
Abstract
In the mid 1990s when Leroy Hood reintroduced the term “Systems Biology”, the fusion of ideas gave rise to confusion to such an extent that there used to be special talks on ‘what is systems biology’? Over the last decade, Systems Biology has undergone directed evolution leading to the emergence of personalized versions of this term. Irrespective of this, strong computational dependency and a significant increase in the scale of investigation often appear as constant features in the systems biology background. In our opinion, Systems Biology is an approach that involves the following (a) experimental and computational studies describing collective behavior of molecules in relation to the pathway and networks, and with the higher-level physiological outcome (b) new experimental and mathematical methods important to study group behavior of interacting components. This chapter describes the origin and evolution of systems biology, as a formal discipline, steps and challenges in building models and their potential applications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The traditional approach of doing science has mainly centered around the twin strategy of observation and classification i.e., observe some measurable quantity, say flower color, height of plant and so on, collect data from a large number of plants and try to find some non-obvious pattern. At least in biology, the role of analytical techniques has rarely been pursued as a serious scientific discipline. This is due to the fact that in the traditional setting biological data was easily countable and available to human analysis and interpretation. The science of taxonomy was built upon the foundation of finding common patterns among a large number of samples and categorizing them hierarchically. The strategy was that a higher-level abstraction should be shared by all the members of the group, which can be further sub-sorted into various bins based on some additional parameter. Thus, you see kingdom, families, genus and species as a top-down flow of information in taxonomy. Charles Darwin stretched the idea of ‘finding patterns from external observations’ further, and ended up his long and careful study by proposing the theory of Natural selection. Lamarck and other scientists extended the story further and tried to make his story predictive.
However, in all these situations classifying organisms did not explain how they worked. There was a need to adopt a different approach. Mendel made the first bold attempts to look beyond a horizontal (population-based) plane of vision and vertically move down from phenotype to causal elements. He assumed a linear correlation between a causal element and a phenotypic observation. It was a groundbreaking work. In absence of any high-resolution physical device, he could generate accurate rules and predictions of inheritance simply by looking at the external phenotypes.
After Mendelian era, the science of biology got predominately biochemical and microscopic. Technological developments helped scientists move from external phenotype to cell interiors. However, due to technical complexity and cost of data generation, biological data was mostly qualitative, studied at the level of human analysis and did not require special mathematical techniques and computational infrastructure for interpretation.
As the technological tools got more sophisticated, scientists moved from external observations to the study of cells, chromosomes, DNA, protein and so on. Having seen so many parts co-existing in a small cellular space, there was a natural curiosity—how are these parts created, used, retired, recycled. What is the role of these parts in determining higher order behavior?
Two parallel efforts were aggressively pursued: (i) uncover as many parts and modules (collection of parts employed for a single purpose) as possible, and (ii) find the role of each part in determining a given phenotype. We call this strategy as ‘reductionist biology’ i.e., reduce a system to a set of components and study each component separately. The Human Genome Sequencing Project was started precisely keeping the first aim of reductionist biology in mind i.e., if we know our genetic blueprint, we will figure out everything about ourselves. In parallel to this, a large body of mutations and chromosomal aberrations was collected from diseased tissue to correlate abnormal physiological/morphological conditions with the underlying genetic cause.
However, soon people realized that reductionist approach was unhelpful beyond a point. There were so many incidences where a visible genetic variation/mutation did not lead to a corresponding change in the phenotype. Worse still, in many instances a so-called important gene when knocked-out did not result in the expected outcome. Organisms employed even unrelated genes take over the function of a missing one. Thus, to learn biological decisions there was a need to invent a novel approach.
The trigger for paradigm shift came when microarray technology was invented in the early 1990s. Suddenly huge real-time data was generated. There was no direct way to understand this data, the underlying hidden patterns and correlations. Instead of focusing on one gene, people could now study hundreds of gene expression events together. The impact of even one gene knock-out could be studied in relation to hundreds of unrelated genes. The point of focus moved from sequence level to the expression level. From low throughput human readable data, the scientific community moved to automated, high throughput, machine analyzable data. This was a real phase shift in biology. One could ask questions about the whole system and not about just few parts. By mid 1990s, Systems Biology had truly arrived. This is not to suggest that Systems Biology started in the mid 1990s. The original seeds of thoughts were sown much earlier.
In 1944, Norbert Weiner foresaw the need for systems approach. Unfortunately, the time was not ripe for Systems Level analysis due to data scarcity. Even if all the data were available at that time, the lack of sufficient computational resources would have still precluded scientists to make best use of it. The idea of systems analysis slowly moved from theoretical to practical realm. In the mid 1960s and 1970s, metabolic control analysis gained prominence. The hope was to study the flow of metabolites through the network and find steps that exerted maximal control over metabolic flux in the network. This came to be known as Biochemical Systems Theory. A number of key concepts we use today in flux and control analysis can be traced back to the earlier work (on computational analysis of metabolic networks) by Michael Savageau and co-workers.
Probably the situation wouldn’t have changed much, but for a new technology invented in the early 1990s. Dr.Stephen Fodor (later Chairman and CEO of Affymetrix) and his colleagues published a ground-breaking work in Science in 1991. Biology suddenly underwent a paradigm shift, from low-throughput to high-throughput science. At the same time, computer technology got more advanced, the microprocessors got faster and the storage got cheaper. Time was ripe to collect large amounts of data and store it in computers for analysis.
In the background of technological developments, Leroy Hood formalized this new integrated biology approach and called it ‘systems biology’. For several years people were confused (and probably still are) about: what is systems biology? The community has gone through significant brainstorming on how to define Systems Biology? Though Leroy Hood projected it a specialized field of science, generally people like to view Systems Biology as an “approach” than an independent discipline (Hao et al. 2003). Given the existence of so many flavors of systems biology, probably it is best to describe the properties of Systems Biology than to give it a rigid definition.
2 Systems Biology—A Primer
2.1 What Is Systems Biology?
First, we need to define the term ‘System’. A System is composed of several elements and is defined by the scope of investigation. For example, to study photosynthesis as a systems biology problem, one would need to describe all the genes and molecular networks involved in the process of photosynthesis. It is not necessary for example to model lipid synthesis, if one is investigating photosynthesis as a systems biology problem. Likewise, one can omit photosynthetic pathways if one is modeling lipid metabolism. In other words, the boundary conditions of a system are determined by the components that are directly involved in the process under study. This is not to say that a system is a space constrained by rigid boundary conditions. In reality, a system is a flexible term, described by the availability of data and by the kind of questions.
Systems Biology is a formal approach to understand higher-level behavior as a result of group interaction of the constituent elementary components. As it involves a large variety and scale of data, computational modeling and analysis is frequently employed to store, understand and find meaningful correlations. Systems Biology starts from experiments, goes through computational route and ends at experiments i.e., experimental data → Statistical treatment and modeling → Correlations → Predictions → Experiments. The key difference between systems biology and traditional biology is the focus on group behavior of molecules as against single molecular correlation in the latter.
2.2 Why Is Systems Biology Necessary?
In physical sciences, modeling and simulation, in addition to theoretical and experimental studies, is the third indispensable approach because not all hypotheses are amenable for confirmation or rejection by experimental observations. In biology, researchers are facing the same or maybe even worse situation. On one hand experimental study is unable to produce enough data for theoretical interpretation; on the other hand, due to data insufficiency and inaccuracy, theoretical research cannot provide substantial guidance and insights for experimentation. To meet this need, computational modeling takes a more important role in biology.
2.3 What Is a Model?
A model is a formal or abstract representation of a system, usually in the form of a set of objects and the relations between them. It is a skeleton of the real system but not a replica, built with key components based on a combination of assumptions and existing knowledge. The key to modeling is the identification of elements that can reflect key global properties with incomplete information. Modeling is an iterative process that repeats until a model reaches its final stage and is validated by experiments. In the process, different prototypes are often developed for validation. A model may be formal, with mathematical representation, or conceptual, with diagrams or even concepts only. It may be mechanistic (cause-effect relationship), or phenomenological i.e., based upon a combination of observed phenomena and expert knowledge. Mathematical models are commonly divided into deterministic (responses to given inputs are predictable) and stochastic (responses are picked up based on probability distribution), quantitative and qualitative, and linear and non-linear.
2.4 Is Modeling in Biology New?
Biological modeling is both old and new. Originating from modeling concepts in physical systems, it has a history of several decades. However, due to the distinctive differences between biological systems and physical systems, biological modeling presents itself with additional challenges and calls for new strategies and tools. To model biological systems at various levels i.e., molecular, cell, tissue and organ different strategies and techniques are needed.
Modeling and simulation appeared on the scientific horizon much before the emergence of molecular and cellular biology. Early on the objective of modeling was to explore the features of black boxes e.g., heart, brain, and circulation system, a concept borrowed from physical systems. In such scenario, the main challenge was to understand and predict the behavior of a system without knowing the microscopic details. The strategy was to reproduce observed phenomena at high level with simplified description of internal structures. Though inferring microscopic details was necessarily a major goal, one needed to know how to understand the system as a whole and utilize this understanding in clinical practice. The cases in point are: the inverse modeling of cardioelectrical (Gulrajani et al. 1988) whose simulation results were used to improve diagnosis of heart and brain diseases.
Two interesting methodological features emerged at this stage. First, since biological systems were treated as physical systems or even structure-less systems, many methods and tools were directly borrowed from engineering fields such as FEM (finite element method) and BEM (boundary element method) to compute biological systems (Bradley et al. 2001). Electrical activity of cardiac cells was abstracted to dipoles with different moment and direction. The second feature was high-level abstraction based on inverse approach. Cellular electrical activity was abstracted as an attribute of dipoles [6]. Consequentially, complex numerical techniques for ODE (ordinary differential equation) and PDE (partial differential equation) solution were developed. Both black box assumption and inverse modeling, though suitable for modeling mechanical systems, suffer from major problems when applied to biological systems. The first one is that many inverse problems are mathematically ill-posed. Even if the available data are adequate and precise, unique solution is not always guaranteed and special techniques like regularization are employed (Johnston and Gulrajani 1997). The second assumes that that the internal structure is static, does hold true when a system evolves with time. Thus, this method cannot describe growth process with gene regulation, for the system undergoes state transition while an inverse solution is searched for. Complex internal structure and evolution are key features that differentiate biological systems from mechanical systems. The top down approach doesn’t work very well in biological systems due to absence of information at various levels. Even the bottom up approach (from molecular modeling to organs) encounters the same problem. The solution is to start at an information level and expand vertically upwards/downwards.
3 Modeling Pathways and Networks
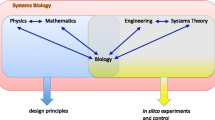
Mendel used simple elementary mathematics of addition and division to obtain laws of inheritance. However, with the arrival of large amount of biochemical and molecular data, mathematical treatments and computer applications got more and more sophisticated (Fig. 1.1). Currently, the predominant phase in biology is process analysis and systems engineering. Process analysis is what we know as Systems Biology and Systems Engineering is commonly referred to as Synthetic Biology.

Predominant computational approaches in biology
Table 1.1 describes some of the commonly used resources and tools in computational systems biology. Modeling is one of the activities in systems biology. It is easy to understand why? Modeling helps address “what-if” questions, facilitate rejection of false hypothesis, and predict future system state in response to a perturbation. Good models are experimentally validated, analyzable and open for manipulation and optimization.
4 Steps in Model Building
Step One
Make a parts list (collect data from literature and experiments). Take into consideration the measurements made, protocols followed, perturbations applied, constraints during experiment and error bar. Was the data independently confirmed? In case of conflicting results, pick up the data from the most reliable group and iterate with the next.
Step Two
Draw an interaction map. The pathway representation should be robust and represent events like translocation, transformation and binding. A pathway map typically consists of nodes (molecules) and edges (interactions). In a standard textbook diagram all the interactions drawn on a uniform background canvas, may (a) belong to different cellular compartments and also (b) occur at different time points. Thus, in reality a standard metabolic/signaling map represents spatially and temporally overlapped data.
Step Three
Converting map into a model. Actually, map itself is a model—a connectivity model. However, to understand dynamic nature of the system a connectivity representation must be converted to a quantitative model. Gene expressions are stochastic and may be modeled with stochastic equations. Metabolic pathways are modeled with Ordinary Differential Equations. Even though Michaelis Menton kinetics is the most accepted way of modeling metabolic events, the MM equation is itself based on assumptions, some of them are not true e.g., well mixed reaction chamber. Figure 1.2 describes a general modeling scheme. Based on the questions asked and system under investigation, distinct modeling approaches are used (Table 1.2).

General modeling scheme
Step Four
Animate the static model. A large number of tools developed for free are available currently (http://www.sbml.org). Most of them offer exchange of results based on the standard SBML output (SBML—Systems Biology Markup Language).
Some of the desirable features of an effective software tool for Systems Biology from both computational and software viewpoints are presented below.
-
a.
Algorithmic Support. Algorithms form the core of any tool. We have seen that there are a number of formalisms and algorithms each with its own strengths and weaknesses. Flexibility to quickly use different algorithms from the same environment would be critical for reducing the cycle time of building large and complex models. We further classify algorithmic support into three divisions:
-
b.
Modeling and Simulation Support. Abstractions of different cellular processes require different information about the target systems such as Gene Regulatory Network, Signal Transduction Network or spatial diffusion. These are based on system specific inputs and implementation of the underlying algorithms. Table I lists details of some of the processes. The whole cell modeling tool must eventually provide support for handling and processing this information.
-
c.
Analysis Support. An important aspect of a typical modeling project in Systems Biology is analysis of the qualitative and quantitative features of the network. Parameter estimation, network optimization, flux balance analysis, bifurcation analysis, extreme pathways and metabolic control analysis are some of the strategies being used currently. Figure 1.3 shows the kind of data used in quantitative model. Parameter estimation algorithms are indispensable for complementing the limited knowledge that can be obtained from experimentation. These algorithms can be used for estimating the unknown rate constants for reproducing an experimentally observed time series. Flux Balance Analysis and Metabolic Control Analysis have a long history of application to metabolic networks. Stoichiometric Network analysis and Extreme Pathways are used to extract qualitative information about a network such as the critical paths.
Fig. 1.3 
shows typical quantities used in a kinetic model

-
d.
Visualization. Powerful visualization tools are necessary for improving the efficiency of the modeling process and understanding the output of the simulation. Some of the desirable features of a visualization tool are:
-
Graphical User Interface for constructing the network and entering various input parameters. A text-based input does not give a good idea of the network topology. Graphical interface becomes particularly desirable for representing spatial features of a model such as compartmentalization and localization. Visualization is required for monitoring the dynamics of a model such as evolution of the network topology through a change in the network layout or the relative concentration of the species through a color code.
-
Powerful graph plotters. The outputs of most of the simulation algorithms are some form of time series. As a result in-built support for powerful plotters is very important for analysis of the output.
-
-
e.
Software Architecture. Simulation and analysis of large-scale models are invariably computationally expensive and often need high performance distributed computing.
Some tasks, amenable to and can benefit from distributed computing, are genetic algorithms based parameter estimation, multiple simulations for parameter sweep and parallel PDE solvers for spatial simulation.
-
f.
Modeling Language. Model building is complex activity requiring collaboration between various research groups, both experimentalists and theorists. Thus development of a common language for smoother information exchange is imperative. Some of the ongoing efforts in this direction are BioPAX, SBML and CellML.
4.1 Challenges in Building Reliable Models
-
Lack of accurate and adequate biological data
-
A general lack of quality control with respect to strain, culture conditions and protocols
-
A cell is a gel, shows gradients, non-uniform distribution of substances in compartments. Frequently, a model does not consider these variables.
-
Parameter values are often inaccurate or taken in special culture/harvesting conditions. To fill in the gap, deterministic and stochastic parameter estimation methods have been developed. However, none of the methods guarantees an accurate answer. Also, given that good data is often less frequently available, the parameter search space is almost always significantly large. The larger the search space, the lesser the possibility of finding an accurate answer.
-
Unknown reaction kinetics
-
Temporal inactivation/degradation of enzymes in generally left out during modeling process
-
Metabolic channeling effects
-
Emergent phenomena
5 Capturing Biological Complexity
The grand challenge of twenty first century is to understand and model complexity of biological systems. Though complexity has been extensively discussed subjects at different levels (Lynch and Conery 2003; Yang et al. 2003), there is no operational definition of complexity for the biological systems (Adami 2002). Some hallmarks of complexity, e.g., linearity and non-linearity, number of parameters, order of equations and evolution of network, come into existence only when a system is formalized in specific ways. Furthermore, from what has been clear, there are two kinds of complexity in biological systems: functional and structural, or dynamic and static; both encountered by modelers. The identification and measurement of biological complexity is a very big task for experimental biologists.
As Adami pointed out, the popular measure of complexity for dynamical systems, computational complexity (for example, the complexity of a sequence can be inferred from what finite state machine can produce), is unsuitable for biological systems. Even though it characterizes the amount of information necessary to predict the future state of the machine it fails to address their meaning in a complex world. Yet the meaning or semantics of molecular interaction really makes sense in signaling processes. An alternative approach may be to think about the complexity issue at higher level and in much larger scope. Recently, the complexity of networks has attracted interests of researchers with different background (Bhalla and Iyenger 1999; Strogatz 2001; Wagner and Fell 2001). Since the topological structure of molecular network, consisting of active genes and proteins, undergoes significant evolution within cells in biological development, to measure complexity of molecular systems, both static and dynamic, according to such evolution may be a practical way, because it is easier to identify and abstract information from it (Bornholdt 2001).
Features in topological structure are also helpful in identifying modularity of molecular interaction. In a large, multicellular landscape, the speed and scope of parallel network evolution in cells, if measured properly, can effectively reflect the complexity of biological systems. Another widely used index of complexity in both physical and biological systems is non-linearity, including parameter sensitivity and initial value sensitivity (Savageau 1971). In evolvable systems, it often implicates the speed of evolution and the appearance of emergent events. Last but not least, the existence of stochasticity and noise increase the complexity of the system even further by introducing issues of robustness, noise resonance and bi-model behaviour.
5.1 Computational Challenges in Building Stochastic Models
Experiments have conclusively proved that molecular activity, including gene regulation, are stochastic (Elowitz 2002). The intrinsic stochasticity of biochemical processes such as transcription and translation generated intrinsic noise; and the fluctuations in the amounts or states of other cellular components lead indirectly to variation in the expression of a particular gene and thus represented extrinsic noise (Swain 2002). There are also opinions that the stochasticity contributes much to system complexity.
To describe the stochasticity, intrinsic and/or extrinsic, two strategies have been developed. The first is to design specific stochastic simulation algorithms that can cut down the computational burden; the second is to use stochastic differential equations, which are modified ODE with stochastic flavor. We first describe these two approaches, then, turn to methods of reducing time consumption of stochastic modeling.
The CME formalism employs an equation for every possible state transition and solves all equations simultaneously. Generating one state transition trajectory is straightforward. However, when the dimensionality of a system increases, the possible trajectories of the state transition, or the state space, explode combinatorially, rendering the system intractable. In view of this serious limitation, Gillespie devised a more efficient algorithm to generate all trajectories (Gillespie 1977). Instead of writing all the master equations explicitly, he generated trajectories by picking up reactions and time intervals according to correct probability distributions so that the probability of generating a given trajectory is exactly the same as the solution of the master equation. For a homogeneous, well-mixed chemical system, Gillespie has proposed two exact Stochastic Simulation Algorithms (SSA), namely the Direct Reaction Method and First Reaction Method to solve the chemical master equations.
Although Gillespie algorithm solves the master equation exactly, it requires substantial amount of computational effort to simulate even a small system. Each of following three factors contributes to a considerable increase of time consumption:
-
Increase in the number of reaction channel
-
Increase in the number of molecules for the species
-
Faster reaction rate of the reaction channels
These factors cause scalability problem, which is similar to the stiffness problem in usual ODE description i.e., whenever reaction rates between different reaction channels vary in magnitude, computation slows down considerably. In the stochastic algorithms, whenever the complexity of a system increases through the augmentation of any of the abovementioned factors, a smaller should be adopted to reflect the true nature of the system, i.e., to maintain the exactness of simulation. The difference in time scale between different reaction channels is a cause for its large computational complexity.
In 1998, Morton-Firth and Bray developed Stochsim algorithm, treating bio- logical components, for examples, enzymes and proteins, as individual interactive objects based on probability distribution derived from experimental data. In this scheme, in each round of computation, a pair of molecules is checked for potential reaction. Due to the probabilistic treatment of interactions between molecules, Stochsim is capable of reproducing realistic stochastic phenomena in biological systems. Though both Gillespie algorithm and Stochsim algorithm are based on the identical, fundamental physical assumptions, an important feature of the latter is the concept of “pseudo-molecules”, which serves as a numerical treatment to maintain the accuracy of the algorithm. Furthermore, in this algorithm, the number of pseudo-molecules can be optimized to overcome the stiffness problem.
In contrast to the variable time step in Gillespie algorithm, Stochsim algorithm uses fixed time step that can be optimized to the desired accuracy. However, the convenience of this measure comes with an additional burden of using empty time step i.e., a time step in which zero events occur. Another limitation of the Gillespie algorithm is its computational infeasibility for multi-state molecules. For example, a protein with ten binding sites will have a total of 210 states and it requires the same amount of reaction channels to simulate this multi-state protein. Considering the scaling feature of Gillespie algorithm with the number of reaction channels, it is impossible to perform such a simulation on with available computational power. Stochsim algorithm can be modified to overcome this problem by associating states to molecules without introducing much computational burden.
Several strategies have been adopted to improve the efficiency of stochastic modeling. Gillespie and Gibson (2001) were the first to modify the SSA to improve efficiency of the algorithms.
Gibson proposed the Next Reaction Method as a revised approach to Gillespie’s First Reaction Method for simulation efficiency. The algorithm has been applied for simulation of the Bacteriophage Lambda model. In 2001, Gillespie presented the Tau-Leap Method to produce significant gains in the computational speed with acceptable loss in accuracy (Gillespie 2001). In the original version of Gillespie Algorithm, master equations were solved exactly to produce precise temporal behavior of systems by generating the exact timing of the firing of each reaction channel. However, it is sometimes unnecessary to obtain so much detail from simulation. Instead of finding out which reaction happens at which time step, one may like to know, how many of each reaction channels are fired at certain time intervals. If the time interval is large enough for many reactions to happen, one can expect substantial gain in the computational speed.
However, the method still possess the inherit disadvantages of supressing stochasticity in fast reaction and the computational efficiency of Implicit Tau Leap method is still unexamined for a large biological pathway model. Another way of improving efficiency of SSA is to adopt multi-scale integration.
5.2 The Rise of Hybrid Modeling
Pure stochastic modeling deals with biological systems as physical systems without biological semantics. Besides the huge burden of time consumption, specific semantic of gene/protein interaction is often buried under low level biochemical reactions.
Hybrid modeling can have multiple meanings. First of all, a model containing metabolic and signaling networks is a hybrid model. Actually these two networks are not independent of each other. For example, in Type II diabetes, the weakened transduction of insulin signal and the changed metabolism activity in cells are closely coupled. In such model, very often, different description methods should be employed to disclose different aspects or parts of a biological system, because, when ODEs are used to describe deterministic events, the basic assumption on continuity and determinism in ODE methods hamper the true representation of noise and stochastic events in cellular environment [64]. Finally, different cellular processes, like gene expression and biochemical reaction and different biochemical reactions, ask for description not only different in methods but also at different time-scales. For a successful simulation, various techniques should be implemented to ensure the feasibility of computation, including the multiple time-scale integration of different equations like ODE, SSA, and SDE [62].
Biological systems in nature undeniably involve multi-scale activities. Algorithms discussed earlier tackle the problem by obtaining solution for the scale of interest while eliminating the other scales in the problem. However, these algorithms produce results of less fidelity in the situation when different scales are heavily coupled together. Furthermore, these algorithms may not be computationally feasible for the scenario as well. One of the methods to reduce simulation time of these algorithms will be to combine different algorithms that handle different scales (Welnan and Engquist ?). The idea of mixing different algorithms to handle hybrid system is not new and has been first adopted in ODE system of equations. Anders [66] presents multi-adaptive-galerkin methods for solving stiff ODE system. The method showcases the possibilty of applying different time-steps and algorithms for different equations in the system and highlights the potential of hybrid methods. However, the method is derived for solving ODE system only and therefore insufficient in tackling the problem in computational cell biology. Recently, Haseltine and Rowlings (2002) presented a method for performing mixed ODE/SSA calculation to approximate system dynamics. The approach are theoretically based on the the equivalence of stochastic and deterministic assumption at the thermodynamic limits, where N and V become infinite. The methods offer insight into integration of the mesoscopic and macroscopic timescale but fail in providing a robust control mechanism and exact mathematical solutions. In addition to that, the methods adopt switches to partition the system into either stochastic or deterministic regime which resulted in sharp transition of the dynamics. This is unnature and unrealistic as compared to the dynamics in the cells which exhibit smooth transition of states from microscopic scale to macroscopic scale.
Integration of diffusion and biochemical pathway has been attempted recentlym (Stundzia and Lumsden 1996). The method derives the reaction-diffusion master equation and simulate the system with SSA. These approaches produce interesting insight about the dynamics between diffusion and chemical reactions. However, the computational requirement is enormous and not feasible for realistic model. Furthermore, the methods do not consider concentration gradient and therefore are not accurate in simulating diffusion processes.
A recent version of Stochsim algorithms includes a 2- dimensional lattice to model the interaction among neighboring molecules. In this approach, spatial information is added as an attribute of each molecular species. The algorithm has been applied for studying the dynamics of signaling proteins associated with the chemotactic receptors of coliform bacteria. MCell [59] has also introduced another way of simulating stochastic diffusion by directly approximate the Brownian movement of individual molecules. In MCell, random numbers are used to determine the motion and direction of molecules during simulation. Due to the incorporation of Monte Carlo simulation and the individual treatment of each molecular species, the results from MCell contain realistic stochastic noise based on the spatial arrangement and number of participating molecules.
Unlike metabolic networks, signaling networks can undergo significant temporospatial changes in embryonic development to endow cells specific identities and to fulfill particular functions within them. For example, a fly is different from a mouse because the molecular interactions within cells of the former produce different signals from the molecular interactions within cells of the latter in body plan development. Since recent progress in developmental biology has indicated that the pathways controlling embryonic development are highly conserved in different animals in both composition and function [82–85], to reveal how slightly different pathways, following what rules, lead to distinctively dissimilar morphogenesis is a great challenge. This, therefore, raises issues of modeling parallel, interactive molecular networks. We list some, but not all, issues here.
First, signaling in a cell is not autonomous in cell fate determination. In development, a cell does not know when to divide, when to die, and when to differentiate. It also does not know, in the absence of environmental messages, whether to differentiate into a myocyte or a neuron. Thus, single cell modeling may not be enough to reveal what we want to know.
Second, various variations can occur, which can be normal and abnormal. In fact, cancer has been seen as aberrant developmental events. To simulate only the normal case is insufficient to understand the properties of signaling networks.
Third, relevant to but different from context dependency is gene function polymorphism. Not like enzymes in metabolic networks showing high specificity, genes in signaling networks can produce and transfer different signals. These constitute basic features of tissue scope molecular level signaling modeling. Considering a small 100 × 100 × 100 tissue cube contains 1 million cells, these issues cannot readily be solved by available modeling platforms.
5.3 Re-Programming Signaling Process in a Cell
One aspect that signaling modeling can make contribute to is the re-programmability of molecular networks, which has been an important research topic (Tada et al. 2001; Hakelien 2002). Carina Dennis, Natures Australia correspondent, describes the technique of turning an adult human cell back to an embryonic state as cellular alchemy [88]. Usually, from state A, an embryonic state, to state B, a state of a fully differentiated myocyte, more than one network configurations mush be underwent. Among explosive combinatorial conditions, how to find a feasible path, consisting of a series of molecular switches, really make sense for experimenters. A wealth of knowledge on dynamics of molecular interaction is very helpful for correct re-programming.
6 Practical Applications of Systems Biology
Systems Biology offers possibility of creating new opportunities for drug target selection based on predictive models. For example, pathway based disease models can be very helpful at the preclinical stage to identify potential toxic effects of lead compounds. If a compound targets network hub, the possibility that such a drug will give rise to a number of side effects is quite high. However, if drug targets turn out to be (a) non-hubs or (b) multiple weak binders in the network collectively bringing about the effect, such lead compounds will be preferred over the rest. Also, the disease and population based drug response models can help lower R&D costs. A prior assessment of side effects/toxic effects can result in speeding up drug discovery, leading to significant savings.
By producing detailed route maps of molecular circuitry in the cell, it is possible, in theory, to develop smarter therapeutic strategies. However, the success of this strategy depends upon completeness and accuracy of relevant data. Systems biology approaches have played a key role in understanding AstraZeneca’s Iressa (gefitinib) Lliver abnormalities were identified by Pfizer, and Johnson & Johnson identified a kinase inhibitor mechanism (extracted from Rubenstein 2008). Dr.Rubenstein’s recent book also includes examples describing nanosystems studies to construct a predictive model for transcription control, ChIP-on-chip technology for global transcription factor identification, and methylation-specific polymerase chain reaction (PCR) for global DNA methylation detection as an entry point to epigenetics.
Identifying systems, building biologically accurate models, with appropriate parameters, performing sensitivity analysis provides a robust ecosystem for carrying out drug development studies. In our experience, the community will increasingly focus on building virtual cell (e.g., virtual E. coli, virtual Pseudomonas) and whole organ (virtual heart, virtual multi-organ diabetic model) in the near future. Professor Dennis Nobel’s group already has significant contribution in this direction. Prof. Nobel is one of the pioneers of Systems Biology and developed the first viable mathematical model of the working heart in 1960. His research focuses on using computer models of biological organs and organ systems to interpret function from the molecular level to the whole organism. Together with international collaborators, his team has used supercomputers to create the first virtual organ, the virtual heart.
The impact of systems biology is also visible through the work of Dr. Jasin A. Papin of the University of Virginia. Recently, his group constructed the first Leishmania major metabolic network that accounts for 560 genes, 1,112 reactions, 1,101 metabolites, and eight unique subcellular localizations. Also, the same group was involved in building a genome-scale constraint-based model of the Pseudomonas aeruginosa strain PAO1, mapping 1,056 genes whose products correspond to 833 reactions and connect 879 cellular metabolites.
7 Conclusion
A system is not equal to the sum of its components. This is especially true of biological systems that show robustness and emergent properties. Due to dynamic and complex interaction among components within and between different levels (Fig. 1.4), the biophysical and biochemical laws that describe these components cannot explain the collective behavior of a system. A grand challenge in systems biology is to identify these rules at the interface and expand in either direction. It is easy to model energy transactions as the energy transfer reactions have been well studied in physical and chemical systems. The more challenging task is to simulate collaborative interactions among molecules that produce and transfer signals.

Different levels when connected give a reasonably accurate picture
As always, new challenges demand new strategies. Signaling pathways, the most difficult to model due to a heterogenous mix of activities involved, can be seen as a kind of molecular body language. We argue that to simulate these molecular activities using a language at a level that matches the molecular body language is a preferable approach. The language should have following minimum features: time-dependent and molecular behavior features, a switchable link between molecules, explicitly defined semantics of interaction, dynamic logging of molecular interaction, hardwiring cellular events with molecular events, and an extension to multicellular modeling capability. We are currently working on building such a language, though its effectiveness hasn’t yet been determined.
One of the challenges in Systems Biology is to identify a complete parts list of a cell and tie them by way of equations, conditional statements that are context dependent. The purpose is to move from structural knowledge to functional knowledge of the system. One of the unsolved mysteries of science is how does the behaviour of a cell at different scales relate to the physiological phenomenon. Constructing a cell from its bare components calls for excellent engineering knowledge, not only for integrating small cell parts into pathways and networks, but also for reverse engineering of the parts from experimental data. The construction of a detailed cell map has to be aided by novel experimental and computational approaches. The future of experimental system biology lies in the invention of novel approaches that generate high throughput and noise free data. In addition, advancement of computational systems biology depends on invention of truly integrated algorithms that are adaptive, robust and capable of simulating multi-scale system. The algorithms will fully integrate different levels of abstractions and reconcile the basic assumptions involved in different timescale and time-span involved. Last but not least, algorithms should also model the smooth transition of a model from mesoscopic to macroscopic scale.
Key: Terms Commonly Used in Systems Biology
Modules are subnetworks with a specific function and which connect with other modules often only at one input node and one output node.
Robustness describes how a network is able to maintain its functionality despite environmental perturbations that affect the components. Robustness also reduces the range of network types that researchers must consider, because only certain types of networks are robust.
Network motifs Patterns of subgraph that recur within a network more often than expected at random.
Path An unbroken series of linear steps. A path has one entry (input) and one exit (output) point.
Pathway A collection of convergent, divergent and cyclic paths. A pathway may have one entry point and many side branches as exit points. The side branches connect a pathway with other pathways. Often, energy-consuming pathways are coupled to energy generating pathways to maintain the overall energy budget.
Network. A set of interacting pathways. A network has multiple entries and multiple exits. Traditionally, pathway was more used for describing metabolic processes and network for gene regulation and signal transduction. Yet there can be metabolic networks, signaling networks, and hybrid networks comprising both metabolic and signaling pathways. The topology of networks reflects some fundamental properties of biological systems involved, and it can be reprogrammed in cells in response to external signals.
Module. A module is a relatively independent functional unit in a cell, which may comprise one or several cross-interacting pathways and autonomously performs a specific function. A functional module can have different structural organization in different cells and at different time, reflecting the substitutability and overlap of gene function. Some biological activities like feedback and amplifier can be explained better in terms of module rather than of pathways or molecules.
Modularity describes the extent to which a system is divided into modules.
Complexity. Biological complexity can be gauged in different dimension. It may cover structural and functional interaction among elements, and the evolution of the systems and subsystem they create. Many mathematical concepts and tools, such as self-organization theory, nonlinear equations, cellular automata and chaos, are used to describe complex biological phenomena.
Robustness. The property of system which indicates the resistance to internal errors and external perturbations
Model. A model is a formal or abstract representation of a system, usually in the form of a set of objects and the relations between them.
System. Consisting of more than one component physically that can be sub systems at lower level, a system possesses more attributes and behaves more complex than any of its component.
Systems Biology. An approach to link the constituent elements of a system with its higher level behavior.
Systems Engineering is a methodology developed in engineering areas but applied in biological modeling to build complex systems from a raw material of components.
Forward Engineering follows a bottom-up approach to model a system and its functional process with known information about its elements.
Reverse engineering is a top-down process, inferring the internal structure and components according to systems behavior.
Systems Theory is a mechanical understanding of system structure behavior.
References
Adami C (2002) What is complexity. Bioessays 24:1085–1094
Bhalla U, Iyengar R (1999) Emergent properties of networks of biological signaling pathways. Science 283:381–387
Bornholdt S (2001) Modeling genetic networks and their evolution: a complex dynamical systems perspective. Biol Chem 382:1289–1299
Bradley C, Harris G, Pullan A (2001) The computational performance of a high-order coupled fem/bem procedure in electropotential problems. IEEE Trans Biomed Eng 48:1238–1250
Elowitz MB (2002) Stochastic gene expression in a single cell. Science 297:1183–1186
Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, Sola D (1991) Light-directed, spatially addressable parallel chemical synthesis. Science 251:767–773
Gillespie DT (1977) Exact stochastic simulation of coupled chemical reactions. J Phys Chem 81:2340–2361
Gillespie DT (2001) Approximate accelerated stochastic simulation of chemically reacting systems. J Chem Phys 115:1716–1733
Gulrajani R, Savard P, Roberge F (1988) The inverse problem in electrocardiography: solutions in terms of equivalent sources. Crit Rev Biomed Eng 16:171–214
Hakelien AM et al (2002) Reprogramming fibroblasts to express t-cell functions using cell extracts. Nat Biotechnol 20:460–466
Hao Z, Huang S, Dhar P ( 2003) The next step in systems biology: Simulating temporo-spatial dynamics of the molecular networks. BioEssays 26:68–72
Haseltine EL, Rawlings JB (2002) Approximate simulation of coupled fast and slow reactions for stochastic chemical kinetics. Chem Phys 117:6959–6969
Johnston P, Gulrajani R (1997) A new method for regularization parameter determination in the inverse problem of electrocardiography. IEEE Trans Biomed Eng 44:19–39
Lynch M, Conery JS (2003) The origins of genome complexity. Science 302:1401–1404
Morton-Firth CJ, Bray D (1998) Predicting temporal fluctuations in an intracellular signalling pathway. J Theor Biol 192:117–128
Rubenstein K (2008) Systems biology: a disruptive technology. CHI insight pharma reports. Ed. 156 pages
Savageau P (1971) Parameter sensitivity as a criterion for evaluating and comparing the performance of biochemical systems. Nature 229:542–544
Strogatz S (2001) Exploring complex networks. Nature 410:268–276
Stundzia AB, Lumsden CJ (1996) Stochastic simulation of coupled reaction-diffusion processes. J Comput Phys 127:196–207
Swain PS (2002) Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc Natl Acad Sci 99:12795–12800
Tada M et al (2001) Nuclear reprogramming of somatic cells by in vitro hybridization with es cells. Curr Biol 11:1553–1558
Wagner A, Fell D (2001) The small world inside large metabolic networks. Proc R Soc Lond B Biol Sci. 268:1803–1810
Yang J, Lusk R, WH Li (2003) Organismal complexity, protein complexity, and gene duplicability. Proc Natl Acad Sci U S A 100:15661–15665
Further Reading
de Jong H (2002) Modeling and simulation of genetic regulatory systems: a literature review. J Comput Biol 9:67–103
Hasty J and Issacs F (2001) Designer gene networks: towards fundamental cellular control. CHAOS 11:207–220
Hlavacek WS, Faeder JR et al (2006) Rules for modeling signal-transduction systems. Sci STKE 344:re6
Hartwell LH, Hopfield JJ, Leibler S, Murray AW (1999) From molecular to modular cell biology. Nature 409:247–252
Iyengar R (2009) Computational biochemistry: systems biology minireview series. J Biol Chem 284:5425–5426
Nurse P (1997) Reductionism: the ends of understanding. Nature 387:657–657
Sauro HM, Bergmann FT (2008): Standards and ontologies in computational systems biology. Essays Biochem 45:211–222
Schadt EE, Zhang B, Zhu J (2009) Advances in systems biology are enhancing our understanding of disease and moving us closer to novel disease treatments. Genetica 136:259–269
Schulze WX, Deng L et al (2005) Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol 1:0008
Smolen P, Baxter DA, Byrne JH (2000) Modeling transcriptional control in gene networks–methods, recent results, and future directions. Bull Math Biol 62:247–292
Yu RC, Resnekov O, Abola AP et al (2008) The Alpha Project: a model system for systems biology research. IET Syst Biol 2:222–233
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Hu, B., Dhar, P. (2015). Introduction to Systems Biology. In: Singh, V., Dhar, P. (eds) Systems and Synthetic Biology. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9514-2_1
Download citation
DOI: https://doi.org/10.1007/978-94-017-9514-2_1
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9513-5
Online ISBN: 978-94-017-9514-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)