
Abstract
The epigenome is the collection of all epigenetic modifications occurring on a genome. To properly generate, analyze, and understand epigenomic data has become increasingly important in basic and applied research, because epigenomic modifications have been broadly associated with differentiation, development, and disease processes, thereby also constituting attractive drug targets. In this chapter, we introduce the reader to the different aspects of epigenomics (e.g., DNA methylation and histone marks, among others), by briefly reviewing the most relevant underlying biological concepts and by describing the different experimental protocols and the analysis of the associated data types. Furthermore, for each type of epigenetic modification we describe the most relevant analysis pipelines, data repositories, and other resources. We conclude that any epigenomic investigation needs to carefully align the selection of the experimental protocols with the subsequent bioinformatics analysis and vice versa, as the effect sizes can be small and thereby escape detection if an integrative design is not well considered.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Epigenomics
In eukaryotes, the DNA is stored in the nucleus through mechanisms allowing DNA packaging in condensed structures. This packaging allows a level of compression such that the DNA of a human diploid cell – which would linearly span for about 2 m – can be condensed efficiently in the space of a cell nucleus, typically 2–10 μm. The uncovering of the minimal unit of such condensation (Kornberg 1974), the nucleosome, showed DNA is tightly packed around a protein octamer (histones), with a left-handed superhelical turn of approximately 147 base pairs. The histone octamer consists of two copies of four histones: H2A, H2B, H3 and H4 and A fifth histone (H1) binds the nucleosome and the linker DNA region and increases the stability. Higher-order packaging structures contribute to the final level of compression.
The nucleosome structure is inherently linked to gene expression, as it is intuitive that nucleosomes have to be displaced to allow gene expression to occur. The structure of the chromatin fulfills the role of condensing and protecting the DNA but it also preserves genetic information and controls gene expression. Therefore, this mechanism represents a process control and the accessibility of the DNA is regulated by chemical modifications that occur at the chromatin level, both for DNA and proteins. In this sense, nucleosomes contribute to regulatory mechanisms because they forbid or allow access for essential processes such as gene transcription or DNA replication (Fyodorov and Kadonaga 2001). For instance, DNA located near entry or exit points of the nucleosome are more accessible than those located centrally (Anderson and Widom 2000). Additionally, nucleosomes regulate DNA breathing (or fraying), that is, the spontaneous local conformational fluctuations within DNA of exit and entry points of nucleosome (Fei and Ha 2013), depending on the sequence wrapped around the histones and the covalent histone modifications. On the other hand, the DNA itself may be chemically modified, without associated changes in its sequence, generating important marks for regulation of gene expression, including DNA methylation. The collection of covalent changes to the DNA and histone proteins in the chromatin is called “epigenome.” Changes in the epigenome are observed during development and differentiation and can be mitotically stable, modulate gene expression patterns in a cell and preserve cellular states. However, we have started to understand that also environmental factors can contribute to reshape the epigenome, potentially providing a mechanism to alter the gene expression program of a cell both in normal and disease conditions.
High-throughput technologies, including next-generation sequencing, offer the unprecedented opportunity of assaying epigenetic alteration usually in a hypothesis-free approach, by looking at multiple sites in the genome and verifying their association with the biological phenomenon observed. Therefore, bioinformatics and biostatistics represent key disciplines for obtaining solid results and are required in each phase of an epigenomics project, from study design to data analysis, visualization, and storage. In this chapter, we will give an overview of the two most studied epigenetic modifications, namely, DNA methylation and histone modifications; we will present the major steps in their respective experimental and data analysis pipelines, briefly discussing the associated challenges and opportunities.
2 DNA Methylation
2.1 Introduction to DNA Methylation
DNA methylation results from the addition of a methyl group to cytosine residues in the DNA to form 5-methylcytosine (5-mC) and in mammals it is predominantly restricted to the context of CpG dinucleotides, although other sequences might be methylated in some tissues (Lister et al. 2009, 2013; Ziller et al. 2013). CpG methylation has not only been observed during development or differentiation and in association with diseases, but has also been proposed as a prerequisite to understand disease pathogenesis in complex phenotypes (Petronis 2010). DNA methylation was initially identified as an epigenetic mark for gene repression (Riggs 1975; Holliday and Pugh 1975). Currently, although the silencing mechanism remains valid, we know that methylation in CpG-rich promoter regions is associated with gene repression, while CpG-poor regions show a less simple connection with transcription (Jones 2012). Therefore, the relationship between DNA methylation and transcriptional activation/repression is more complex than initially portrayed and dependent on the genomic and cellular context.
In the human genome, 70–80 % of CpG sequences are methylated (Ziller et al. 2013); however, both the distribution of CpG dinucleotides and the DNA methylation mark are not evenly distributed. CpG islands (CGI) are sequences with high C + G content that are generally unmethylated and colocalize with more than half of the promoters of human genes (Illingworth and Bird 2009). Housekeeping genes generally contain a CGI in the neighborhood of their TSS (Transcription Start Site), concordantly with the notion that chromatin at promoter with CGI shows a transcriptionally permissive state (Deaton and Bird 2011).
In addition to CGIs and TSSs, methylation at other classes of genomic elements has gained further attention over time. For example, CpG shores are genomic regions up to 2 kb distant from CGI, which show lower CpG density but increased variability in DNA methylation, and are found “to be among the most variable genomic regions” (Ziller et al. 2013). Most of tissue-specific DNA methylation in fact, as well as methylation differences between cancer and normal tissue, occur at CpG shores (Irizarry et al. 2009). DNA methylation at enhancers is also highly dynamic (Ziller et al. 2013; Stadler et al. 2011), has been shown to vary in physiological and pathological contexts (Aran and Hellman 2013; Lindholm et al.2014; Rönnerblad et al. 2014), and methylation levels at enhancers are more closely associated with gene expression alterations than promoter methylation in cancer (Aran et al. 2013). Enhancers represent crucial determinants of tissue-specific gene expression and their identification methods include the analysis of epigenomic data (ChIP-seq, DNase-seq), since enhancer chromatin shows characteristic marks (Calo and Wysocka 2013). Moreover, DNA methylation at enhancer elements can influence the binding of Transcription Factors (TFs) (Stadler et al. 2011; Wiench et al. 2011), providing a direct link between CpG hypomethylation and target gene expression. However, it remains unsolved how this complex interplay is regulated and whether DNA methylation changes are a consequence of TF binding or whether they drive enhancer activity through exclusion of TF.
2.2 The Axes of DNA Methylation Variability
The role of DNA methylation variation has been investigated in many different contexts. Below we will give a brief overview of the phenotypes, settings, and major domains that together constitute the “axes” along which variability in DNA methylation has been studied.
Development
Early studies proposed DNA methylation as a mechanism involved in X-chromosome inactivation and developmental programs (Riggs 1975; Holliday and Pugh 1975). Since then, the dynamics of DNA methylation during developmental changes has been studied extensively, and technological advances now render possible the study of methylomes of single cells (Smallwood et al. 2014; Guo et al. 2014), with manifest implications for the study of early embryos.
Imprinting and X Chromosome Inactivation
Through the phenomenon of imprinting, genes that are expressed in allele-specific manner have regions showing parent-of-origin specific DNA methylation. When measured at an imprinted region, methylation is expected to approach a theoretical 50 % level. X-chromosome inactivation in females is also achieved through methylation, in order to transcriptionally silence the inactivated X chromosome, which is random in humans, and obtain gene dosage similar to males. Therefore, measured levels of DNA methylation differ by gender at X chromosome.
Disease
The study of DNA methylation variability in common complex diseases is the focus of Epigenome-Wide Association Studies (EWAS), which aim at associating phenotypic traits to interindividual epigenomic variation, and in particular DNA methylation. A notable example is represented by cancer EWAS, which not only aim at understanding the molecular changes of tumorigenic pathways and disease risk, but also exploit DNA methylation profiling for disease diagnosis and prognosis. It is also thought that a combination of environmental, genetic, and epigenetic interactions contribute to the problem of the “missing heritability” (Eichler et al. 2010; Feinberg 2007).
Space and Time
When designing and analyzing EWAS, it should be carefully considered that CpG methylation is subjected to spatial and temporal variability. One could consider the genome space as the main axis of variability, because different genomic elements have different methylation levels and show different degree of inter-sample variability. Alternatively, the tissue/cell type space represents another important axis of variation, as it is extensively established that different cell types possess their characteristic methylome. Other cases illustrate perfectly the extent of temporal variability. Monozygotic twins, for example, accumulate variability over time in their epigenome, such that older monozygous twins have higher differences in CpG methylation than younger twins (Fraga et al. 2005). Moreover, an “epigenetic drift” has been generally observed during aging (Bjornsson et al. 2008; Teschendorff et al. 2013b), confirming that both hypomethylation and hypermethylation are occurring over time, with acceleration dependent on disease or tissue factors (Horvath 2013; Horvath et al. 2014; Hannum et al. 2013).
Genotype
Genotype is a strong source of interindividual variability in DNA methylation (Bell et al. 2011). Such genetic variants are defined as methylation quantitative trait loci (meQTLs) and they have been described in blood and other tissues (Bell et al. 2011; Drong et al. 2013; Shi et al. 2014). It is possible that some genotype-dependent CpGs mediate the genetic risk of common complex diseases (Liu et al. 2013).
Environment
Accumulating evidence shows that several environmental factors can influence DNA methylation. For example, dietary factors have the potency to alter the degree of DNA methylation in different tissues (Feil and Fraga 2011; Lim and Song 2012). Cigarette smoking and pollution represent other known epigenetic modifiers (Lee and Pausova 2013; Feil and Fraga 2011). Moreover, short- or long-term physical exercise have also been proposed as physiological stimuli which can cause changes in DNA methylation (Barrès et al. 2012; Rönn et al. 2013; Lindholm et al. 2014).
2.3 Methods for DNA Methylation Profiling
Classically, methods for measuring DNA methylation have been divided into three major classes, including enrichment-based methods, digestion with methylation-sensitive restriction enzymes, and methods using bisulfite (BS) treatment. When coupled with DNA sequencing, affinity-based enrichment of methylated DNA fragments allows the interrogation of methylation of genomic regions with a methyl-binding protein (MBD-seq) (Serre et al. 2010) or an antibody (MeDIP-seq) (Down et al. 2008). These measurements do not give an absolute estimation of the methylation levels, but rather a relative enrichment that is dependent on the CpG density and the quality of the affinity assay (i.e., immunoprecipitation). Furthermore, the length of the DNA fragments determines their resolution. Similarly, methods based on restriction enzymes measure the relative enrichment after digesting the DNA with endonucleases that are sensitive to cytosine methylation (MRE-seq) (Maunakea et al. 2010), and they are therefore influenced by the genomic frequency of the recognition site for the selected enzyme. In this chapter, we will focus on methods using bisulfite conversion to assay the cytosine methylation status. After treatment with sodium bisulfite, unmethylated cytosines (C) in the genomic DNA are selectively converted to uracil (U), which are replaced by thymine (T) following PCR amplification (Fig. 1.1b). Methylated Cs are however protected from being converted. Afterward, the methylation levels can be quantified using microarrays or sequencing. Bisulfite treatment may be combined with digestion using methylation-insensitive restriction enzymes, in a technique called Reduced-Representation Bisulfite Sequencing (RRBS) (Meissner et al. 2008), to reduce the amount of reads to a fraction of the genome and thus reduce the cost. As opposed to Whole Genome Bisulfite Sequencing (WGBS) (Lister et al. 2009), this approach has reduced genome-wide coverage, but the coverage is higher for CpG islands (Harris et al. 2010). It is alternatively possible to capture targeted DNA fragments, in order to restrict the sequencing to specific regions (Lee et al. 2011). Both sequencing and microarray technology offer single-base resolution. While microarray platforms have a lower cost per sample and limited genome-wide coverage, WGBS has the most comprehensive genome-wide coverage but at a higher cost. In the next sections we will better elucidate microarray- and sequence-based approaches, together with an overview of the analysis pipelines and softwares.

Overview of experimental methods and bisulfite treatment for the analysis of DNA methylation. (a) The experimental methods to assay are shown, as further explained in the text. (b) The bisulfite treatment and PCR reactions will result in the conversion of unmethylated cystosines (C) into thymines (T), while 5-methylcitosine (mC) will be protected from the bisulfite-induced conversion
3 Bisulfite Microarrays
Commercially available microarray platforms conditioned the growing availability of EWAS, allowing a large sample size at an affordable cost. The sample size issue is relevant since, in many cases, changes in DNA methylation are mild and the biological variability may be high. Illumina Infinium HumanMethylation27 (27k) and HumanMethylation450 BeadChip (450k) are the most common types of oligonucleotide microarrays used for DNA methylation studies; at the date of writing (December 2014) >16,000 27k and >21,000 450k samples are deposited on GEO (Gene Expression Omnibus) database. The 450k arrays are based on the Infinium chemistry and contain 485,512 probes, targeting 99 % of genes and 96 % of CpG island regions (Bibikova et al. 2011). Oligonucleotide probes are attached to beads and deposited on an array, where the detection of the methylation status occurs through fluorescence reading. They represent an extension of the previous 27k platform, which was biased toward promoter regions. This extension resulted in wider coverage (but still limited compared to sequencing methods), specially toward other genomic regions like gene bodies and CpG shores (Bibikova et al. 2011; Sandoval et al. 2011). However, this also resulted in the introduction of two different bead types associated to two different chemical assays, Infinium I and Infinium II. Infinium I consists of two bead types (Methylated and Unmethylated) for the same CpG locus, both sharing the same color channel, whereas Infinium II utilizes a single bead type and two color channels (green and red) (Bibikova et al. 2011). Infinium II assays have larger variance and are less sensitive for the detection of extreme methylation values, which is probably associated to the dual-channel readout, thus rendering the Infinium I assay a better estimator of the true methylation state (Dedeurwaerder et al. 2011; Teschendorff et al. 2013a; Marabita et al. 2013). Moreover, different genomic elements (promoters, CpG islands, gene bodies, etc.) have different relative fraction of type I or type II probes (Dedeurwaerder et al. 2011). Methods have been introduced to correct for probe-type bias (see below for discussion).
The C methylation status for single CpG sites at each allele is always binary (0 or 1); however, the measured methylation levels can, in principle, take any value between 0 and 1 when averaging over many cells, or when the methylation status differs between the two alleles (imprinting, X-chromosome inactivation). For bisulfite microarrays, the methylation level is usually measured in two different scales, the β-value and the M-value. The β-value is calculated from signal intensities and can be interpreted as the percentage of methylation (it ranges from 0 to 1). It is related to the M-value through a logistic transformation. See reference (Du et al. 2010) for a detailed description of the two quantities. Even if M-values cannot be directly interpreted as methylation percentages, they offer several advantages, including the possibility of employing downstream association models that rely on the assumption of Gaussianity, as β-values appear compressed in the high and low range and often display heteroscedasticity. Moreover, when the sample size is relatively large the use of β- or M-values has been shown to give similar results, but with a limited sample size, M-values allow more reliable identification of true positives (Zhuang et al. 2012). However, from a pragmatic point of view and to allow biological interpretation, it is always advisable to report the final effect size in terms of median or mean β-value change, even if the feature selection step has been performed in the M-value space.
Independently of the scale used, the methylation profile for each sample shows a bimodal distribution, with two peaks corresponding to the unmethylated and methylated CpG positions. Because of the technical differences in probe design, a correction method is advisable. It could be argued that for CpG-level methylation difference analysis, the comparison will involve only probes of the same type. However, several indications suggest that it is advantageous to perform probe-type correction: (a) when the fold change (or effect size) is used in combination with the p-value for feature selection, as otherwise a bias may result from the dissimilar range between probes of different type (Marabita et al. 2013); (b) when dimensionality reduction or clustering algorithms are used, the pattern of variability between probe types may bias the grouping of CpG sites; (c) when DMR identification is anticipated, the methylation estimates along subsequent genomic positions will be dependent on probe type.
Methods for reducing the probe-type bias include a peak-based correction (Dedeurwaerder et al. 2011), SWAN method (Maksimovic et al. 2012), subset quantile normalization (Touleimat and Tost 2012), and BMIQ (Teschendorff et al. 2013a). In a benchmarking work (Marabita et al. 2013), BMIQ resulted as the best algorithm for reducing probe design bias. BMIQ, which employs a beta-mixture and quantile dilation intra-array normalization strategy, is available through several R packages (ChAMP (Wei et al. 2014), RnBeads (Assenov et al. 2014), WateRmelon (Schalkwyk et al. 2013)). Briefly, it first applies a beta-mixture model to assign probes of a given design type to methylation states and subsequently and uses state-membership probabilities to reassign the quantiles of the type2 probes according to the type1 distribution. Finally, for the probes with intermediate methylation values (which are not well described by a beta-distribution), a methylation-dependent dilation transformation is used, which also preserves the monotonicity and continuity of the data.
While probe-type normalization is a form of within-array normalization, between-array normalization is intended to remove part of the technical variability that is not associated with any biological factor, but which can be considered as caused by experimental procedures. For 450k data, there is no consensus on the best approach (Wilhelm-Benartzi et al. 2013; Dedeurwaerder et al. 2014), although a comparison of different normalization pipelines has been performed in recent works (Marabita et al. 2013; Pidsley et al. 2013). Many of the proposed approaches employ a form of quantile normalization (QN), which has been shown to perform well for gene expression studies (Irizarry et al. 2003). The goal of QN is to produce identical distribution of probe intensities for all the arrays and it has been applied to 450k data in several forms (Dedeurwaerder et al. 2014). While forcing the distribution of the methylation estimates to be the same for all the samples is a reasonably too strong an assumption for many biological comparisons, normalizing signal intensities appears a valid alternative in reducing technical variability in several contexts (Marabita et al. 2013; Dedeurwaerder et al. 2014). However, examination of the signal intensities and the study design should guide the application of this level of between-samples normalization, in order not to harm the integrity of the biological signal. A recent extension of QN, termed functional normalization (Fortin et al. 2014), uses control probes from the array to remove unwanted variation, assuming that summarized control probes function as surrogates of the nonbiological variation, which may include batch effects (see below).
Several comprehensive R packages have been developed for the processing and the analysis of 450k data (such as lumi (Du et al. 2010), methylumi (Davis et al. 2014), minfi (Aryee et al. 2014), wateRmelon (Schalkwyk et al. 2013), ChAMP (Wei et al. 2014), and RnBeads (Assenov et al. 2014)), and the reader is referred elsewhere for detailed discussion on popular pipelines and packages (Morris and Beck 2014; Wilhelm-Benartzi et al. 2013; Marabita et al. 2013; Dedeurwaerder et al. 2014).
Another type of unwanted variation in 450k data is represented by batch effects, which contaminate many high-throughput experiments including 450k arrays (Leek et al. 2010; Sun et al. 2011). We define a batch as a subgroup of samples or experiments exhibiting a systematic nonbiological difference that is not correlated with the biological variables under study. For example, different batches are represented by groups of samples that are processed separately, on different days or by a different operator. However, the definition of a batch results from careful examination of the data set, in order to identify what is an appropriate batch variable other than the processing group, as the slide or the position on the slide (i.e., the array), which represent known sources of batch effect for 450k arrays (Sun et al. 2011; Marabita et al. 2013; Harper et al. 2013).
Batch effects can only affect a subset of probes instead of generating artifacts globally; therefore, many normalization methods fail in eliminating or reducing batch effects. Specific methods have been developed to deal with this source of variability, including ComBat (Johnson et al. 2006), SVA (Surrogate Variable Analysis) (Leek and Storey 2007), ISVA (Independent Surrogate Variable Analysis) (Teschendorff et al. 2011), RUV (Remove Unwanted Variation) (Gagnon-Bartsch and Speed 2012; Fortin et al. 2014). The above methods aim at removing the unwanted variation that remains in high-throughput assays despite the application of between-sample normalization procedures. They rely on the explicit specification of the experimental design, in order to maintain the variability associated to a biological factor, while removing variability associated to either known or unknown batch covariates. For example, the ComBat method directly removes known batch effects and returns adjusted methylation data, by using an empirical Bayes procedure. However, when the sources of unwanted variation are unknown, surrogate variables can be identified by SVA directly from the array data. This method does not directly adjust the methylation data; however, in a second step, the latent variables can be included as covariates into a statistical model, in order to identify differential methylation while correcting for batch effect. Similarly, ISVA, an extension of SVA, does not adjust data but identifies features associated with the phenotype of interest in the presence of potential confounding factors. However, the methods indicated above may still fail or be inapplicable. Therefore, it is important to remember that the best safeguard against problematic batch effects is a careful experimental design (Leek et al. 2010), coupled with a random assignment of the samples to the arrays, the inclusion of a method to account for batch effect and possibly the presence of technical replicates, one for each processing subgroup, if the samples cannot be processed together in the case of large cohorts.
Whole blood is one of the most extensively used tissues for EWAS studies because it is easily accessible and minimally invasive, allowing large cohorts to be characterized prospectively and retrospectively, in contrast to most disease-relevant tissues that are hard to collect. However, cellular heterogeneity is an important factor to consider in the analysis of 450k data, particularly when blood is the source of DNA. In fact, cellular composition can explain a large fraction of the variability in DNA methylation (Reinius et al. 2012; Jaffe and Irizarry 2014). It can thus represent an important confounder in the association analysis when the phenotype under study alters cellular composition in blood, therefore resulting in spurious associations. Statistical methods are available to adjust for cellular composition. The popular Houseman method (Houseman et al. 2012) requires the availability of reference data measuring DNA methylation profiles for individual cell types in order to estimate cell proportions, which can be used to adjust a regression model (Liu et al. 2013). Alternatively, reference-free approaches (Zou et al. 2014; Houseman et al. 2014) can be employed to deconvolute DNA methylation when a reference data set is not available or extremely difficult to obtain.
A critical goal of most experimental designs is to identify DNA methylation changes that correlate with the phenotype of interest, for example, by comparing cases and controls. A detailed discussion of the available methods is beyond the scope of this chapter; however, we will briefly describe some of the most popular methods. We will first consider the identification of Differentially Methylated Positions (DMPs). The first and very simple approach consists in the calculation of a Δβ as the difference between the median β-values of two experimental groups, and selecting probes whose absolute Δβ exceeds a threshold. A |Δβ| > 0.2 corresponds to the recommended difference that can be detected with 99 % confidence according to Bibikova et al. (Bibikova et al. 2011). Many works identify DMPs using a threshold on a p-value from a statistical test (t-test, Mann–Whitney test), including a correction method for multiple hypothesis testing (Bonferroni or False Discovery Rate correction). Moreover, to decrease the false positive rate, a second threshold on the effect size is recommended (Marabita et al. 2013; Dedeurwaerder et al. 2014). For example, a minimal fold change could be considered (if working with log-ratios), or a minimal difference in the β-values (5–10 %). Another popular method is represented by the moderated statistical tests as implemented in limma (Smyth 2004), which uses a moderated t-statistic and an empirical Bayes approach to shrink the estimated sample variances toward a pooled estimate across sites, resulting in better inference when the number of samples is small. In this latter case, M-values are appropriate, as limma expects log-ratios and the Gaussiainty assumption is violated by the bounded nature of β-values.
An alternative approach for feature selection consists in assessing differential variability between sites instead of using statistics based on differential methylation. In epigenomics of common diseases, this notion has been proposed to be relevant for understanding and predicting diseases (Feinberg et al. 2010; Feinberg and Irizarry 2010), by assuming that common disease involves a combination of genetic and epigenetic factors and that DNA methylation variability could either mediate genetic effects or be mediators of environmental effects. Methods are available to analyze differential variability and associate it with a phenotype of interest (Teschendorff and Widschwendter 2012; Jaffe et al. 2012a).
While the best approach for the identification of Differentially Methylated Regions (DMRs) is today represented by bisulfite sequencing, 450k arrays are a possible alternative and methods have been developed to deal with their characteristics, including Probe Lasso (Butcher and Beck 2015), Bump hunting (Jaffe et al. 2012b), DMRcate (Peters and Buckley 2014), and A-clustering (Sofer et al. 2013). The genomic coverage of 450k arrays is uneven, with a bias toward CpG islands, promoters, and genic regions; moreover, neighboring CpG sites have highly correlated methylation levels. These characteristics complicate the application of fixed window-based approaches for the identification of DMRs, and methods like Probe Lasso apply a flexible window based on probe density to call DMR and calculate a p-value by combining individual p-values, weighting by the underlying correlation structure of methylation level. The Bump hunting method (which is not restricted to 450k arrays) is another approach that was developed to deal with the spatial correlation of CpG positions, and which finds genomic regions where there is statistical evidence of an association.
4 Bisulfite Sequencing
Bisulfite sequencing (BS) has been thoroughly compared with other sequence- and array-based approaches (Bock et al. 2010; Harris et al. 2010; Li et al. 2010) and it currently represents the gold-standard technology for a quantitative and accurate genome-wide measurement of DNA methylation at single base-pair resolution. Although a less cost-attractive option, sequencing technologies and experimental protocols have advanced recently and it is becoming advantageous to use BS in many settings. For example, the profiling of the methylome in single cells has been recently achieved (Smallwood et al. 2014; Guo et al. 2014).
The use of next-generation sequencing has not only represented a technological improvement, but it has also contributed conceptual developments in our understanding of the biological role of DNA methylation (Rivera and Ren 2013). For example, the traditional view of DNA methylation favored a mitotically stable modification, characteristic of repressed chromatin. However, sequencing technologies have expanded our view on DNA methylation, and we have started to understand the complexity of this epigenetic modification and its dynamical patterns, the relationship with other marks (including 5-hydroxymethylcytosine (5hmC) or 5-formylcytosine (5fC)), and the distribution of non-CpG methylation in embryonic or adult tissues, for example.
Several protocol variants exist for performing genome-wide BS (Lister and Ecker 2009; Laird 2010), and here we focus on two of the most widely used, namely, WGBS and RRBS. The two strategies use bisulfite treatment to infer the methylation status of the Cs in the genome; however, they noticeably differ for their genome-wide coverage and costs. RRBS libraries are prepared by digesting genomic DNA with the methylation-insensitive restriction enzyme MspI, which cut at the CCGG sites. After end-repair and adapter ligation, DNA is size-selected and treated with sodium bisulfite. Then, purified DNA is PCR-amplified and sequenced. RRBS provides single-base resolution measurements of DNA methylation, with good coverage for CpG-rich regions (as CpG islands), but low genome-wide coverage. Therefore, this method increases the depth and reduces the cost per CpG for cytosines in CpG islands (Harris et al. 2010). Instead, WGBS has larger genome-wide coverage, but increased cost. WGBS libraries are generated from fragmented genomic DNA, which is adapter-ligated, size-selected, bisulfite-converted, and finally amplified by PCR amplification. However, modifications of this experimental workflow have been introduced in order to expand the applicability of this approach to many settings. For example, Post-Bisulfite Adaptor Tagging (PBAT) has been developed to reduce the loss of amplifiable DNA caused by degradation during bisulfite conversion, and therefore to reduce the amount of input DNA (Miura et al. 2012). Alternatively, a “tagmentation” protocol (Tn5mC-seq) allows the production of libraries from reduced amount of starting DNA (Adey and Shendure 2012).
The recent work by Ziller et al. (2013) observed that roughly only 20 % of CpG methylation in the genome can be considered “dynamic,” and that therefore a substantial part of WGBS reads are potentially uninteresting, resulting in a combined loss of around 80 % of sequencing depth due to noninformative reads and static regions. Therefore, capture protocols that sequence target regions would appear to be advantageous if a flexible design could allow one to focus on representative, dynamic, or regulatory regions only. For example, the Agilent SureSelect platform allows BS on a selected panel of regions using hybridization probes (Ivanov et al. 2013; Miura and Ito 2015). The predefined regions include 3.7 M CpGs on CpG islands and promoters, cancer and tissue DMRs, DNAseI hypersensitive sites, and other regulatory elements.
The percentage of methylation after sequencing is calculated by counting the reads supporting a methylated or unmethylated C, and this is achieved by aligning reads to a reference genome. However, the bisulfite treatment converts unmethylated Cs into Ts, resulting in libraries of reduced complexity and reads that do not exactly match the reference genome sequence. Therefore, a method is needed to incorporate the possible conversion into the alignment procedure. Several alternative strategies and aligners have been proposed and their different features have been reviewed elsewhere (Bock 2012; Krueger et al. 2012; Tran et al. 2014). Bismark (Krueger and Andrews 2011), for example, represents one of the most popular mapping tools. It converts in silico all the Cs both in the reads and in the reference genome; then a standard aligner (Bowtie or Bowtie2) is used to map the reads to each strand of the genome. This method therefore uses only three letters for alignment, and the reduced complexity is compensated by the lack of bias toward methylated regions. To avoid decreased mapping efficiency, special care should be taken by an initial quality control and it is recommended to perform both sequence adapter trimming and adaptive quality trimming at the read 3′ end (Krueger et al. 2012). Indeed, some libraries may show both reduced quality scores and the presence of adapter sequences at the end of the reads (if the read length is longer than the DNA fragment), causing a dramatic decrease in the percentage of mapped reads.
After mapping, the DNA methylation levels are calculated from the aligned reads, counting the number of reads containing a C or a T in the genome, for each C independently of the context. Usually, only CpG methylation is further retained for downstream analysis; however, non-CpG methylation can be analyzed as well using BS, if required in the biological context (Lister et al. 2009, 2013). At this stage, M-bias plots (Hansen et al. 2012) can help in identifying any bias in methylation levels toward the beginning or the end of the reads. For example, many library preparation protocols include an end-repair step after DNA fragmentation. This enzymatic reaction will introduce unmethylated Cs, which will align to the genome, but without preserving their original methylation. Therefore, if detected with the M-bias plot, this effect should be removed by excluding the biased positions from the methylation call. If desired, the Bis-SNP package (Liu et al. 2012) can perform base quality recalibration, indel calling, genotyping, and methylation extraction from BS data.
Fragments aligning exactly to the same genomic position could be the result of PCR amplification. However, the execution of de-duplication step is dependent on the exact experimental protocol. For example, in RRBS libraries it is expected that a higher fraction of fragments will all start at the same genomic location, given the initial MspI enzymatic digestion, and therefore the de-duplication step could remove large fraction of valid reads. For other protocols, including WGBS and target enrichment, de-duplication is suitable to prevent multiple counting of the same fragment, which will cause methylation bias.
Similar to microarrays, the analysis of BS data allows site- and region-level differential methylation analysis. While some aspects are common to all DNA methylation studies, specific considerations and statistical tools apply only to BS data. The simplest test for assessing differential methylation is Fisher’s exact test. This method uses read counts to assess statistical significance; however, it is not able to completely model the biological variability. If biological replicates are present, the counts are pooled together to apply this method, thus removing the within-group variation that is a requisite to evaluate significant difference given the observed biological differences between samples of the same group. Therefore, logistic (MethylKit (Akalin et al. 2012)) or beta-binomial (methylSig (Park et al. 2014)) models have been used to account for sampling (read coverage) and biological variability.
For the identification of DMR, the abovementioned Bump hunting method could be extended to deal with sequencing data (Jaffe et al. 2012b). Similarly, BSmooth (Hansen et al. 2012) (available through bbseq) identifies regions as groups of consecutive CpGs where an absolute score (similar to t-statistics) is above a selected threshold. The approach is based on the application of a local regression to smooth the methylation profiles using weights that are also influenced by the coverage. In this way, the algorithm improves the precision and allows the use of a lower coverage threshold, by assuming that the methylation estimates vary smoothly along the genome. This method is therefore mainly applicable for WGBS in the presence of biological replicates, from which variability is modeled. Local smoothing is also used by another algorithm, BiSeq (Hebestreit et al. 2013), which instead was developed for targeted BS approaches such as RRBS. BiSeq first finds clusters of CpGs and applies local smoothing before testing for differential methylation, using a beta regression model and a Wald test. The algorithm also provides a hierarchical method for calculating an FDR on clusters and sites, and therefore allows defining DMR boundaries.
In order to functionally annotate the discovered DMPs/DMRs, pathway or gene ontology analysis is commonly used. In a typical enrichment analysis, DMRs are first mapped to their nearest genes and then the fraction of annotated genes with a DMR for a given pathway/ontology is compared to the total fraction of genes annotated with that category in the genome. To this purpose, numerous tools are available, which use different algorithms to define enrichment (Huang et al. 2009). Otherwise, a region-based enrichment analysis for cis-regulatory regions is possible through the GREAT tool (http://great.stanford.edu/). This software defines gene regulatory domains with an adjustable “association rule” to connect a TSS (transcriptional start site) of a gene with its cis-regulatory region, such that all DMRs (or other noncoding sequences) that lie within the regulatory domain are assumed to regulate that gene. Then, a genomic region-based enrichment significance test is performed, accounting for the length of gene regulatory domains. Thus, the functional enrichment is carried using regions as input, instead of genes. This approach has been shown to improve the functional interpretation of regulatory regions (McLean et al. 2010). However, even assuming the mapping problem has been solved, it is important to remember that for regions changing in DNA methylation, there is no absolute and unequivocal link between the direction of change and the corresponding change in gene expression. For example, for promoter regions with CpG islands, a methylation event corresponds to gene repression; however, opposite examples have been reported for other regions (Jones 2012). Moreover, when the probes on the array are not evenly distributed across the genome, the use of the proper background is important not to bias the pathway/ontology enrichment analysis. For all the abovementioned reasons, care should be included in performing and interpreting functional enrichment analysis with DNA methylation data, in order to avoid potential biases.
5 Histone Modifications
While DNA methylation was the first uncovered epigenetic regulatory mechanism, several other mechanisms have been uncovered. Arguably, “histone modification” is among the most relevant epigenetic marks. In this third section, we provide an introduction to histone modifications, an overview of profiling experimental protocols and data analysis.
5.1 Introduction to Histone Modifications
Histones are key players because, through covalent modifications of their residuals (e.g., lysine), they have a crucial role in the regulation of transcription, DNA repair, and replication. These modifications are dynamically regulated by chromatin-modifying enzymes (Kouzarides 2007); an enzyme first recognizes available docking sites in histones and then recruits additional chromatin modifiers and remodeling enzymes. Enzymes are associated with specific histone modifications. During the last decades, major efforts have been devoted to the experimental, functional, and regulatory characterization of the different covalent modifications (Tollefsbol 2010). Most relevant experimental protocols and data analysis procedures are described in the next subsection. Table 1.1 summarizes the most relevant types of histone modifications such as methylation, sumoylation, ubiquitination, and acetylation.
The histone modifications selected in the ENCODE project are among the most well characterized (Consortium et al. 2012) and include H2A.Z, H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K9me1, H3K9me3, H3K27ac, H3K27me3, H3K36me3, H3K29me2, H4K20me1. For the interested reader we also recommend to consider chromatin modifications associated with nucleosome regulation (Tessarz and Kouzarides 2014; Becker and Workman 2013), the role of histone variants (Henikoff and Smith 2015) and its association to disease (Maze et al. 2014).
5.2 Profiling Histone Modifications: Experimental Protocol and Data Analysis
5.2.1 Protocol
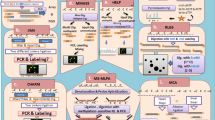
The idea behind genome-wide histone modification profiling is the generation of DNA fragments enriched with the selected histone mark of interest. Once the DNA fragments are obtained, microarray-based or sequencing-based technologies can be applied to quantify the histone marks. The widely accepted protocol for histone mark DNA fragment enrichment detection is Chromatin Immunoprecipitation (ChIP) (Solomon et al. 1988).
ChIP is a powerful tool for studying protein–DNA interactions. Briefly, ChIP consists of two experimental parts:
-
1.
DNA–protein fragment generation. First protein–DNA complexes are cross-linked in living cells. This is usually achieved by the addition of formaldehyde. Next, cells are lysed and chromatin is mechanically sheared in order to obtain fragments of 0.2–2 kb depending on later requirements (array or sequencing). In the context of histone modification, DNA digestion without cross-linking or sonication is preferred for fragmenting the DNA.
-
2.
Enrichment for selected marker. Antibodies are used to immunoprecipitate cross-linked protein–DNA complexes enriched with a selected epitope. Then cross-links are reversed and DNA is recovered.
The DNA recovered can be then processed in two different ways:
-
(a)
Array-based profiling: this technique is named ChIP-on-chip and consists in the labeling and hybridization of enriched DNA fragments to tiling DNA microarrays. ChIP-on-chip allowed the first genome-wide study of DNA–protein binding interactions (Ren et al. 2000; Blat and Kleckner 1999). Before Next-Generation Sequencing (NGS) became widely affordable, ChIP-on-chip was the standard methodology for genome-wide histone profiling. However, with the advent of sequencing technologies ChIP-on-chip has been replaced by ChIP-seq because the latter produces better signal-to-noise ratios, allows a better detection of marks (Ho et al. 2011), has higher resolution, fewer artifacts, greater coverage, and larger dynamic range (Park 2009). For this reason, for the rest of the chapter we will discuss mainly sequencing-based analysis.
-
(b)
Sequencing-based profiling: similar to DNA methylation, NGS provided novel and better tools for histone genome-wide profiling. Interestingly, ChIP-seq was one of the earliest applications of NGS (Johnson et al. 2007; Barski et al. 2007). Nowadays, sample preparation kits for ChIP-seq are commercially available, thus facilitating the preparation of libraries for sequencing.
In Fig. 1.2, the ChIP-seq protocol is detailed. The outcome of ChIP-seq is a set of (millions of) DNA sequences that require processing in order to identify the regions associated to the mark of interest. ChIP-seq has been widely used for profiling histone marks, transcription factor binding, and DNA methylation; in each case, the experimental and data analysis procedures are adapted accordingly. When doing ChIP-seq, it is critical to generate a control ChIP-seq experiment (Landt et al. 2012), which is necessary to account for possible biases, because DNA digestion may not be completely uniform. Two methods for the generation of control libraries are considered: (1) “Input”: DNA from the same sample is processed as any ChIP-seq library but without the immune-precipitation step; and (2) “mock ChIP-seq”: DNA from the same sample is processed similarly but using instead a “control antibody” expected to react only with an irrelevant nonnuclear antigen. Several works have shown the benefits of using control libraries (Landt et al. 2012; Liang and Keleş 2012); interestingly, the possibility of using immunoprecipitation of histone H3 as a background has been proposed (Flensburg et al. 2014).

ChIP-seq protocol. The figure depicts the different experimental steps of ChIP-seq as described in the text (Figure generated by Jkwchiu under CreativeCommons3.0)
5.2.2 Data Analysis
The aim of data analysis is to identify the genomic regions associated with the mark of interest. In the case of ChIP-on-chip, Negre et al. (2006) and Huebert et al. (2006) provide an integrated overview of experimental procedures and data analysis methods while Benoukraf et al. (2009) provide an analysis suite for ChIP-on-chip data analysis.
In the case of ChIP-seq, the starting material is a set of (millions of) DNA sequences and for each sequence, a string of quality score for each base. The analysis of ChIP-seq data involves several steps, some of which are shared among several NGS-based data analysis pipelines:
Step (1) Quality Control
The first step of the analysis is to assess the quality of the data from the set of sequences. Several tools do exist, but arguably FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) from Babraham Institute and Picard (http://broadinstitute.github.io/picard/) from the Broad Institute are two of the most common. The most relevant quality measures are shown in Fig. 1.3 and briefly described here. We highly recommend the reader to visit the online tutorial material of mentioned tools; some of the measures explained below are estimated differently by the different tools.

Example of FastQC output. (a) Per sequence GC content: expected (blue) versus observed (red). Y-axis presents the number of reads and X-axis shows the mean GC content. (b) Quality scores across all bases. Y-axis presents the quality score and X-axis shows position in the read. (c) Quality score distribution over sequences. Y-axis presents the number of reads and X-axis shows the mean sequence quality. (d) K-mer enrichment. Y-axis presents the relative enrichment of a k-mer and X-axis shows the position in the read
-
1.
Percentage of duplications: percentage of sequences that are not unique in the set of DNA sequences. An elevated duplication level may argue for PCR artifacts or DNA contamination.
-
2.
Per sequence GC content: the level of GC content is expected to be similar to that of the entire genome. This is not true when considering DNA methylation but is commonly considered valid when doing histone mark analysis. An example is provided in Fig. 1.3a.
-
3.
Quality scores across all bases: we observe that the base quality score is degraded in the last bases and this is expected because sequencing chemistry degrades with increased read length. However, when the quality is below a certain threshold (e.g., median for any base below 25) the quality of the sequences is under question. Figure 1.3b provides the distribution of quality score along reads, while Fig. 1.3c provides the density of the median quality score.
-
4.
k-mer content: investigate if a k-mer (a sequence of length k) is overrepresented at different locations of the sequences. It is usual to investigate if the initial part of the sequences contains an overrepresentation of adapter sequences, because those will require trimming. Figure 1.3d provides an overview.
FastQC provides certain thresholds to raise warnings on the different quality controls; however, as important as those thresholds is that quality measures are homogeneous among all samples under consideration. In addition, software that provides comprehensive quality controls on ChIP-seq data includes CHANCE (Diaz et al. 2012) or even user-friendly tools such as CLCbio software (CLCbio 2014). Additionally, ChIPQC Bioconductor package provides an R-based tool for quality metrics generation of ChIP-seq data (Carroll et al. 2014b).
Step (2) Mapping to a reference genome
The next task is to map reads (sequences) to the genome of reference. To this end, several softwares exist, with one of the most widely used tools being, arguably, bowtie2 (Langmead and Salzberg 2012). The general output of a mapper assigns each read to a genomic location and a quality of the mapping; a summarized example is depicted in Fig. 1.4a, where reads are mapped to genomic regions, either to the positive or negative strand. It is always recommended to, at least, visually investigate selected regions; among the visualization tools UCSC Genome Browser (Karolchik et al. 2014) and Broad Institute’s IGV (Robinson et al. 2011) are commonly used; we also recommend the use of Bioconductor package tracktables (Carroll et al. 2014a) to generated customized visualizations and dynamic IGV reports. Figure 1.4b provides an example of a UCSC Genome Browser histone mark summary visualization where for each base the enrichment is provided. Since ChIP-seq reads are sequenced from both ends of a signal, the positive and negative strands will enrich each one at different ends; for this reason, the signal is to be considered bimodal when considering positive and negative strands simultaneously (Zhang et al. 2008). We denote the distance between those ends as d.

ChIP-seq mapped data. The figure presents an example of mapped reads into the genome. (a) Short genomic region where it is superposed the mapping of several reads to be positive (blue) or negative (red) strand. (b) USCS display of a genomic region where the enrichment of H3K27Ac enrichment score is shown at a base resolution
Step (3) Peak Identification
Histone marks are identified across the genome as “peaks.” Figure 1.4b provides an intuitive idea of the signal as a peak: we are interested in finding genomic regions where several consecutive bases show significant signal enrichment. In Fig. 1.4b, the left part of the signal (pink) shows low enrichment while to the right there are several regions (peaks) with higher enrichment. Many algorithms, usually called peak-finders, have been developed in order to identify significant enriched regions (peaks) from sequencing data. It is out of the scope of this chapter to present a comprehensive review of them, but we shortly characterize them:
-
Generic peak-finders: among the first peak-finder algorithms, the most successful one was the Model-based analysis of ChIP-seq data (MACS) algorithm from Zhang et al. (2008), which uses some concepts inherited from algorithms developed for ChIP-on-chip data analysis. Briefly, MACS first performs a linear scaling of the control library to be the same as the signal ChIP-seq library. Subsequently, MACS models the distribution of the number of reads per base as a Poisson distribution and then considers all reads a d/2 number of bases across the genome. Finally, a search for significantly enriched regions is conducted through a sliding window of 2*d size. The use of a control library allows FDR estimation. In Bailey et al. (2013), a discussion among current methodologies is provided.
-
Histone-specific peak-finder: many of the methodologies developed considered the peaks of interest to be narrow, such as those observed from most Transcription Factor (TF) ChIP-seq data. However, in the case of histone marks, histone modification enzymes, chromatin remodeling complexes, or RNAPII, we expect a spreading of the signal over larger regions; those are defined as broad-source factors by Landt et al. (2012). For this reason, methodologies such as SICER (Zang et al. 2009), which aims at the identification of statistically significant spatial cluster of signals, were developed. Methods aiming at uncovering both broad and narrow peaks also exist (Peng and Zhao 2011), which would be optimal for mixed-source factors, that is, marks that can be broad or narrow.
The output of most peak-finders provides similar type of information. Most common outputs include the following:
-
Genomic location: chromosome, start and ending site.
-
Summit: in many cases, the base of the peak with the highest enrichment is also identified.
-
Signal strength: number of reads or number of reads per million are also usually provided.
-
Statistical significance: p-values and FDRs are provided. This allows the use of different thresholds during follow-up analyses.
In Landt et al. (2012), and as part of the ENCODE consortia, the authors recommend the use of ChIP-seq replicates; it is recommended to generate a control library for every chromatin preparation and sonication batch. When more than one library is prepared and analyzed against the control, we will obtain a set of peaks for every replication; in those cases, irreproducible discovery rate (IDR) (Li et al. 2011) allows assessing agreement between replicates and also provides FDR estimates for peaks.
Step (4) Peak analysis
Once signals have been uncovered, many follow-up analyses are possible. We enumerate the most common ones:
-
Motif discovery: denotes the identification of transcription factor binding sites in peaks. When applied to TF ChIP-seq, it allows the uncovering of associated TF motifs; however, when applied to histone marks, the identification of motifs and its characterization through motif databases such as TRANSFAC (Matys et al. 2006) or JASPAR (Portales-Casamar et al. 2010) may provide insights into histone-associated TFs for the system under study. MD tools are Homer (Heinz et al. 2010), MEME suite (Bailey et al. 2009), CisFinder (Sharov and Ko 2009), and rGADEM (Droit et al. 2014) among many others. Tran and Huang (2014) is a recent survey on MD web tools.
-
Pathway enrichment analysis: similarly to gene expression analysis, it is important to reveal if the signal from the peaks can be associated, for instance, with specific pathways, diseases, or gene ontology terms. Mapping peaks to genes and then applying classical gene set analyses is an option. However, this option may not be optimal because biases are introduced by gene length (higher probability of having peaks) or by peaks from intergenic regions (such peaks may be associated with genes 10–20 kb away, and which are therefore possibly not closest to the peak itself). ChIP-Enrich (Welch et al. 2014) was developed to correct for gene lengths, while GREAT (McLean et al. 2010) introduces different definitions of gene domains to correct for the uncertainty of the gene-peak mappings.
-
Mapping to genes: because different histone marks may act at different genomic locations, the characterization of peaks as being intergenic, or associated with promoter, gene body, intron, exon, or start/end of the gene (among others) may also provide insights into a histone’s genomic location preferences and association mechanisms. ENCODE provided relevant examples of such characterization in (ENCODE Project Consortium et al. 2007).
6 Repositories and Other Resources
A common task in current data analysis is represented by the integration of different public available data with own experimental data. A first use is, for instance, the overlap of a given histone mark, that is, H3K4me1, in a specific system, that is, CD4 T cells, with H3K4me1 profiles of other cell or tissue types. A second possible usage is to conduct integrative analysis with different epigenetic marks in order to gain functional insight into the regulatory network that is active in the studied biological process.
Typically, histone marks are analyzed in combination with gene expression or DNA methylation data. Furthermore, the researcher has now the availability of a growing selection of epigenomic data (Table 1.2), produced by several international consortia and projects. The size of the epigenomic data sets and publications has grown a lot in recent years, resulting in the availability of different data types that are essential to define the function of the regions under study, and which can be visualized using online or local tools (Table 1.3).
Large data sets allow the research of regulatory mechanisms, impossible to perform in smaller samples. For instance, the idea that histone marks act in a combinatorial manner was considered by different researchers when ChIP-on-chip experiments were first generated; the ENCODE’s pilot project (Thurman et al. 2007) identified higher-order patterns of active and repressed functional domains in human chromatin, through the integration of histone modifications, RNA output, and DNA replication timing. Only when Zho’s laboratory generated ChIP-seq data for several histones and for the same system (CD4+ T cells) was it possible to obtain more robust insights into the cooperation among histone marks (Wang et al. 2008). Interestingly, Karlic et al. (2010) showed that specific combination of histone marks was predictive of gene expression; later the prediction of gene expression was also conducted in new ENCODE data by Dong et al. (2012). Histone acetylation dynamics were also investigated by Zho’s laboratory by profiling HDACs and HATs again in CD4+ T cells.
Over the years, the ENCODE project has generated larger sets of histone mark profiles for several histone marks and several cell types. Interestingly, the generation of such large data sets motivated the use of unsupervised learning methods (Hoffman et al. 2013; Ernst and Kellis 2010; Ernst et al. 2011) in order to identify functional regions and classify them into a small number of labels. In the analysis, data from histone modifications, DNase-seq, FAIRE, RNA polymerase 2, and CTCF were considered. Labels were annotated in a post hoc analysis step; those were further summarized into summary states (Transcription Start Site, Promoter Flanking, Enhancer, Weak Enhancer, CTCF binding, Transcribed Region, and Repressed or Inactive Region).
7 Conclusions
We have presented a brief overview of epigenomics and provided the newcomer with information of available tools for the analysis of epigenomic data sets. However, the methodologies are in continuous development especially in the context of data integration.
References
Adey A, Shendure J. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 2012;22(6):1139–43.
Akalin A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):R87.
Anderson JD, Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol. 2000;296(4):979–87.
Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154(1):11–3.
Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013;14(3):R21.
Aryee MJ, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics (Oxford, Engl). 2014;30(10):1363–9.
Assenov Y, et al. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods. 2014;11:1138–40.
Bailey T, et al. Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput Biol. 2013;9(11):e1003326.
Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(Web Server issue):W202–8.
Barrès R, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15(3):405–11.
Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–37. doi:10.1016/j.cell.2007.05.009.
Becker PB, Workman JL. Nucleosome remodeling and epigenetics. Cold Spring Harb Perspect Biol. 2013;5(9). pii: a017905.
Bell JT, et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12(1):R10.
Benoukraf T, et al. CoCAS: a ChIP-on-chip analysis suite. Bioinformatics (Oxford, Engl). 2009;25(7):954–5.
Bibikova M, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98(4):288–95.
Bjornsson HT, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299(24):2877–83.
Blat Y, Kleckner N. Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell. 1999;98(2):249–59.
Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012;13(10):705–19.
Bock C, et al. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol. 2010;28(10):1106–14.
Butcher LM, Beck S. Probe Lasso: A novel method to rope in differentially methylated regions with 450 K DNA methylation data. Methods. 2015;72:21–8.
Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49(5):825–37.
Carroll T, et al. tracktables: build IGV tracks and HTML reports. R package version 1.0.0; 2014a.
Carroll TS, et al. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front Genet. 2014b;5:75.
CLCbio, CLC shape-based peak caller. White paper. 2014. http://www.clcbio.com/files/whitepapers/whitepaper-chip-seq-analysis.pdf.
Consortium, T.E.P. et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;488(7414):57–74.
Davis S, et al. methylumi: Handle Illumina methylation data. R package version 2.12.0; 2014.
Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–22.
Dedeurwaerder S, et al. Evaluation of the Infinium Methylation 450 K technology. Epigenomics. 2011;3(6):771–84.
Dedeurwaerder S, et al. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief Bioinform. 2014;15:929–41.
Diaz A, Nellore A, Song JS. CHANCE: comprehensive software for quality control and validation of ChIP-seq data. Genome Biol. 2012;13(10):R98.
Dong X, et al. Modeling gene expression using chromatin features in various cellular contexts. Genome Biol. 2012;13(9):R53.
Down TA, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;26(7):779–85.
Droit A, et al. rGADEM: de novo motif discovery. R package version 2.14.0; 2014.
Drong AW, et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue. PLoS One. 2013;8(2):e55923.
Du P, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587.
Eichler EE, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11(6):446–50.
ENCODE Project Consortium, et al. Identification and analysis of functional elements in 1 % of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816.
Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28(8):817–25.
Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–9.
Fei J, Ha T. Watching DNA breath one molecule at a time. Proc Natl Acad Sci U S A. 2013;110(43):17173–4.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2011;13(2):97–109.
Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–40.
Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci U S A. 2010;107 Suppl 1:1757–64.
Feinberg AP, et al. Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci Transl Med. 2010;2(49):49ra67.
Flensburg C, et al. A comparison of control samples for ChIP-seq of histone modifications. Front Genet. 2014;5:329.
Fortin J-P, et al. Functional normalization of 450 k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15(11):503.
Fraga MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–9.
Fyodorov DV, Kadonaga JT. The many faces of chromatin remodeling: SWItching beyond transcription. Cell. 2001;106(5):523–5.
Gagnon-Bartsch JA, Speed TP. Using control genes to correct for unwanted variation in microarray data. Biostatistics. 2012;13(3):539–52.
Guo H, et al. The DNA methylation landscape of human early embryos. Nature. 2014;511(7511):606–10.
Hannum G, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–67.
Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13(10):R83.
Harper KN, Peters BA, Gamble MV. Batch effects and pathway analysis: two potential perils in cancer studies involving DNA methylation array analysis. Cancer Epidemiol Biomarkers Prev. 2013;22(6):1052–60.
Harris RA, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010;28(10):1097–105.
Hebestreit K, Dugas M, Klein H-U. Detection of significantly differentially methylated regions in targeted bisulfite sequencing data. Bioinformatics (Oxford, Engl). 2013;29(13):1647–53.
Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–89.
Henikoff S, Smith MM. Histone variants and epigenetics. Cold Spring Harb Perspect Biol. 2015;7(1):a019364.
Ho JWK, et al. ChIP-chip versus ChIP-seq: lessons for experimental design and data analysis. BMC Genomics. 2011;12:134.
Hoffman MM, et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41(2):827–41.
Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science (New York, NY). 1975;187(4173):226–32.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115.
Horvath S, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A. 2014;111(43):15538–43.
Houseman EA, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86.
Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics (Oxford, Engl). 2014;30(10):1431–9.
Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13.
Huebert DJ, et al. Genome-wide analysis of histone modifications by ChIP-on-chip. Methods. 2006;40(4):365–9.
Illingworth RS, Bird AP. CpG islands–‘a rough guide’. FEBS Lett. 2009;583(11):1713–20.
Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64.
Irizarry RA, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–86.
Ivanov M, et al. In-solution hybrid capture of bisulfite-converted DNA for targeted bisulfite sequencing of 174 ADME genes. Nucleic Acids Res. 2013;41(6):e72.
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15(2):R31.
Jaffe AE, Feinberg AP, et al. Significance analysis and statistical dissection of variably methylated regions. Biostatistics. 2012a;13(1):166–78.
Jaffe AE, Murakami P, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012b;41(1):200–9.
Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2006;8(1):118–27.
Johnson DS, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science (New York, NY). 2007;316(5830):1497–502.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–92.
Karlic R, et al. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 2010;107(7):2926–31.
Karolchik D, et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42(Database issue):D764–70.
Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science (New York, NY). 1974;184(4139):868–71.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705.
Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics (Oxford, Engl). 2011;27(11):1571–2.
Krueger F, et al. DNA methylome analysis using short bisulfite sequencing data. Nat Methods. 2012;9(2):145–51.
Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nat Rev Genet. 2010;11(3):191–203.
Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–31.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Lee KWK, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4:132.
Lee E-J, et al. Targeted bisulfite sequencing by solution hybrid selection and massively parallel sequencing. Nucleic Acids Res. 2011;39(19):e127.
Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3(9):1724–35.
Leek JT, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–9.
Li N, et al. Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods. 2010;52(3):203–12.
Li Q, et al. Measuring reproducibility of high-throughput experiments. Ann Appl Stat. 2011;5(3):1752–79.
Liang K, Keleş S. Normalization of ChIP-seq data with control. BMC Bioinformatics. 2012;13:199.
Lim U, Song M-A. Dietary and lifestyle factors of DNA methylation. In: Methods in molecular biology. Methods in molecular biology. Totowa: Humana Press; 2012. p. 359–76. Available at: http://springerlink.bibliotecabuap.elogim.com/10.1007/978-1-61779-612-8_23.
Lindholm ME, et al. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics. 2014;9(12):1557–69.
Lister R, Ecker JR. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res. 2009;19(6):959–66.
Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–22.
Lister R, et al. Global epigenomic reconfiguration during mammalian brain development. Science (New York, NY). 2013;341(6146):1237905.
Liu Y, et al. Bis-SNP: combined DNA methylation and SNP calling for Bisulfite-seq data. Genome Biol. 2012;13(7):R61.
Liu Y, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31(2):142–7.
Maksimovic J, Gordon L, Oshlack A. SWAN: subset quantile within-array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13(6):R44.
Marabita F, et al. An evaluation of analysis pipelines for DNA methylation profiling using the illumina HumanMethylation450 BeadChip platform. Epigenetics. 2013;8(3):333–46.
Matys V, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34(Database issue):D108–10.
Maunakea AK, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–7.
Maze I, et al. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat Rev Genet. 2014;15(4):259–71.
McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501. pp.nbt.1630–9.
Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205):766–70.
Miura F, Ito T. Highly sensitive targeted methylome sequencing by post-bisulfite adaptor tagging. DNA Res. 2015;22:13–8.
Miura F, et al. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40(17):e136.
Morris TJ, Beck S. Analysis pipelines and packages for Infinium Human Methylation 450 BeadChip (450 k) data. Methods. 2015;72:3–8.
Negre N, et al. Mapping the distribution of chromatin proteins by ChIP on chip. Methods Enzymol. 2006;410:316–41.
Park PJ. ChIP–seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669–80.
Park Y, et al. methylSig: a whole genome DNA methylation analysis pipeline. Bioinformatics (Oxford, Engl). 2014;30:2414–22.
Peng W, Zhao K. An integrated strategy for identification of both sharp and broad peaks from next-generation sequencing data. Genome Biol. 2011;12(7):120.
Peters T, Buckley M. DMRcate: illumina 450 K methylation array spatial analysis methods. R package version 1.2.0; 2014.
Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465(7299):721–7.
Pidsley R, et al. A data-driven approach to preprocessing illumina 450 K methylation array data. BMC Genomics. 2013;14(1):293.
Portales-Casamar E, et al. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38(Database issue):D105–10.
Reinius LE, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility A. H. Ting, ed. PLoS One. 2012;7(7):e41361.
Ren B, et al. Genome-wide location and function of DNA binding proteins. Science (New York, NY). 2000;290(5500):2306–9.
Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14(1):9–25.
Rivera CM, Ren B. Mapping human epigenomes. Cell. 2013;155(1):39–55.
Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6.
Rönn T, et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue J. M. Greally, ed. PLoS Genet. 2013;9(6):e1003572.
Rönnerblad M, et al. Analysis of the DNA methylome and transcriptome in granulopoiesis reveals timed changes and dynamic enhancer methylation. Blood. 2014;123(17):e79–89.
Sandoval J, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6(6):692–702.
Schalkwyk LC, et al. wateRmelon: Illumina 450 methylation array normalization and metrics. R package version 1.5.1; 2013.
Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38(2):391–9.
Sharov AA, Ko MSH. Exhaustive search for over-represented DNA sequence motifs with CisFinder. DNA Res. 2009;16(5):261–73.
Shi J, et al. Characterizing the genetic basis of methylome diversity in histologically normal human lung tissue. Nat Commun. 2014;5:3365.
Smallwood SA, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11(8):817–20.
Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in 1053 microarray experiments. Stat Appl Genet Mol Biol. 2004;3(1):1–25.
Sofer T, et al. A-clustering: a novel method for the detection of co-regulated methylation regions, and regions associated with exposure. Bioinformatics. 2013;29:2884–91.
Solomon MJ, Larsen PL, Varshavsky A. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell. 1988;53(6):937–47.
Stadler MB, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480(7378):490–5.
Sun Z, et al. Batch effect correction for genome-wide methylation data with Illumina Infinium platform. BMC Med Genomics. 2011;4(1):84.
Teschendorff AE, Widschwendter M. Differential variability improves the identification of cancer risk markers in DNA methylation studies profiling precursor cancer lesions. Bioinformatics (Oxford, Engl). 2012;28(11):1487–94.
Teschendorff AE, Zhuang J, Widschwendter M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics (Oxford, Engl). 2011;27(11):1496–505.
Teschendorff AE, Marabita F, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics (Oxford, Engl). 2013a;29(2):189–96.
Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet. 2013b;22(R1):R7–15.
Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15(11):703–8.
Thurman RE, et al. Identification of higher-order functional domains in the human ENCODE regions. Genome Res. 2007;17(6):917–27.
Tollefsbol T, editor. Handbook of epigenetics. San Diego: Academic; 2011.
Touleimat N, Tost J. Complete pipeline for Infinium ®Human Methylation 450 K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4(3):325–41.
Tran NTL, Huang C-H. A survey of motif finding Web tools for detecting binding site motifs in ChIP-Seq data. Biol Direct. 2014;9:4.
Tran H, et al. Objective and comprehensive evaluation of bisulfite short read mapping tools. Adv Bioinformatics. 2014;2014:472045.
Wang ZB, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903.
Wei G, Morris TJ, et al. ChAMP: 450 k chip analysis methylation pipeline. Bioinformatics. 2014;30:428–30. http://www.ncbi.nlm.nih.gov/pubmed/18552846.
Welch RP, et al. ChIP-Enrich: gene set enrichment testing for ChIP-seq data. Nucleic Acids Res. 2014;42(13):e105.
Wiench M, et al. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011;30(15):3028–39.
Wilhelm-Benartzi CS, et al. Review of processing and analysis methods for DNA methylation array data. Br J Cancer. 2013;109(6):1394–402.
Zang C, et al. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics (Oxford, Engl). 2009;25(15):1952–8.
Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137.
Zhuang J, Widschwendter M, Teschendorff AE. A comparison of feature selection and classification methods in DNA methylation studies using the Illumina Infinium platform. BMC Bioinformatics. 2012;13(1):59.
Ziller MJ, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500(7463):477–81.
Zou J, et al. Epigenome-wide association studies without the need for cell-type composition. Nat Methods. 2014;11(3):309–11.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Marabita, F., Tegnér, J., Gomez-Cabrero, D. (2015). Introduction to Data Types in Epigenomics. In: Teschendorff, A. (eds) Computational and Statistical Epigenomics. Translational Bioinformatics, vol 7. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9927-0_1
Download citation
DOI: https://doi.org/10.1007/978-94-017-9927-0_1
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9926-3
Online ISBN: 978-94-017-9927-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)