
Abstract
The HSP70 family of molecular chaperones consists of at least eight members that are highly evolutionarily conserved. Whereas more than one member of this family is implicated in cancer, the most compelling and abundant data point to the involvement of the predominant stress-inducible form of this protein in cancer etiology and progression. High levels of HSP70 staining in tumors emerged as a significant marker of poor prognosis in human tumors over 20 years ago. Since that time, the important role of this protein in cellular transformation, viral infection, immune function, and the cellular stress response has come to be appreciated and understood. In the past 10 years, the findings that many different types of human tumors are addicted to this protein for survival, and that silencing HSP70 is cytotoxic to tumor but not normal cells, have led to the emergence of the first specific inhibitors of this family of molecular chaperones for cancer therapy. Here-in we review the pro-tumorigenic function(s) of this protein, our understanding of how HSP70 mediates protein quality control, and the current efforts to target and inhibit this protein for cancer therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- HSP70
- HSP90
- Apoptosis
- Apoptosome
- Chaperone
- Co-chaperone
- Drug resistance
- HSP70 inhibitors
- Allosteric inhibitors of HSP70
- Proteostasis
- Senescence
- Lysosome
- Autophagy
- Cancer therapy
10.1 HSP70 Expression in Cancer
The stress-inducible isoform of HSP70 (also called HSP701A1, HSP70-1, or HSP72) is a molecular chaperone that is overexpressed in the majority of human cancers, of various histological origins. Conversely, this protein is nearly undetectable in normal, unstressed cells (Daugaard et al. 2007; Murphy 2013). Elevated basal expression of HSP70 in cancer cells is believed to help these cells to maintain protein homeostasis in the high stress environment of cancer. This stress is derived from both extrinsic (for example, the exterior tumor microenvironment) and intrinsic factors within the cell. Specifically, the tumor microenvironment subjects cancer cells to excess reactive oxygen species, acidosis, hypoxia, and nutrient deprivation; these signals in turn activate the stress-inducible transcription factor heat shock factor 1 (HSF1), the primary transcription factor regulating HSP70 expression (Zorzi and Bonvini 2011). Additionally, intrinsic factors such as aneuploidy, the presence of mutant oncoproteins and high metabolic stress can lead to increased HSF1 activation and HSP70 expression in tumor cells. Finally, there are also some regulators of HSF1, such as the key metabolic controller mTOR, which are frequently activated in cancers, and lead to overexpression of HSP70 in tumor cells (Chou et al. 2012). HSP70 expression promotes growth and survival of tumors in the presence of such adverse conditions (Leu et al. 2011; Juhasz et al. 2013; Lee et al. 2013). Overexpression of HSP70 in cancer has been related to tumor growth, differentiation, drug resistance, metastasis, apoptosis, and poor patient prognosis (Stankiewicz et al. 2005; Juhasz et al. 2013; Kang et al. 2013; Lee et al. 2013).
10.2 High Levels of HSP70 Are Associated with Poor Prognosis
The majority of evidence suggests that HSP70 overexpression correlates with adverse prognosis, decreased differentiation, and increased proliferation (Ciocca and Calderwood 2005). HSP70 expression has been correlated with both early and late stages of cancer. For example, high levels of HSP70 correlates with early stages of disease in prostate (Abe et al. 2004) and hepatocellular (Chuma et al. 2003) carcinomas. Correspondingly, studies involving the ectopic expression of HSP70 in untransformed cells have shown that HSP70 confers tumorigenicity in vitro and in vivo, suggesting that induction of high HSP70 levels is an early event in tumor formation (Seo et al. 1996; Barnes et al. 2001; Meng et al. 2011). There are also multiple studies correlating HSP70 expression with advanced stages of disease. For example, overexpression of HSP70 serves as a marker of advanced disease in melanoma (Lazaris et al. 1995), oral squamous cell carcinoma (Kaur et al. 1998), bladder cancer (Syrigos et al. 2003), intestinal gastric cancer (Lee et al. 2013), colorectal cancer (Hwang et al. 2003), and uterine squamous cell carcinoma (Ralhan and Kaur 1995). Importantly, overall and disease-free survival is significantly reduced when HSP70 is overexpressed in several cancers including endometrial carcinoma (Nanbu et al. 1998), acute myeloid leukemia (Thomas et al. 2005), and breast cancer (Ciocca et al. 1993; Thanner et al. 2003). Furthermore, high levels of HSP70 have been associated with drug resistance. Chemotherapeutic drugs cause increase expression of HSP70 as the cells launch a cytoprotective response. As two examples, an in vitro study of prostate cancer found that treatment with cisplatin increased the expression of HSP70 and mediated resistance to apoptosis (Ren et al. 2008). Similarly, imatinib-resistant chronic myeloid leukemia cells demonstrated a threefold increase in HSP70 expression compared to imatinib-naive cells, and HSP70 knockdown significantly reduced cell viability in drug-resistant cells (Pocaly et al. 2007).
10.3 Cancer-Relevant Pathways Controlled by HSP70
10.3.1 Apoptosis
The accumulated evidence indicates that cancer cells become addicted to HSP70 due to the ability of this protein to serve as a master regulator of several cancer-relevant pathways. For example, HSP70 is able to inhibit extrinsic and intrinsic apoptosis at several points in the apoptotic cascade by binding and modulating stress kinases, and by interfering with the function of the BCL-2 family of apoptotic proteins (Arya et al. 2007; Zorzi and Bonvini 2011). This protein also inhibits the extrinsic pathway of cell death by inhibiting receptor-mediated activation of apoptosis. It does this by binding directly to the death receptors DR4 and DR5 that are activated by TNF-related apoptosis-inducing ligand (TRAIL), which inhibits the formation of the death-inducing signaling complex (DISC) (Guo et al. 2005) (Fig. 10.1a). In general, activation of c-Jun N-terminal kinase (JNK) under stress normally results in the simultaneous phosphorylation and inhibition of the anti-apoptotic proteins BCL-2 and BCL-XL, and phosphorylation and activation of the pro-apoptotic proteins BID and BAX. However, HSP70 has been shown to inhibit stress-induced cell death by binding and inhibiting apoptosis signal-regulated kinase 1 (ASK1), JNK, and p38 mitogen activated protein kinase (Zorzi and Bonvini 2011; Juhasz et al. 2013). Therefore, in the presence of HSP70, BCL-2 and BCL-XL remain active and stabilize the mitochondria, BID and BAX remain inactive and do not translocate to the mitochondria, and SMAC and cytochrome c are not released (Stankiewicz et al. 2005) (Fig. 10.1a). Downstream of the mitochondria, HSP70 binds to the caspase recruitment domain of apoptosis protease-activating factor-1 (APAF-1), and thereby prevents the recruitment of procaspase-9 and the formation of the apoptosome (Ravagnan et al. 2001) (Fig. 10.1a). Correspondingly, HSP70 depletion with small interfering RNA (siRNA) in pancreatic cancer cells led to increased Annexin V-positive cells and caspase-3 and caspase-9 activation, which is consistent with observations that HSP70 can prevent caspase-9 recruitment to the apoptosome (Aghdassi et al. 2007). Given these multiple avenues of controlling cancer cell apoptosis, it is perhaps not surprising that infection of cancer cells with an adenovirus expressing antisense HSP70 results in loss of viability in vitro and tumor reduction in vivo (Nylandsted et al. 2002).

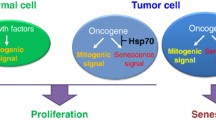
Overexpression of HSP70 affects multiple cancer-relevant pathways. (a) HSP70 inhibits the extrinsic apoptosis pathway by binding to the death receptors DR4 and DR5, and preventing the formation of the DISC. HSP70 inhibits the intrinsic pathway by inhibiting ASK1 and JNK, thereby preventing BAX and BID translocation to the mitochondria. HSP70 also prevents apoptosis downstream of the mitochondria by binding to APAF-1 and preventing formation of the apoptosome. In addition, HSP70 stabilizes lysosome membranes and inhibits lysosome membrane permeabilization (LMP). (b) HSP70 inhibits both p53-dependent and -independent senescence. (c) As an obligate co-chaperone for HSP90, HSP70 is essential for the proper functioning of HSP90, and proper folding of HSP90 client proteins
10.3.2 Senescence, Lysosome Function
In addition to suppressing key proteins in the apoptotic pathway, evidence suggests that HSP70 can also inhibit senescence. For example, siRNA-mediated depletion of HSP70 results in a senescent phenotype, including flattened cell morphology, increased phosphorylation on serine 15 of p53, decreased proliferation, and increased senescence-associated β-galactosidase staining (Yaglom et al. 2007). Additionally, expression of oncogenic forms of PI3K and RAS in breast epithelial cells showed dependency on HSP70 for transformation, in both a p53-dependent and -independent manner, respectively (Fig. 10.1b) (Gabai et al. 2009). Lysosome stability is important for tumor growth, and lysosomal membrane permeabilization (LMP) induced by various stresses results in cell death, while extracellular release of lysosomal proteases can promote tumor invasion (Bivik et al. 2007; Doulias et al. 2007; Dudeja et al. 2009; Zorzi and Bonvini 2011). Specifically in cancer but not normal cells, HSP70 has been found bound to the lysosomal membrane, stabilizing it in the face of stressors such as tumor necrosis factor (TNF), etoposide, UV radiation, and hydrogen peroxide; notably, when HSP70 is depleted, these stresses result in increased cell death (Nylandsted et al. 2004; Bivik et al. 2007; Doulias et al. 2007). In addition, HSP70 interacts with proteins involved in lysosomal membrane permeabilization (LMP), including BAX, JNK, and p53, and this protein can prevent LMP and the release of proteolytic hydrolases, and to thus maintain cellular integrity.
10.3.3 HSP90
The chaperones HSP70 and HSC70 are obligate co-chaperones for HSP90. Not surprisingly then, depletion or inhibition of HSP70 results in reduced solubility of HSP90 client proteins, which become sequestered and inactivated in detergent insoluble compartments in the cell (Leu et al. 2011; Murphy 2013). In a well-controlled study by Workman and colleagues, simultaneous silencing of HSP70 and heat shock cognate 70 (HSC70) resulted in proteasome-dependent degradation of the HSP90 cancer-critical client proteins, C-RAF and CDK4, and initiation of apoptosis in cancer cells, but not in normal non-transformed cells (Powers et al. 2008). These data argue that simultaneous targeting of both HSP70, and the constitutively expressed family member HSC70, may be required for effective cancer therapy.
10.4 HSP70 Chaperone Structure
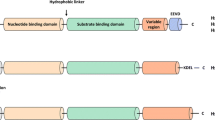
As indicated above, HSP70 is implicated in the pathogenesis of cancer (Ciocca and Calderwood 2005; Powers et al. 2009) and neurodegenerative diseases (Patury et al. 2009; Evans et al. 2010). Therefore, targeting HSP70 function represents a novel therapeutic strategy for these diseases. This has led to increased interest in understanding the structure and mechanism of allostery of this chaperone. The Hsp70 family of proteins (HSP70 in humans, DnaK in E. coli) has a highly conserved domain structure. Each contains a 45 kDa N-terminal nucleotide binding domain (NBD) that has ATPase activity and a 25 kDa C-terminal protein substrate binding domain (SBD), joined by a short flexible linker (Bukau and Horwich 1998; Mayer 2010; Zuiderweg et al. 2013) (Fig. 10.2a). The interdomain linker is only 12 residues in length and is highly conserved (Vogel et al. 2006). Because these two domains are rarely seen together in crystal structures, their detailed structures were described separately in the 1990s. The NBD consists of two flexible lobes, I and II, and each is further divided into subdomains A and B, which form a deep nucleotide-binding cleft (Bork et al. 1992). Structural data suggest that the nucleotide-binding site is located at the bottom of the cleft (Flaherty et al. 1990). This structure was first reported for the NBD isolated from bovine ADP-bound HSC70, and subsequently has been confirmed by others (Flaherty et al. 1990; Harrison et al. 1997; Sriram et al. 1997; Chang et al. 2008).

Structure and allosteric cycle of HSP70. (a) The proposed confirmations of the two ‘end-point’ states of HSP70: the closed state is illustrated by the crystal structure of ADP-bound DnaK (Escherichia coli HSP70; PDB 2KHO; right panel); the open state is illustrated by the crystal structure of the ATP-bound state of Sse1 (yeast Hsp110; PDB 2QXL; left panel). (b) ATP binding to the nucleotide binding domain (NBD) facilitates low affinity binding of substrates, such as those presented to HSP70 via the HSP40 co-chaperone. HSP40 stimulates nucleotide hydrolysis and increases the affinity for substrate, by mediating the closure of the SBD α–helical lid. Nucleotide Exchange Factors (NEFs) assist with ADP release, which causes substrate release
The substrate binding domain (SBD) consists of two functionally relevant subdomains: SBDβ, a 15 kDa β-sandwich subdomain comprised of two four-stranded β sheets with a hydrophobic pocket for peptide binding between them, and SBDα, a 10 kDa mobile α-helical C-terminal region that is believed to function as a “lid” that closes over the substrate (Wang et al. 1998) (Fig. 10.2a). This structure was first described in E. coli for the SBD of DnaK in complex with a short peptide (Zhu et al. 1996). C-terminal to the “lid” is the least conserved region amongst HSP70 family members, which terminates with an EEVD (amino acid designations) motif, capable of interacting with tetratricopeptide repeat (TPR) domain-containing proteins. Sequence variation of this region is believed to allow different HSP70 family members to interact with distinct co-chaperones.
10.5 The HSP70 Functional Cycle
HSP70 assists the folding of its clients using repeated cycles of binding and releasing of misfolded proteins. The ability of the SBD to preferentially bind to exposed hydrophobic polypeptide sequences allows this domain to recognize the non-native states of many proteins (Chiti and Dobson 2006; Eichner and Radford 2011). In some cases, members of the DnaJ/HSP40 family of co-chaperones recognize and bind to such substrates first, and then present them to the HSP70 protein (Bukau and Horwich 1998; Han and Christen 2003). Similar to other ATP-dependent chaperones, the activity of HSP70 requires the energy of ATP binding and hydrolysis and involves critical allostery between the NBD and SBD. Specifically, ADP-ATP exchange in the NBD, which is catalyzed by nucleotide exchange factors (NEFs) like BAG-1 (Liu and Hendrickson 2007; Schuermann et al. 2008), triggers the attachment of the interdomain linker and the α-helical “lid” to the NBD, thereby opening up the peptide binding pocket (this is referred to as an “open” domain-docked conformation of HSP70) (Fig. 10.2b). Subsequently, this results in release of bound substrate from the SBD. Released unfolded peptides are believed to then spontaneously collapse into their native state in free solution (Sharma et al. 2010), or to re-bind to HSP70 for further unfolding, or to transfer to downstream chaperones and possibly be targeted for degradation. Binding of a new peptide to the SBD induces a conformational change that is propagated back to the NBD, increasing the rate of ATP hydrolysis. Finally, hydrolysis of ATP is signaled back to the SBD, resulting in closing of the α-helical “lid” and enhancement of substrate affinity (that is, the “closed” domain-undocked conformation of HSP70) (Fig. 10.2b).
In the attempt to understand the allosteric regulation of HSP70, researchers have focused on comparing the crystal structures of ADP- and ATP-bound DnaK, an HSP70 orthologue from E. coli that shares about 60 % sequence homology with human HSP70 and has a similar domain structure. These studies revealed that in ADP-bound DnaK, the NBD and SBD behave as independent units, and the structures observed show no stable communication between them (Swain et al. 2007; Bertelsen et al. 2009) (Fig. 10.2a, left panel). However, upon ATP binding, the protein undergoes large conformational changes in both domains, which come together to form an intimately packed domain-docked structure (Fig. 10.2a, right panel) (Kityk et al. 2012; Zhuravleva et al. 2012; Qi et al. 2013). It was shown that the interdomain linker of DnaK binds between the IA and IIA subdomains of the isolated NBD, and this alone is necessary and sufficient for ATPase activation (Vogel et al. 2006; Swain et al. 2007). Similarly, the recently described structure of an HSP70 homolog, yeast HSP110, Sse1, shows that the linker is hidden between the NBD and SBD when HSP70 is in the ATP-bound state. This suggests a tight NBD-SBD interaction and is consistent with previously observed ATP-mediated conformational changes (Buchberger et al. 1995; Rist et al. 2006; Liu and Hendrickson 2007; Mapa et al. 2010). These structural data highlight the role of the interdomain linker in the docking of the SBD onto the NBD; however they do not explain how binding of ATP induces allosteric “lid” opening and subsequent loss of affinity for substrates (Flynn et al. 1989; Schmid et al. 1994). To understand the mechanism of this allosteric signal transmission between the NBD and SBD, Mayer and colleagues captured and characterized a normally transient conformation of human HSP70 bound to ATP by engineering disulfide bonds between SBDα and the subdomain IB of the NBD. They showed that ATP binding to the N-terminus of HSP70 induces a clockwise rotation of NBD lobe II, leading to closure of the nucleotide-binding cleft between subdomains IB and IIB. This conformational change widens a space between subdomains IA and IIA, creating a binding site for the interdomain linker, which then docks SBDβ to the ATP-stabilized NBD. The intimate interaction between the NBD and SBDβ interfaces results in SBDα displacement from SBDβ and binding to subdomain IB of the NBD (“lid” opening). Conformational changes of the SBD also propagate to the other side of SBDβ (loop L3,4 of the β-domain), where the substrate-binding cleft opens up, allowing bound substrates to rapidly dissociate. These dramatic allosteric rearrangements in both domains triggered by ATP binding lead to conversion of HSP70 into the stable, open domain-docked state (Kityk et al. 2012; Zhuravleva et al. 2012; Qi et al. 2013). This study reveals that open (ATP-bound) conformation of HSP70 differs from the closed (ADP-bound) not only in the orientation of the α-helical lid, but also in the conformation of the peptide-binding pocket of SBDβ. It also identifies the NBD-linker interaction as a first critical step in interdomain communication. Combined with the knowledge that mutations in the linker abolish chaperone function of HSP70 (Han and Christen 2001), these data point to a role for the linker as a key site in allosteric regulation.
10.6 Opportunities to Interfere with the HSP70 Function
Increased understanding of how ligands modulate the HSP70 allosteric cycle facilitates the design of small allosteric inhibitors of HSP70 for therapeutic targeting of this chaperone. ATP hydrolysis plays a significant role in regulating the HSP70 chaperone activity. Therefore, inhibition of the enzymatic activity of HSP70 with competitive nucleotide analogs that disrupt HSP70-ATP interaction represent a logical approach to interfere with the function of this chaperone. While this strategy has been successful for HSP90 (Messaoudi et al. 2008), compounds that bind directly to the ATPase domain of HSP70 were not identified until recently. A lack of progress in targeting this domain can be explained by its unique structural and chemical characteristics. HSP70 has a 300-fold higher affinity for nucleotide binding when compared to HSP90 (Borges and Ramos 2006; Williamson et al. 2009; Massey 2010). In addition, analysis of the ATP-site, combining structure-based design and computational modeling, revealed that the ATP-binding site of HSP70 consists predominantly of hydrophilic residues, and that the interaction energy between the nucleotide and the chaperone is mostly mediated by the phosphate groups (Hurley 1996; Liu and Hendrickson 2007). This is generally associated with poor druggability of the enzyme active site because it does not allow the formation of hydrogen bonds and hydrophobic interactions between most inhibitors and the ATP-binding pocket (Halgren 2009). Despite these barriers, the ATP-binding site of HSP70 has a favorable size and is well enclosed, indicating that it remains a potentially druggable site. Indeed, recently novel competitive inhibitors of the HSP70 ATPase activity were identified, and these inhibitors were shown to inhibit cancer cell viability in vitro (Williamson et al. 2009).
Another way to modify HSP70 activity is by inhibiting its interactions with important co-chaperones, such as J proteins, nucleotide exchange factors (NEFs) and TPR domain-containing proteins. This can be achieved by blocking protein-protein interactions (PPIs) between HSP70 and co-chaperones or by targeting allosteric sites that disrupt these interactions. Normally, binding of a particular co-chaperone to HSP70 results in activation of a specific function in chaperone biology such as protein trafficking and translocation, protein folding or degradation (Table 10.1). Consequently, the rationale for developing chemical modulators of these interactions is to target a specific subset of HSP70 biology without impacting global proteostasis. Although PPIs are very difficult to inhibit (Arkin and Wells 2004; Gestwicki and Marinec 2007), several molecules have already been reported to specifically disrupt the interaction of HSP70 with particular co-chaperones and lead to distinct effects on cellular homeostasis (Yi and Regan 2008; Roodveldt et al. 2009; Dorard et al. 2011). Taken together, the available structural data suggest there are many opportunities to chemically target HSP70. Recent promising advances in developing HSP70 inhibitors are discussed below.
10.7 HSP70 Inhibitors
Recently, several inhibitors of HSP70 have been reported. Some of the most promising ones are discussed below, and their structures are depicted in Fig. 10.3a. For a more detailed comparative analysis of all the HSP70 inhibitors, readers are directed to some excellent recent reviews on this subject (Patury et al. 2009; Evans et al. 2010; Powers et al. 2010; Goloudina et al. 2012; Murphy 2013).

Small molecule inhibitors of HSP70. (a) Chemical structures of HSP70 inhibitors. (b) Schematic representation of HSP70 domain architecture and the possible sites targeted by the various described inhibitors
10.7.1 Deoxyspergualin/Dihydropyrimidines
One of the first compounds identified to bind and inhibit HSP70 function was the antibiotic 15-deoxyspergualin (DSG) (Plowman et al. 1987). DSG binds to the C-terminus of HSP70 (Fig. 10.3b) with a K D of 4 μM and hinders its function by interfering with its steady-state ATPase activity (Nadler et al. 1995; Brodsky 1999). Although this compound showed promise as an antitumor agent in a mouse leukemia model (Plowman et al. 1987), its efficacy as an anticancer drug in a clinical trial involving metastatic breast cancer was shown to be limited (Dhingra et al. 1994). A subsequent search for compounds with a structural similarity to DSG led to the identification of a class of dihydropyrimidines that block HSP70 ATPase activity (Fewell et al. 2001, 2004). This second-generation of dihydropyrimidines, called the MAL3 series of compounds, were also developed through a screening process (Fewell et al. 2004). Among them, MAL3-101 deserves special mention. It interferes with the co-chaperone HSP40’s ability to stimulate the ATPase activity of HSP70 and prevents its function (Rodina et al. 2007). MAL3-101 blocks cancer cell proliferation (Braunstein et al. 2011) and was shown to be efficacious in a xenograft model of multiple myeloma (Rodina et al. 2007; Braunstein et al. 2011). This series of compounds awaits further testing as prospective anti-cancer drugs.
10.7.2 MKT-077
MKT-077, a rhodocyanine compound, was discovered to accumulate selectively in mitochondria of tumor cells and shown to exert an anti-proliferative effect on cancer cells (Koya et al. 1996). Interestingly, this compound binds an allosteric negatively charged pocket close to the ATP-binding site on HSP70 (Fig. 10.3b), and inhibits the ADP-bound state of this protein, interfering with the HSP70 folding cycle (Rousaki et al. 2011). MKT-077 is primarily toxic to tumor cells and its mechanism of action involves blocking the mitochondrial form of HSP70, HSP70-9 (also called Grp75) (Wadhwa et al. 2000; Rousaki et al. 2011). Mouse xenograft studies demonstrated time- and concentration-dependent antitumor activity for this compound (Chiba et al. 1998). In addition, MKT-077 is cytotoxic to different tumor cell lines, inhibits autophagy and reduces the level of HSP90 client proteins, such as CDK4 and HER-2 (Budina-Kolomets et al. 2014). However, analysis of this compound in a Phase I clinical trial led to severe renal impairment and toxicity in both animal and human subjects, thus limiting further testing and use of this compound (Propper et al. 1999). Recently, a derivative of MKT-077, called YM-1 was developed and shown to be selectively toxic to a variety of tumor cell lines, and to restore tamoxifen sensitivity to tamoxifen-resistant MCF-7 cells (Koren et al. 2012). Thus YM-1 has promising therapeutic potential.
10.7.3 Allosteric Inhibitors of HSP70: Compounds YK-5, 17a, 20a and 27c
Chiosis and colleagues recently identified a previously unrecognized allosteric site in HSP70 using a computational approach (Rodina et al. 2013). Analysis of the geometry and the environment of this computationally-identified allosteric site provided an initiation point for the development of ligands that would bind to this site. The first allosteric inhibitor designed and tested in this series was YK-5, which was found to block both HSP70 and HSC70 function (Rodina et al. 2011). YK-5 blocks HSP70 function by altering the interactions between HSP70, HSP90 and HOP, thus inhibiting the formation of this chaperone complex. This leads to the destabilization of the HSP90 client proteins HER2, RAF-1 and AKT, thereby leading to their degradation. YK-5 reportedly blocks cell proliferation, induces apoptosis and prevents HSF-1 activation or feedback heat shock response in cancer cells. However, YK-5 was found to be an irreversible covalent modifier of HSP70; therefore, this group further developed this series of inhibitors, leading to the identification of two irreversible derivatives, 17a and 20a, which selectively bind HSP70 in cancer cells. Addition of 17a and 20a at low micromolar doses led to a reduction in the steady state levels of HSP70-HOP-HSP90 chaperone complex and the degradation of HSP90 client proteins, along with the induction of cell cycle arrest and apoptosis (Rodina et al. 2013; Kang et al. 2014).
10.7.4 Peptide Inhibitors
HSP70 has a prominent cytoprotective role and this protein increases the oncogenic potential of cancer cells and blocks apoptosis by interacting directly with, and neutralizing the function of, apoptotic protease activation factor-1 (APAF-1) (Beere et al. 2000) and apoptosis-inducing factor (AIF) (Ravagnan et al. 2001). Garrido and co-workers utilized this anti-apoptotic characteristic of HSP70 and designed the peptide ADD70 (AIF-Derived Decoy for HSP70), which is both nontoxic and cytosolic, and is able inhibit the interaction of HSP70 with AIF and other client proteins. They employed deletion mutants and computer modeling methods to develop this AIF-derived blocking peptide, corresponding to the amino acids 150–228 of AIF, a region necessary for binding the SBD of HSP70 (Fig. 10.3b) and neutralizing its activity (Schmitt et al. 2003). Further investigation by this group showed that expression of ADD70 peptide in tumor cells decreased their tumorigenicity by increasing infiltration of cytotoxic CD8+ T cells into the tumor. In addition, ADD70 sensitized rat colon cancer cells and mouse melanoma cells to the chemotherapeutic agent cisplatin (Schmitt et al. 2006), indicating that it shows promise as an anti-tumor agent. Due to the large size of the ADD70 peptide, introducing it into tumor cells can be sometimes challenging. To eliminate this limiting factor this group developed smaller peptide aptamers, A8 and A17, which bind to the peptide-binding and the ATP-binding domains of HSP70, respectively, and specifically inhibit the chaperone activity of HSP70. A 13-amino acid peptide was further synthesized from the variable region of A17 (called P17) that specifically blocked HSP70 and induced regression of subcutaneous tumors in vivo, via the recruitment of macrophages and T lymphocytes into the tumor bed (Rerole et al. 2011).
10.7.5 Ver-155008
Massey and co-workers developed the first adenosine-derived inhibitor of HSP70 by designing ATP-analogs that bind the ATP-binding pocket of the chaperone by using the crystal structure of HSC70/BAG-1. This led to the development of the ATP-analog Ver-155008, which binds to HSP70 with a K D of 0.3 μM and inhibits its activity (Williamson et al. 2009). Ver-155008 is exclusively cytotoxic to cancer cells and downregulates expression of the HSP90 client proteins CDK4, HER2 and RAF-1 in tumor cells (Massey et al. 2010; Budina-Kolomets et al. 2014). It also has anti-proliferative function in a variety of cancer cells; however its ability to induce apoptotic cell death in cancer cells is somewhat limited (Massey et al. 2010). It remains to be seen if derivatives of Ver-155008 can be developed to increase the efficacy of this compound.
10.7.6 The HSP70 Antibody Cm70.1
HSP70 was first reported by Multhoff and co-workers to be localized in the plasma membrane of tumor cells (Multhoff et al. 1995; Multhoff and Hightower 1996), where it is integrated into lipid rafts (Schilling et al. 2009). Although HSP70 lacks a transmembrane domain, its presence in the plasma membrane is believed to be necessary for the stabilization of tumor cell membranes from damage due to environmental stress. Intriguingly, only tumor cells, but not corresponding normal cells, express HSP70 on the surface and stain positively for the IgG1 mouse monoclonal antibody (mAb) cmHsp70.1 (Multhoff and Hightower 2011). The Cm70.1 antibody specifically recognizes an epitope in the C-terminus of the inducible HSP70 (Fig. 10.3b) in viable tumors of both humans in vitro and in mouse tumors in vivo (Stangl et al. 2011). Extensive screening studies in nearly 1,000 primary human tumor biopsies and the corresponding normal tissues indicate that HSP70 is often expressed on the plasma membrane of tumor cells only, and its presence is linked with decreased overall survival of patients; therefore Cm70.1 serves as a negative prognostic marker (Pfister et al. 2007). Interestingly, injecting tumor bearing mice with the cmHsp70.1 antibody significantly inhibited tumor growth and enhanced overall survival, while injection of the peptide corresponding to C-terminal region (aa 450–461) of HSP70 abrogated this effect. This finding suggests that cmHsp70.1 may be a useful therapeutic tool.
10.7.7 PES and Derivatives
2-phenylethynesulfonamide (PES, also known as pifithrin-μ) was initially discovered by Gudkov and colleagues as an inhibitor that blocks the ability of the p53 tumor suppressor protein to traffic to mitochondria, and induce BAX/BAK-dependent apoptosis (Strom et al. 2006). However, at that time, PES was not found to interact with p53, so the target of this compound, and its activity in cells, was unknown. Leu and colleagues were the first to note that treatment of cells with PES appeared to cause the accumulation of autophagosomes in cancer cells. This group went on to show definitively that treatment with PES caused inhibition of autophagy, a key survival pathway that requires the lysosome, and was considered by some to be the Achilles Heel of cancer cells. Based upon the promise of PES as an autophagy inhibitor, Leu generated biotinylated versions of PES and used these compounds to pull down PES-interacting proteins: the only cellular protein found to interact with PES was HSP70 (Leu et al. 2009). This study thus provided the mechanistic basis for Gudkov’s finding that PES inhibits mitochondrial trafficking of p53, as HSP70 was later shown to be required for the ability of proteins to traffic to mitochondria (Pimkina and Murphy 2011).
Leu showed that PES binds the carboxy-terminal substrate-binding domain (amino acids 386–641) of HSP70 and interferes with its protein-folding function. Further investigations showed that PES binds a region located between the substrate binding domain and the C-terminal helical “lid” of this protein (Fig. 10.3b) (Balaburski et al. 2013). Consistent with this finding, exposure of cells to PES prevents binding of HSP70 to critical chaperones namely CHIP and HOP, and inhibits HSP70 function. PES is able to induce cell death in tumor cells through multiple mechanisms: it suppresses macroautophagy (as HSP70 binds to, and stabilizes, the lysosome membrane), inhibits NFκB activation (primarily because IkB-α is an HSP70 target), inhibits function of HSP90 client proteins (Leu et al. 2009, 2011), induces G2/M cell cycle arrest and inhibits the catalytic activity of the anaphase promoting complex/cyclosome (APC/C) in cell-free systems (Balaburski et al. 2013). Interestingly, Budina-Kolomets and colleagues recently compared the activity of three HSP70 inhibitors: Ver-155008, MKT-077 and a more potent derivative of PES, PES-Cl. They concluded that in tumor cells, although all three inhibitors can inhibit autophagy and cause reduced levels of HSP90 client proteins, only PES-Cl can inhibit the APC/C and induce G2/M arrest (Budina-Kolomets et al. 2014). In pre-clinical studies PES (40 mg/kg) and the derivative PES-Cl (20 mg/kg) were shown to protect mice from lymphoma (PES/PES-Cl given once per week for 20 weeks) without any cytotoxic effects to the liver or kidney (Leu et al. 2009; Balaburski et al. 2013). In addition, other groups have demonstrated that PES is effective in killing leukemia cells, without causing cytotoxicity to normal hematopoietic cells, and that this HSP70 inhibitor synergizes with other anti-cancer compounds (Steele et al. 2009; Kaiser et al. 2011).
Recently it was discovered that both HSP70 and HSP90 assist in the stabilization or assembly of the purinosome complex, a dynamic multi-protein complex of enzymes involved in purine synthesis. PES was shown to disrupt the purinosome complex in tumor cells, and this compound is able to do so synergistically with the antimetabolite methotrexate (French et al. 2013). In addition, both HSC70 and HSP70 form the ribosome-associated chaperone system and facilitate co-translational folding and elongation of nascent polypeptides as it emerges from the ribosome exit tunnel. However during various stress-inducing conditions, misfolded proteins sequester the chaperone proteins and interfere with their folding function and cause translational pausing of early elongating ribosomes. Both PES and VER-155008 are able to recapitulate this condition and cause ribosome pausing including decreased protein translation (Liu et al. 2013).
10.8 Future Perspectives
There is enormous potential for HSP70 inhibition to contribute to cancer therapy. Some barriers to this field exist, however, which must be overcome for the field to move forward. First, the disappointing clinical utility of HSP90 inhibitors has led to some souring of the field on HSP70 inhibitors for cancer therapy. However, one of the reasons that HSP90 inhibitors have poor clinical responses is believed to be because of the substantial upregulation of HSP70 inhibitors in treated cancer cells; this up-regulation is due to the release of the master regulator of heat shock transcriptional response, HSF-1, in response to HSP90 inhibition. One solution to this issue will be to combine HSP90 inhibitors with HSP70 inhibitors; indeed, we have seen that these two inhibitors synergize in the treatment of cancer cells (A. Budina-Kolomets, unpublished data). The second hurdle will be the identification of key “client” proteins for HSP70, and the elucidation of the critical cancer-relevant pathways controlled by this protein. It is now clear that HSP70 protein, like the chaperone HSP90, is subject to extensive post-translational modifications, and the nature and impact of these modifications, and how they may differ in tumor and normal cells, needs to be identified. Finally, because HSP70 undergoes extensive movement associated with ATP/ADP and substrate binding, crystallography of this protein has been challenging: however, the use of NMR and X ray crystallography of domains of this protein is allowing for clearer pictures to emerge. Crossing these hurdles should allow for this promising class of compounds to progress to the clinic.
References
Abe M, Manola JB, Oh WK, Parslow DL, George DJ, Austin CL, Kantoff PW (2004) Plasma levels of heat shock protein 70 in patients with prostate cancer: a potential biomarker for prostate cancer. Clin Prostate Cancer 3:49–53
Aghdassi A, Phillips P, Dudeja V, Dhaulakhandi D, Sharif R, Dawra R, Lerch MM, Saluja A (2007) Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res 67:616–625
Arkin MR, Wells JA (2004) Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov 3:301–317
Arya R, Mallik M, Lakhotia SC (2007) Heat shock genes – integrating cell survival and death. J Biosci 32:595–610
Balaburski GM, Leu JI, Beeharry N, Hayik S, Andrake MD, Zhang G, Herlyn M, Villanueva J, Dunbrack RL Jr, Yen T et al (2013) A modified HSP70 inhibitor shows broad activity as an anticancer agent. Mol Cancer Res 11:219–229
Barnes JA, Dix DJ, Collins BW, Luft C, Allen JW (2001) Expression of inducible Hsp70 enhances the proliferation of MCF-7 breast cancer cells and protects against the cytotoxic effects of hyperthermia. Cell Stress Chaperones 6:316–325
Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR (2000) Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2:469–475
Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER (2009) Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci U S A 106:8471–8476
Bivik C, Rosdahl I, Ollinger K (2007) Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis 28:537–544
Borges JC, Ramos CH (2006) Spectroscopic and thermodynamic measurements of nucleotide-induced changes in the human 70-kDa heat shock cognate protein. Arch Biochem Biophys 452:46–54
Bork P, Sander C, Valencia A (1992) An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc Natl Acad Sci U S A 89:7290–7294
Braunstein MJ, Scott SS, Scott CM, Behrman S, Walter P, Wipf P, Coplan JD, Chrico W, Joseph D, Brodsky JL et al (2011) Antimyeloma effects of the heat shock protein 70 molecular chaperone inhibitor MAL3-101. J Oncol 2011:232037
Brodsky JL (1999) Selectivity of the molecular chaperone-specific immunosuppressive agent 15-deoxyspergualin: modulation of Hsc70 ATPase activity without compromising DnaJ chaperone interactions. Biochem Pharmacol 57:877–880
Buchberger A, Theyssen H, Schroder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B (1995) Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J Biol Chem 270:16903–16910
Budina-Kolomets A, Balaburski GM, Bondar A, Beeharry N, Yen T, Murphy ME (2014) Comparison of the activity of three different HSP70 inhibitors on apoptosis, cell cycle arrest, autophagy inhibition, and HSP90 inhibition. Cancer Biol Ther 15:194–199
Bukau B, Horwich AL (1998) The Hsp70 and Hsp60 chaperone machines. Cell 92:351–366
Chang YW, Sun YJ, Wang C, Hsiao CD (2008) Crystal structures of the 70-kDa heat shock proteins in domain disjoining conformation. J Biol Chem 283:15502–15511
Chiba Y, Kubota T, Watanabe M, Matsuzaki SW, Otani Y, Teramoto T, Matsumoto Y, Koya K, Kitajima M (1998) MKT-077, localized lipophilic cation: antitumor activity against human tumor xenografts serially transplanted into nude mice. Anticancer Res 18:1047–1052
Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366
Chou SD, Prince T, Gong J, Calderwood SK (2012) mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS One 7:e39679
Chuma M, Sakamoto M, Yamazaki K, Ohta T, Ohki M, Asaka M, Hirohashi S (2003) Expression profiling in multistage hepatocarcinogenesis: identification of HSP70 as a molecular marker of early hepatocellular carcinoma. Hepatology 37:198–207
Ciocca DR, Calderwood SK (2005) Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10:86–103
Ciocca DR, Clark GM, Tandon AK, Fuqua SA, Welch WJ, McGuire WL (1993) Heat shock protein hsp70 in patients with axillary lymph node-negative breast cancer: prognostic implications. J Natl Cancer Inst 85:570–574
Daugaard M, Rohde M, Jaattela M (2007) The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett 581:3702–3710
Dhingra K, Valero V, Gutierrez L, Theriault R, Booser D, Holmes F, Buzdar A, Fraschini G, Hortobagyi G (1994) Phase II study of deoxyspergualin in metastatic breast cancer. Invest New Drugs 12:235–241
Dorard C, de Thonel A, Collura A, Marisa L, Svrcek M, Lagrange A, Jego G, Wanherdrick K, Joly AL, Buhard O et al (2011) Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat Med 17:1283–1289
Doulias PT, Kotoglou P, Tenopoulou M, Keramisanou D, Tzavaras T, Brunk U, Galaris D, Angelidis C (2007) Involvement of heat shock protein-70 in the mechanism of hydrogen peroxide-induced DNA damage: the role of lysosomes and iron. Free Radic Biol Med 42:567–577
Dudeja V, Mujumdar N, Phillips P, Chugh R, Borja-Cacho D, Dawra RK, Vickers SM, Saluja AK (2009) Heat shock protein 70 inhibits apoptosis in cancer cells through simultaneous and independent mechanisms. Gastroenterology 136:1772–1782
Eichner T, Radford SE (2011) A diversity of assembly mechanisms of a generic amyloid fold. Mol Cell 43:8–18
Evans CG, Chang L, Gestwicki JE (2010) Heat shock protein 70 (hsp70) as an emerging drug target. J Med Chem 53:4585–4602
Fewell SW, Day BW, Brodsky JL (2001) Identification of an inhibitor of hsc70-mediated protein translocation and ATP hydrolysis. J Biol Chem 276:910–914
Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, Brodsky JL (2004) Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem 279:51131–51140
Flaherty KM, DeLuca-Flaherty C, McKay DB (1990) Three-dimensional structure of the ATPase fragment of a 70K heat-shock cognate protein. Nature 346:623–628
Flynn GC, Chappell TG, Rothman JE (1989) Peptide binding and release by proteins implicated as catalysts of protein assembly. Science 245:385–390
French JB, Zhao H, An S, Niessen S, Deng Y, Cravatt BF, Benkovic SJ (2013) Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proc Natl Acad Sci U S A 110:2528–2533
Gabai VL, Yaglom JA, Waldman T, Sherman MY (2009) Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol Cell Biol 29:559–569
Gestwicki JE, Marinec PS (2007) Chemical control over protein-protein interactions: beyond inhibitors. Comb Chem High Throughput Screen 10:667–675
Goloudina AR, Demidov ON, Garrido C (2012) Inhibition of HSP70: a challenging anti-cancer strategy. Cancer Lett 325:117–124
Guo F, Sigua C, Bali P, George P, Fiskus W, Scuto A, Annavarapu S, Mouttaki A, Sondarva G, Wei S et al (2005) Mechanistic role of heat shock protein 70 in Bcr-Abl-mediated resistance to apoptosis in human acute leukemia cells. Blood 105:1246–1255
Halgren TA (2009) Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model 49:377–389
Han W, Christen P (2001) Mutations in the interdomain linker region of DnaK abolish the chaperone action of the DnaK/DnaJ/GrpE system. FEBS Lett 497:55–58
Han W, Christen P (2003) Mechanism of the targeting action of DnaJ in the DnaK molecular chaperone system. J Biol Chem 278:19038–19043
Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J (1997) Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science 276:431–435
Hurley JH (1996) The sugar kinase/heat shock protein 70/actin superfamily: implications of conserved structure for mechanism. Annu Rev Biophys Biomol Struct 25:137–162
Hwang TS, Han HS, Choi HK, Lee YJ, Kim YJ, Han MY, Park YM (2003) Differential, stage-dependent expression of Hsp70, Hsp110 and Bcl-2 in colorectal cancer. J Gastroenterol Hepatol 18:690–700
Juhasz K, Lipp AM, Nimmervoll B, Sonnleitner A, Hesse J, Haselgruebler T, Balogi Z (2013) The complex function of hsp70 in metastatic cancer. Cancers (Basel) 6:42–66
Kaiser M, Kuhnl A, Reins J, Fischer S, Ortiz-Tanchez J, Schlee C, Mochmann LH, Heesch S, Benlasfer O, Hofmann WK et al (2011) Antileukemic activity of the HSP70 inhibitor pifithrin-mu in acute leukemia. Blood Cancer J 1:e28
Kang Y, Jung WY, Lee H, Jung W, Lee E, Shin BK, Kim A, Kim HK, Kim BH (2013) Prognostic significance of heat shock protein 70 expression in early gastric carcinoma. Kor J Pathol 47:219–226
Kang Y, Taldone T, Patel HJ, Patel PD, Rodina A, Gozman A, Maharaj R, Clement CC, Patel MR, Brodsky JL et al (2014) Heat shock protein 70 inhibitors. 1. 2,5′-Thiodipyrimidine and 5-(Phenylthio)pyrimidine acrylamides as irreversible binders to an allosteric site on heat shock protein 70. J Med Chem 57:1188–1207
Kaur J, Srivastava A, Ralhan R (1998) Expression of 70-kDa heat shock protein in oral lesions: marker of biological stress or pathogenicity. Oral Oncol 34:496–501
Kityk R, Kopp J, Sinning I, Mayer MP (2012) Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol Cell 48:863–874
Koren J 3rd, Miyata Y, Kiray J, O’Leary JC 3rd, Nguyen L, Guo J, Blair LJ, Li X, Jinwal UK, Cheng JQ et al (2012) Rhodacyanine derivative selectively targets cancer cells and overcomes tamoxifen resistance. PLoS One 7:e35566
Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, Shishido CLB (1996) MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res 56:538–543
Lazaris AC, Theodoropoulos GE, Aroni K, Saetta A, Davaris PS (1995) Immunohistochemical expression of C-myc oncogene, heat shock protein 70 and HLA-DR molecules in malignant cutaneous melanoma. Virchows Arch 426:461–467
Lee HW, Lee EH, Kim SH, Roh MS, Jung SB, Choi YC (2013) Heat shock protein 70 (HSP70) expression is associated with poor prognosis in intestinal type gastric cancer. Virchows Arch 463:489–495
Leu JI, Pimkina J, Frank A, Murphy ME, George DL (2009) A small molecule inhibitor of inducible heat shock protein 70. Mol Cell 36:15–27
Leu JI, Pimkina J, Pandey P, Murphy ME, George DL (2011) HSP70 inhibition by the small-molecule 2-phenylethynesulfonamide impairs protein clearance pathways in tumor cells. Mol Cancer Res 9:936–947
Liu Q, Hendrickson WA (2007) Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell 131:106–120
Liu B, Han Y, Qian SB (2013) Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol Cell 49:453–463
Mapa K, Sikor M, Kudryavtsev V, Waegemann K, Kalinin S, Seidel CA, Neupert W, Lamb DC, Mokranjac D (2010) The conformational dynamics of the mitochondrial Hsp70 chaperone. Mol Cell 38:89–100
Massey AJ (2010) ATPases as drug targets: insights from heat shock proteins 70 and 90. J Med Chem 53:7280–7286
Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T, Macias AT, Daniels Z, Geoffroy S, Dopson M et al (2010) A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother Pharmacol 66:535–545
Mayer MP (2010) Gymnastics of molecular chaperones. Mol Cell 39:321–331
Meng L, Hunt C, Yaglom JA, Gabai VL, Sherman MY (2011) Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene 30:2836–2845
Messaoudi S, Peyrat JF, Brion JD, Alami M (2008) Recent advances in Hsp90 inhibitors as antitumor agents. Anticancer Agents Med Chem 8:761–782
Multhoff G, Hightower LE (1996) Cell surface expression of heat shock proteins and the immune response. Cell Stress Chaperones 1:167–176
Multhoff G, Hightower LE (2011) Distinguishing integral and receptor-bound heat shock protein 70 (Hsp70) on the cell surface by Hsp70-specific antibodies. Cell Stress Chaperones 16:251–255
Multhoff G, Botzler C, Wiesnet M, Muller E, Meier T, Wilmanns W, Issels RD (1995) A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int J Cancer 61:272–279
Murphy ME (2013) The HSP70 family and cancer. Carcinogenesis 34:1181–1188
Nadler SG, Eversole AC, Tepper MA, Cleaveland JS (1995) Elucidating the mechanism of action of the immunosuppressant 15-deoxyspergualin. Ther Drug Monit 17:700–703
Nanbu K, Konishi I, Mandai M, Kuroda H, Hamid AA, Komatsu T, Mori T (1998) Prognostic significance of heat shock proteins HSP70 and HSP90 in endometrial carcinomas. Cancer Detect Prev 22:549–555
Nylandsted J, Wick W, Hirt UA, Brand K, Rohde M, Leist M, Weller M, Jaattela M (2002) Eradication of glioblastoma, and breast and colon carcinoma xenografts by Hsp70 depletion. Cancer Res 62:7139–7142
Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, Weber E, Multhoff G, Rohde M, Jaattela M (2004) Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 200:425–435
Patury S, Miyata Y, Gestwicki JE (2009) Pharmacological targeting of the Hsp70 chaperone. Curr Top Med Chem 9:1337–1351
Pfister K, Radons J, Busch R, Tidball JG, Pfeifer M, Freitag L, Feldmann HJ, Milani V, Issels R, Multhoff G (2007) Patient survival by Hsp70 membrane phenotype: association with different routes of metastasis. Cancer 110:926–935
Pimkina J, Murphy ME (2011) Interaction of the ARF tumor suppressor with cytosolic HSP70 contributes to its autophagy function. Cancer Biol Ther 12:503–509
Plowman J, Harrison SD Jr, Trader MW, Griswold DP Jr, Chadwick M, McComish MF, Silveira DM, Zaharko D (1987) Preclinical antitumor activity and pharmacological properties of deoxyspergualin. Cancer Res 47:685–689
Pocaly M, Lagarde V, Etienne G, Ribeil JA, Claverol S, Bonneu M, Moreau-Gaudry F, Guyonnet-Duperat V, Hermine O, Melo JV et al (2007) Overexpression of the heat-shock protein 70 is associated to imatinib resistance in chronic myeloid leukemia. Leukemia 21:93–101
Powers MV, Clarke PA, Workman P (2008) Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 14:250–262
Powers MV, Clarke PA, Workman P (2009) Death by chaperone: HSP90, HSP70 or both? Cell Cycle 8:518–526
Powers MV, Jones K, Barillari C, Westwood I, van Montfort RL, Workman P (2010) Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 9:1542–1550
Propper DJ, Braybrooke JP, Taylor DJ, Lodi R, Styles P, Cramer JA, Collins WC, Levitt NC, Talbot DC, Ganesan TS et al (1999) Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann Oncol 10:923–927
Qi R, Sarbeng EB, Liu Q, Le KQ, Xu X, Xu H, Yang J, Wong JL, Vorvis C, Hendrickson WA et al (2013) Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat Struct Mol Biol 20:900–907
Ralhan R, Kaur J (1995) Differential expression of Mr 70,000 heat shock protein in normal, premalignant, and malignant human uterine cervix. Clin Cancer Res 1:1217–1222
Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jaattela M, Penninger JM, Garrido C et al (2001) Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol 3:839–843
Ren A, Yan G, You B, Sun J (2008) Down-regulation of mammalian sterile 20-like kinase 1 by heat shock protein 70 mediates cisplatin resistance in prostate cancer cells. Cancer Res 68:2266–2274
Rerole AL, Gobbo J, De Thonel A, Schmitt E, Pais de Barros JP, Hammann A, Lanneau D, Fourmaux E, Deminov O, Micheau O et al (2011) Peptides and aptamers targeting HSP70: a novel approach for anticancer chemotherapy. Cancer Res 71:484–495
Rist W, Graf C, Bukau B, Mayer MP (2006) Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J Biol Chem 281:16493–16501
Rodina A, Vilenchik M, Moulick K, Aguirre J, Kim J, Chiang A, Litz J, Clement CC, Kang Y, She Y et al (2007) Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat Chem Biol 3:498–507
Rodina E, Vorobieva N, Kurilova S, Mikulovich J, Vainonen J, Aro EM, Nazarova T (2011) Identification of new protein complexes of Escherichia coli inorganic pyrophosphatase using pull-down assay. Biochimie 93:1576–1583
Rodina A, Patel PD, Kang Y, Patel Y, Baaklini I, Wong MJ, Taldone T, Yan P, Yang C, Maharaj R et al (2013) Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein. Chem Biol 20:1469–1480
Roodveldt C, Bertoncini CW, Andersson A, van der Goot AT, Hsu ST, Fernandez-Montesinos R, de Jong J, van Ham TJ, Nollen EA, Pozo D et al (2009) Chaperone proteostasis in Parkinson’s disease: stabilization of the Hsp70/alpha-synuclein complex by Hip. EMBO J 28:3758–3770
Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER (2011) Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol 411:614–632
Schilling D, Gehrmann M, Steinem C, De Maio A, Pockley AG, Abend M, Molls M, Multhoff G (2009) Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J 23:2467–2477
Schmid D, Baici A, Gehring H, Christen P (1994) Kinetics of molecular chaperone action. Science 263:971–973
Schmitt E, Parcellier A, Gurbuxani S, Cande C, Hammann A, Morales MC, Hunt CR, Dix DJ, Kroemer RT, Giordanetto F et al (2003) Chemosensitization by a non-apoptogenic heat shock protein 70-binding apoptosis-inducing factor mutant. Cancer Res 63:8233–8240
Schmitt E, Maingret L, Puig PE, Rerole AL, Ghiringhelli F, Hammann A, Solary E, Kroemer G, Garrido C (2006) Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res 66:4191–4197
Schuermann JP, Jiang J, Cuellar J, Llorca O, Wang L, Gimenez LE, Jin S, Taylor AB, Demeler B, Morano KA et al (2008) Structure of the Hsp110: Hsc70 nucleotide exchange machine. Mol Cell 31:232–243
Seo JS, Park YM, Kim JI, Shim EH, Kim CW, Jang JJ, Kim SH, Lee WH (1996) T cell lymphoma in transgenic mice expressing the human Hsp70 gene. Biochem Biophys Res Commun 218:582–587
Sharma SK, De los Rios P, Christen P, Lustig A, Goloubinoff P (2010) The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat Chem Biol 6:914–920
Sriram M, Osipiuk J, Freeman B, Morimoto R, Joachimiak A (1997) Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure 5:403–414
Stangl S, Gehrmann M, Dressel R, Alves F, Dullin C, Themelis G, Ntziachristos V, Staeblein E, Walch A, Winkelmann I et al (2011) In vivo imaging of CT26 mouse tumours by using cmHsp70.1 monoclonal antibody. J Cell Mol Med 15:874–887
Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD (2005) Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem 280:38729–38739
Steele AJ, Prentice AG, Hoffbrand AV, Yogashangary BC, Hart SM, Lowdell MW, Samuel ER, North JM, Nacheva EP, Chanalaris A et al (2009) 2-Phenylacetylenesulfonamide (PAS) induces p53-independent apoptotic killing of B-chronic lymphocytic leukemia (CLL) cells. Blood 114:1217–1225
Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, Bosykh DA, Burdelya LG, Macklis RM, Skaliter R et al (2006) Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol 2:474–479
Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM (2007) Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell 26:27–39
Syrigos KN, Harrington KJ, Karayiannakis AJ, Sekara E, Chatziyianni E, Syrigou EI, Waxman J (2003) Clinical significance of heat shock protein-70 expression in bladder cancer. Urology 61:677–680
Thanner F, Sutterlin MW, Kapp M, Rieger L, Kristen P, Dietl J, Gassel AM, Muller T (2003) Heat-shock protein 70 as a prognostic marker in node-negative breast cancer. Anticancer Res 23:1057–1062
Thomas X, Campos L, Mounier C, Cornillon J, Flandrin P, Le QH, Piselli S, Guyotat D (2005) Expression of heat-shock proteins is associated with major adverse prognostic factors in acute myeloid leukemia. Leuk Res 29:1049–1058
Vogel M, Mayer MP, Bukau B (2006) Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem 281:38705–38711
Wadhwa R, Sugihara T, Yoshida A, Nomura H, Reddel RR, Simpson R, Maruta H, Kaul SC (2000) Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res 60:6818–6821
Wang H, Kurochkin AV, Pang Y, Hu W, Flynn GC, Zuiderweg ER (1998) NMR solution structure of the 21 kDa chaperone protein DnaK substrate binding domain: a preview of chaperone-protein interaction. Biochemistry 37:7929–7940
Williamson DS, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Foloppe N, Francis GL, Graham CJ, Howes R et al (2009) Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J Med Chem 52:1510–1513
Yaglom JA, Gabai VL, Sherman MY (2007) High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res 67:2373–2381
Yi F, Regan L (2008) A novel class of small molecule inhibitors of Hsp90. ACS Chem Biol 3:645–654
Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272:1606–1614
Zhuravleva A, Clerico EM, Gierasch LM (2012) An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 151:1296–1307
Zorzi E, Bonvini P (2011) Inducible hsp70 in the regulation of cancer cell survival: analysis of chaperone induction, expression and activity. Cancers (Basel) 3:3921–3956
Zuiderweg ER, Bertelsen EB, Rousaki A, Mayer MP, Gestwicki JE, Ahmad A (2013) Allostery in the Hsp70 chaperone proteins. Top Curr Chem 328:99–153
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Budina-Kolomets, A., Basu, S., Belcastro, L., Murphy, M.E. (2015). The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities. In: Wondrak, G. (eds) Stress Response Pathways in Cancer. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9421-3_10
Download citation
DOI: https://doi.org/10.1007/978-94-017-9421-3_10
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9420-6
Online ISBN: 978-94-017-9421-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)