in particular tissues and how this could contribute to metabolic impairment, several tissue-specific SIRT1 deficient mouse models have been generated. In the sections below we will discuss the hypothesized roles for SIRT1 in different tissues and how the different tissue-specific models have validated or not these possible actions .
4.3.2 SIRT1 Functions in the Liver
4.3.2.1 SIRT1 and Hepatic Glucose Production
Liver is the major gluconeogenic organ in mammalian organisms. Formation of glucose from noncarbohydrate sources such as lactate , glycerol or amino acids is called gluconeogenesis . Initial hypothesis suggested that SIRT1 could potentiate gluconeogenesis by directly deacetylating and enhancing the transcriptional activity of PGC-1α or FoxO1, which are considered key positive controllers of the gluconeogenic transcriptional program (Brunet et al. 2004; Erion et al. 2009; Frescas et al. 2005; Rodgers et al. 2005; Rodgers and Puigserver 2007). Experiments using tail-vein injection of adenoviruses demonstrated that the activation of PGC1α by SIRT1 overexpression induced gluconeogenic genes expression , potentiated glucose production and repressed glycolytic genes mRNA levels (Rodgers and Puigserver 2007). Conversely, shRNA mediated inhibition of SIRT1 in liver was enough to decrease glycemia and improve both glucose and pyruvate tolerance (Rodgers and Puigserver 2007). Moreover, gluconeogenic capacity was almost completely defective upon SIRT1 reduction. Accordingly, the injection of antisense oligonucleotides against SIRT1 induces an increase of FoxO1 and PGC-1α acetylation , leading to decreased glycemia and hepatic glucose production through the down regulation of gluconeogenic gene expression (Erion et al. 2009).
Given the above observations, it would be expected that the genetic ablation of SIRT1 in the liver would lead to compromised gluconeogenic function and decreased basal glycemia . To date, a number of independent liver-specific SIRT1 knockout mice have been generated. Surprisingly, none of them displayed reduced basal glycemia (Chen et al. 2008; Purushotham et al. 2009; Wang et al. 2011; Wang et al. 2010). In fact, one of them had a tendency to have higher glucose levels even on chow diet and displayed enhanced glucose production upon fasting (Wang et al. 2011). Conversely, the initial work using adenoviral vectors suggested that higher SIRT1 levels in the liver should promote hyperglycemia (Rodgers and Puigserver 2007). These results, however, have been challenged recently by findings indicating that liver overexpression of SIRT1 actually attenuates hyperglycemia in insulin resistant mouse models (Li et al. 2011). In line with the latter observation, mice with a whole body overexpression of SIRT1 do not show signs of hyperglycemia and are protected against glucose intolerance (Banks et al. 2008; Pfluger et al. 2008).
The above results indicate that there are certain discrepancies between the results obtained in the initial experiments using adenoviral vectors and the transgenic mouse models. Therefore, either the adenoviral shRNA delivery is causing additional effects or SIRT1 transgenic models suffer from some sort of compensation. To shed some light onto these discrepancies it might be worth also considering a number of molecular and physiological observations. First of all SIRT1 activation in the liver does not seem to take place in the initial phases of gluconeogenesis (Liu et al. 2008). Hepatic glucose production is controlled during the early fasting stages by the cAMP response element binding protein (CREB) regulated transcription coactivator 2 (CRTC2). Detailed time-course analyses revealed that SIRT1 activation occurs during later phases, leading to the deacetylation and degradation of CRTC2, which attenuates the gluconeogenic rate (Liu et al. 2008). Similarly pharmacological activation of SIRT1 by resveratrol does not lead to enhanced hepatic glucose production (Baur et al. 2006; Lagouge et al. 2006), despite leading to a marked deacetylation of SIRT1 targets (Baur et al. 2006). Instead, it normalizes glycemia in insulin resistant mice (Lagouge et al. 2006). Altogether, these observations indicate that SIRT1 activation is not per se linked to enhanced hepatic glucose production, even if in some scenarios this might be the case. Rather, SIRT1 activation generally leads to an attenuation of the gluconeogenic rate, which constitutes a valuable therapeutic strategy in situations of insulin resistance and type 2 diabetes .
4.3.2.2 SIRT1 and Hepatic Lipid Metabolism
As described next, most studies to date agree that SIRT1 activation enhances oxidative metabolism in liver. The knock-down or genetic ablation of SIRT1 in liver induces hepatic lipid accumulation by upregulating the expression of lipogenic genes and reducing fatty acid oxidation capacity (Purushotham et al. 2009; Rodgers and Puigserver 2007; Wang et al. 2010; Xu et al. 2010). This renders SIRT1 deficient livers more sensitive to high-fat diet-induced hepatic steatosis (Purushotham et al. 2009). Conversely, SIRT1 overexpressing mice are protected against hepatic lipid accumulation and inflammation when fed a western diet (Li et al. 2011). Strikingly, one of the liver-specific knock-out models displayed the unusual feature of being protected against hepatic steatosis (Chen et al. 2008). The reasons for such a discordant observation in this particular model are not yet clear
So, how does SIRT1 enhance oxidative metabolism and prevent hepatic lipid accumulation? Rodgers et al., observed an increase in fatty acid oxidation genes and of cholesterol transport and the decrease in lipogenic gene expression induced by SIRT1 was dependent of PGC1α (Rodgers and Puigserver 2007). This is not surprising, as PGC-1α is a key downstream deacetylation target of SIRT1 in the regulation of mitochondrial and fatty acid oxidation gene expression. Complementarily, Purushotham et al. suggested that SIRT1 positively controls fatty acid oxidation though peroxisome proliferator-activated receptor α (PPARα) activation (Purushotham et al. 2009). The nuclear receptor PPARα regulates lipid metabolism , and, more particularly, gene expression implicated in β-oxidation . In SIRT1 deficient livers, no activation of PPARα target genes expression in presence of PPARα synthethic ligands was observed (Purushotham et al. 2009). Mechanistically, the authors elegantly demonstrated that SIRT1 binds to the ligand binding domain (LBD) and DNA binding domain (DBD) of PPARα. Consistently, SIRT1 was found to be present on the promoter of PPARα-target genes (Purushotham et al. 2009). In turn, SIRT1 allows proper deacetylation of PGC-1α, which then can coactivate PPARα. In the absence of SIRT1, PGC-1α remains associated in a constitutively hyperacetylated state , which dampens PGC-1α coativating activity (Rodgers et al. 2005) and, hence, blunts PPARα transcriptional activation.
SIRT1 inhibits lipogenic gene expression, most likely by acting as a negative regulator of Sterol Regulatory Element Binding Protein (SREBP)-1c (Ponugoti et al. 2010; Rodgers and Puigserver 2007; Walker et al. 2010). SREBP-1c is a transcription factor activated in situations of nutrient abundance. It promotes the expression of lipogenic and cholesterogenic genes in order to facilitate fat storage. Walker et al. have demonstrated that deacetylation of SREBP-1c by SIRT1 makes the protein prone to ubiquitin-mediated degradation (Walker et al. 2010). Hence, SIRT1 activation leads to decreased SREBP-1c protein levels. This is manifested in a decreased abundance of SREBP-1c on the promoter of lipogenic target genes upon SIRT1 activation (Ponugoti et al. 2010; Walker et al. 2010).
4.3.2.3 SIRT1 and Cholesterol Metabolism
Several observations, such as decreased expression of genes involved in cholesterol transport in SIRT1 liver-specific KO mice or the reduction of blood cholesterol levels in SIRT1 overexpressing mice, suggest that SIRT1 could also modulate cholesterol metabolism (Bordone et al. 2007; Rodgers and Puigserver 2007). Indeed, SIRT1 have been shown to modulate cholesterol metabolism though a positive control of Liver X Receptors (LRX) , LXRα and LXRβ. To do so, SIRT1 deacetylates LXRα on Lys432 and LXRβ on Lys433, promoting their activation (Li et al. 2007). In mouse embryonic fibroblasts (MEFs) from SIRT1 knockout mice, LXR target genes expression is decreased (Li et al. 2007). Similarly, LXR targets are not fully activated in SIRT1 liver-specific knockout mice mice fed with high-caloric diet, a condition where LXRs are highly active (Chen et al. 2008). Upon ligand binding, LXRs interact with SIRT1, which then mediates its deacetylation and activation. This deacetylation event, however, also makes LXRs more susceptible to ubiquitination and degradation (Li et al. 2007). The knock-down of SIRT1 in the liver also leads to decreased expression of CYP7A1 , a key LXR target, even though whether this happens in a LXR dependent fashion is unclear (Rodgers and Puigserver 2007). It is worth mentioning that LXRs are also a potent inducer of lipid anabolism by increasing SREBP-1c activity (Kalaany and Mangelsdorf 2006). However, SIRT1 can deacetylate SREBP-1c and lead it to proteasomal degradatation (Walker et al. 2010). Therefore, SIRT1 activation might prompt the beneficial effects of LXR activity on cholesterol homeostasis while preventing the detrimental effects on lipid anabolism by deacetylating SREBP-1c . This scenario is perfectly aligned with the data obtained in mouse models, where SIRT1 transgenesis improves cholesterol metabolism and prevents hepatic steatosis, while SIRT1 deletion in the liver favours lipid accumulation .
The bile acid sensing Farnesoid X-receptor (FXR) is another key player in the regulation of cholesterol and lipid metabolism that can be directly deacetylated on Lys157 and Lys217 by SIRT1 (Kemper et al. 2009). Down regulation of hepatic SIRT1 increases FXR acetylation, which inhibits its heterodimerization with RXRα (Kemper et al. 2009). Indeed, high FXR acetylation levels are observed in a mouse model of metabolic disease (Kemper et al. 2009). In line with these observations, SIRT1 deletion in the liver is enough to deregulate FXR transcriptional program and lead to the formation of cholesterol gallstones (Purushotham et al. 2012b). Activation of SIRT1 might, hence, become an attractive strategy to activate FXR and induce its target genes expression, which could contribute to the better cholesterol homeostasis .
4.3.3 SIRT1 Functions in Skeletal Muscle
Sketetal muscle is a key player in whole body metabolic homeostasis . A major feature of skeletal muscle is its plasticity. It can dramatically increase glucose uptake upon insulin stimulation or contraction and accounts for up to 80 % of total post-pandrial glucose disposal (Hawley et al. 2006). Similarly it can regulate oxidative metabolism and mitochondrial biogenesis in response to a number of stimuli. For example, skeletal muscle switches from glucose to fatty acid utilization during fasting so to spare glucose for other tissues. Therefore, muscle exquisitely fine tunes fuel utilization to environmental cues.
Initial hints on the possible roles of SIRT1 in skeletal muscle were obtained when SIRT1 was identified as a negative myogenic regulator . Overexpression of SIRT1 impairs myotube formation (Fulco et al. 2003). Conversely, decreased SIRT1 triggers premature differentiation (Fulco et al. 2003). Mechanistically this effect could be explained because SIRT1 represses the muscle transcriptional regulator MyoD , which acts as a critical determinant of skeletal muscle differentiation (Fulco et al. 2003). SIRT1 is also a key mediator by which nutrient restriction impairs muscle differentiation. Upon glucose depletion, AMPK is activated. In turn, AMPK enhanced Nampt expression, which increased the NAD+ availability SIRT1 activity (Fulco et al. 2008).
As mentioned above, muscle is a very plastic tissue, and has the ability to regulate oxidative metabolism and mitochondrial function in response to contraction or changes in nutrient availability. PGC-1α is a key master regulator of mitochondrial biogenesis in mammals. PGC-1α overexpression is enough to trigger mitochondrial biogenesis and oxidative metabolism in skeletal muscle (Lin et al. 2002). SIRT1 deacetylates PGC-1α in skeletal muscle during fasting and this deacetylation is required for PGC-1α-mediated induction of mitochondrial and fatty acid oxidation gene expression (Gerhart-Hines et al. 2007). Similarly, PGC-1α becomes deacetylated after an exercise bout (Canto et al. 2009). AMPK seems to play a key role in triggering SIRT1 activity during energy stress. Indeed, various labs have demonstrated that the activation of SIRT1 in response to nutrient or energy deprivation depends on AMPK activation (Canto et al. 2010; Fulco et al. 2008). The link between AMPK and SIRT1 activities can be explained by, at least, two non-exclusive mechanisms. The first link proposed consists in the modulation of NAD+ bioavailabily. The shift from glucose to fat oxidation induced by AMPK allows an increase in NAD+ that is enough to activate SIRT1 in a relatively short time frame (Canto et al. 2009). Additionally, AMPK triggers Nampt expression, which helps maintaining a more protracted increase in NAD+ (Canto et al. 2009; Fulco et al. 2008). As an alternative possibility, it has been recently proposed that AMPK could phosphorylate SIRT1 and abrogate this way its interaction with DBC1 (Nin et al. 2012). However, the phosphorylation of SIRT1 by AMPK has not been observed previously by other labs (Canto et al. 2009; Greer et al. 2007), and the residues reported are far from conserved or from matching AMPK target consensus sequences. At this level, it is also interesting to note that resveratrol , which is generally considered as a SIRT1 activator, also requires AMPK in vivo to activate SIRT1 and achieve its beneficial metabolic effects (Canto et al. 2010; Um et al. 2010). However, it has been recently noticed that this might only be the case when resveratrol is used at high doses (Price et al. 2012).
Considering the above data, one would expect that SIRT1 transgenesis would increase mitochondrial biogenesis in skeletal muscle. In line with this speculation, global SIRT1 transgenic mice have been shown to display higher mitochondrial content (Price et al. 2012). Conversely, deletion of SIRT1 in adulthood led to impaired mitochondrial function (Price et al. 2012). Similarly, muscle-specific deletion of SIRT1 leads to a slight impairment in mitochondrial function (Menzies et al. 2013), even though this was not clearly observed in another study in a similar mouse model (Philp et al. 2011). The reason for this discrepancy might derive from the different Cre lines used to ablate the Sirt1 gene in muscle. In both mouse models, however, the deletion of SIRT1 in skeletal muscle, however, did not seem to have a major effect on metabolic homeostasis of mice on regular diet (Menzies et al. 2013; Schenk et al. 2011).
A second extrapolation derived from above mentioned the cell-based assays would be that mice deficient in SIRT1 would display impaired adaptation to nutrient and energy stress. In line with this, muscle-specific SIRT1 knock-out mice failed to become more insulin sensitive upon caloric restriction (CR) (Schenk et al. 2011). CR increases SIRT1 deacetylase activity in skeletal muscle, in parallel with enhanced insulin-stimulated PI3K signaling and glucose uptake (Schenk et al. 2011). These adaptations in skeletal muscle insulin action triggered by CR were completely abrogated in mice lacking SIRT1 deacetylase activity in muscle (Schenk et al. 2011). This could be mechanistically explained by various reasons. Firstly, SIRT1 was found to be required for the deacetylation and inactivation of the transcription factor Stat3 during CR, which resulted in decreased gene and protein expression of the p55α/p50α subunits of PI3K , thereby promoting more efficient PI3K signaling during insulin stimulation (Schenk et al. 2011). Alternatively, SIRT1 has also been demonstrated to be a repressor of PTP1b, a major tyrosine phosphatase for the insulin receptor and the insulin receptor substrate proteins , IRS1 and IRS2 (Sun et al. 2007). Therefore, it is likely that SIRT1 deficient muscles also display higher PTP1b activity, which would also prevent the enhancement of insulin signaling in response to calorie restriction. These results clearly support the notion that SIRT1 is key for metabolic adaptations triggered by nutrient deprivation in skeletal muscle. Given this premise, it was surprising to see that mice lacking SIRT1 in skeletal muscle could perfectly adapt and enhance oxidative metabolism in response to exercise training (Philp et al. 2011). However, exercise is a complex stimuli, affecting multiple pathways with likely redundant functions. Strikingly, PGC-1α was normally deacetylated in response to exercise, despite the lack of SIRT1 (Philp et al. 2011). To solve this paradox, it was proposed that muscle contraction decreases the interaction of PGC-1α with the acetyltransferase enzyme, GCN5 (Philp et al. 2011). This way, PGC-1α deacetylation upon exercise would not be a consequence of enhanced deacetylation but of decreased acetylation rates. Importantly, it was recently found that resveratrol had synergistic effects with exercise on muscle mitochondrial biogenesis (Menzies et al. 2013). While the effect of exercise on mitochondrial biogenesis was independent of SIRT1, the synergy of resveratrol and exercise was lost on SIRT1 muscle-specific knock-out mice (Menzies et al. 2013).
Altogether, these results indicate that SIRT1 activation can improve mitochondrial function in mice and influence insulin sensitivity. However, they also demonstrate that many physiological stimuli prompting enhanced oxidative metabolism in muscle may rely in additional complementary effectors to induce such adaptations .
4.3.4 SIRT1 Functions in Adipose Tissues
4.3.4.1 White Adipose Tissue
White adipose tissue (WAT) is an important tissue for the regulation of metabolic homeostasis. WAT is the major site for fat storage in mammalian organisms. Fat storages are dynamically regulated in WAT, and lipolytic or lipogenic processes can be activated in response to nutrients and hormones. In obesity and type 2 diabetes (T2D) , circulating free fatty acid (FFA) levels are high, generally correlating with insulin resistance in both conditions. Importantly, the WAT also has critical actions as an endocrine tissue, by secreting hormones and cytokines , such as leptin, adiponectin or TNFα, that affect insulin sensitivity, inflammation and, therefore, have major consequences on metabolic homeostasis (Rosen and Spiegelman 2006).
One of the critical regulators of fat storage in WAT is the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) , whose activity promotes adipocyte differentiation and lipid anabolism (Rosen and Spiegelman 2006). A possible role of SIRT1 in WAT homeostasis was evidenced when it was found that SIRT1 could act as a PPARγ repressor (Picard et al. 2004). During fasting SIRT1 associated with PPARγ and promoted the binding of the corepressor NCoR1 (Picard et al. 2004). This favored fat mobilization instead of storage. A complementary study demonstrated that SIRT1 could repress PPARγ transcriptional activity on target lipogenic genes through a direct inhibitory-deacetylation (Qiang et al. 2012). The ablation of SIRT1 in adipose tissue promotes body weight gain, mostly due to an increase in fat mass. The size of adipocyte was bigger than in control mice, even on chow diet (Chalkiadaki and Guarente 2012). Altogether, this renders the adipocyte-specific SIRT1 KO mice prone to develop insulin resistance. Importantly, it has been described that obesity results in decreased SIRT1 in rodent and human adipose tissues (Chalkiadaki and Guarente 2012; Costa Cdos et al. 2010; Gillum et al. 2011). The reason for this decrease might rely on the fact that obesity triggers the cleavage of SIRT1 via a caspase 1 dependent mechanism (Chalkiadaki and Guarente 2012). This cleavage renders SIRT1 prone to degradation, therefore, decreasing global SIRT1 activity.
Two models of SIRT1 overexpression in mice have shown to be protected against HFD-induced insulin resistance (Banks et al. 2008; Pfluger et al. 2008). However, none of them observed decreased fat mass. Interestingly, a third model of global SIRT1 overexpression displayed a decrease in fat mass (Bordone et al. 2007), in line with the observations derived from the use of pharmacological activators of SIRT1, such as resveratrol or SRT1720 (Feige et al. 2008; Lagouge et al. 2006).
Adipose tissue inflammation is believed to be a hallmark of whole body insulin resistance. All animal models examined to date suggest a protective role of SIRT1 in adipose tissue inflammation (Chalkiadaki and Guarente 2012; Gillum et al. 2011). SIRT1 represses the expression of genes implicated in inflammation in adipocytes (Yoshizaki et al. 2009). Adipose-specific SIRT1 knockout mice displayed an increase of macrophage recruitment to adipose tissue (Gillum et al. 2011). In line with these studies, overexpression of SIRT1 in mice (SirBACO mice) prevents against adipose tissue macrophage accumulation caused by HFD (Gillum et al. 2011). Importantly, it has also been shown in humans how SIRT1 mRNA levels are inversely related to adipose tissue macrophage infiltration in human sub-cutaneous fat (Gillum et al. 2011).
4.3.4.2 Brow Adipose Tissue
Brown adipose tissue (BAT) has a remarkable abundance of mitochondria and contributes positively to energy expenditure, at least in mice. BAT is characterized by the expression of the mitochondrial uncoupling protein UCP1 , which allows dissipation of energy as heat for thermogenesis (Rosen and Spiegelman 2006). In response to adrenergic stimulation or cold exposure, white adipocytes can also obtain brown adipocyte-like characteristics (Orci et al. 2004; Puigserver et al. 1998; Rosen and Spiegelman 2006; Tiraby et al. 2003; Wu et al. 2012a). The binding of PGC1α to PPARγ promotes brown adipocyte-like features in white adipocytes though an up-regulation of brown-adipocyte specific genes, such as UCP1, and a down-regulation of white-adipocyte specific genes (Puigserver et al. 1998). Adipose tissue specific SIRT1 knockout mice display both enhanced WAT and BAT mass, due to enhanced fat accumulation (Chalkiadaki and Guarente 2012). Given the ability of SIRT1 to increase PGC-1α activity and lipid oxidation, SIRT1 activation might prevent excessive accumulation of lipids in BAT by boosting fat consumption and enhancing thermogenic function. Studies on indirect SIRT1 activation models would support this hypothesis. First, deletion of PARP-1 in mice increases NAD+ content and SIRT1 activity in BAT. PARP-1-/- mice can retain body temperature much better upon cold exposure, testifying for enhanced thermogenic function (Bai et al. 2011b). The BAT of these mice display higher mitochondrial content and a transcriptional up-regulation of genes implicated in mitochondrial respiration and fatty acid oxidation, as well as UCP1 (Bai et al. 2011b). Second, the enhancement of SIRT1 activity promoted by nicotinamide riboside also results in a better ability to maintain their body temperature during cold exposure (Canto et al. 2012). These two studies illustrate that activation of SIRT1 could benefit BAT function.
A recent study has demonstrated a role for SIRT1 in the “browning” of WAT. Overexpression of SIRT1 enhances a down-regulation of WAT specific genes in a white adipose depots and up-regulates BAT characteristics, while SIRT1 deletion has the opposite effect (Qiang et al. 2012). To do so, SIRT1 deacetylates PPARγ by SIRT1. The deacetylation of PPARγ favors the recruitment of PRDM16, a transcriptional coregulator that drives the BAT adipogenic program (Seale et al. 2007). This way, mice overexpressing SIRT1 have a more potent induction of a BAT-like phenotype of the subcutaneous WAT upon cold exposure (Qiang et al. 2012).
Altogether, while we are just beginning to understand the influence of SIRT1 on adipose tissue homeostasis, it seems clear that SIRT1 promotes lipid mobilization and enhances BAT-like characteristics. Therefore, SIRT1 activation could protect against metabolic diseases by enhancing energy expenditure and favoring thermogenic function .
4.3.5 SIRT1 Functions in the Pancreas
Pancreatic β-cells play a central position in the regulation of glucose homeostasis by secreting insulin in response to elevated glucose levels. Initial data from transgenic models proposed that SIRT1 positively controls glucose stimulated insulin secretion (GSIS) . The overexpression of SIRT1 specifically in pancreatic β-cells is enough to improve glucose tolerance and insulin secretion in response to glucose or KCl stimulation (Moynihan et al. 2005). In line with this model, GSIS is blunted in islets from SIRT1 knockout mice or in β-cells where SIRT1 has been knocked down by siRNAs (Bordone et al. 2006). Both studies converge proposing that SIRT1 improves GSIS though a negative control of the mitochondrial uncoupling protein UCP2. This would favor ATP production in response to higher glucose levels. Along this line, β-cell specific overexpression of SIRT1 suffices to prevent glucose intolerance upon high-fat feeding. The beneficial effects of SIRT1 on insulin secretion, however, are lost upon aging (Ramsey et al. 2008), to be completely lost at 18 -24 months of life. This was explained by a decrease of NAD+ availability in aged tissues, which could potentially limit the activation of SIRT1. Supporting this hypothesis, increasing NAD+ by NMN supplementation is enough to recover the benefits of β-cell SIRT1 overexpression in aged mice. Highlighting the relevance of NAD+ availability for SIRT1 activity and pancreatic function, the Slow Wallerian Degeneration (WldS) spontaneous mutant mice, overexpressing a chimeric protein that contains the full length NMNAT1 protein, display enhanced NAD+ availability and are protected against streptozotocin- and dietary-induced glucose intolerance in a SIRT1 dependent manner (Wu et al. 2011). Further supporting the beneficial effects of SIRT1 on pancreatic function, Wu et al. have shown that SIRT1 activation could be a promising strategy to prevent the deleterious effects of palmitate on insulin secretion and β-cell function (Wu et al. 2012b).
Pancreatic β-cell function can be also influenced by controlling β-cell mass. β-cell mass is determined by the balance between apoptotic, proliferative and neogenic processes. Pancreatic β-cell mass and β cell area are unchanged in β-cell specific SIRT1 overexpressing mice as well as in heterozygous and homozygous SIRT1 knockout mice (Moynihan et al. 2005) (Bordone et al. 2006). The lack of effect of SIRT1 on pancreatic β-cell mass, however, has been challenged recently by a few observations. First, the deletion of the PARP-2 gene leads to a constitutive increase in SIRT1 expression in many tissues, including pancreas (Bai et al. 2011a). PARP-2 -/- mice display marked glucose intolerance despite being more insulin sensitive (Bai et al. 2011a). Explaining this, GSIS is dramatically impaired in PARP-2 deficient mice. Upon close examination β-cell mass was significantly reduced in PARP-2 -/- mice, and failed to proliferate upon high-fat feeding, further magnifying HFD-induced glucose intolerance (Bai et al. 2011a). Mechanistically, it was proposed that higher SIRT1 activity led to a constitutive deacetylation and activation of FOXO1 , a negative regulator of β-cell proliferation (Kitamura et al. 2002). In line with this, the expression of pdx1, a FOXO1 target and a critical regulator of β-cell proliferation and differentiation (Kitamura et al. 2002), was dramatically reduced in PARP-2 deficient mice, as well as that of PDX1 target genes (Bai et al. 2011a). In line with this, GLP-1 positively influences β-cell proliferation by disrupting the association between FoxO1 and SIRT1 (Bastien-Dionne et al. 2011). This promotes FoxO1 hyperacetylation and nuclear exclusion, therefore relieving the repression of pancreatic β-cell proliferation. This constitutes another example where SIRT1 is also regarded as a negative regulator of β-cell mass and where SIRT1 inhibition might actually be positive to enhance β-cell function.
While seemingly opposite, these dichotomy of effects might have an explanation. While chronic activation of SIRT1 could be deleterious, SIRT1 might be implicated in the protection and adaptation of β-cells to oxidative stress and inflammation. In line with this, SIRT1 have been shown to be protective against cytokine induced β-cell toxicity (Lee et al. 2009). Several mechanisms might contribute to do so. First, SIRT1 negatively controls the pro-inflammatory NF-kB signaling pathway though nuclear deacetylation of the subnit p65 which prevent the DNA binding and, consequently, the transcriptional activity of NF-kB (Lee et al. 2009; Yeung et al. 2004). Similarly, the control of FoxO1 activity by SIRT1 might critical for the protection against oxidative stress in many cell types and tissues, including β-cells (Brunet et al. 2004; Hughes et al. 2011; Kitamura et al. 2005). This way, SIRT1 might have both protective and detrimental roles on β-cell function, depending on the timing and flexibility of its activation. Therefore, therapeutic approaches aimed to increase SIRT1 activity in β-cells should take into account this delicate balance .
4.3.6 SIRT1 and Food Intake Behaviour
The hypothalamus has a key role in the control of food intake, glucose homeostasis and energy balance. Hypothalamic neurons are able to detect changes in circulating hormones and nutrients and to respond to these changes by secreting several hunger/satiety hormones such as a-melanocyte-stimulating hormone (α-MSH) or agouti-related protein (AgRP) . The proopiomelanocortin (POMC) expressing neurons and the AgRP expressing neurons in the hypothalamus constitute central nodes in the regulation of feeding behaviour and energy expenditure. POMC neurons negatively controls food intake principally through the release of the α-MSH, which is a ligand for melanocortin-4 receptor (MC4R) neurons . This way, α-MSH acts as an agonist of the MC4R to promote satiety. Conversely, the AgRP is an antagonist of MC4R and has the opposite effect, that is promoting food intake in response to fasting or caloric restriction (Gao and Horvath 2008).
SIRT1 is highly expressed in the arcuate nucleus (ARC) , where we find the AgRP and POMC neurons, and in the vendromedial nuclei (VMN) , where we find the MC4R neurons (Ramadori et al. 2008; Satoh et al. 2010). Intracerebroventicular (ICV) injection of Ex527, a SIRT1 inhibitor, or SIRT1 siRNAs leads to reduced food intake in rodents (Cakir et al. 2009; Dietrich et al. 2010). In fact, the selective deletion of SIRT1 in AgRP neurons is enough to decrease food intake due to impaired MC4R antagonism from this neurons (Dietrich et al. 2010). This result suggests that SIRT1 might be required to increase food intake in situations of nutrient deprivation. Strikingly, no effects on food intake where observed when SIRT1 gene was deleted in POMC neurons (Ramadori et al. 2010). Interestingly, mice lacking SIRT1 in POMC neurons were prone to obesity upon high-fat feeding. This effect, however, does not seem to stem from the control of food intake behaviour but, rather by indirectly decreasing the metabolic rate of peripheral tissues (Ramadori et al. 2010).
Whole brain overexpression of SIRT1 (BRASTO mice ) promotes physical activity in response to CR (Satoh et al. 2010). This observation suggests SIRT1 could play a role in the control of pituitary hormones and metabolic peripherical effects in response to different dietetary. Again, this points out how SIRT1 might be activated upon nutrient scarcity and promotes adaptations aimed to enhance food foraging behavior. In apparent discrepancy, whole-body overexpression or deletion of SIRT1 does not seem to lead to major changes in food intake (Banks et al. 2008; Pfluger et al. 2008; Price et al. 2012). However, these mice undergo very significant metabolic changes, which might feedback and interfere with the natural regulation and effects of SIRT1. Indeed, the regulation of endogenous SIRT1 activity in the hypothalamus is still nebulous. While it seems clear that SIRT1 expression and activity might be modulated by food intake, a couple of studies have led to apparently opposite conclusions. On one hand, it has been shown that fasting increases SIRT1 activity, inducing the deacetylation of FoxO. This, in turn, represses POMC neurons and enhances AgRP expression, therefore promoting food intake (Cakir et al. 2009). Another study, however, demonstrates that SIRT1 protein level decreases during fasting in hypothalamus (Sasaki et al. 2010). In this case, shockingly, the authors argue that SIRT1 inhibits FoxO1-dependent expression of AgRP and consequently leads to the cessation of feeding (Sasaki et al. 2010). While these two studies illustrate the relevant nature of the SIRT1-FoxO1 axis for the regulation of food intake, the intricacy of the system is still far from unraveled.
Other studies have tried to clarify SIRT1 actions during feeding cycles by analyzing a possible role of SIRT1 in circadian food intake behavior. Indeed, SIRT1 has been found to act as a key regulator of the core circadian clock molecular machinery (Asher et al. 2008; Nakahata et al. 2008). The regulation of NAD+ bioavailability might constitute an attractive mechanism tying the circadian fluctuations of SIRT1 activity. Essentially, the expression levels of Nampt, the critical rate limiting enzyme in the mammalian NAD+ salvaging pathway, display a robust diurnal oscillation, with a peak around the beginning of the dark period in mice, in line with the maximal peak for the circadian fluctuation of SIRT1 activity (Nakahata et al. 2009; Ramsey et al. 2009). SIRT1 negatively regulates CLOCK:BMAL-1 transcriptional activity, which is a key positive controller of Nampt expression (Nakahata et al. 2009; Ramsey et al. 2009). Hence, the activation of SIRT1 shuts down Nampt expression. This will likely promote a decrease in NAD+ levels low enough to limit SIRT1. Once SIRT1 activity is low enough, CLOCK:BMAL-1 activity will be increased, and Nampt expression will be slowly recovered, reaching full circle. This way, SIRT1 constitutes an attractive mechanism by which metabolism can be tightly interconnected with circandian food intake behavior.
4.4 Conclusions and Future Perspectives
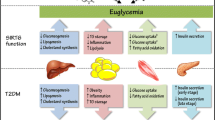
In this chapter we have provided an overview on the myriad of functions that SIRT1 exerts on metabolic regulation, based on the lessons learnt from transgenic mouse models (Fig. 4.1). Most data support the notion that SIRT1 contributes to an efficient adaptation to couple cellular and organismal metabolism to the nutritional and energy status. The activation of SIRT1 can be intimately linked to cellular metabolism by the rate-limitation imposed by NAD+ bioavailability. In general, most animal models demonstrate that SIRT1 activation is generally linked to a more efficient use of lipid energy sources and respiratory metabolism.

Roles of SIRT1 in tissues implicated in metabolism regulation
Despite the initial claims of SIRT1 as a “longevity” gene are debatable, SIRT1 can certainly impact on health and age-related decline in a more pleiotropic manner. In this sense, the complexity of SIRT1 physiology, largely conserved throughout evolution, involves an intricate network of downstream substrates, whose activation is only partially controlled via SIRT1. This can explain why initial findings on cultured cell models of SIRT1 overexpression or downregulation have sometimes not clearly mirrored into mouse physiology, where changes of SIRT1 activity might be more subtle or temporarily controlled. Another caveat on our understanding of SIRT1 comes from the knowledge inferred through the use of resveratrol or other so-called small molecule SIRT1 activators, whose specificity of action is far from clear.
Altogether, the collection of mouse models generated to date, have largely clarified the true role and limitations of SIRT1 actions. First, they established SIRT1 as a key gene for proper early organismal development. Tissue or temporatilly-controlled transgenic models all converge in the key role of SIRT1 for metabolic efficiency. Therefore, SIRT1 constitutes an extremely attractive target to improve oxidative metabolism and mitochondrial function, generally impaired in insulin resistant and aged population. However, a fine-tuned SIRT1 activity might be key to fully provide metabolic advantages. This is exemplified on the pancreatic regulation of SIRT1 activity, where constitutive activation of SIRT1 has been reported to be detrimental for global glucose tolerance. Similarly, enhanced SIRT1 activity in the heart can lead to cardiac failure by promoting dilated cardiomyopathy (Oka et al. 2011). Indeed, too much of a good thing might not always be that desirable and critical balances between metabolic benefits and side-effects might need to be balanced upon pharmacological approaches aimed to increase SIRT1 activity.
References
Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, Chini EN (2006) Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem Biophys Res Commun 349:353–359
Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423:181–185
Araki T, Sasaki Y, Milbrandt J (2004) Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305:1010–1013
Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U (2008) SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134:317–328
Bai P, Canto C (2012) The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab 16:290–295
Bai P, Canto C, Brunyanszki A, Huber A, Szanto M, Cen Y, Yamamoto H, Houten SM, Kiss B, Oudart H et al (2011a) PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab 13:450–460
Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH et al (2011b) PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13:461–468
Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, Accili D (2008) SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab 8:333–341
Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN (2007) The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. Faseb J 21:3629–3639
Bastien-Dionne PO, Valenti L, Kon N, Gu W, Buteau J (2011) Glucagon-like peptide 1 inhibits the sirtuin deacetylase SirT1 to stimulate pancreatic beta-cell mass expansion. Diabetes 60:3217–3222
Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K et al (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337–342
Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci U S A 101:12130–12135
Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L (2006) C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell 125:1165–1177
Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA (2002) Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem 277:45099–45107
Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, Moffat C, Crawford S, Saliba S, Jardine K et al (2008) SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One 3:e1759
Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J et al (2007) SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 6:759–767
Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A et al (2006) Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol 4:e31
Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R (2011) Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS One 6:e19194
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY et al (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015
Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvari M, Piper MD, Hoddinott M, Sutphin GL, Leko V, McElwee JJ et al (2011) Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477:482–485
Cakir I, Perello M, Lansari O, Messier NJ, Vaslet CA, Nillni EA (2009) Hypothalamic Sirt1 regulates food intake in a rodent model system. PLoS One 4:e8322
Calnan DR, Brunet A (2008) The FoxO code. Oncogene 27:2276–2288
Canto C, Auwerx J (2009) Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab 20:325–331
Canto C, Auwerx J (2012) Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev 64:166–187
Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458:1056–1060
Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P et al (2012) The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 15:838–847
Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J (2010) Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle. Cell Metab 11:213–219
Caton PW, Kieswich J, Yaqoob MM, Holness MJ, Sugden MC (2011) Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia 54:3083–3092
Chalkiadaki A, Guarente L (2012) High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab 16:180–188
Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L (2008) Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev 22:1753–1757
Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB (2005) Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123:437–448
Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF (2003) Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A 100:10794–10799
Chiang MC, Cheng YC, Lin KH, Yen CH (2013) PPARgamma regulates the mitochondrial dysfunction in human neural stem cells with tumor necrosis factor alpha. Neuroscience 229:118–129
Costa Cdos S, Hammes TO, Rohden F, Margis R, Bortolotto JW, Padoin AV, Mottin CC, Guaragna RM (2010) SIRT1 transcription is decreased in visceral adipose tissue of morbidly obese patients with severe hepatic steatosis. Obes Surg 20:633–639
Coste A, Louet JF, Lagouge M, Lerin C, Antal MC, Meziane H, Schoonjans K, Puigserver P, O’Malley BW, Auwerx J (2008) The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1{alpha}. Proc Natl Acad Sci U S A 105:17187–17192
Costford SR, Bajpeyi S, Pasarica M, Albarado DC, Thomas SC, Xie H, Church TS, Jubrias SA, Conley KE, Smith SR (2010) Skeletal muscle NAMPT is induced by exercise in humans. Am J Physiol Endocrinol Metab 298:E117–E126
Dietrich MO, Antunes C, Geliang G, Liu ZW, Borok E, Nie Y, Xu AW, Souza DO, Gao Q, Diano S et al (2010) Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: roles for Sirt1 in neuronal firing and synaptic plasticity. J Neurosci 30:11815–11825
Erion DM, Yonemitsu S, Nie Y, Nagai Y, Gillum MP, Hsiao JJ, Iwasaki T, Stark R, Weismann D, Yu XX et al (2009) SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proc Natl Acad Sci U S A 106:11288–11293
Escande C, Chini CC, Nin V, Dykhouse KM, Novak CM, Levine J, van Deursen J, Gores GJ, Chen J, Lou Z et al (2010) Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J Clin Invest 120:545–558
Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J (2008) Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab 8:347–358
Fjeld CC, Birdsong WT, Goodman RH (2003) Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci U S A 100:9202–9207
Frescas D, Valenti L, Accili D (2005) Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem 280:20589–20595
Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V (2008) Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell 14:661–673
Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V (2003) Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell 12:51–62
Gao Q, Horvath TL (2008) Neuronal control of energy homeostasis. FEBS Lett 582:132–141
Gerhart-Hines Z, Dominy JE Jr, Blattler SM, Jedrychowski MP, Banks AS, Lim JH, Chim H, Gygi SP, Puigserver P (2011) The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol Cell 44:851–863
Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P (2007) Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. Embo J 26:1913–1923
Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, Muise ES, Hsiao JJ, Frederick DW, Yonemitsu S et al (2011) SirT1 regulates adipose tissue inflammation. Diabetes 60:3235–3245
Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A (2007) The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem 282:30107–30119
Han L, Zhou R, Niu J, McNutt MA, Wang P, Tong T (2010) SIRT1 is regulated by a PPAR{gamma}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res 38:7458–7471
Hawley JA, Hargreaves M, Zierath JR (2006) Signalling mechanisms in skeletal muscle: role in substrate selection and muscle adaptation. Essays Biochem 42:1–12
Hayashida S, Arimoto A, Kuramoto Y, Kozako T, Honda S, Shimeno H, Soeda S (2010) Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARalpha in mice. Mol Cell Biochem 339:285–292
Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M (2010) Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun 1:3
Houtkooper RH, Canto C, Wanders RJ, Auwerx J (2010) The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31:194–223
Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13:225–238
Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, Riera TV, Lee JE, E SY, Lamming DW et al (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339:1216–1219
Hughes KJ, Meares GP, Hansen PA, Corbett JA (2011) FoxO1 and SIRT1 regulate beta-cell responses to nitric oxide. J Biol Chem 286:8338–8348
Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000a) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795–800
Imai S, Johnson FB, Marciniak RA, McVey M, Park PU, Guarente L (2000b) Sir2: an NAD-dependent histone deacetylase that connects chromatin silencing, metabolism, and aging. Cold Spring Harb Symp Quant Biol 65:297–302
Ivy JM, Klar AJ, Hicks JB (1986) Cloning and characterization of four SIR genes of Saccharomyces cerevisiae. Mol Cell Biol 6:688–702
Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13:2570–2580
Kalaany NY, Mangelsdorf DJ (2006) LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol 68:159–191
Kemper JK, Xiao Z, Ponugoti B, Miao J, Fang S, Kanamaluru D, Tsang S, Wu SY, Chiang CM, Veenstra TD (2009) FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab 10:392–404
Kim EJ, Kho JH, Kang MR, Um SJ (2007) Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell 28:277–290
Kim JE, Chen J, Lou Z (2008) DBC1 is a negative regulator of SIRT1. Nature 451:583–586
Kim MY, Kang ES, Ham SA, Hwang JS, Yoo TS, Lee H, Paek KS, Park C, Lee HT, Kim JH et al (2012) The PPARdelta-mediated inhibition of angiotensin II-induced premature senescence in human endothelial cells is SIRT1-dependent. Biochem Pharmacol 84:1627–1634
Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH 3rd, Wright CV, White MF, Arden KC, Accili D (2002) The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 110:1839–1847
Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D (2005) FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab 2:153–163
Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P et al (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127:1109–1122
Lee J, Padhye A, Sharma A, Song G, Miao J, Mo YY, Wang L, Kemper JK (2010) A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J Biol Chem 285:12604–12611
Lee JH, Song MY, Song EK, Kim EK, Moon WS, Han MK, Park JW, Kwon KB, Park BH (2009) Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes 58:344–351
Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L (2007) SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28:91–106
Li Y, Xu S, Giles A, Nakamura K, Lee JW, Hou X, Donmez G, Li J, Luo Z, Walsh K et al (2011) Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J 25:1664–1679
Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN et al (2002) Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418:797–801
Lin SJ, Ford E, Haigis M, Liszt G, Guarente L (2004) Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev 18:12–16
Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J 3rd et al (2008) A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 456:269–273
Lombard DB, Pletcher SD, Canto C, Auwerx J (2011) Ageing: longevity hits a roadblock. Nature 477:410–411
McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, Lansdorp PM, Lemieux M (2003) The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol 23:38–54
Menzies KJ, Singh K, Saleem A, Hood DA (2013) Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J Biol Chem 288(10):6968–6979
Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S et al (2006) International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev 58:726–741
Michan S, Sinclair D (2007) Sirtuins in mammals: insights into their biological function. Biochem J 404:1–13
Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 16:4623–4635
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB et al (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450:712–716
Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, Abdelmohsen K, Shin YK, Canto C, Scheibye-Knudsen M et al (2011) SRT1720 improves survival and healthspan of obese mice. Sci Rep 1:70
Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S (2005) Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab 2:105–117
Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P (2008) The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 134:329–340
Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P (2009) Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324:654–657
Nasrin N, Kaushik VK, Fortier E, Wall D, Pearson KJ, de Cabo R, Bordone L (2009) JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One 4:e8414
Neilson JR, Sharp PA (2008) Small RNA regulators of gene expression. Cell 134:899–902
Nemoto S, Fergusson MM, Finkel T (2004) Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 306:2105–2108
Nin V, Escande C, Chini CC, Giri S, Camacho-Pereira J, Matalonga J, Lou Z, Chini EN (2012) Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J Biol Chem 287:23489–23501
Noriega LG, Feige JN, Canto C, Yamamoto H, Yu J, Herman MA, Mataki C, Kahn BB, Auwerx J (2011) CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep 12:1069–1076
Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, Sadoshima J (2011) PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab 14:598–611
Okazaki M, Iwasaki Y, Nishiyama M, Taguchi T, Tsugita M, Nakayama S, Kambayashi M, Hashimoto K, Terada Y (2010) PPARbeta/delta regulates the human SIRT1 gene transcription via Sp1. Endocr J 57:403–413
Orci L, Cook WS, Ravazzola M, Wang MY, Park BH, Montesano R, Unger RH (2004) Rapid transformation of white adipocytes into fat-oxidizing machines. Proc Natl Acad Sci U S A 101:2058–2063
Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B et al (2010) SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem 285:8340–8351
Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, Ke H, Rehmann H, Taussig R, Brown AL et al (2012) Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148:421–433
Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E et al (2008) Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab 8:157–168
Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH (2008) Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A 105:9793–9798
Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC et al (2011) Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J Biol Chem 286:30561–30570
Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L (2004) Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429:771–776
Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao J, Zang M, Wu SY, Chiang CM, Veenstra TD, Kemper JK (2010) SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem 285:33959–33970
Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS et al (2012) SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 15:675–690
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92:829–839
Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X (2009) Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 9:327–338
Purushotham A, Xu Q, Li X (2012a) Systemic SIRT1 insufficiency results in disruption of energy homeostasis and steroid hormone metabolism upon high-fat-diet feeding. FASEB J 26:656–667
Purushotham A, Xu Q, Lu J, Foley JF, Yan X, Kim DH, Kemper JK, Li X (2012b) Hepatic deletion of SIRT1 decreases hepatocyte nuclear factor 1alpha/farnesoid X receptor signaling and induces formation of cholesterol gallstones in mice. Mol Cell Biol 32:1226–1236
Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, Rosenbaum M, Zhao Y, Gu W, Farmer SR et al (2012) Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell 150:620–632
Ramadori G, Fujikawa T, Fukuda M, Anderson J, Morgan DA, Mostoslavsky R, Stuart RC, Perello M, Vianna CR, Nillni EA et al (2010) SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell Metab 12:78–87
Ramadori G, Lee CE, Bookout AL, Lee S, Williams KW, Anderson J, Elmquist JK, Coppari R (2008) Brain SIRT1: anatomical distribution and regulation by energy availability. J Neurosci 28:9989–9996
Ramsey KM, Mills KF, Satoh A, Imai S (2008) Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 7:78–88
Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C et al (2009) Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324:651–654
Revollo JR, Grimm AA, Imai S (2004) The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 279:50754–50763
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118
Rodgers JT, Puigserver P (2007) Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A 104:12861–12866
Rosen ED, Spiegelman BM (2006) Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444:847–853
Sakamoto J, Miura T, Shimamoto K, Horio Y (2004) Predominant expression of Sir2alpha, an NAD-dependent histone deacetylase, in the embryonic mouse heart and brain. FEBS Lett 556:281–286
Sasaki T, Kim HJ, Kobayashi M, Kitamura YI, Yokota-Hashimoto H, Shiuchi T, Minokoshi Y, Kitamura T (2010) Induction of hypothalamic Sirt1 leads to cessation of feeding via agouti-related peptide. Endocrinology 151:2556–2566
Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT, Minor W, Scrable H (2008) Phosphorylation regulates SIRT1 function. PLoS One 3:e4020
Satoh A, Brace CS, Ben-Josef G, West T, Wozniak DF, Holtzman DM, Herzog ED, Imai S (2010) SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J Neurosci 30:10220–10232
Schenk S, McCurdy CE, Philp A, Chen MZ, Holliday MJ, Bandyopadhyay GK, Osborn O, Baar K, Olefsky JM (2011) Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. J Clin Invest 121:4281–4288
Schmidt MT, Smith BC, Jackson MD, Denu JM (2004) Coenzyme specificity of Sir2 protein deacetylases: implications for physiological regulation. J Biol Chem 279:40122–40129
Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, Tavernier G, Langin D, Spiegelman BM (2007) Transcriptional control of brown fat determination by PRDM16. Cell Metab 6:38–54
Shore D, Squire M, Nasmyth KA (1984) Characterization of two genes required for the position-effect control of yeast mating-type genes. EMBO J 3:2817–2823
Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, Zhai Q (2007) SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab 6:307–319
Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y (2007) Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem 282:6823–6832
Tiraby C, Tavernier G, Lefort C, Larrouy D, Bouillaud F, Ricquier D, Langin D (2003) Acquirement of brown fat cell features by human white adipocytes. J Biol Chem 278:33370–33376
Um JH, Park SJ, Kang H, Yang S, Foretz M, McBurney MW, Kim MK, Viollet B, Chung JH (2010) AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 59:554–563
van Roermund CW, Elgersma Y, Singh N, Wanders RJ, Tabak HF (1995) The membrane of peroxisomes in Saccharomyces cerevisiae is impermeable to NAD(H) and acetyl-CoA under in vivo conditions. Embo J 14:3480–3486
Viswanathan M, Guarente L (2011) Regulation of Caenorhabditis elegans lifespan by sir-2.1 transgenes. Nature 477:E1–E2
Viswanathan M, Kim SK, Berdichevsky A, Guarente L (2005) A role for SIR-2.1 regulation of ER stress response genes in determining C. elegans life span. Dev Cell 9:605–615
Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, Boss O, Hirsch ML, Ribich S, Smith JJ et al (2010) Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev 24:1403–1417
Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX (2011) Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest 121:4477–4490
Wang RH, Li C, Deng CX (2010) Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int J Biol Sci 6:682–690
Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G et al (2012a) Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150:366–376
Wu J, Zhang F, Yan M, Wu D, Yu Q, Zhang Y, Zhou B, McBurney MW, Zhai Q (2011) WldS enhances insulin transcription and secretion via a SIRT1-dependent pathway and improves glucose homeostasis. Diabetes 60:3197–3207
Wu L, Zhou L, Lu Y, Zhang J, Jian F, Liu Y, Li F, Li W, Wang X, Li G (2012b) Activation of SIRT1 protects pancreatic beta-cells against palmitate-induced dysfunction. Biochim Biophys Acta 1822:1815–1825
Xiong S, Salazar G, Patrushev N, Alexander RW (2011) FoxO1 mediates an autofeedback loop regulating SIRT1 expression. J Biol Chem 286:5289–5299
Xu F, Gao Z, Zhang J, Rivera CA, Yin J, Weng J, Ye J (2010) Lack of SIRT1 (Mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/- mice: a role of lipid mobilization and inflammation. Endocrinology 151:2504–2514
Yamakuchi M (2012) MicroRNA Regulation of SIRT1. Front Physiol 3:68
Yamakuchi M, Ferlito M, Lowenstein CJ (2008) miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A 105:13421–13426
Yang Y, Fu W, Chen J, Olashaw N, Zhang X, Nicosia SV, Bhalla K, Bai W (2007) SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat Cell Biol 9:1253–1262
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW (2004) Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J 23:2369–2380
Yoshino J, Mills KF, Yoon MJ, Imai S (2011) Nicotinamide Mononucleotide, a Key NAD(+) Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab 14:528–536
Yoshizaki T, Milne JC, Imamura T, Schenk S, Sonoda N, Babendure JL, Lu JC, Smith JJ, Jirousek MR, Olefsky JM (2009) SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol Cell Biol 29:1363–1374
Yuan J, Luo K, Liu T, Lou Z (2012) Regulation of SIRT1 activity by genotoxic stress. Genes Dev 26:791–796
Zannini L, Buscemi G, Kim JE, Fontanella E, Delia D (2012) DBC1 phosphorylation by ATM/ATR inhibits SIRT1 deacetylase in response to DNA damage. J Mol Cell Biol 4:294–303
Zhang Q, Wang SY, Fleuriel C, Leprince D, Rocheleau JV, Piston DW, Goodman RH (2007) Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc Natl Acad Sci U S A 104:829–833
Zhang T, Berrocal JG, Frizzell KM, Gamble MJ, DuMond ME, Krishnakumar R, Yang T, Sauve AA, Kraus WL (2009) Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J Biol Chem 284:20408–20417
Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W (2008) Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451:587–590
Acknowledgements
We thank the members of the Canto lab for inspiring discussions on SIRT1. We specially thank Dr. Sameer Kulkarni for carefully editing the manuscript. MB and CC are employees of the Nestlé Institute of Health Sciences S.A. The authors declare no financial interest on the research discussed in this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Boutant, M., Cantó, C. (2016). SIRT1 in Metabolic Health and Disease. In: Houtkooper, R. (eds) Sirtuins. Proteins and Cell Regulation, vol 10. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-0962-8_4
Download citation
DOI: https://doi.org/10.1007/978-94-024-0962-8_4
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-0961-1
Online ISBN: 978-94-024-0962-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)