
Abstract
As a pathogen of plague, Yersinia pestis caused three massive pandemics in history that killed hundreds of millions of people. Yersinia pestis is highly invasive, causing severe septicemia which, if untreated, is usually fatal to its host. To survive in the host and maintain a persistent infection, Yersinia pestis uses several stratagems to evade the innate and the adaptive immune responses. For example, infections with this organism are biphasic, involving an initial “noninflammatory” phase where bacterial replication occurs initially with little inflammation and following by extensive phagocyte influx, inflammatory cytokine production, and considerable tissue destruction, which is called “proinflammatory” phase. In contrast, the host also utilizes its immune system to eliminate the invading bacteria. Neutrophil and macrophage are the first defense against Yersinia pestis invading through phagocytosis and killing. Other innate immune cells also play different roles, such as dendritic cells which help to generate more T helper cells. After several days post infection, the adaptive immune response begins to provide organism-specific protection and has a long-lasting immunological memory. Thus, with the cooperation and collaboration of innate and acquired immunity, the bacterium may be eliminated from the host. The research of Yersinia pestis and host immune systems provides an important topic to understand pathogen-host interaction and consequently develop effective countermeasures.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
10.1 Interactions of Yersinia pestis and Immune Cells
Most human plague cases present clinically as one of three primary forms: bubonic, septicemic, or pneumonic plague. The most common form is bubonic plague, which is transmitted from rodent reservoirs to humans via the biting of infected fleas. Patients with primary bubonic plague can develop secondary septic or pneumonic infections, the latter of which can then be spread from person to person via respiratory droplets generated from severe sneezing and coughing of patients. Pneumonic plague is nearly always fatal unless treated with effective antibiotics within 20 h post symptom onset. All types of infection will cause a response of the host immune system against the invading Yersinia pestis.
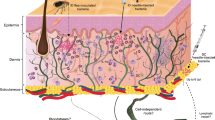
During the early stages of infection, Y. pestis can enter both macrophages and neutrophils through either active or passive entry mechanisms [1]. Although early researches showed Y. pestis is typically killed in neutrophils, whereas in macrophages, it can survive and acquire antiphagocytic capabilities, which enables its extracellular survival in vivo. Recent studies indicated that a small percent of Y. pestis can also survive and replicate in neutrophils and send a PS (a marker of early apoptosis) to macrophages [2, 3]. This progress may provide Y. pestis with a less inflammatory route of entry into macrophages, resulting in decreased proinflammatory signaling. To survive inside the host and maintain a persistent infection, Y. pestis uses a variety of different mechanisms to evade or overcome the host immune system, especially the innate immune system, as shown in Fig. 10.1.

Interactions between Yersinia pestis and the host immune system
10.2 Yersinia pestis Overcomes the Immune Response
Y. pestis uses many different virulence factors to resist host immune responses, facilitate cellular attachment or invasion, subvert endocytic trafficking, block phagocytosis, modulate apoptotic pathways, and manipulate innate immunity and host responses as part of the initial infection process.
10.2.1 Dampening of the Inflammatory Response
Many studies have demonstrated that infection by Y. pestis elicits a notably delayed inflammatory response [4–6]. Type III secretion system (T3SS), which is one of the pathogens’ major virulence factors, plays a crucial role in dampening host inflammatory responses. Multiple signaling pathways are repressed when the host is infected by Y. pestis by various Yersinia outer proteins (Yops). YopH inactivates the phosphatidylinositol-3 kinase (PI3K)-AKT cascade in macrophages, which correlates with the downregulation of mRNA coding for monocyte chemoattractant protein 1 (MCP-1) [7]. YopJ/P suppresses the mitogen-activated protein kinase (MAPK) signaling pathway by an unknown mechanism [8]. In vitro experiments indicated that YopE strongly inhibits nuclear factor-kappa B (NF-kB) activation and JNK and ERK activation, whereas YopT only moderately inhibits these responses [9]. YopK and YopM both inhibit inflammasomes [10, 11]. In addition, Y. pestis can suppress the production of cytokines and chemokines. LcrV, which is the needle tip protein that assembles into the needle tip complex, is associated with suppression of tumor necrosis factor alpha (TNFα) and interferon gamma (IFNγ) and the induction of immunosuppressive interleukin (IL)-10 in mice [12]. YopE can also prevent the production of IL-8 [9]. YopM interacts with protein kinase C-like 2 and ribosomal protein S6 kinase, which are also involved in proinflammatory signaling [13]. Recently, the N terminus of YscF was also found to decrease cytokine induction [14]. Inhibition of signaling pathways and suppression of cytokines not only reduces the activation of natural killer (NK) cells and phagocytes but also destroys the inflammatory environment necessary for adaptive immunity.
Lipopolysaccharide (LPS) is a major component of the outer membrane in Gram-negative bacteria and a ligand for toll-like receptor 4 (TLR4). In different host-specific environments, the expression and formation of LPS in Y. pestis change accordingly [15, 16]. When the bacteria grow in the flea gut (21–26 °C), they produce a typical hexa-acylated LPS, which activates TLR4-mediated immune signaling to induce the expression of proinflammatory cytokines (TNFα, IL-1, IL-6, and IL-8). However, after the temperature transition from the flea (26 °C) to the mammalian host (37 °C), Y. pestis instantly begins to produce tetra-acylated LPS, which is nonstimulatory for TLR4 and an antagonist for the stimulatory hexa-acylated form of LPS. Importantly, a Y. pestis strain that produces hexa-acylated lipid A at 37 °C was found to be more than 100,000-fold attenuated in a mouse model of bubonic plague [17]. The attenuated phenotype is accompanied by increased production of TNFα and depends upon host expression of TLR4. It is thus clear that Y. pestis evades innate immunity, at least in part, by avoiding TLR-mediated activation of innate immunity.
10.2.2 Resistance to Phagocytosis
Because Y. pestis proliferates extracellularly, it is essential to block phagocytosis after contact with host cells. At least four Yop effectors (YopH, YopE, YopT, and YopO) are involved in inhibiting the phagocytosis of yersiniae; however, their mechanisms are all different. YopH is a tyrosine phosphatase, which acts on several cytoskeletal proteins including p130Cas [18–20]. Dephosphorylation of p130Cas disrupts its interaction with Crk and subsequent Rac activation required for Yersinia internalization [21]. YopE, which acts as a GTPase-activating protein for the RhoA family of GTPases (RhoA, Rac and Cdc42) [22], inhibits Rac-dependent actin polymerization either directly or through inactivation of the upstream RhoG [23, 24]. YopT is a protease that specifically cleaves RhoA, Rac, and Cdc42 near their carboxyl termini [25], which irreversibly inactivates host cell proteins. YopO interferes with the host cell regulation of Rho GTPases by actin [26–30]. Using various models, several different cellular activities have been attributed to YopJ, including deubiquination of Ikβ and TNF receptor-associated factors (TRAFs), as well as acetylation of MAPK kinases. However, precisely how YopJ exerts its activity during infection is presently unclear. In summary, the translocated Yop effectors are exotoxins that disable the phagocytic machinery by (1) disrupting the host cell cytoskeleton, (2) suppressing cytokine production, and (3) interfering with cell signal pathways.
Another crucial antiphagocytic factor is the F1 capsule protein (Caf1), which is solely produced by Y. pestis. However, the mechanism of resistance to phagocytosis induced by this capsule protein is different from that of T3SS and presumably by preventing interactions with receptors that could potentially recognize and result in the uptake of pathogens [31]. PsaA was also shown to inhibit phagocytosis, most likely by binding lipoproteins that prevent recognition by host cells [32, 33].
10.2.3 Resistance to Complement-Dependent Bacteriolysis
Complement-dependent bacterial killing is one of the first lines of innate immunity against pathogens. Y. pestis must survive in blood to cause disease and be transmitted from host to host by fleas. Thus, resistance to serum complement is an essential phenotype for bacterial survival in blood. Unlike the enteropathogenic yersiniae (Y. enterocolitica and Y. pseudotuberculosis), which are fully resistant to complement when they are grown at 37 °C, but not when they are grown at 26 °C [5, 30], Y. pestis is constitutively resistant to complement at both 26 and 37 °C. In Y. enterocolitica, resistance to complement has been shown to involve YadA, Ail, OmpR, and LPS O antigen [34]. By binding to the complement regulatory proteins factor H and the C4b-binding protein, Ail and YadA provide resistance against complement-mediated killing [35, 36]. OmpR might alter the susceptibility of Y. enterocolitica to complement-mediated killing through remodeling of the outer membrane [37]. However, Y. pestis does not express YadA; therefore, Y. pestis LPS may mediate serum resistance [38], although there is no evidence of a direct role for LPS in serum resistance. Thus, Ail was found to play the crucial role in the inhibition of the bactericidal properties of complement by Y. pestis. Notably, there are four Ail/OmpX homologues encoded by the Y. pestis genome, but only y1324 (in KIM strain, corresponding to YPO2506 in CO92, ompX) confers resistance to human serum. Deletion of this gene results in a rapid, essentially 100 % loss of serum resistance [39, 40]. This loss is attributed to the action of complement because heat-inactivated serum does not have lethal properties. Although currently the exact mechanisms of resistance to complement-mediated killing conferred by Y. pestis Ail are still unknown, based on the studies from two other yersiniae (Y. enterocolitica and Y. pseudotuberculosis), it is likely that the protein binds to negative regulators of alternative (factor H) and classical and lectin [C4b-binding protein (C4BP)] complement pathways, thus preventing complement-mediated attack of the pathogen [36, 41–43].
10.2.4 Other Impacts on Host Immunity
Many bacteria have evolved means to convert host plasminogen to plasmin, a protease that degrades extracellular matrix. In Y. pestis plasminogen activator (Pla), encoded by the Y. pestis-specific plasmid pPCP1, plays an important role. In comparison with the wild type, Y. pestis lacking Pla has been reported to have greatly reduced virulence when introduced via the intradermal and subcutaneous routes but produced equivalent or nearly equivalent virulence when introduced by the intravenous route [5, 44, 45].These results partly different from early studies, which indicated even lack of plasmid pPst (also call pPCP1), did not lead to an increase in LD50 with either by subcutaneously or by respiratory route challenge [46, 47]. In the pneumonic plague model, the dissemination of Pla-deficient Y. pestis to the circulatory system was found to be unaffected, whereas bacterial growth in the lung was greatly reduced [44, 48]. Further studies are required to discern the specific mechanisms by which Pla impacts Y. pestis virulence. In addition, Pla has been reported to be a ligand for a macrophage and DC surface receptor, DEC-205, which is related to antigen presentation [49].
The 2-component regulatory system (2CS) OmpR-EnvZ is required to resist innate immunity during the early and late stages of the plague [50]. Different from Y. pestis’ 23 other 2CSs, OmpR-EnvZ is the only one required for production of bubonic, septicemic, and pneumonic plague. In in vitro studies, OmpR-EnvZ was required to counter serum complement and leukocytes but was not required for the secretion of antiphagocytic exotoxins. In contrast, in in vivo studies, Y. pestis lacking OmpR-EnvZ did not induce an early immune response in the skin and was fully virulent in neutropenic mice.
10.3 Innate Immunity in Plague
Normally, once a bacterium infects the host, the innate immune response provides immediate protection and 4–5 days post infection, the T or B cell-mediated adaptive immune responses begins to provide organism-specific protection. In 2005, using Yop beta-lactamase hybrids and fluorescent staining of live cells from plague-infected animals; Marketon et al. found that Y. pestis selected immune cells for injection. Further research in vivo showed that macrophages, dendritic cells, and granulocytes/neutrophils are early targets for injection, whereas B and T lymphocytes were rarely selected. Thus, it appears that Y. pestis appears to destroy cells with innate immune functions that represent the first line of defense [51]. A similar study identified the pulmonary cells targeted by Y. pestis during primary pneumonic plague using a FRET-based probe to quantitate injection of effector proteins by the Y. pestis T3SS [52]. They found that these bacteria target alveolar macrophages early during infection of mice, followed by a switch in host cell preference to neutrophils. In addition to mouse models, the Gunnison’s prairie dog, an important natural host of plague, was investigated [53]. This study highlights the importance of innate immunity against plague in wild Gunnison’s prairie dogs.
10.3.1 Macrophages
Y. pestis has long been considered to be a facultative intracellular pathogen [54]. Macrophages are often regarded as permissive sites for survival and replication of Y. pestis at early stages of infection. Bubonic plague is typically initiated as an intradermal infection following the bite of an infected flea. Then, Y. pestis may invade the host directly through the skin and encounter phagocytes such as neutrophils and macrophages at the site of invasion. Most Y. pestis bacilli initially present are likely killed by neutrophils; however, the bacteria phagocytosed by macrophages can survive. Y. pestis preferentially infects host macrophages, probably via recognition of specific surface-associated CCR5 molecules [55]. This intracellular growth is essential for the pathogenesis of Y. pestis in three ways. First, macrophages provide a niche, allowing bacteria to proliferate and acquire the ability to evade phagocytosis. Second, intracellular growth in macrophages provides a protected environment for the bacteria to avoid contact with other components of the host immune system such as complement. Third, Y. pestis in macrophages can express various virulence determinants [56, 57]. Furthermore, macrophages may provide the bacterium with a vehicle for transport from the initial site of infection to deeper lymph tissues [58, 59]. The late stage of Y. pestis infection is characterized by a rapid increase in the number of bacteria within the spleen and escape of bacteria from macrophages into the extracellular compartment of the spleen. The cause of this escape is likely to be related to the macrophage necrosis or apoptosis observed during in vitro studies [57, 60]. Once Y. pestis replicates and expresses various virulence determinants in macrophages, they can be released into the extracellular compartment and spread systemically with the acquisition of phagocytosis resistance. Although Y. pestis can survive and replicate in macrophages during the early stage of infection, this macrophage compliance can be overcome in vitro by stimulation with a combination of IFNγ and TNFα [1].
Although macrophages provide a niche for Y. pestis survival and replication in the early stages of infection, Y. pestis can also cause macrophage death. Two distinct processes, corresponding to the inflammatory crescendo, are observed in vivo. Y. pestis causes apoptosis in naïve macrophages, but cell death in activated macrophages by inflammatory pyroptosis [61]. During macrophage apoptosis, Y. pestis YopJ is necessary. When the bacterial LPS contacts host macrophages, proapoptotic signaling is activated [62, 63]. LPS also upregulates cell survival genes and inflammatory cytokine production controlled by MAPK and NF-kB [62, 64, 65]; however, YopJ inhibits their activation [66, 67], and, therefore, apoptotic signaling predominates [62, 68]. Apoptotic cells are cleared by phagocytes, and this encounter triggers production of the anti-inflammatory cytokines TGFα and IL-10 [69, 70], making the process noninflammatory. Pyroptosis results from the activation of caspase-1, which is functionally distinct from the structurally related apoptotic caspases [36]. Y. pestis-induced pyroptosis requires plasmid-encoded T3SS, but not YopJ or any of the other known effector molecules [61]. Caspase-1 stimulates maturation and secretion of multiple inflammatory cytokines such as IL-1 and IL-18 [71]. Thus, pyroptosis causes inflammation. Why are two kinds of macrophage death necessary in Y. pestis infection? In bubonic plague, the infections are obviously biphasic: bacteria initially replicate without a measurable host response for periods up to 36–48 h, with a noticeable lack of inflammation, but, eventually, phagocyte influx into infected tissues and lymph nodes results in inflammation, cytokine production, and tissue necrosis [6, 72–74]. The change in mode of macrophage death is partly explained by observation of the host responses to Y. pestis infection. Apoptosis (noninflammatory death) [75–77] of naïve macrophages is consistent with initial bacterial growth in the relative absence of inflammation. Pyroptosis (inflammatory death) [61, 78] in activated macrophages corresponds to later stages of infection, accompanied by enhanced cytokine production and tissue damage. Induction of both apoptosis and pyroptosis in macrophages may be a mechanism by which pathogens preferentially trigger immune cell death, resulting in bacterial dissemination and disruption of host innate immune signaling.
The available data strongly suggest that Y. pestis growth within macrophages plays an important, perhaps critical, pathogenic role during plague [58]. During bubonic plague, macrophages may provide a protected intracellular niche that allows time for flea-transmitted Y. pestis bacilli to adjust to growth within mammals, in part by upregulating expression of capsular F1 protein, LcrV, and Yops, thus enabling subsequent growth as extracellular, phagocyte-resistant bacteria. However, in pneumonic plague the role of macrophages seems different from that in bubonic plague. Dr. Goldman’s group found that depletion of roughly 92 % of alveolar macrophages had little or no effect on bacterial burden in the lungs [52]. They gave two possible reasons: macrophages are not involved in limiting yersiniae survival in the lungs or that Y. pestis is able to neutralize the antibacterial effects of sentinel alveolar macrophages, presumably as a result of expressing antiphagocytic/anti-inflammatory factors including the Yops, F1 capsular protein, and pH 6 antigen.
10.3.2 Neutrophils
Histological, flow cytometric, and laser confocal microscopy evidence indicates that Y. pestis phagocytosed by neutrophils are killed [1, 54]. However, Y. pestis primarily targets neutrophils early after inoculation in the lung, presumably to limit host innate immune mechanisms aimed at bacterial killing and clearance. Thus, it seems that the interaction between Y. pestis and host neutrophils may have a strong bearing on the outcome of infection [79].
Researchers often assess bubonic plague through needle-inoculated Y. pestis. However, Shannon et al. found that the innate cellular host responses to flea-transmitted Y. pestis differed from and were more variable than responses to needle-inoculated bacteria [80]. They therefore developed new tools allowing for intravital microscopy of Y. pestis in the dermis of an infected mouse after transmission by its natural route of infection, the bite of an infected flea. They found that uninfected flea bites typically induced minimal neutrophil recruitment. The magnitude of neutrophil response to flea-transmitted Y. pestis varied considerably and appeared to correspond to the number of bacteria deposited at the bite site.
Dr. Goldman’s group first identified the initial host cell targets of fully virulent Y. pestis during pulmonary infection. Using flow cytometry to monitor injection of a YopE-TEM fusion protein by the T3SS, they showed that Y. pestis initially targets CD11c high alveolar macrophages and neutrophils in the lungs. However, in contrast to the bubonic plague, during the first 24 h after pulmonary infection with a fully virulent Y. pestis strain, no significant changes were observed in the lungs in the levels of neutrophil infiltrate, the expression of adhesion molecules, or the expression of the major neutrophil chemoattractant CXCL1 (also known as keratinocyte cell-derived chemokine, KC) [4]. These results indicate that Y. pestis can slow the rate of neutrophil influx to the lungs by delaying the onset of chemokine and cytokine release. Moreover, neutrophils were observed to be “tightly packed” within spaces 72 h post infection during pneumonic plague [6]. However, in mice infected with an avirulent Y. pestis strain, early induction of chemokines, rapid neutrophil infiltration, and reduced bacterial burden were observed in the lungs of mice [81]. These results indicate that strain virulence may determine the host immune responses. Moreover, it seems that prevention of the early influx of neutrophils to the lungs is of major importance for Y. pestis virulence.
A growing volume of literature highlights interactions between Y. pestis and neutrophils, but the deletion of neutrophils during pneumonic plague shows significantly different results in various studies. Dr. Goldman’s group showed that depletion of neutrophils had little to no effect on bacterial burden in the lungs [52]. This is in contrast to others studies, such as by Laws et al. who reported a modest increase in bacterial titers in lungs of infected mice early after neutrophil depletion [79]. Additionally, our group found neutrophil deletion with anti-Ly6G antibodies decreased survival in an intranasal Y. pestis mouse model [82]. Notably, a fully virulent Yersinia pestis strain, CO92, was used in Dr. Goldman’s research; however, our study used a Y. pestis strain (strain 201) virulent to mice but nonvirulent to humans. Thus, the disparity in findings may come from differences in mouse lines and Y. pestis strains.
The mechanisms used by Y. pestis for targeting neutrophils have also been described. The Y. pestis adhesin Ail is required for efficient targeting of neutrophils in vivo [83]. The Ail protein was previously reported to inhibit the innate immune response, in particular the recruitment of a protective polymorphonuclear leukocyte (PMN) responses to the infected lymph node [84]. To identify factors conferring specificity to neutrophil targeting, a study investigated the role of serum [83]. They found that neutrophil targeting is mediated by complement receptor 3 (CR3) and, to a lesser extent, CD14. However, the exact nature of the receptor-ligand interactions and their contributions toward target cell selection remain unknown.
10.3.3 Other Innate Immune Cells
10.3.3.1 Dendritic Cells
Dendritic cells (DCs) are potent and specialized antigen-presenting cells, which help to generate more effective T helper cells. Therefore, an effective immune evasion strategy by a pathogen would be to target DCs and induce them to become tolerogenic, thus priming a regulatory IL-10 response that blocks inflammation and allows the pathogen to multiply without restraint. Furthermore, impairing DC maturation and promoting apoptosis of DCs can also help pathogens to disarm host defenses. During Y. pestis infection, DCs are one of the early targets of T3SS effectors [51]. Shannon et al. observed minimal interaction between Y. pestis and DCs; however, DCs consistently migrate toward flea-bitten sites containing Y. pestis [80]. The most pronounced effect of Y. pestis on DCs appears to be the paralysis of DC movement by impairing the cytoskeleton rearrangement function, attenuating the presentation of Y. pestis antigens by DCs [85]. Richard et al. found that the ability of Y. pestis to initiate DC activation is determined by its lipid A structure and depends on the pattern recognition receptor TLR4 [86]. In the bubonic plague model, IL-10 and TLR6 deficient mice are protected from plague infection, the mechanism of which was that TLR6 drove differentiation of tolerogenic DC and contributed to LcrV-mediated plague pathogenesis [87]. In plague vaccine studies, the interaction between vaccine and DCs also plays an important role. The protective mechanisms induced by rF1 + rV probably involve the activation of DCs, which initiate a primary immune response in naïve T cells [88]. Further study proved that LcrV targeting of DCs elicits combined humoral and cellular immunity and induces protection in a mouse model of pneumonic plague [89].
10.3.3.2 Natural Killer Cell
NK cells are a subset of lymphocytes that arrive at inflammatory sites and directly kill pathogen-infected cells without the recognition of antigenic peptides. Notably, Y. pestis can cause a global depletion of NK cells and decrease the secretion of IFNγ, resulting in reduced production of reactive nitrogen intermediates by macrophages. The cause of these anti-NK effects is the effector YopM, possibly by affecting the expression of IL-15 and its receptor IL-15Rα [90]. To elucidate whether NK1.1+ cells were critical for the virulence effect of YopM, Ye et al. continued to test the effects of NK cell depletion on bacterial growth and found no effect of ablation on viable bacterial numbers of either the wild type or the YopM mutant strain in either the liver or spleen [91]. Thus, they concluded that NK cells are redundant for YopM’s pathogenic mechanism.
10.4 Adaptive Immunity in Plague
In addition to innate immunity, Y. pestis infection can induce adaptive immunity. Adaptive immunity is characterized by the expansion, differentiation, and persistence of antigen-specific B and T cells. The primary function of B cells is to produce antibodies, thereby facilitating humoral defense, while the primary function of T cells is to produce phagocyte-activating cytokines, thereby facilitating cellular defense. However, during Y. pestis infection, humoral immunity and cellular immunity are not separated, and indeed they cooperate and collaborate with each other against the plague. For example, antibodies can protect T cell-deficient mice [92], and conversely T cells can protect antibody-deficient mice [93]. Therefore, characterization of Y. pestis-specific adaptive immune response in the host will provide a wealth of information for illustrating bacterial virulence and promoting the development of specific vaccines.
10.4.1 T Cell-Mediated Immune Responses to Yersinia pestis
A growing body of evidence demonstrates the crucial roles of T cell-mediated immunity against Y. pestis. A function of expanded pathogen-specific T cells is to secrete IFNγ and TNFα, and depletion of these proinflammatory cytokines prior to the passive transfer of the LcrV antibody into mice completely abrogated the protective effect observed in undepleted mice [94]. This indicates that a cellular proinflammatory response provides critical protective functions during humoral defense against lethal pulmonary Y. pestis infection. Direct evidence for the importance of the cellular response in protection against the plague came from studies in μMT mice, which are functional B cell-deficient mice and cannot produce antibodies. Transfer of Y. pestis-primed T cells into naïve μMT mice protected them against lethal intranasal Y. pestis challenge [93]. In another study, anti-F1 IgG transferred passively into T cell receptor knockout mice (TCR−/−) at 24 h after infection was unable to rescue them, whereas it fully protected μMT mice, indicating that T cells play a critical role in the protection mediated by antibody to F1-antigen[95]. Thus, identification of Y. pestis antigens that stimulate a protective T cell response is one of the major goals for vaccine development. Using silico computer analysis and an in vitro IFNγ assay, we identified potential T cell antigens. In all, 34 individual proteins that stimulated a strong IFNγ response from splenocytes of mice immunized with Y. pestis live attenuated vaccine EV76 have been identified. Furthermore, in addition to LcrV, nine proteins may provide partial protection against challenge with a low dose of Y. pestis [96].
CD4+ T helper (Th) cells include different subtypes based on their cytokine and transcription factor signatures [97]: Th1 cells produce IFNγ as their “signature” cytokine; Th2 cells produce IL-4, IL-5, and IL-13; Th17 cells produce IL-17; and regulatory T cells (Tregs) express the transcription factor FoxP3. Different T cell lineages play different roles in Y. pestis infection. Signal transducer and activator of transcription (STAT)-4 knockout mice, which have a reduced Th1 response when immunized with F1/V, were poorly protected against Y. pestis challenge. However, STAT-6 knockout mice with intact Th1 responses and diminished Th2 responses were fully protected [98]. These results indicate that Th1-mediated immune mechanisms, activated following Stat 4 phosphorylation, are essential in protection against the plague. Therefore, one potential way to improve the efficacy of plague vaccines would be to increase the Th1 response. Dinc et al. assessed the efficacy of the novel SA-4-1BBL costimulatory molecule as a Th1 adjuvant to improve cellular responses generated by the rF1-V vaccine [99]. They proved that addition of SA-4-1BBL improves the efficacy of the subunit vaccine by generating a strong Th1 cellular immune response without significant impact on the generation of Ab responses. Additionally, Smiley’s group demonstrated that vaccination with live attenuated Y. pestis induces Th17 cells [100]. IL-17 was found to contribute to defense against pulmonary Y. pestis challenge [82], although this IL-17-mediated protection does not appear to result entirely from enhanced bacterial clearance. These results indicate that plague vaccines aiming to induce mixed Th1 and Th17 cellular responses would provide more powerful and comprehensive protection.
In addition to CD4+ T cells, specific CD8+ T cell responses also contribute to defense against pulmonary Y. pestis infection, although alone they may be insufficient to combat fully virulent Y. pestis strains. YopE of Y. pestis was found to contain a dominant CD8 T cell epitope, which can be recognized by nearly 20 % of pulmonary CD8 T cells. Moreover, immunizing mice with a single peptide, YopE69–77, suffices to confer protection from lethal pulmonary challenge [101]. A further study investigated the effector functions of YopE69–77-specific CD8 T cells during pulmonary Y. pestis infection [102]. They concluded that specific CD8 T cell-mediated protection against pneumonic plague is dependent on TNFα and IFNγ, but not on perforin. In addition, CD4+ and CD8+ T cells were found to synergistically protect against pneumonic plague in this mouse model [103].
10.4.2 Antibody-Mediated Defense Against Yersinia pestis
Because of the complex antigen structure of Y. pestis, a number of antibodies are found in Y. pestis-infected patients and model animals. Serum samples collected from plague convalescent patients can transfer passive protection to naïve mice, indicating that antibody-mediated defense plays an important role against Y. pestis challenge. In previous studies, F1 and LcrV were proven to provide a high degree of protection, and the corresponding vaccines also showed efficacy in small animal models [104–107]. Using an antigen microarray, which contained more than 140 Y. pestis virulence-associated proteins, we screened the antibody responses of plague patients [108]. Apart from F1, YopD, YopE, and pH6 antigens, which have been described previously as immunogens, ten other novel immunogenic proteins were found. Antibody titers and persistence are correlated with vaccine efficacy. Previous studies confirmed that the F1 antibody could persist for 1–4 years in humans [109]. Further studies in our lab explored the antibody profile from 65 plague patients who were in remission for more than 10 years using a protein microarray [110]. Results showed that antibody to F1 can persist in recovered patients for more than 10 years, while antibodies to LcrV and YopD were present for an even longer period of time. As artificial passive immunization has been demonstrated to be effective against Y. pestis infection in animals, how about maternal antibodies? We carried out a study evaluating the kinetics, protective efficacy, and transmission modes of maternal antibodies, using mice immunized with plague subunit vaccine [111]. The results indicated that maternal antibodies induced by the plague subunit vaccine in mother mice can be transferred to newborn mice via both the placenta and lactation were sustained for more than 10 weeks and provide early protection against plague for newborn mice.
Long-lived plasma cells and memory B cells are responsible for the long-term humoral immunity elicited by vaccination. Memory B cells are in charge of driving the rapid anamnestic antibody response that occurs after re-exposure to antigen. The serum antibody level is maintained by long-lived plasma cells. Thus, studies in our lab investigated the kinetics of memory B cell and plasma cell response in mice immunized with plague subunit vaccine F1 or the live attenuated vaccine EV76 [112]. The number of memory B cells in the spleens was significantly higher than that in the bone marrow, which is consistent with a previous study that found that the majority of memory B cells are present in the spleen [113]. We also found that the boost of antibody titer after revaccination may be dependent on the existence of memory B cells and an excess of antigen.
Although passive transfer of specific antibody can provide protection in rodents against pneumonic plague, in nonhuman primates vaccinated with F1/LcrV, high-titer specific antibody at the time of challenge cannot protect primates against pneumonic plague [114, 115]. These observations strongly suggest that antibody titer alone, at least as measured by standard ELISA, do not suffice in predicting the efficacy of pneumonic plague vaccines. Therefore, which factors are involved in antibody-mediated protection? First, neutrophils were found to contribute to antibody-mediated defense against pneumonic plague because neutrophil depletion abrogates serotherapy-mediated protection from pneumonic plague in a mouse model. Similar results were reported in a mouse model of septicemic plague, where neutrophil depletion abrogates protection mediated by polyclonal anti-LcrV [116]. Second, cytokines from phagocytes enhances antibody-mediated protection. Whether genetic deficiency or antibody neutralization of IFNγ and TNFα, cytokine depletion significantly impairs serotherapy-mediated protection [98, 117]. In addition, passive protection with antibody to F1 in infected mice was reduced in C3−/− mice that lack a functional complement system, indicating the contribution of Fc-effector mechanisms to antibody-mediated protection [95].
References
Lukaszewski RA, Kenny DJ, Taylor R, Rees DG, Hartley MG, Oyston PC. Pathogenesis of Yersinia pestis infection in BALB/c mice: effects on host macrophages and neutrophils. Infect Immun. 2005;73(11):7142–50.
Fukuto HS, Bliska JB. Editorial: Yersinia pestis survives in neutrophils and sends a PS to macrophages: bon appetit! J Leukoc Biol. 2014;95(3):383–5.
Spinner JL, Winfree S, Starr T, Shannon JG, Nair V, Steele-Mortimer O, Hinnebusch BJ. Yersinia pestis survival and replication within human neutrophil phagosomes and uptake of infected neutrophils by macrophages. J Leukoc Biol. 2014;95(3):389–98.
Bubeck SS, Cantwell AM, Dube PH. Delayed inflammatory response to primary pneumonic plague occurs in both outbred and inbred mice. Infect Immun. 2007;75(2):697–705.
Lathem WW, Price PA, Miller VL, Goldman WE. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science. 2007;315(5811):509–13.
Lathem WW, Crosby SD, Miller VL, Goldman WE. Progression of primary pneumonic plague: a mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci U S A. 2005;102(49):17786–91.
Sauvonnet N, Lambermont I, van der Bruggen P, Cornelis GR. YopH prevents monocyte chemoattractant protein 1 expression in macrophages and T-cell proliferation through inactivation of the phosphatidylinositol 3-kinase pathway. Mol Microbiol. 2002;45(3):805–15.
Orth K, Palmer LE, Bao ZQ, Stewart S, Rudolph AE, Bliska JB, Dixon JE. Inhibition of the mitogen-activated protein kinase kinase superfamily by a Yersinia effector. Science. 1999;285(5435):1920–3.
Viboud GI, Mejia E, Bliska JB. Comparison of YopE and YopT activities in counteracting host signalling responses to Yersinia pseudotuberculosis infection. Cell Microbiol. 2006;8(9):1504–15.
Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe. 2010;7(5):376–87.
LaRock CN, Cookson BT. The Yersinia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe. 2012;12(6):799–805.
Brubaker RR. Interleukin-10 and inhibition of innate immunity to Yersiniae: roles of Yops and LcrV (V antigen). Infect Immun. 2003;71(7):3673–81.
McDonald C, Vacratsis PO, Bliska JB, Dixon JE. The yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. J Biol Chem. 2003;278:18514–23.
Osei-Owusu P, Jessen Condry DL, Toosky M, Roughead W, Bradley DS, Nilles ML. The N terminus of type III secretion needle protein YscF from Yersinia pestis functions to modulate innate immune responses. Infect Immun. 2015;83(4):1507–22.
Rebeil R, Ernst RK, Jarrett CO, Adams KN, Miller SI, Hinnebusch BJ. Characterization of late acyltransferase genes of Yersinia pestis and their role in temperature-dependent lipid A variation. J Bacteriol. 2006;188(4):1381–8.
Tan L, Darby C. Yersinia pestis is viable with endotoxin composed of only lipid A. J Bacteriol. 2005;187(18):6599–600.
Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, Conlon JE, Fukase K, Kusumoto S, Sweet C, Miyake K, et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol. 2006;7(10):1066–73.
de la Puerta ML, Trinidad AG, del Carmen Rodriguez M, Bogetz J, Sanchez Crespo M, Mustelin T, Alonso A, Bayon Y. Characterization of new substrates targeted by Yersinia tyrosine phosphatase YopH. PLoS One. 2009;4(2):e4431.
Hamid N, Gustavsson A, Andersson K, McGee K, Persson C, Rudd CE, Fallman M. YopH dephosphorylates Cas and Fyn-binding protein in macrophages. Microb Pathog. 1999;27(4):231–42.
Persson C, Carballeira N, Wolf-Watz H, Fallman M. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Cas and FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO J. 1997;16(9):2307–18.
Weidow CL, Black DS, Bliska JB, Bouton AH. CAS/Crk signalling mediates uptake of Yersinia into human epithelial cells. Cell Microbiol. 2000;2(6):549–60.
Von Pawel-Rammingen U, Telepnev MV, Schmidt G, Aktories K, Wolf-Watz H, Rosqvist R. GAP activity of the Yersinia YopE cytotoxin specifically targets the Rho pathway: a mechanism for disruption of actin microfilament structure. Mol Microbiol. 2000;36(3):737–48.
Roppenser B, Roder A, Hentschke M, Ruckdeschel K, Aepfelbacher M. Yersinia enterocolitica differentially modulates RhoG activity in host cells. J Cell Sci. 2009;122(Pt 5):696–705.
Andor A, Trulzsch K, Essler M, Roggenkamp A, Wiedemann A, Heesemann J, Aepfelbacher M. YopE of Yersinia, a GAP for Rho GTPases, selectively modulates Rac-dependent actin structures in endothelial cells. Cell Microbiol. 2001;3(5):301–10.
Shao F, Dixon JE. YopT is a cysteine protease cleaving Rho family GTPases. Adv Exp Med Biol. 2003;529:79–84.
Rosqvist R, Forsberg A, Rimpilainen M, Bergman T, Wolf-Watz H. The cytotoxic protein YopE of Yersinia obstructs the primary host defence. Mol Microbiol. 1990;4(4):657–67.
Andersson K, Carballeira N, Magnusson KE, Persson C, Stendahl O, Wolf-Watz H, Fallman M. YopH of Yersinia pseudotuberculosis interrupts early phosphotyrosine signalling associated with phagocytosis. Mol Microbiol. 1996;20(5):1057–69.
Iriarte M, Cornelis GR. YopT, a new Yersinia Yop effector protein, affects the cytoskeleton of host cells. Mol Microbiol. 1998;29(3):915–29.
Aepfelbacher M, Heesemann J. Modulation of Rho GTPases and the actin cytoskeleton by Yersinia outer proteins (Yops). Int J Med Microbiol. 2001;291(4):269–76.
Aepfelbacher M, Zumbihl R, Heesemann J. Modulation of Rho GTPases and the actin cytoskeleton by YopT of Yersinia. Curr Top Microbiol Immunol. 2005;291:167–75.
Du Y, Rosqvist R, Forsberg A. Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect Immun. 2002;70(3):1453–60.
Payne D, Tatham D, Williamson ED, Titball RW. The pH 6 antigen of Yersinia pestis binds to beta1-linked galactosyl residues in glycosphingolipids. Infect Immun. 1998;66(9):4545–8.
Makoveichuk E, Cherepanov P, Lundberg S, Forsberg A, Olivecrona G. pH6 antigen of Yersinia pestis interacts with plasma lipoproteins and cell membranes. J Lipid Res. 2003;44(2):320–30.
Anisimov AP, Dentovskaya SV, Titareva GM, Bakhteeva IV, Shaikhutdinova RZ, Balakhonov SV, Lindner B, Kocharova NA, Senchenkova SN, Holst O, et al. Intraspecies and temperature-dependent variations in susceptibility of Yersinia pestis to the bactericidal action of serum and to polymyxin B. Infect Immun. 2005;73(11):7324–31.
Bliska JB, Falkow S. Bacterial resistance to complement killing mediated by the Ail protein of Yersinia enterocolitica. Proc Natl Acad Sci U S A. 1992;89(8):3561–5.
Kirjavainen V, Jarva H, Biedzka-Sarek M, Blom AM, Skurnik M, Meri S. Yersinia enterocolitica serum resistance proteins YadA and ail bind the complement regulator C4b-binding protein. PLoS Pathog. 2008;4(8):e1000140.
Skorek K, Raczkowska A, Dudek B, Mietka K, Guz-Regner K, Pawlak A, Klausa E, Bugla-Ploskonska G, Brzostek K. Regulatory protein OmpR influences the serum resistance of Yersinia enterocolitica O:9 by modifying the structure of the outer membrane. PLoS One. 2013;8(11):e79525.
Porat R, McCabe WR, Brubaker RR. Lipopolysaccharide-associated resistance to killing of yersiniae by complement. J Endotoxin Res. 1995;2:91–7.
Kolodziejek AM, Sinclair DJ, Seo KS, Schnider DR, Deobald CF, Rohde HN, Viall AK, Minnich SS, Hovde CJ, Minnich SA, et al. Phenotypic characterization of OmpX, an Ail homologue of Yersinia pestis KIM. Microbiology. 2007;153(Pt 9):2941–51.
Bartra SS, Styer KL, O’Bryant DM, Nilles ML, Hinnebusch BJ, Aballay A, Plano GV. Resistance of Yersinia pestis to complement-dependent killing is mediated by the Ail outer membrane protein. Infect Immun. 2008;76(2):612–22.
Biedzka-Sarek M, Jarva H, Hyytiainen H, Meri S, Skurnik M. Characterization of complement factor H binding to Yersinia enterocolitica serotype O:3. Infect Immun. 2008;76(9):4100–9.
Biedzka-Sarek M, Salmenlinna S, Gruber M, Lupas AN, Meri S, Skurnik M. Functional mapping of YadA- and Ail-mediated binding of human factor H to Yersinia enterocolitica serotype O:3. Infect Immun. 2008;76(11):5016–27.
Ho DK, Riva R, Kirjavainen V, Jarva H, Ginstrom E, Blom AM, Skurnik M, Meri S. Functional recruitment of the human complement inhibitor C4BP to Yersinia pseudotuberculosis outer membrane protein Ail. J Immunol. 2012;188(9):4450–9.
Sebbane F, Jarrett CO, Gardner D, Long D, Hinnebusch BJ. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc Natl Acad Sci U S A. 2006;103(14):5526–30.
Sodeinde OA, Subrahmanyam YV, Stark K, Quan T, Bao Y, Goguen JD. A surface protease and the invasive character of plague. Science. 1992;258(5084):1004–7.
Drozdov IG, Anisimov AP, Samoilova SV, Yezhov IN, Yeremin SA, Karlyshev AV, Krasilnikova VM, Kravchenko VI. Virulent non-capsulate Yersinia pestis variants constructed by insertion mutagenesis. J Med Microbiol. 1995;42(4):264–8.
Samoilova SV, Samoilova LV, Yezhov IN, Drozdov IG, Anisimov AP. Virulence of pPst + and pPst- strains of Yersinia pestis for guinea-pigs. J Med Microbiol. 1996;45(6):440–4.
Laudisoit A, Leirs H, Makundi RH, Van Dongen S, Davis S, Neerinckx S, Deckers J, Libois R. Plague and the human flea Tanzania. Emerg Infect Dis. 2007;13(5):687–93.
Zhang SS, Park CG, Zhang P, Bartra SS, Plano GV, Klena JD, Skurnik M, Hinnebusch BJ, Chen T. Plasminogen activator Pla of Yersinia pestis utilizes murine DEC-205 (CD205) as a receptor to promote dissemination. J Biol Chem. 2008;283(46):31511–21.
Reboul A, Lemaitre N, Titecat M, Merchez M, Deloison G, Ricard I, Pradel E, Marceau M, Sebbane F. Yersinia pestis requires the 2-component regulatory system OmpR-EnvZ to resist innate immunity during the early and late stages of plague. J Infect Dis. 2014;210(9):1367–75.
Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O. Plague bacteria target immune cells during infection. Science. 2005;309(5741):1739–41.
Pechous RD, Sivaraman V, Price PA, Stasulli NM, Goldman WE. Early host cell targets of Yersinia pestis during primary pneumonic plague. PLoS Pathog. 2013;9(10):e1003679.
Busch JD, Van Andel R, Stone NE, Cobble KR, Nottingham R, Lee J, Versteeg M, Corcoran J, Cordova J, Van Pelt W, et al. The innate immune response may be important for surviving plague in wild gunnison’s prairie dogs. J Wildl Dis. 2013;49(4):920–31.
Cavanaugh DC, Randall R. The role of multiplication of Pasteurella pestis in mononuclear phagocytes in the pathogenesis of flea-borne plague. J Immunol. 1959;83:348–63.
Elvin SJ, Williamson ED, Scott JC, Smith JN, De Lema Perez G, Chilla S, Clapham P, Pfeffer K, Schlondorff D, Luckow B. Evolutionary genetics: ambiguous role of CCR5 in Y. pestis infection. Nature. 2004;430(6998):417.
Pujol C, Bliska JB. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis. Infect Immun. 2003;71(10):5892–9.
Straley SC, Harmon PA. Yersinia pestis grows within phagolysosomes in mouse peritoneal macrophages. Infect Immun. 1984;45(3):655–9.
Pujol C, Bliska JB. Turning Yersinia pathogenesis outside in: subversion of macrophage function by intracellular yersiniae. Clin Immunol. 2005;114(3):216–26.
Zhou D, Han Y, Yang R. Molecular and physiological insights into plague transmission, virulence and etiology. Microbes Infect. 2006;8(1):273–84.
Weeks S, Hill J, Friedlander A, Welkos S. Anti-V antigen antibody protects macrophages from Yersinia pestis-induced cell death and promotes phagocytosis. Microb Pathog. 2002;32(5):227–37.
Bergsbaken T, Cookson BT. Macrophage activation redirects yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 2007;3(11):e161.
Ruckdeschel K, Pfaffinger G, Haase R, Sing A, Weighardt H, Hacker G, Holzmann B, Heesemann J. Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-beta, but not MyD88, in bacteria-infected murine macrophages. J Immunol. 2004;173(5):3320–8.
Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424(6950):743–8.
Zhang Y, Ting AT, Marcu KB, Bliska JB. Inhibition of MAPK and NF-kappa B pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J Immunol. 2005;174(12):7939–49.
Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–7.
Palmer LE, Hobbie S, Galan JE, Bliska JB. YopJ of Yersinia pseudotuberculosis is required for the inhibition of macrophage TNF-alpha production and downregulation of the MAP kinases p38 and JNK. Mol Microbiol. 1998;27(5):953–65.
Schesser K, Spiik AK, Dukuzumuremyi JM, Neurath MF, Pettersson S, Wolf-Watz H. The yopJ locus is required for Yersinia-mediated inhibition of NF-kappaB activation and cytokine expression: yopJ contains a eukaryotic SH2-like domain that is essential for its repressive activity. Mol Microbiol. 1998;28(6):1067–79.
Denecker G, Declercq W, Geuijen CA, Boland A, Benabdillah R, van Gurp M, Sory MP, Vandenabeele P, Cornelis GR. Yersinia enterocolitica YopP-induced apoptosis of macrophages involves the apoptotic signaling cascade upstream of bid. J Biol Chem. 2001;276(23):19706–14.
Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2(12):965–75.
Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390(6658):350–1.
Fantuzzi G, Dinarello CA. Interleukin-18 and interleukin-1 beta: two cytokine substrates for ICE (caspase-1). J Clin Immunol. 1999;19(1):1–11.
Sebbane F, Gardner D, Long D, Gowen BB, Hinnebusch BJ. Kinetics of disease progression and host response in a rat model of bubonic plague. Am J Pathol. 2005;166(5):1427–39.
Balada-Llasat JM, Mecsas J. Yersinia has a tropism for B and T cell zones of lymph nodes that is independent of the type III secretion system. PLoS Pathog. 2006;2(9), e86.
Guinet F, Ave P, Jones L, Huerre M, Carniel E. Defective innate cell response and lymph node infiltration specify Yersinia pestis infection. PLoS One. 2008;3(2):e1688.
Mills SD, Boland A, Sory MP, van der Smissen P, Kerbourch C, Finlay BB, Cornelis GR. Yersinia enterocolitica induces apoptosis in macrophages by a process requiring functional type III secretion and translocation mechanisms and involving YopP, presumably acting as an effector protein. Proc Natl Acad Sci U S A. 1997;94(23):12638–43.
Monack DM, Mecsas J, Ghori N, Falkow S. Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc Natl Acad Sci U S A. 1997;94(19):10385–90.
Ruckdeschel K, Roggenkamp A, Lafont V, Mangeat P, Heesemann J, Rouot B. Interaction of Yersinia enterocolitica with macrophages leads to macrophage cell death through apoptosis. Infect Immun. 1997;65(11):4813–21.
Shin H, Cornelis GR. Type III secretion translocation pores of Yersinia enterocolitica trigger maturation and release of pro-inflammatory IL-1beta. Cell Microbiol. 2007;9(12):2893–902.
Laws TR, Davey MS, Titball RW, Lukaszewski R. Neutrophils are important in early control of lung infection by Yersinia pestis. Microbes Infect. 2010;12(4):331–5.
Shannon JG, Bosio CF, Hinnebusch BJ. Dermal neutrophil, macrophage and dendritic cell responses to Yersinia pestis transmitted by fleas. PLoS Pathog. 2015;11(3), e1004734.
Vagima Y, Zauberman A, Levy Y, Gur D, Tidhar A, Aftalion M, Shafferman A, Mamroud E. Circumventing Y. pestis Virulence by Early Recruitment of Neutrophils to the Lungs during Pneumonic Plague. PLoS Pathog. 2015;11(5):e1004893.
Bi Y, Zhou J, Yang H, Wang X, Zhang X, Wang Q, Wu X, Han Y, Song Y, Tan Y, et al. IL-17A produced by neutrophils protects against pneumonic plague through orchestrating IFN-gamma-activated macrophage programming. J Immunol. 2014;192(2):704–13.
Merritt PM, Nero T, Bohman L, Felek S, Krukonis ES, Marketon MM. Yersinia pestis targets neutrophils via complement receptor 3. Cell Microbiol. 2015;17(5):666–87.
Hinnebusch BJ, Jarrett CO, Callison JA, Gardner D, Buchanan SK, Plano GV. Role of the Yersinia pestis Ail protein in preventing a protective polymorphonuclear leukocyte response during bubonic plague. Infect Immun. 2011;79(12):4984–9.
Velan B, Bar-Haim E, Zauberman A, Mamroud E, Shafferman A, Cohen S. Discordance in the effects of Yersinia pestis on the dendritic cell functions manifested by induction of maturation and paralysis of migration. Infect Immun. 2006;74(11):6365–76.
Robinson RT, Khader SA, Locksley RM, Lien E, Smiley ST, Cooper AM. Yersinia pestis evades TLR4-dependent induction of IL-12(p40)2 by dendritic cells and subsequent cell migration. J Immunol. 2008;181(8):5560–7.
Depaolo RW, Tang F, Kim I, Han M, Levin N, Ciletti N, Lin A, Anderson D, Schneewind O, Jabri B. Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe. 2008;4(4):350–61.
Kingston R, Burke F, Robinson JH, Bedford PA, Jones SM, Knight SC, Williamson ED. The fraction 1 and V protein antigens of Yersinia pestis activate dendritic cells to induce primary T cell responses. Clin Exp Immunol. 2007;149(3):561–9.
Do Y, Koh H, Park CG, Dudziak D, Seo P, Mehandru S, Choi JH, Cheong C, Park S, Perlin DS, et al. Targeting of LcrV virulence protein from Yersinia pestis to dendritic cells protects mice against pneumonic plague. Eur J Immunol. 2010;40(10):2791–6.
Kerschen EJ, Cohen DA, Kaplan AM, Straley SC. The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect Immun. 2004;72(8):4589–602.
Ye Z, Kerschen EJ, Cohen DA, Kaplan AM, van Rooijen N, Straley SC. Gr1+ cells control growth of YopM-negative Yersinia pestis during systemic plague. Infect Immun. 2009;77(9):3791–806.
Green M, Rogers D, Russell P, Stagg AJ, Bell DL, Eley SM, Titball RW, Williamson ED. The SCID/Beige mouse as a model to investigate protection against Yersinia pestis. FEMS Immunol Med Microbiol. 1999;23(2):107–13.
Parent MA, Berggren KN, Kummer LW, Wilhelm LB, Szaba FM, Mullarky IK, Smiley ST. Cell-mediated protection against pulmonary Yersinia pestis infection. Infect Immun. 2005;73(11):7304–10.
Lin JS, Park S, Adamovicz JJ, Hill J, Bliska JB, Cote CK, Perlin DS, Amemiya K, Smiley ST. TNFalpha and IFNgamma contribute to F1/LcrV-targeted immune defense in mouse models of fully virulent pneumonic plague. Vaccine. 2010;29(2):357–62.
Levy Y, Flashner Y, Tidhar A, Zauberman A, Aftalion M, Lazar S, Gur D, Shafferman A, Mamroud E. T cells play an essential role in anti-F1 mediated rapid protection against bubonic plague. Vaccine. 2011;29(40):6866–73.
Li B, Zhou L, Guo J, Wang X, Ni B, Ke Y, Zhu Z, Guo Z, Yang R. High-throughput identification of new protective antigens from a Yersinia pestis live vaccine by enzyme-linked immunospot assay. Infect Immun. 2009;77(10):4356–61.
Bi Y, Liu G, Yang R. Reciprocal modulation between TH17 and other helper T cell lineages. J Cell Physiol. 2011;226(1):8–13.
Elvin SJ, Williamson ED. Stat 4 but not Stat 6 mediated immune mechanisms are essential in protection against plague. Microb Pathog. 2004;37(4):177–84.
Dinc G, Pennington JM, Yolcu ES, Lawrenz MB, Shirwan H. Improving the Th1 cellular efficacy of the lead Yersinia pestis rF1-V subunit vaccine using SA-4-1BBL as a novel adjuvant. Vaccine. 2014;32(39):5035–40.
Lin JS, Kummer LW, Szaba FM, Smiley ST. IL-17 contributes to cell-mediated defense against pulmonary Yersinia pestis infection. J Immunol. 2011;186(3):1675–84.
Lin JS, Szaba FM, Kummer LW, Chromy BA, Smiley ST. Yersinia pestis YopE contains a dominant CD8 T cell epitope that confers protection in a mouse model of pneumonic plague. J Immunol. 2011;187(2):897–904.
Szaba FM, Kummer LW, Duso DK, Koroleva EP, Tumanov AV, Cooper AM, Bliska JB, Smiley ST, Lin JS. TNFalpha and IFNgamma but not perforin are critical for CD8 T cell-mediated protection against pulmonary Yersinia pestis infection. PLoS Pathog. 2014;10(5):e1004142.
Philipovskiy AV, Smiley ST. Vaccination with live Yersinia pestis primes CD4 and CD8 T cells that synergistically protect against lethal pulmonary Y. pestis infection. Infect Immun. 2007;75(2):878–85.
Anderson Jr GW, Leary SE, Williamson ED, Titball RW, Welkos SL, Worsham PL, Friedlander AM. Recombinant V antigen protects mice against pneumonic and bubonic plague caused by F1-capsule-positive and -negative strains of Yersinia pestis. Infect Immun. 1996;64(11):4580–5.
Andrews GP, Heath DG, Anderson Jr GW, Welkos SL, Friedlander AM. Fraction 1 capsular antigen (F1) purification from Yersinia pestis CO92 and from an Escherichia coli recombinant strain and efficacy against lethal plague challenge. Infect Immun. 1996;64(6):2180–7.
DeBord KL, Anderson DM, Marketon MM, Overheim KA, DePaolo RW, Ciletti NA, Jabri B, Schneewind O. Immunogenicity and protective immunity against bubonic plague and pneumonic plague by immunization of mice with the recombinant V10 antigen, a variant of LcrV. Infect Immun. 2006;74(8):4910–4.
Leary SE, Griffin KF, Garmory HS, Williamson ED, Titball RW. Expression of an F1/V fusion protein in attenuated Salmonella typhimurium and protection of mice against plague. Microb Pathog. 1997;23(3):167–79.
Li B, Zhou D, Wang Z, Song Z, Wang H, Li M, Dong X, Wu M, Guo Z, Yang R. Antibody profiling in plague patients by protein microarray. Microbes Infect. 2008;10(1):45–51.
Rasoamanana B, Leroy F, Boisier P, Rasolomaharo M, Buchy P, Carniel E, Chanteau S. Field evaluation of an immunoglobulin G anti-F1 enzyme-linked immunosorbent assay for serodiagnosis of human plague in Madagascar. Clin Diagn Lab Immunol. 1997;4(5):587–91.
Li B, Du C, Zhou L, Bi Y, Wang X, Wen L, Guo Z, Song Z, Yang R. Humoral and cellular immune responses to Yersinia pestis infection in long-term recovered plague patients. Clin Vaccine Immunol. 2012;19(2):228–34.
Qi Z, Zhao H, Zhang Q, Bi Y, Ren L, Zhang X, Yang H, Yang X, Wang Q, Li C, et al. Acquisition of maternal antibodies both from the placenta and by lactation protects mouse offspring from Yersinia pestis challenge. Clin Vaccine Immunol. 2012;19(11):1746–50.
Zhang X, Wang Q, Bi Y, Kou Z, Zhou J, Cui Y, Yan Y, Zhou L, Tan Y, Yang H, et al. Kinetics of memory B cell and plasma cell responses in the mice immunized with plague vaccines. Scand J Immunol. 2014;79(3):157–62.
Shenoy GN, Chatterjee P, Kaw S, Mukherjee S, Rathore DK, Bal V, Rath S, George A. Recruitment of memory B cells to lymph nodes remote from the site of immunization requires an inflammatory stimulus. J Immunol. 2012;189(2):521–8.
Williamson ED, Flick-Smith HC, Waters E, Miller J, Hodgson I, Le Butt CS, Hill J. Immunogenicity of the rF1 + rV vaccine for plague with identification of potential immune correlates. Microb Pathog. 2007;42(1):11–21.
Bashaw J, Norris S, Weeks S, Trevino S, Adamovicz JJ, Welkos S. Development of in vitro correlate assays of immunity to infection with Yersinia pestis. Clin Vaccine Immunol. 2007;14(5):605–16.
Cowan C, Philipovskiy AV, Wulff-Strobel CR, Ye Z, Straley SC. Anti-LcrV antibody inhibits delivery of Yops by Yersinia pestis KIM5 by directly promoting phagocytosis. Infect Immun. 2005;73(9):6127–37.
Parent MA, Wilhelm LB, Kummer LW, Szaba FM, Mullarky IK, Smiley ST. Gamma interferon, tumor necrosis factor alpha, and nitric oxide synthase 2, key elements of cellular immunity, perform critical protective functions during humoral defense against lethal pulmonary Yersinia pestis infection. Infect Immun. 2006;74(6):3381–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Bi, Y. (2016). Immunology of Yersinia pestis Infection. In: Yang, R., Anisimov, A. (eds) Yersinia pestis: Retrospective and Perspective. Advances in Experimental Medicine and Biology, vol 918. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-0890-4_10
Download citation
DOI: https://doi.org/10.1007/978-94-024-0890-4_10
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-0888-1
Online ISBN: 978-94-024-0890-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)