
Abstract
In this chapter, the key role of fuel cells and regenerative fuel cell technologies, in the future energetic scenarios based on renewable energy sources, is described. The fuel cell technology is reviewed; its working principles are summarized and mathematically described by means of a simple analytical model. It is found that the limits of fuel cell competitiveness can be overcome only through a specific design of its key component materials: the electrolyte and the electrocatalyst. Examples of material design research achievements are reported for both fuel cells and regenerative fuel cells.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The first commercial application of fuel cells (FC) goes back to the years 1965–1966 and to the Project Gemini. Since that time, although the energy production–storage–consumption issue became a worldwide critical issue, both economically and ecologically, and despite the many progresses in the field, fuel cells are still “searching to find a killer application that allows their penetration into the market” (Winter and Brodd 2004).
Fuel cells belong to the class of electrochemical devices in which the atomic bond energy is directly converted to electrical energy. To the same class belong batteries and electrochemical capacitors. All these systems have two electrodes in contact with an electrolytic solution, and all energy production processes are happening at the interface electrode–electrolyte solution. Fuel cells are open systems in which reactants have to be provided from outside (storage outside), while batteries are closed systems containing the reactant in their interior (storage inside). Electrochemical capacitors, which store energy in electrical double layer at the electrode–electrolyte interface, may deliver energy also in the absence of redox reactions.
Fuel cells have been considered as a replacement to the internal combustion engines and combustion power plants, due to a possible lower ecological impact and comparable, or higher, energy conversion efficiency.
The best way to compare between each other all these devices, and to understand their domains of applicability, is via the so-called Ragone plot, in which the specific energy (Wh/kg) is reported in the abscissa and the specific power (W/kg) is reported in the ordinate. The Ragone plot of Fig. 1 shows that fuel cells are high energy systems of quite low power, while supercapacitors and capacitors can deliver the low energy content at a high rate (high power). Batteries have an intermediate behavior, but the combustion energy is at the upper right part of the plot, showing that, at the moment, “no single electrochemical power source can match the characteristics of the internal combustion engine” (Winter and Brodd 2004).

Ragone plot with the energy/power storage domains of electrochemical devices as compared to that of combustion engines. Abscissa: specific energy, Wh/kg; ordinate: specific power, W/kg (Adapted from Winter and Brodd 2004)
Then, fuel cells alone are best suited for specific stationary energy applications and to replace batteries and super capacitors in electronic devices. In addition, and from life cycle assessment, FC are expected to have a very low ecological impact.
To enhance at the same time the specific power and the specific energy, and also to try to bridge the gap with the internal combustion engines, R&D activities are running in the direction of hybridization, in which, for example, carbon nanotube or graphene-enhanced ultracapacitors are coupled with fuel cells. Transition metal and light metal-decorated graphenes are expected to have a very high specific power (see Figure 1 in Ref. (Tozzini and Pellegrini 2013)), and their hybridization with fuel cells, in devices which can be classified either as electrochemical asymmetric ultracapacitor or as modified FC (Signorelli 2009), can deliver high energy at high power.
Aside from these specific applications, fuel cells have a key role in all energetic scenarios based on renewable energy sources. In such scenarios, CO2 is strongly reduced and the energy is produced/distributed on small scale. The reduction of CO2 is achieved by the production of electricity, heat, and “solar fuels,” mainly hydrogen, from renewable sources, while the small scale is achieved by scalable power and storage systems managed by smart grids. In the production schemes of hydrogen, fuel cells are coupled with a sun-powered thermo-catalytic reactor able to split the water molecule or with a system producing electricity from renewables plus an electrolyzer. In both schemes, the renewable energy is transferred to the hydrogen obtained from the water splitting, and water is re-obtained from a hydrogen fuel cell able to deliver back part of the renewable energy as electricity.
The coupling of a fuel cell with an electrolyzer is realized via the regenerative fuel cells (RFC) or, better, via the unitized RFC (URFC) in which a single device is able to work as an electrolyzer and as a fuel cell (Andrews and Doddathimmaiah 2008; Mitlitsky et al. 1998).
The aim of this paper is to present the basic definitions, the basic role and properties, and the basic mechanisms of transport in all the components making a fuel cell. The same presentation is given for the URFCs. It is only from this starting point that deeper analysis aimed to optimize and to enhance fuel cell, and hybrid fuel cell performances can be understood and developed.
The paper is organized as follows. The next Section, Sect. 2, introduces the FC working principle, the role of each FC component, and the classification scheme of the known FCs. Sect. 3 introduces the quantities which enter in the description of the FC performance curve and the FC potential as a function of the FC current density, and it explains, through a simple analytical model, the overall shape of the curve, from low to high current density. Section 4 is devoted to the main materials used as electrolyte in the membrane region and as catalyst in the reactive regions. The section points out the phenomena happening in the involved materials. The final section, Sect. 5, deals with regenerative and unitized regenerative FCs. Also in this case, the working principle, the basic roles and properties of involved materials, and the transport mechanisms are presented and explained.
2 The Fuel Cell Technology: Working Principle
Redox reactions involve the transfer of electrons between species. By separating oxidation and reduction half-reactions and by leading the electron transfer through an external circuit, chemical energy is transformed directly into electrical energy.
In an electrochemical cell, oxidation and reduction reactions are separated in space and connected by an external electric circuit. The two separated regions, where the two half-reactions take place, are called the electrodes: the anode for the oxidation and the cathode for the reduction reaction. The separator, capable to transport the ionic species to provide a complete electric circuit, is called the electrolyte.
Fuel cells are electrochemical cells, which use a fuel as reductant and oxygen as oxidant (see Fig. 2). Hydrogen is the most common fuel, but hydrocarbons and alcohols are sometimes used.

FC working principle
In principle, fuel cell operation can be reversed, and the FC can work as an electrolyzer producing oxygen and hydrogen molecules from water and electricity. In the reversible fuel cell concept, the same physical cell is used for both operation modes.
In the next subsection, we schematically list all the main components of a fuel cell, their functions and phenomena, and the constituting materials. Afterwards, we will give some more details on the key FC components, the electrolyte, and the reactive regions of the electrodes.
2.1 The FC Components
2.1.1 Flow Channels and Current Collectors
Main function: (1) The flow channels must drive the fuel to the anode and the oxidant to the cathode and allow the removal of the reaction products (H2O, CO2). (2) The current collector must collect the electrons produced by the electrochemical reaction at the anode and, through the external circuit, bring them at the cathode.
Auxiliary functions: In polymer electrolyte membrane fuel cells (PEMFC), the current collectors have also a mechanical support function and a thermal control function.
Material properties: Good electronic and thermal conductor
Phenomena: Reactant flow conduction, thermal and electronic conduction
2.1.2 Electrode Diffusion Layers
Main functions: The function of the electrode diffusion layers is to drive the reactants from the flow channels to the electrochemically reactive regions and the electrons from the electrochemically reactive regions to the current collectors.
Auxiliary functions: In solid oxide fuel cells (SOFC), electrodes can have also a mechanical support function and can catalyze the methane reforming reaction.
Material properties: Porous material, good electronic conductor
Phenomena: Reactant diffusion, electronic conduction, nonelectrochemical reactions (shift reaction, reforming reaction)
2.1.3 Electrodes Reactive Layer
Main functions: The function of the electrode reactive layers is to catalyze the electrochemical reactions.
Material properties: Porous material, electronic and ionic conductor, high catalytic activity
Phenomena: Electrochemical reaction, catalysis, electronic and ionic conduction, reactant diffusion
2.1.4 The Electrolyte
Main functions: The function of the electrolyte is to conduct an ionic current between the two electrodes while constraining the electrons to pass through the external circuit.
Auxiliary function: In most of the cells, the electrolyte should keep the fuel separated from the oxidant in order to avoid direct (nonelectrochemical) reactions.
Phenomena: Ionic conduction, reactant crossover
Material properties: Good ionic conductor, electronic insulator, minimum permeability to reactant
2.2 The FC Types
As schematically described in the previous section, the main physical phenomena occurring inside the fuel cells are transport of ions, electrons and neutral species, and electrochemical reactions. While chemical diffusion and electronic conduction phenomena are common to a very wide number of applications, electrocatalysis and ionic conduction are much more specific and complex. Therefore, the key FC components are the electrolyte and the reactive regions of the electrodes.
Different strategies have been studied to make these components efficient, robust, and economically affordable, but none of them has shown a clear and general advantage. Thus, different types of FC have been developed, or are still under development, and are classified following the operating temperature range and the electrolyte material.
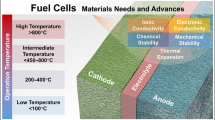
2.2.1 Temperature Classification
The reaction kinetics should be as fast as possible in order to minimize activation losses. Three main factors influence the reaction kinetics: the catalyzers quality, the temperature, and the availability of reaction sites.
In practice, a good structural design of the reactive layer should guarantee the simultaneous presence of ions, electrons, and reactant fluxes on the catalyst surface (triple phase boundaries). Beside this, either very efficient catalyzer or very high temperature is needed to minimize the activation losses. As a consequence, a first main FC classification is based on the operating temperature range.
At low temperatures (≤100 °C), precious metals and, in particular, platinum are the catalyzers of choice. On the other hand, at such “low” temperatures, the fuel cell is easy to manage, with quick start-up times and no thermal breaking risk. Moreover, liquid water can be used to transport ions through the electrolyte. Low-temperature fuel cells (LTFC) include PEMFC and alkaline fuel cells (AFC).
At very high temperatures (>600 °C), platinum is not necessary anymore and less costly catalyzers (e.g., nickel) can be used. Moreover, catalyzer sensibility to poisoning substances is strongly reduced and a larger variety of fuels can be used. However, such high temperature brings strong material issues and long start-up times. High-temperature fuel cells (HTFC) include SOFC and molten carbonate fuel cells (MCFC).
Intermediate temperature FC exists as well, which share the main disadvantages of the other two classes, including the need of precious metal catalysts and long start-up times. Among this class, we mention the phosphoric acid fuel cells (PAFC).
2.2.2 Electrolyte Classification
The function of the electrolyte is to conduct an ionic current between the two electrodes while constraining the electrons to pass through the external circuit. The main issue is to find a manageable ionic conductor material, with a good ionic conductivity in order to minimize ohmic losses.
Different ionic conduction mechanisms can be used, including:
-
Water electrolytic solutions (acid or basic) (PEMFC, AFC) (low temperatures—not much larger than the water boiling temperature)
-
Anhydrate liquids as phosphoric acid (PAFC) or molten salts (MCFC) (intermediate to high temperatures)
-
Conduction in solids (SOFC) (high temperatures)
A number of different ionic species can be used as charge carriers. For example, in hydrogen fuel cells which separate the hydrogen oxidation reaction 2H2 + O2 → 2H2O, the following ions are commonly used:
Protons H+ (PEMFC, PAFC, and proton SOFC):
Hydroxide OH− (AFC, anion electrolyte membrane FC, AEMFC):
Oxygen ions O− (SOFC):
Carbonate ions CO2− 3 (MCFC):
3 FC Performances: Analysis and Optimization
The fuel cell operation is characterized by the cell potential versus current density I: the V(I) performance curve. This curve provides direct information on:
-
Electric power density, which can be obtained as V(I) × I. The maximum of this function has an important role in dimensioning the FC system for applications requiring high power peaks (e.g., automotive applications).
-
Energy conversion efficiency, which can be obtained as V(I)/V 0, where V 0 is the theoretical potential. Efficiency is a fundamental parameter for all applications.
-
Maximum current density, I lim.
-
Dissipated power, which can be obtained as (V 0 − V(I)) × I. This quantity is the heat generated by the fuel cell.
In the next subsections, we will derive a simple but quite accurate analytical expression for V(I), from which all the above information can be easily derived and analyzed.
3.1 The Fuel Cell Potentials
The theoretical potential, V 0, is a thermodynamic quantity that depends on the specific electrochemical reaction, temperature, and chemical concentrations. It corresponds to the maximum potential achievable with a 100 % conversion efficiency. Potential losses inside a fuel cell originate either at the interfaces between ionic and electronic conducting phases, where all the electrochemistry takes place, or internally to each single phase. The single phase losses, η, are essentially of Ohmic origin, linear with the cell current density, and, therefore, they have a trivial effect on the performance curves:
where R is the fuel cell resistance.
More complex is the I-behavior of the interface overpotentials, η int, which, as illustrated in Fig. 3, originate in the small electrode reactive region, where the two phases coexist.

I-behavior of the potentials in the FC regions
In the Figure, we have arbitrarily represented the theoretical potential V 0 as the sum of an anodic, E A, and a cathodic, E C, contributions.
Therefore, we write the cell potential as made up of zero-current term, E; a potential drop term of Ohmic origin, η ohm; and potential drop terms originated by the interface electrochemistry in the anode and cathode reactive regions, η int:
3.2 The Interface Phenomena
At open circuit, ions and electrons produced by the electrochemical reactions are accumulated around the electrode–electrolyte interface. The reaction goes on until the electrostatic potential of this “double electric layer” equals the reaction potential, E A or E C (see Fig. 3).
When the circuit is closed, electronic and ionic current tends to cancel polarizations, while reactions tend to restore them. When reactions are not fast enough, depolarizations, or overpotentials, are created which contribute to accelerate reactions. Equilibrium is reached that, in general, is well described by the Butler–Volmer formula, which links the current density production to the depolarization η int:
in which a is the catalyst area per unit volume, i 0,ref the reference current density, c RL the reactant concentration in the reactive layer, c ref the reference concentration, α and γ the kinetic parameters, and β = 1/k B T.
The cell current density can be obtained by integrating the Butler–Volmer equation within the reactive layer thickness, L RL:
from which we can get η int(I).
3.3 The Interface Overpotential
By using the assumptions:
-
1.
The electrochemical reaction follows the phenomenological Butler–Volmer equation.
-
2.
The potentials and the reactant concentration are approximately constant inside the reactive layer.
-
3.
The electrolyte does not conduct electronic current.
It follows that the crossover current is zero and η int and c RL are constant. The integral in Eq. 8 becomes trivial:
Equation 9 can be easily inverted to get η int as a function of I:
In the Tafel limit αβη int >> 1, the second exponential term in Eq. 9 can be neglected and the expression becomes much simpler:
The equations above express η int as a function of the current density I and of the reactant concentration inside the reactive layer c RL, which, on its hand, depends on the current density. In the next section, in order to derive the performance curve, we make such dependency explicit.
3.4 Reactant Concentration
Reactant concentrations can be obtained by using the following assumptions:
-
1.
The gradient of the reactant concentration is proportional to the molar flux, N.
-
2.
The molar flux is proportional to the current density.
-
3.
The fluxes are 1D in the direction perpendicular to the FC plane.
The first assumption implies that the reactant diffuses through the electrode with a constant diffusion coefficient D. This can be written as
which is a phenomenological diffusion equation. The second assumption implies that all the reactant flux is consumed by the electrochemical reaction and no flux crosses through the electrolyte. In this condition, the molar flux, N, can be expressed through the Faraday relation:
where F is the Faraday constant and n is the number of electrons exchanged in the electrochemical reaction for each reactant molecule.
Substituting this expression of N in Eq. 12 and integrating through the electrode diffusion layer, we easily get
where I lim represents the value of current density at which the reactant concentration in the reactive layer becomes 0, c in is the value of reactant concentration in the gas channel, and L D is the thickness of the diffusion layer.
By inserting this expression of c RL in Eq. 10 or 11, we get explicit expressions for the interface overpotential; in the Tafel limit:
3.5 Analytical Expression for the Performance Curve
By using Eqs. 5, 6, and 15, we finally get
In Fig. 4, the different terms of the above expression are associated to the various characteristic behaviors of the performance curve.

The performance curve of a FC
The term aL RL reflects the effective catalyzer volume and i 0,ref its quality. It is seen that changing the catalyzer, the entire performance curve is shifted vertically by a constant amount.
The quantity R reflects the electrolyte conductivity and is responsible for the slope of the performance curve in its quasi-linear portion.
The value I lim reflects the quality of oxygen or fuel transport. Its effect on the performance curve is a sharp voltage fall at high current densities.
Equation 16 is, in fact, the result of the simplest model representation describing all the main characteristics of the performance curve. Note that this model is general, while most, if not all, of the literature models are specific to particular FC types. A large number of more accurate and complex models exist in the literature, but their use and description lay beyond the scope of this chapter (e.g., Bernardi and Verbrugge 1992; Pisani et al. 2002a, b).
The present model can be easily and effectively used to perform parametric studies, optimizations, and cost analysis. It can also be used as a “semiempirical” function, by fitting the parameters to experimental performance curves and by using their physical meaning to analyze the FC working behavior and identify possible failures.
In the next section, Eq. 16 is analyzed to provide some general information on the fuel cell performances.
3.6 A Simple Analysis of the Performance Curve
The potential in Eq. 16 is a monotone decreasing function of the current density. The fuel cell conversion efficiency, which is proportional to V, is, as well, a decreasing function of I. Therefore, maximal efficiency is obtained for I close to 0. On the other hand, the electric power P = V(I) × I is 0 for both I = 0 and V(I) = 0, while it is positive in between, where, obviously, it reaches a maximum value P = P max. Optimal operating conditions, in general, depend on the particular application and range between I = 0 and I = I(P max), depending on the relative importance of efficiency versus produced power. For current density larger than I(P max), both power and efficiency decrease and operating in that range should be avoided.
The limiting current density depends mainly on the reactant transport and the concentration overpotential is relevant only near I lim. A good design of the cell should guarantee I lim >> I(P max). In such conditions, indeed, concentration overpotential plays a marginal role and can be neglected.
The maximal efficiency, near I = 0, depends only on the reaction kinetics, while the maximal power density depends both on kinetics and on the cell resistance R.
In summary, it can be stated that efficiency and power losses depend mainly on electrolyte and electrocatalyst materials. The research on the optimization and design of materials for these crucial components is very much alive as exemplified in the next Section.
4 FC Materials
The function of the electrolyte is to conduct an ionic current between the two electrodes while constraining the electrons to pass through the external circuit. The main issue is to find a manageable ionic conductor material, with a good ionic conductivity in order to minimize ohmic losses.
Different ionic conduction mechanisms can be used, including:
-
Conduction in water electrolytic solutions (acid or basic)
-
Conduction in unhydrated ionic liquids as phosphoric acid or molten salts
-
Conduction in solids
The materials allowing the above ion transport mechanisms are very different and range from polymeric membranes to ceramic materials. None of the known materials possessed the characteristics of conductivity, durability, and cheapness required to make the fuel cell technology competitive in large-scale applications, and, thus, a large amount of scientific work has been devoted in the development of “new” electrolytic materials optimized for the fuel cell technology.
In this section, as an example of such “material design,” we focus, in general, on the LTFC technology and, in particular, on the polymer electrolyte membranes. Similar efforts and improvements have been obtained in the field of ceramic materials for the SOFC technology.
The modern state of the art of functional materials for application in LTFCs consists in:
-
(a)
Proton-conducting membranes based on pristine perfluorinated ionomers operating at a high hydration degree at ca. 80 °C
-
(b)
Supported platinum electrocatalysts
These FCs must be fueled with very pure reactants to achieve the optimal performance. As of today, there are significant obstacles for the implementation of FC technology in practical applications. In particular (Di Noto et al. 2012d):
-
Operation at 80 °C in humidified conditions adds significantly to the complexity and cost of the FC power plant, which must include bulky and expensive heat and water management modules.
-
Functional materials are very expensive; large amounts of scarce elements (i.e., platinum-group metals, PGMs, such as Pt) are needed, or a very complex synthetic procedure (e.g., in the case of perfluorinated ionomers) must be followed.
-
Durability should be improved, to match the requirements of applications (up to ca. 5,000 h for the automotive sector and 40,000 h for stationary systems).
-
Very pure reactants are expensive, but are a necessity since state-of-the-art electrocatalysts are characterized by a low tolerance to common contaminants (e.g., CO found in H2 fuel obtained by steam-reforming processes).
To address these issues, one of the main approaches is to increase the operating temperature of the FC up to 120 °C and higher, without humidifying the reactant streams. In these conditions, the management of heat and water in the FC power plant becomes much easier, yielding much cheaper and compact systems. Furthermore, at T > 120 °C, the tolerance of the electrocatalysts to the contaminants increases dramatically, and the FC can run directly on cheap hydrogen obtained from steam-reforming processes. Another possibility is to devise FC systems based on anion-exchange membranes, AEMs, capable to carry OH− anions; indeed, in an alkaline environment, it is possible to prepare highly efficient electrocatalysts that do not require PGMs. However, significant efforts are still to be spent in this area as AEM performance is compromised by the exposure to the traces of CO2 found in air; in addition, AEMs are not as durable as state-of-the-art perfluorinated ionomers.
As of today, significant efforts are also spent in the development of oxygen reduction reaction (ORR), electrocatalysts for application in an acid environment (i.e., in PEMFCs, and high-temperature PEMFCs, HT-PEMFCs) which are not based on PGMs. Very promising results have been achieved, even if the durability and current density yielded by the electrodes mounting these PGM-free electrocatalysts are still to be increased to be of interest for practical applications.
4.1 Electrolyte Membranes for FCs
The electrolyte membranes for application in FCs must ensure a facile and selective transport of the ions involved in the operation of the device.
To maximize the FC performance, as a function of the operative temperature and of the ion type involved in the transport (e.g., in PEMFC or AEMFC), several types of widely different materials are used in the fabrication of the electrolyte membranes. However, the vast majority of these materials share some common fundamental features, the most important of which is a clear phase separation between highly polar domains (where the ions actually migrate) and a supporting matrix, providing the electrolyte membrane with its mechanical properties.
Perfluorinated ionomers are the reference materials to manufacture proton-conducting membranes for application in PEMFCs. These materials include a perfluorocarbon backbone and perfluoroethereal side chains tipped with −SO3H groups, giving rise to the polar domains (see Fig. 5).

Some common matrices used in the fabrication of electrolyte membranes for PEMFCs and HT-PEMFCs (Adapted from Di Noto et al. 2012d)
The various ionomers are differentiated by the exact chemical structure and linear density of side chains along the backbone. As of today, most research has been devoted to Nafion™; recently, other ionomers have attracted attention such as the 3 M ionomers (Giffin et al. 2012a). The latter systems are characterized by shorter side chains in comparison with Nafion, including only one ether linkage (see Fig. 5); this has significant repercussions on the domain separation and proton conduction mechanism of the material (Giffin et al. 2012a). In particular, it is observed that the short side chains of 3 M ionomers yield an improved proton conductivity to the final electrolyte membrane in comparison with Nafion.
Water plays a crucial role in the phase separation and proton conduction mechanism of regular perfluorinated ionomers. However, if water is not present (e.g., in a PEMFC operating at T > 90 °C, or if the reactant feeds are not humidified properly), the drop in proton conductivity is dramatic, compromising the operation of the PEMFC. One way to address this issue is to dope a perfluorinated ionomer with a proton-conducting ionic liquid, PCIL (Di Noto et al. 2010). In the resulting systems, the PCIL substitutes water as the ion-conducting medium in the polar domains (see Fig. 6), ensuring remarkable proton conductivity even at high temperatures and in dry conditions.

Typical morphology of a perfluorinated ionomer doped with a PCIL
While perfluorinated ionomers show an excellent proton conductivity in hydrated conditions and a very good chemical and electrochemical stability, they are also very expensive owing to a complex synthetic process involving very hazardous intermediates. One way to address these issues is to develop other ionomers based on much cheaper polyaromatic backbones such as polyethersulfone (PSU), polyphenylenesulfone (PPSU), and polyetheretherketone (PEEK). The proton conductivity is bestowed by functionalizing these polymeric backbones with suitable amounts of –SO3H groups (see Fig. 5). The phase separation properties and proton-conducting mechanisms of these materials are significantly different in comparison with standard perfluorinated ionomers (Di Noto et al. 2012b).
Another possibility is to devise hybrid materials, where a polymeric matrix characterized by a high chemical and electrochemical stability (e.g., PTFE blended with silicone rubber) is used to disperse an inorganic component bearing sulfonated groups, which allow the hybrid to conduct protons (see Fig. 7 ) (Di Noto et al. 2012c).

Structure of a proton-conducting hybrid membrane consisting of PTFE, silicone rubber, and functionalized silica (Adapted from Di Noto et al. 2012c)
The electrolyte membranes for application in HT-PEMFCs show a completely different chemical composition and structure in comparison with those adopted in PEMFCs. In general, the HT-PEMFC membranes consist of (a) a polymer matrix able to tolerate high temperatures and bearing a large concentration of functionalities bestowing to the system a high Lewis basicity (e.g., polybenzimidazole or its derivatives; see Fig. 6) and (b) phosphoric acid, which is used to soak the polymer matrix and acts as the proton-conducting medium.
Acid–base proton exchange reactions are known to take place between the phosphoric acid and the imidazole moieties in the polymer chains. This gives rise to a perturbation in the extended conjugated systems of the resulting protonated diimidazolium cations, allowing rotation of the benzimidazole monomer units along the polymer chain.
Finally, the ionomers used to fabricate the anion-exchange membranes (AEMs) for application in AEMFCs are characterized by backbone chains, which may be comprised of block copolymers, which are functionalized with the anion-exchange groups allowing the system to carry ions. Very little information is available on the details of the ion conduction mechanism of these systems. However, it was demonstrated that in the case of the commercial Selemion AMV™ anion-exchange membrane, the dynamics of the membrane are not significantly involved in the mechanism of long-range conduction, unlike most of the ionomers used in PEMFCs (Giffin et al. 2012b).
One of the most fruitful avenues of research in the field of electrolyte membranes for application in FCs is the development of hybrid materials, obtained by doping a pristine ionomer with one or more additional components (e.g., ceramic oxide nanoparticles, ionic liquids). This approach gives rise to the formation of additional interfaces in the hybrid material, where a variety of new interactions between the host ionomer and the guest component(s) take place. Consequently, it becomes possible to modulate the properties of the host ionomer, so that the final hybrid electrolyte membrane is optimized for the intended application. In general, the introduction of a filler based on oxide nanoparticles improves the mechanical properties of the host ionomer, owing to the formation of dynamic cross-links between the two components (see Fig. 8) (Di Noto et al. 2009).

Effect of the doping with a “core–shell” inorganic nanofiller on the mechanical properties of hybrid inorganic–organic Nafion-based nanocomposite membranes. (a) Trends of storage and loss moduli versus T in {Nafion/[(ZrO2)(Ta2O5)0.119]Ψ} membranes. (b) Samples after DMA measurements
The large body of research carried out in this area yielded, until today, an improved understanding of (a) the complex interplay between the composition, morphology, thermomechanical properties and electric response and (b) the long-range charge transport mechanism of ion-conducting membranes. This information is of fundamental importance to devise new electrolyte membranes for application in energy conversion and storage devices other than FCs, e.g., dye-sensitized solar cells, DSSCs, and redox flow batteries, RFBs.
4.2 ORR Electrocatalysts for Application in FC
The kinetics of the electrochemical processes involved in the operation of PEMFCs must be promoted by suitable electrocatalysts to achieve a performance level compatible with the applications. Today’s state-of-the-art electrocatalysts for PEMFCs are characterized by platinum nanocrystals supported on active carbons featuring a large surface area such as Vulcan XC-72R (see Fig. 9).

Morphology of a “core–shell” carbon nitride nano-electrocatalyst (Adapted from Di Noto et al. 2012a)
The hydrogen oxidation reaction, HOR, is a relatively facile process; on the other hand, the oxygen reduction reaction (ORR) is much more sluggish, giving rise to significantly higher overpotentials. Thus, most of the Pt loading in a PEMFC fueled with H2 is located at the cathode electrode. For these reasons, most of today’s research efforts are focused to devise new ORR electrocatalysts characterized by a higher activity, a lower loading of Pt or other scarce elements, and a better durability in comparison with the state of the art.
In the last few years, a large number of innovative preparation methods are described in the literature (see, e.g., Ref. (Di Noto and Negro 2010)), allowing to introduce in the final electrocatalysts the desired concentration of metal atoms. The latter become embedded in metal alloy nanoparticles, which bear on their surface the active sites of the electrocatalyst. The best performance in the ORR is achieved with bi-/plurimetal active sites, comprising a platinum-group metal (PGM), e.g., Pt or Pd, together with one or more first-row transition metals such as Fe, Co, or Ni (Di Noto and Negro 2010). The PGM must be in its (0) oxidation state, allowing an easy adsorption of the incoming oxygen molecules. The other metals must be in their most stable oxidized state (e.g., III in the case of Fe) and act as cocatalysts; the nitrogen atoms of the coordination nests bind them to the surface of the active sites, preventing their facile removal during FC operation. The cocatalysts are strong Lewis acids; thus, they facilitate the protonation of ORR intermediates on the active sites, prompting the removal of ORR products. Consequently, the active sites are regenerated more quickly, boosting the kinetics of the ORR process according to a bifunctional mechanism. On the other hand, the introduction of a first-row transition metal in the PGM lattice of the metal alloy nanoparticles of the electrocatalysts also gives rise to a contraction of the unit cell; correspondingly, the center of the d-band is lowered, pushing to higher potentials the blocking of ORR active sites by oxygen-based adsorbates. As a result, the minimum ORR overpotential is lowered, improving the kinetics of the process by means of an electronic mechanism.
A major breakthrough in the development of this family of electrocatalysts was achieved with the ideation of “core–shell” materials (see Fig. 9) (Di Noto and Negro 2010).
The morphology of a “core–shell” electrocatalyst allows a better dispersion of the active sites in comparison with a similar pristine material, facilitating the transport of reactants and products in the electrode layer (Di Noto et al. 2012a). Moreover, very small metal alloy nanoparticles can be formed, maximizing the area of the active sites. Consequently, a much-improved FC performance is achieved.
5 Regenerative FC
A regenerative fuel cell (RFC) is a single device or system capable of functioning as either an electrolyzer or a fuel cell (Andrews and Doddathimmaiah 2008). Usually the reversible reaction employed is the decomposition of water into hydrogen and oxygen and their recombination to form water. Among other reversible reactions studied for RFCs have been hydrogen–halogen reactions (in particular bromine) and the zinc–oxygen reaction (Mitlitsky et al. 1998).
In a discrete regenerative fuel cell (DRFC), the device used to perform the electrolysis is separate to that used as a fuel cell, but these two devices are both integrated into a single system. However, it is only in what is called a unitized regenerative fuel cell (URFC) that the RFC concept is fully manifest, by using the same physical cell in both electrolyzer and fuel cell modes (see Fig. 10). When a DC electricity is applied to the cell in electrolyzer (E) mode, water is dissociated into hydrogen and oxygen gas, which are stored. In fuel cell mode, these gases fed back into the cell to regenerate electricity and reform water. More commonly, just the hydrogen is stored and the RFC draws on oxygen from the air when operating as a fuel cell.

A schematic of a unitized regenerative fuel cell system (URFC) supplied by renewable energy sources of electricity
A URFC offers potential cost, space, and mass savings over the conventional hydrogen-based electrical energy storage system and a DRFC, in that only one electrochemical cell is required rather than two. The concomitant technical challenge is getting a round-trip energy efficiency in a URFC close to or equal to that of a separate electrolyzer and fuel cell. Andrews and Doddathimmaiah’s (Andrews and Doddathimmaiah 2008) review found the highest reported round-trip energy efficiency for a URFC was 38.3 % (E mode, 85.3 %; FC mode, 44.9 %). It was suggested that raising this to 45 % should be achievable, some 5 % points lower than that achievable with the best performing discrete electrolyzer and fuel cell in combination.
The design of URFC stacks involves some special challenges. In particular, similar operating conditions must be maintained in both modes in all cells within the stack in terms of gas flows, water flows, pressure, and temperature, so that each cell output is around the same value. The water management system must in E mode ensure sufficient water is available on the O-side electrodes of each cell in the stack, and remove water that gathers on the H-side electrodes due to osmotic drag of water molecules by H+ ion across the membrane. On switching to FC mode, this system must remove any excess water in O-side GDBs and flow channels, remove water vapor produced by the overall electrochemical reaction, and ensure the membrane remains hydrated.
Functioning URFC stacks with up to seven cells in series have recently been reported in the literature (e.g., Ref. (Su et al. 2009; Grigoriev et al. 2011)).
5.1 Materials
Most URFCs to date have employed a proton exchange membrane (PEM) as a solid electrolyte, since they work at relatively low temperatures up to 80 ºC. However, high-temperature (600–1,000 °C) solid oxide cells have also been used.
In the most common type of URFC, the electrode from which hydrogen is evolved in E mode and consumed in FC mode stays the same (see Fig. 11). In E mode, the catalyst layer in the oxygen electrode must facilitate the water-molecule splitting reaction, and in FC mode the reverse water-formation reaction. So this layer must be bifunctional and usually contains a mix of catalysts: Pt for use in FC mode with the addition of other elements and/or their oxides to enhance the water splitting reaction. The catalyst layer on the hydrogen side typically contains just Pt, which functions equally well in the hydrogen molecule formation and dissociation reactions.

The electrolyzer (E) mode (top) and fuel cell (FC) mode (bottom) reactions in a URFC in which the oxygen and hydrogen electrodes remain the same in the two modes
The main catalysts reported as being used on URFCs on the oxygen side over the past few years are still Pt plus Ir, IrO2, RuO2, IrO2/RuO2, Pt/TiO2, and Ir/TiO2 (see, e.g., Ref. (Jung et al. 2009; Cruz et al. 2012)). A lot of the emphasis in research on these has been on fabricating porous structures of these mixed catalysts to maximize the effective reaction areas.
The support medium for the catalysts on the oxygen side is also ideally porous and must be resistant to the water and oxidizing environment. Materials employed include the usual Nafion and carbon cloth and paper, graphitized carbon (see Ref. (Pai and Tseng 2012)), titania (Huang et al. 2012), Sb-doped SnO2 (ATO), plasma-assisted deposition of nanoparticles of catalyst (Pt and Pd) on carbon powder, nanotubes and nanofibers (Fedotov et al. 2013), titanium carbide, TiC, and TiCN (Garcia et al. 2013; Sui et al. 2011).
Materials being investigated for the GDL on the oxygen side to prevent degradation and corrosion include a conventional carbon substrate with a protective microporous layer of iridium–titanium nitride, titanium felt (Ito et al. 2012), and metallic ceramics such as titanium carbide (TiC) (Chen et al. 2010).
5.2 Challenges
A key challenge in future URFC design is to obtain a round-trip energy efficiency very close that of a system with a separate electrolyzer and fuel cell, with an oxygen-side catalyst layer that performs equally well in both E and FC modes remaining a focus in research. The development of long-lived cells resistant to corrosion or other degradation, and retention of structural integrity and strength after repeated cycling and mode switching, is another important goal. There is still a lack of practical designs and operating experience for URFC stacks with more than six or seven cells. URFC stacks must be able to operate in E mode at pressures up to 10 or 20 bar, so that hydrogen can be stored as compressed gas or metal hydrides in vessels of practical size in stand-alone power supplies, without the need for an external compressor.
If these technical challenges can be overcome, the potential cost, space, and mass savings that a URFC offers compared to conventional hydrogen-based electrical energy storage system open up numerous applications in space vehicles and satellites, submarines, aircraft, remote or distributed terrestrial energy supply systems, electricity storage in central grids or at a local level when there is variable energy input from solar or wind power, electricity storage at a local level, and possibly in hydrogen electric cars capable of generating some of their own hydrogen fuel when parked.
References
Andrews, J., Doddathimmaiah, A.: Regenerative fuel cells. In: Fuel Cell Materials, pp. 344–385. Woodhead Publishing, Cambridge, UK (2008)
Bernardi, D.M., Verbrugge, M.: A mathematical model of the solid-polymer-electrolyte fuel cell. J. Electrochem. Soc. 139, 2477 (1992)
Chen, G., Zhang, H., Zhong, H., Na, H.: Gas diffusion layer with titanium carbide for a unitized regenerative fuel cell. Electrochem. Acta 55, 8801 (2010)
Cruz, J., Baglio, V., Siracusano, S., Ornelas, R., Arriaga, L., Antonucci, V., Arico, A.: Nanosized Pt/IrO2 electrocatalyst prepared by modified polyol method for application as dual function oxygen electrode in unitized regenerative fuel cells. Int. J. Hydrog. Energy 37, 5508 (2012)
Di Noto, V., Negro, E.: Development of nano-electrocatalysts based on carbon nitride supports for the ORR processes in PEM fuel cells. Electrochem. Acta 55, 7564 (2010)
Di Noto, V., Lavina, S., Negro, E., Vittadello, M., Conti, F., Piga, M.: Hybrid inorganic-organic proton conducting membranes based on nafion and 5 wt% of MxOy(M = Ti, Zr, Hf, Ta and W). Part ii: relaxation phenomena and conductivity mechanism. J. Power Sources 187, 57 (2009)
Di Noto, V., Negro, E., Sanchez, J.Y., Iojoiu, C.: Structure-relaxation interplay of a new nanostructured membrane based on tetraethylammonium trifluoromethanesulfonate ionic liquid and neutralized nafion 117 for high-temperature fuel cells. J. Am. Chem. Soc. 132, 2183 (2010)
Di Noto, V., Negro, E., Polizzi, S., Agresti, F., Giffin, G.A.: Synthesis-structure-morphology interplay of bimetallic “core-shell” carbon nitride nano-electrocatalysts. Chem. Sus. Chem. 5, 2451 (2012a)
Di Noto, V., Piga, M., Giffin, G.A., Pace, G.: Broadband electric spectroscopy of proton conducting SPEEK membranes. J. Membr. Sci. 58, 390–391 (2012b)
Di Noto, V., Piga, M., Giffin, G., Negro, E., Furlan, C., Vezzù, K.: New nanocomposite hybrid inorganic–organic proton-conducting membranes based on functionalized silica and PTFE. Chem. Sus. Chem. 5, 1758–1766 (2012c)
Di Noto, V., Zawodzinski, T.A., Herring, A.M., Giffin, G.A., Negro, E., Lavina, S.: Polymer electrolytes for a hydrogen economy. Int. J. Hydrog. Energy 37, 6120 (2012d)
Fedotov, A., Grigoriev, S., Millet, P., Fateev, V.N.: Plasma-assisted Pt and Pt–Pd nano-particles deposition on carbon carriers for application in PEM electrochemical cells. Int. J. Hydrog. Energy 38, 8568 (2013)
Garcia, G., Roca-Ayats, M., Lillo, A., Galante, J.L., Pena, M.A., Martinez-Huerta, M.V.: Catalyst support effects at the oxygen electrode of unitized regenerative fuel cells. Catal. Today 210, 67 (2013)
Giffin, G.A., Haugen, G.M., Hamrock, S.J., Di Noto, V.: Interplay between structure and relaxations in perfluorosulfonic acid proton conducting membranes. J. Am. Chem. Soc. 135, 822 (2012a)
Giffin, G.A., Lavina, S., Pace, G., Di Noto, V.: Interplay between the structure and relaxations in selemion amv hydroxide conducting membranes for AEMFC applications. J. Phys. Chem. C 116, 23965 (2012b)
Grigoriev, S., Millet, P., Porembsky, V., Fateev, V.N.: Development and preliminary testing of a unitized regenerative fuel cell based on PEM technology. Int. J. Hydrog. Energy 36, 4164 (2011)
Huang, S.-Y., Ganesan, P., Jung, H.-Y., Popov, B.N.: Development of supported bifunctional oxygen electrocatalysts and corrosion-resistant gas diffusion layer for unitized regenerative fuel cell applications. J. Power Sources 198, 23 (2012)
Ito, H., Maeda, T., Nakano, A., Hwang, C.M., Ishida, M., Kato, A., Yoshida, T.: Int. J. Hydrog. Energy 37, 7418 (2012)
Jung, H.-Y., Park, S., Popov, B.N.: Electrochemical studies of an unsupported PtIr electrocatalyst as a bifunctional oxygen electrode in a unitized regenerative fuel cell. J. Power Sources 191, 357 (2009)
Mitlitsky, F., Myers, B., and Weisberg, A.H.: Regenerative fuel systems R & D. Proceedings of the U.S. DOE Hydrogen Program Review, Arlington (1998)
Pai, Y.-H., Tseng, C.-W.: Preparation and characterization of bifunctional graphitized carbon-supported Pt composite electrode for unitized regenerative fuel cell. J. Power Sources 202, 28 (2012)
Pisani, L., Murgia, G., Valentini, M., D’Aguanno, B.: A new semi-empirical approach to performance curves of polymer electrolyte fuel cells. J. Power Sources 108, 192 (2002a)
Pisani, L., Murgia, G., Valentini, M., D’Aguanno, B.: A working model of polymer electrolyte fuel cells: comparison between theory and experiments. J. Electrochem. Soc. 149, A898 (2002b)
Signorelli, R. High energy and power density nanotube-enhanced ultracapacitor design, modeling, testing, and predicted performance. PhD thesis, MIT (2009)
Su, H.N., Liao, S.J., Xu, L.M.: Design, fabrication and preliminary study of a mini power source with a planar six-cell PEM unitised regenerative fuel cell stack. Fuel Cells 9, 522 (2009)
Sui, S., Ma, L., Zhai, Y.: Tic supported Pt-Ir electrocatalyst prepared by a plasma process for the oxygen electrode in unitized regenerative fuel cells. J. Power Sources 196, 5416 (2011)
Tozzini, V., Pellegrini, V.: Prospects for hydrogen storage in graphene. Phys. Chem. Chem. Phys. 15, 80–89 (2013)
Winter, M., Brodd, R.: What are batteries, fuel cells, and supercapacitors? Chem. Rev. 104, 4245–4269 (2004)
Acknowledgements
The financial support of the Sardinia Region Authority is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Pisani, L., D’Aguanno, B., Di Noto, V., Andrews, J. (2014). Fuel Cell Technology and Materials. In: Cao, G., Orrù, R. (eds) Current Environmental Issues and Challenges. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-8777-2_4
Download citation
DOI: https://doi.org/10.1007/978-94-017-8777-2_4
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-8776-5
Online ISBN: 978-94-017-8777-2
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)