
Abstract
Systemic inflammation associated with diverse clinical conditions is a vital problem in critical care medicine with significant morbidity and mortality. In this chapter, we describe exclusively on systemic inflammation caused by sepsis, ischemia and reperfusion injury, and trauma hemorrhagic shock. Despite all efforts in the clinical arena treatment for these indications remain limited. The only FDA approved drug as a treatment for sepsis, Xigris (drotrecogin alfa [activated]) has recently been voluntarily withdrawn by Eli Lily. There is an unmet and urgent clinical need exists for novel therapies for these conditions. There are pathological similarities as well as differences exist among these conditions. Even though all three pathologies are initiated by different means, all leads to exaggerated inflammatory response and multi-organ failure. Therefore, therapies developed to dampen the exaggerated systemic inflammation could be beneficial for all three pathologies. Milk fat globule-epidermal growth factor-factor 8 (MFG-E8) is first identified as a bridging molecule that accelerated the interaction between apoptotic cells and phagocytes and facilitates the engulfment of apoptotic cells. We then, demonstrated that MFG-E8 plays a significant role in sepsis, ischemia and reperfusion injury, and trauma hemorrhagic shock. In this chapter, we will briefly review the different systemic inflammatory conditions and describe the key evidence for the role of MFG-E8 and highlight the notion that MFG-E8 could be developed as a potential therapeutic for these indications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Systemic inflammation caused by sepsis, ischemia/reperfusion injury and trauma/hemorrhage is a critical problem causing significant morbidity and mortality [1]. Sepsis and septic shock is the second leading cause of death in the non-coronary intensive care units and is in the top 10 leading causes of deaths overall in the United States [2]. The mortality rate for severe sepsis and septic shock is about 30 % annually, with over 200,000 deaths per year [3] which is similar to the number of people dying with acute myocardial infarction. Similar to sepsis, ischemia and reperfusion (I/R) injury also results in high morbidity and mortality. Gut or mesenteric ischemia remains a critical clinical condition, resulting in mortality as high as 60 % [4]. Hepatic ischemia-reperfusion (I/R) damage occurs in diverse clinical settings including liver transplantation and liver resection [5, 6]. Acute renal failure (ARF) caused by renal I/R injury is quite common in hospitalized patients, affecting 3–7 % of general admissions, and as much as 25–30 % of patients in intensive care units [7, 8]. Trauma is the fifth leading cause of death overall and the number one cause of death for patients between the ages of 1 and 40 [9, 10]. As such, development of therapies for such systemic inflammatory conditions is an unmet need exists for efficient patient care.
There are pathological similarities as well as differences exist among these conditions. Sepsis is defined as a systemic host response to an infectious origin which eventually leads to systemic inflammatory response and multi-organ failure (MOF) [11, 12]. Ischemia and reperfusion (I/R) injury is a pathological condition characterized by an initial restriction of blood supply to a specific organ followed by subsequent restoration of perfusion and reoxygenation. I/R injury is manifested as an initial tissue hypoxia due to occlusion of the arterial blood supply and a subsequent tissue injury and exacerbation of inflammatory response as a consequence of the reperfusion. The major organs that are affected by I/R injuries due to varied clinical conditions are the gut, the liver and the kidneys. I/R injury typically occurs in a sterile environment and eventually leads to exaggerated inflammation and MOF [13]. While exsanguinations and head injury continue to account for a large number of early trauma deaths, the majority of late trauma deaths occur as a result of infection and/or MOF [9, 10]. Even though all three pathologies are initiated by different means, all leads to exaggerated inflammatory response and MOF. Therefore, therapies developed to dampen the exaggerated systemic inflammation could be beneficial for all three pathologies.
Milk fat globule-epidermal growth factor-factor 8 (MFG-E8) was first identified by Hanayama et al. [14] as a bridging molecule that accelerated the interaction between apoptotic cells and phagocytes and facilitates the engulfment of apoptotic cells. We then, demonstrated that MFG-E8 is decreased in sepsis and the reduction in its expression leads to impairment of apoptotic cell clearance resulting in increased mortality. Administration of exogenous MFG-E8 in septic animals increased apoptotic cell clearance, reduction in inflammatory response, and improved survival [15–17]. We also showed beneficial effect of MFG-E8 in a number of other organ injury conditions [18–21]. In this chapter, we will briefly review the different systemic inflammatory conditions and describe the key evidence for the role of MFG-E8 in these indications. With the available data, we highlight the notion that MFG-E8 could be developed as a potential therapeutic agent for sepsis, ischemia and reperfusion injury, and trauma hemorrhagic shock.
2 MFG-E8 and Sepsis
Sepsis is a critical problem causing significant morbidity and mortality [1]. It continues to be the second leading cause of death in non-coronary intensive care units, and is in the top 10 leading causes of deaths overall in the United States [2]. It is estimated that there are more than 1,000,000 cases of sepsis among hospitalized patients each year in the US. The incidence of sepsis among hospitalized patients is increasing by 8.7 % per year. Numerous reports have shown that the incidence of sepsis and severe sepsis is increasing in excess of the growth of the population [12]. The mortality rate for severe sepsis and septic shock is about 30 % annually, with over 200,000 deaths per year [3] which is similar to the number of people dying with acute myocardial infarction. Sepsis is defined as a systemic host response to an infection caused by bacteria, virus or fungi [11]. Sepsis tends to occur from specific and consistent sources such as respiratory infections, genitourinary and abdominal sources of infection with primary bacteremia, and other unknown sources. The occurrence of severe sepsis is related to the source of infection, as in the cases of patients with respiratory infection who are at high risk for developing respiratory related organ dysfunction. Regardless of the time and the organisms, the treatment of infection is the primary antisepsis therapy. From a clinical perspective, antimicrobial therapy is the chosen method of treatment. However, the choice of antibiotics, and the timing of their administration are extremely critical for successful outcome. Thus, there has been a substantial amount of work ranging from analyzing triage decisions made for intensive care unit admissions [22] to evaluating cortisol as a potential treatment against sepsis [23]. Despite these efforts, the treatment of sepsis however, has remained elusive. The only FDA approved drug as a treatment for sepsis, Xigris (drotrecogin alfa [activated]) [24, 25] has recently been voluntarily withdrawn by Eli Lily. There is an unmet and urgent clinical need exists for a sepsis therapy.
During inflammation and sepsis, systemic increases in pro-inflammatory cytokines have been shown to increase mortality [26]. During sepsis and other states with systemic inflammatory response, several cell types (e.g., B cells, CD4 T cells, dendritic cells (DCs), vascular endothelial cells and enteric epithelial cells) undergo apoptosis [27–31]. Apoptotic cells that are not cleared are likely to undergo secondary necrosis [32], thereby continuing to release harmful and toxic mediators and worsening sepsis. Studies have shown that phagocytic function of macrophages is impaired in late sepsis [33, 34]. Hanayama et al. [35] have discovered that lack of clearance of apoptotic B cells in the spleen potentially leads to autoimmune diseases which underscores the importance of clearing apoptotic cells from organism [36]. MFG-E8, a 64-kDa secretory protein that is mainly produced by the spleen, was responsible for removal of apoptotic cells. Without MFG-E8, engulfment and removal of apoptotic cells were impaired which led to the release of autoantibodies [35]. MFG-E8 was originally identified as a component of milk-fat globules [37] but secreted by activated macrophages and immature dendritic cells [38]. The most remarkable function of MFG-E8 is its ability to promote the clearance of apoptotic cells by forming a tether between phagocytes and apoptotic cells [14, 39]. One unique characteristic of apoptotic cells is to expose their phosphatidylserine (PS) from its inner leaflet membrane to the outer surface. This is termed “eat me” signal which can allure distinct opsonins (i.e., MFG-E8), to recognize and bring apoptotic cells to the close vicinity of phagocytes [40]. MFG-E8 has a strong binding affinity to the exposed PS of apoptotic cells and facilitates phagocytic engulfment via αVβ3 or αVβ5 integrins. This triggers a conformational change in the integrin receptor that signals the recruitment of various signaling cascade proteins and transforms the macrophage into a phagocyte capable of engulfment [41, 42]. Thus, MFG-E8 promotes the engulfment of apoptotic cells by working as a bridging molecule between those cells and phagocytes [14].
In an animal model of cecal ligation and puncture (CLP)-induced sepsis, we showed that MFG-E8 levels were decreased by 45 % in the blood during late sepsis (i.e., 20 h after CLP) indicating the systemic scale of its depletion under septic conditions [16]. A 48–70 % reduction was observed in the spleen and liver tissues [15]. This decrease in the MFG-E8 expression in late sepsis was associated with impaired phagocytosis of apoptotic cells or apoptotic cell clearance [15]. Splenic macrophages from MFG-E8 deficient (Mfge8-/-) mice showed a dramatically decreased ability to phagocytose apoptotic cells under normal conditions as compared to wild type mice, suggesting a critical role for MFG-E8 in this process. Interestingly, Mfge8-/- mice accumulated higher amounts of apoptotic cells as compared to the WT mice during late sepsis. These data clearly demonstrated that the clearance of apoptotic cells is directly regulated by MFG-E8 [16]. Endotoxemia also reduced splenic MFG-E8 expression in a dose dependent manner and the downregulation of MFG-E8 expression in CLP-induced sepsis was attenuated by the LPS inhibitor, polymyxin B. The CLP-induced suppression was not observed in either CD14−/− or TLR4-mutated mice. These studies indicated that MFG-E8 production is down-regulated in sepsis by LPS-CD14 dependent fashion, leading to a reduction of phagocytosis of apoptotic cells [43].
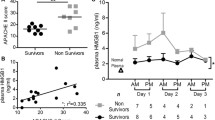
MFG-E8 is secreted from DCs in exosomes that resemble milk fat globules in size and membrane lipid composition [38, 44]. These tiny vesicles (50–100 nm in diameter) are derived from multivesicular bodies, intermediates in the endosome maturation between endosome and endolysosome [44, 45]. Fusion of these multivesicular bodies with the plasma membrane leads to the release of MFG-E8 containing exosomes. In this regard, we isolated MFG-E8 containing exosomes from rat bone marrow immature DCs. Treatment of rats with MFG-E8 containing exosomes at the time of CLP, reduced the presence of apoptotic cells by 33 %. Peritoneal macrophages from exosome-treated rats displayed a 2.8-fold increased ability to phagocytose apoptotic thymocytes [16]. Thus, the reduced presence of apoptotic cells in exosome-treated septic rats could have been due to the increase in apoptotic clearance. Treatment also reduced plasma tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) levels and improved survival from 44 % in the saline treated animals to 81 % in the treatment ones [15]. Similarly, treatment of septic rats with recombinant murine MFG-E8 (rmMFG-E8) attenuated the inflammatory response during sepsis, increased apoptotic cell clearance, and improved survival [16]. To develop MFG-E8 as a therapeutic agent against sepsis, recombinant human MFG-E8 (rhMFG-E8) was expressed in bacterial system, purified and confirmed of having biological activity similar to the mouse counterpart in abilities of mediating the phagocytosis of apoptotic cells by macrophages [46]. Treatment with the purified rhMFG-E8 in septic rats significantly reduced, organ injury indicators (AST, ALT, creatinine, lactate), serum IL-6 and TNF-α, and plasma HMGB-1 levels [17]. In a 10-day survival study in septic rats, vehicle-treated rats produced 36 % survival rate, while rhMFG-E8 treatment significantly improved survival rate to 68–72 %. Treatment with rhMFG-E8 significantly reduced the number of apoptotic cells detected suggesting increased apoptotic cell clearance. In addition to its role in apoptotic cell clearance, a recent study showed that MFG-E8-mediated potential therapeutic benefits in sepsis and intestinal injury were not solely dependent on the enhanced clearance of apoptotic cells, but also due to diverse cellular events to maintain epithelial integrity and healing of the injured mucosa [47]. In this regard, we have shown that the pre-treatment with rmMFG-E8 followed by endotoxemia showed significant attenuation of TNF-α levels in circulation and in splenic tissues suggesting an anti-inflammatory role of MFG-E8. In contrast, endotoxemia in the Mfge8-/- mice caused greater increase in TNF-α than those in WT mice [48]. Aziz et al. [48] further demonstrated that MFG-E8-mediated decrease in TNF-α is regulated by pSTAT3/SOCS3 leading to downregulation of NF-kB and subsequent decrease in TNF-α. Nevertheless, these findings taken together clearly provided evidence to develop rhMFG-E8 as a therapy for patients suffering from sepsis (Fig. 7.1).

Potential mechanism of MFG-E8 in sepsis and I/R injury. Sepsis and I/R injury is caused by a number of factors including neutrophil infiltration, complement activation, ROS, mitochondrial dysfunction and apoptosis leading to increase in proinflammatory mediators and organ injury. During sepsis, downregulation of the endogenous MFG-E8 in the tissues, i.e., spleen, liver and kidneys, attenuates apoptotic cell clearance (phagocytosis) and exacerbates organ injury. Administration of recombinant MFG-E8 (rMFG-E8) enhances the phagocytic activity and attenuates inflammation and decreases organ injury. In macrophages, rMFG-E8 upregulates pSTAT3/SOC3 signaling pathway and attenuates proinflammatory cytokines and decrease organ injury (Schematic illustration of data compiled from references [15, 16, 18–20, 48])
3 MFG-E8 and Ischemia/Reperfusion Injury
3.1 MFG-E8 and Gut I/R
Gut or mesenteric ischemia remains a critical clinical condition, resulting in mortality as high as 60 % [4]. Intestinal ischemia and subsequent reperfusion are encountered in a variety of clinical conditions, including acute mesenteric ischemia, intestinal obstruction, incarcerated hernia, small intestine volvulus and necrotizing colitis. The consequences of mestenteric ischemia are devastating to the patient and usually results in severe diarrhea, malabsorption, short bowel syndrome, and death. The pathophysiology of gut ischemia/reperfusion (I/R) involves tissue ischemia followed by cellular damage due to resumption of blood (reperfusion). Tissue ischemia initiates a series of events that can ultimately lead to cellular dysfunction and necrosis, and subsequent reperfusion causes more tissue damage including remote organ injury and subsequent death [13, 49–58]. A common complication of gut I/R is acute lung injury (ALI) and it contributes to the high mortality rate observed in gut I/R injuries. ALI is caused by a systemic inflammatory response due to the release of proinflammatory cytokines and bacteria-derived endotoxins from reperfused ischemic tissue [59, 60]. The mechanism of ALI involves a complex cross-talk among various cellular components of the alveolar microenvironment, their secretory products, and leukocyte recruitment from the vascular bed in regions of inflammation. Activated neutrophils release proteolytic enzymes, such as elastase and myeloperoxidase (MPO) and reactive oxygen species, including hydrogen peroxide and superoxide. Excessive production of these factors not only destroys invaded pathogens, but also engages in the disruption of the endothelial barrier and promotes tissue damage. These events lead to ALI that is clinically manifested as acute respiratory distress syndrome followed by multiple organ dysfunction syndrome [61–64]. Even though numerous treatment modalities have been implicated in reducing ALI-induced mortality, none have been successful [65]. Thus, the development of novel and effective therapies for ALI is crucial for the improvement of patient outcome.
The gut is one of the most sensitive organs to I/R injury [55, 66]. Ischemia initiates a series of events that can ultimately lead to cell dysfunction and necrosis, and resumption of blood (reperfusion) causes more tissue damage [49–58]. The lungs are among the organs that are most severely affected by gut I/R-induced injury [67]. Ischemia or I/R induces apoptosis in various organs [68–70]. Apoptosis has been considered as the principal mode of cell death during I/R [66, 71–73]. Apoptotic cells stimulate inflammatory responses if they are not removed by phagocytes [74]. Deficient clearance of apoptotic cells leads to inflammation and tissue injury [39, 75]. MFG-E8 plays a crucial role for the engulfment of apoptotic cells by phagocytes [14, 35]. In this regard, we have shown that in a mouse model of gut I/R induced by superior mesenteric artery occlusion followed by reperfusion, as compared to the WT mice, Mfge8-/- mice produced much severe ALI after gut I/R [18]. MFG-E8 levels were markedly reduced in the spleen, gut and lungs by 50–70 %, suggesting impaired apoptotic cell clearance [76]. Treatment with rmMFG-E8 in gut I/R-induced WT mice significantly decreased lung apoptosis, improved lung morphology, and reduced neutrophil infiltration into the lungs. Treatment also suppressed tissue injury and inflammation as evidenced by reduction in liver enzymes (AST, ALT), lactate and creatinine, decreased proinflammatory cytokines (TNF-α, IL-6, IL-1β), and improved survival. Thus, MFG-E8 may serve as a novel treatment option for gut I/R-induced ALI.
3.2 MFG-E8 and Hepatic I/R
Hepatic ischemia-reperfusion (I/R) damage, which occurs in diverse clinical settings including liver transplantation, trauma, hemorrhagic shock, or liver surgery, is a serious clinical complication that may compromise liver function because of extensive hepatocellular loss. I/R injury represents a complex series of events that result in cellular and tissue damage. It involves the transient deprivation of blood flow and oxygen, and the return of blood flow during reperfusion with concomitant release of reactive oxygen species (ROS), inflammatory mediators, adhesion molecules, adenosine triphosphate (ATP) depletion, and derangements in calcium homeostasis. Finally, these functional changes induce cell death due to apoptosis as well as necrosis [5, 6]. Despite the fact that hepatic injury is a major clinical problem, no reliable therapies have been established. The development of a therapy for hepatic I/R would indeed benefit patients undergoing liver surgery and liver transplantation.
It has been shown that programmed cell death or apoptosis of liver sinusoidal cells and hepatocytes is a prominent feature of liver I/R injury, in both experimental models and clinical transplantation [5, 77, 78]. Historically, apoptosis has been seen as an ordinary process of cell suicide that, unlike necrosis, does not elicit inflammation [32]. Studies have shown that if the removal process of apoptotic cells fails, apoptotic cells undergo secondary necrosis, which enables to release potentially cytotoxic intracellular contents, followed by inflammation and impaired tissue repair [79, 80]. In a rat model of hepatic I/R, liver and plasma levels of MFG-E8 were significantly decreased. Administration of rhMFG-E8 significantly improved liver injury, suppressed apoptosis, attenuated inflammation and oxidative stress, and downregulated the NF-κB signaling pathway. In a survival study conducted using Mfge8-/- mice and WT mice, the survival rate of the Mfge8-/- mice was markedly reduced as compared to that of the WT mice indicating that the Mfge8-/- mice were more susceptible to hepatic I/R-mediated mortality than the WT mice. In contrast, exogenous administration of rhMFG-E8 in WT mice improved the survival rate after hepatic I/R from 31 % in the saline treated animals to 70 % in the treatment ones [19]. Furthermore, it has been demonstrated that MFG-E8-mediated therapeutic potential is not only dependent on enhancement of phagocytosis, but also on multiple cellular events associated with tissue remodeling [47, 81, 82]. MFG-E8-mediated multiple physiological events may represent an effective therapeutic option in tissue injury following an episode of hepatic I/R.
3.3 MFG-E8 and Renal I/R
Acute renal failure (ARF) is a critical clinical problem posing significant economic and financial burden on the society. ARF is quite common in hospitalized patients, affecting 3–7 % of general admissions, and as much as 25–30 % of patients in intensive care units. Renal ischemia-reperfusion (I/R) injury causes ARF in various clinical settings, including kidney transplantation and cardiopulmonary and aortic bypass surgery. Renal I/R injury is associated with high mortality and morbidity [7, 8]. Current strategies used to prevent ARF consist mainly of fluid resuscitation and diuretics, and/or prevention of the insinuating factor. Despite these efforts, the mortality remains unacceptably high and has not improved in several decades. There is an urgent need to develop therapeutics to fight this pathological condition.
During renal I/R injury, renal damage begins immediately from the onset of ischemia. Upon restoration of perfusion, however, the tissues undergo further injury. Reperfusion injury involves the accumulation of neutrophils, generation of free oxygen radicals, and cytokine activation. These changes may also be seen histopathologically, as demonstrated by the loss of the brush border, tubular disruption, and cast formation [83]. Renal damage due to I/R injury occurs as early as 5 h following injury as evidenced by a rising serum lactate, TNF-α, IL-6 and TGF-β levels, as well as decreasing systemic venous oxygen levels [84]. In the clinical setting, serum markers such as blood urea nitrogen and creatinine, are regarded as gold standards for renal compromise, but these markers may not become elevated until 24 h after the initial injury. Studies looking at early biomarkers, such as keratinocyte-derived chemokine (KC) and neutrophil gelatinase-associated lipocalin (NGAL), demonstrate that increases in these markers are associated with the development of ARF [85, 86]. With a better understanding of the pathophysiology of ARF as well as the identification of new biomarkers, one is able to determine the actual time point in the evolution of renal compromise pharmacological or hormonal therapy would be beneficial.
Ischemia typically damages renal tubular epithelial cells and also glomerular cells and is characterized by several hallmark features at the cellular level: Profound intracellular ATP depletion and a fall in tissue oxygen and glucose content with a concomitant rise in intracellular calcium [87, 88]. Although ischemic events alone may lead to necrosis and apoptosis in the kidneys, reperfusion occurs upon restoration of blood flow and is associated with increased apoptosis and necrosis in addition to the production of reactive oxygen species (ROS) and inflammatory mediators [89, 90]. Renal I/R injury can be ameliorated by inhibiting molecules involved in apoptosis, necrosis, or inflammation, suggesting that multiple injury and death mechanism may be involved in renal I/R injury [91]. Among these, the coexistence of apoptosis and necrosis in renal tissues is a characteristic feature of renal I/R injury. Both types of cell death have been implicated significantly in the pathogenesis of ARF, which is marked by a loss of tubular epithelial cells and subsequent renal dysfunction [92, 93]. Moreover, additional mechanisms which contribute to the ongoing pathogenesis of I/R injury-induced ARF has been reported. For instance, renal vascular endothelial injury and dysfunction, due to increases in renal vascular resistance and persistent reductions in renal blood flow, exacerbates hypoxia and play an important part in extending renal tubular epithelial injury and subsequent cell death [94]. In a rat model of bilateral renal ischemia followed by reperfusion (renal I/R) [20], MFG-E8 mRNA and protein expressions were significantly decreased in the kidneys and spleen. Treatment with rmMFG-E8 recovered renal dysfunction, significantly suppressed inflammatory responses, reduced apoptosis and necrosis, and improved capillary functions in the kidneys. In a 60 h survival study, survival rate after renal I/R injury decreased significantly from 44 % in the WT mice to 11 % in the Mfge8-/- mice. Interestingly, the exogenous treatment with rmMFG-E8 in the WT mice showed significant improvement in survival rate to 73 %. These data collectively demonstrated that the protective effect of MFG-E8 is mediated through the enhancement of apoptotic cell clearance and improvement of capillary functions in the kidneys. Thus, MFG-E8 could be developed as a novel treatment for renal I/R injury.
3.4 MFG-E8 and Hemorrhagic Shock
Trauma is the fifth leading cause of death overall and the number one cause of death for patients between the ages of 1 and 40 [9, 10]. In the US, about 90,000 people die annually due to traumatic injuries and complications. It is estimated that 10–20 % of the deaths are potentially preventable and nearly 80 % of these occur due to hemorrhage and it occurs within the first 24 h after injury [9, 95–97]. Immediate hemorrhage control and adequate fluid resuscitation are the key components of early trauma care. While fluid resuscitation decreases the risk of death in severe hemorrhage, it increases the risk of death in less severe hemorrhage. Despite the fact that fluid infusion at a pre-determined rate has shown to reduce organ injury and reduce mortality, the best approach recommended is to avoid unnecessary field interventions and focus on fast and efficient transport of the patient to hospital [98]. Advances in trauma care systems and emergency medical services have resulted in a significantly large percentage of patients who survive to hospital admission [96]. Another strategy implemented is the hypotensive resuscitation that showed some reduction in the risk of death. Hypotensive resuscitation at a fixed rate of 60–80 cc/kg/h generally maintains the systolic blood pressure of 80–90 mmHg and mean arterial pressure of 40–60 mmHg. Although the data suggest this strategy of infusion rates is beneficial in hemorrhagic shock, it requires monitoring of hemodynamic changes which would be difficult to accomplish in the field. Another strategy for resuscitation is the use of hypertonic saline. An number of pre-clinical studies have demonstrated that hypertonic saline modulate the immune response and leads to attenuation of immune mediated cellular injury [99–108]. However, in two recent multicenter clinical trials, hypertonic saline treated patients experienced early high mortality in comparison to normal saline treatment and thus, hypertonic saline is not recommended for resuscitation in trauma patients [109].
Observational data from trauma centers and the battlefield suggest that early administration of component therapy containing fresh frozen plasma and platelets may be beneficial [110, 111]. Based on battlefield experience, US Army instituted a policy of using a 1:1:1 ratio of packed red blood cells:fresh frozen plasma:platelets in the battlefield for those who meet the criteria for massive resuscitation. However, no study has identified the optimal ratios of blood components to be used for resuscitation [109]. In addition, although advance in viral screening have markedly decreased the risk of infectious transmissions, blood transfusion remains to be associated with numerous side effects. Blood transfusion has shown to cause early immune activation resulting in systemic inflammatory response syndrome and immune suppression which predisposes the patients to infection [112–116]. In addition to fluid resuscitation, a wide range of pharmacological agents including neuroendocrine agents, calcium channel blockers, prostaglandins, sex steroids, immune modulators, and histone deacetylase inhibitors have been tested in pre-clinical trials. Although majority of these agents show beneficial effects in animal models, none have been in clinical use as resuscitative agents. Thus, there is an urgent unmet need in the development of novel therapies for hemorrhagic shock.
Exsanguination and head injury continue to account for a large number of early trauma deaths, the majority of late trauma deaths occur as a result of infection and/or multi-organ failure. The clinical association of late trauma deaths and the development of multi-organ failure have been established as early as in the 1970s. However, only within the past few decades that the focus has been directed towards inflammatory response and how it may predisposes the body to infection and multi-organ failure. Hemorrhagic shock induces a surge of inflammatory cytokines including IL-6 and TNF-α which is associated with increased mortality [26, 117]. Prolonged and severe hemorrhagic state leads to tissue hypoxia and the presence of apoptotic cells [32]. If these apoptotic cells are not cleared, they will likely undergo secondary necrosis and release harmful agents and worsens hemorrhagic shock.
Apoptotic cell death is prevalent in gastrointestinal associated intestinal epithelial cells [118–120]. These cells are already prone to apoptosis after noxious stimuli exposure because these cell types normally undergo a rapid physiological turnover that is believed to be a result of apoptosis. Since bowel is the primary organ responsible for inflammatory response in trauma and shock, if accelerated cell death occurs in the intestine of patients with trauma and shock, important pathologic consequences could result. During trauma and shock, the intestinal wall loses its barrier function which results in the leakage of endotoxin and bacteria into the circulation causing a systemic inflammatory response. Apoptosis of intestinal epithelial tissues occur as rapidly as 2–3 h after initial injury and it compromises bowel wall integrity and becomes the primary mode for bacterial or endotoxin translocation into the systemic circulation [121]. In contrast, increased apoptosis of peripheral blood neutrophils is associated with reduced incidence of infection in trauma patients with hemorrhagic shock [122]. Clearance of apoptotic peripheral blood neutrophils by the liver and spleen inhibit inflammatory response thereby sparing the other organs such as the lung, which is among the most common sites of infection following serious trauma that leads to multi-organ failure and death.
Ingestion of apoptotic cells by macrophages results in the release of anti-inflammatory mediators, including TGF-β1 and PGE2 and suppresses the production of pro-inflammatory cytokines such as IL-8, TNF-α and thrombaxane A2 [123, 124]. In this regard, MFG-E8 has been identified as a bridging molecule between professional phagocytes via the αVβ3 or αVβ5 integrins and apoptotic cells via PS, which accelerates the engulfment of apoptotic cells [41, 42]. In a mice model of pressure-controlled (25 ± 5 mmHg) hemorrhagic shock [21], MFG-E8 levels in the plasma, lungs and spleen were significantly decreased at 4 h after hemorrhage. Resuscitation with rhMFG-E8 significantly improved apoptosis at 4 h as evidenced by a reduction in TUNEL positive cells and cleaved caspase-3 expression. Neutrophil infiltration into the lungs and spleen were also blunted. Pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) were reduced significantly in plasma (64–73 %), lungs (24–58 %) and spleen (49–76 %). In a 7 day survival study, a significant improvement (83 % vs. 43 %) with one-time dose of rhMFG-E8 as compared to normal saline treated mice after hemorrhage was observed. These data taken together suggest that rhMFG-E8 could be developed as a treatment for hemorrhagic shock.
4 Future Perspectives
In this chapter, we clearly demonstrated that MFG-E8 could be developed as a novel therapy for sepsis, ischemia and reperfusion injury, and trauma hemorrhagic shock. The data described further indicates that MFG-E8 could be functioning as a tether between apoptotic cells and phagocytes for efficient engulfment of apoptotic cells and thereby reduce inflammation and improve survival. It is also implicated that MFG-E8 could function directly by binding to αVβ3 or αVβ5 integrins and upregulate or downregulate signaling components causing the reduction in inflammation. Regardless of its mechanism of action, it is clear that administration of MFG-E8 is beneficial in attenuating the exaggerated inflammatory response associated with systemic inflammation caused by sepsis, ischemia and reperfusion injury, and trauma hemorrhagic shock. Thus, MFG-E8 treatment could be a potential therapy for patients suffering from complications associated with such conditions.
References
Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC et al (2010) The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med 38:367–374
Dombrovskiy VY, Martin AA, Sunderram J, Paz HL (2007) Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med 35:1244–1250
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J et al (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29:1303–1310
Tendler DA (2003) Acute intestinal ischemia and infarction. Semin Gastrointest Dis 14:66–76
Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Pina E, Geller DA (2008) Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res 147:153–159
Serracino-Inglott F, Habib NA, Mathie RT (2001) Hepatic ischemia-reperfusion injury. Am J Surg 181:160–166
Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW (2005) Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16:3365–3370
Lameire N, Van Biesen W, Vanholder R (2005) Acute renal failure. Lancet 365:417–430
Cothren CC, Moore EE, Hedegaard HB, Meng K (2007) Epidemiology of urban trauma deaths: a comprehensive reassessment 10 years later. World J Surg 31:1507–1511
Heron M, Hoyert DL, Murphy SL, Xu J, Kochanek KD et al (2009) Deaths: final data for 2006. Natl Vital Stat Rep 57:1–134
Martin GS, Mannino DM, Eaton S, Moss M (2003) The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348:1546–1554
Martin GS (2012) Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev Anti Infect Ther 10:701–706
Eltzschig HK, Eckle T (2011) Ischemia and reperfusion–from mechanism to translation. Nat Med 17:1391–1401
Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A et al (2002) Identification of a factor that links apoptotic cells to phagocytes. Nature 417:182–187
Miksa M, Wu R, Dong W, Das P, Yang D et al (2006) Dendritic cell-derived exosomes containing milk fat globule epidermal growth factor-factor VIII attenuate proinflammatory responses in sepsis. Shock 25:586–593
Miksa M, Wu R, Dong W, Komura H, Amin D et al (2009) Immature dendritic cell-derived exosomes rescue septic animals via milk fat globule epidermal growth factor VIII. J Immunol 183:5983–5990
Shah KG, Wu R, Jacob A, Molmenti EP, Nicastro J et al (2012) Recombinant human milk fat globule-EGF factor 8 produces dose-dependent benefits in sepsis. Intensive Care Med 38:128–136
Cui T, Miksa M, Wu R, Komura H, Zhou M et al (2010) Milk fat globule epidermal growth factor 8 attenuates acute lung injury in mice after intestinal ischemia and reperfusion. Am J Respir Crit Care Med 181:238–246
Matsuda A, Jacob A, Wu R, Zhou M, Aziz M et al (2013) Milk fat globule–EGF factor VIII ameliorates liver injury after hepatic ischemia-reperfusion. J Surg Res 180:e37–e46
Matsuda A, Wu R, Jacob A, Komura H, Zhou M et al (2011) Protective effect of milk fat globule-epidermal growth factor-factor VIII after renal ischemia-reperfusion injury in mice. Crit Care Med 39:2039–2047
Zhang F, Shah KG, Qi L, Wu R, Barrera R et al (2012) Milk fat globule epidermal growth factor-factor 8 mitigates inflammation and tissue injury after hemorrhagic shock in experimental animals. J Trauma Acute Care Surg 72:861–869
Sprung CL, Zimmerman JL, Christian MD, Joynt GM, Hick JL et al (2010) Recommendations for intensive care unit and hospital preparations for an influenza epidemic or mass disaster: summary report of the European Society of Intensive Care Medicine’s Task Force for intensive care unit triage during an influenza epidemic or mass disaster. Intensive Care Med 36:428–443
Briegel J, Sprung CL, Annane D, Singer M, Keh D et al (2009) Multicenter comparison of cortisol as measured by different methods in samples of patients with septic shock. Intensive Care Med 35:2151–2156
Bernard GR, Macias WL, Joyce DE, Williams MD, Bailey J et al (2003) Safety assessment of drotrecogin alfa (activated) in the treatment of adult patients with severe sepsis. Crit Care 7:155–163
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF et al (2001) Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 344:699–709
Lotze MT, Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5:331–342
Huber-Lang MS, Younkin EM, Sarma JV, McGuire SR, Lu KT et al (2002) Complement-induced impairment of innate immunity during sepsis. J Immunol 169:3223–3231
Kaufmann I, Hoelzl A, Schliephake F, Hummel T, Chouker A et al (2006) Polymorphonuclear leukocyte dysfunction syndrome in patients with increasing sepsis severity. Shock 26:254–261
Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC et al (2003) Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol 171:909–914
Ayala A, Herdon CD, Lehman DL, DeMaso CM, Ayala CA et al (1995) The induction of accelerated thymic programmed cell death during polymicrobial sepsis: control by corticosteroids but not tumor necrosis factor. Shock 3:259–267
Efron PA, Tinsley K, Minnich DJ, Monterroso V, Wagner J et al (2004) Increased lymphoid tissue apoptosis in baboons with bacteremic shock. Shock 21:566–571
Fink SL, Cookson BT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73:1907–1916
Zhang P, Bagby GJ, Stoltz DA, Summer WR, Nelson S (1998) Enhancement of peritoneal leukocyte function by granulocyte colony-stimulating factor in rats with abdominal sepsis. Crit Care Med 26:315–321
Wysocka M, Robertson S, Riemann H, Caamano J, Hunter C et al (2001) IL-12 suppression during experimental endotoxin tolerance: dendritic cell loss and macrophage hyporesponsiveness. J Immunol 166:7504–7513
Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M et al (2004) Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304:1147–1150
Kim S, Chung EY, Ma X (2005) Immunological consequences of macrophage-mediated clearance of apoptotic cells. Cell Cycle 4:231–234
Stubbs JD, Lekutis C, Singer KL, Bui A, Yuzuki D et al (1990) cDNA cloning of a mouse mammary epithelial cell surface protein reveals the existence of epidermal growth factor-like domains linked to factor VIII-like sequences. Proc Natl Acad Sci U S A 87:8417–8421
Miyasaka K, Hanayama R, Tanaka M, Nagata S (2004) Expression of milk fat globule epidermal growth factor 8 in immature dendritic cells for engulfment of apoptotic cells. Eur J Immunol 34:1414–1422
Hanayama R, Miyasaka K, Nakaya M, Nagata S (2006) MFG-E8-dependent clearance of apoptotic cells, and autoimmunity caused by its failure. Curr Dir Autoimmun 9:162–172
Somersan S, Bhardwaj N (2001) Tethering and tickling: a new role for the phosphatidylserine receptor. J Cell Biol 155:501–504
Akakura S, Singh S, Spataro M, Akakura R, Kim JI et al (2004) The opsonin MFG-E8 is a ligand for the alphavbeta5 integrin and triggers DOCK180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Exp Cell Res 292:403–416
Albert ML, Kim JI, Birge RB (2000) Alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol 2:899–905
Komura H, Miksa M, Wu R, Goyert SM, Wang P (2009) Milk fat globule epidermal growth factor-factor VIII is down-regulated in sepsis via the lipopolysaccharide-CD14 pathway. J Immunol 182:581–587
Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L et al (1999) Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol 147:599–610
Thery C, Boussac M, Veron P, Ricciardi-Castagnoli P, Raposo G et al (2001) Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol 166:7309–7318
Qiang X, Li J, Wu R, Ji Y, Chaung W et al (2011) Expression and characterization of recombinant human milk fat globule-EGF Factor VIII. Int J Mol Med 28:1071–1076
Bu HF, Zuo XL, Wang X, Ensslin MA, Koti V et al (2007) Milk fat globule-EGF factor 8/lactadherin plays a crucial role in maintenance and repair of murine intestinal epithelium. J Clin Invest 117:3673–3683
Aziz M, Jacob A, Matsuda A, Wu R, Zhou M et al (2011) Pre-treatment of recombinant mouse MFG-E8 downregulates LPS-induced TNF-alpha production in macrophages via STAT3-mediated SOCS3 activation. PLoS One 6:e27685
Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW et al (2001) Post-injury multiple organ failure: the role of the gut. Shock 15:1–10
Pierro A, Eaton S (2004) Intestinal ischemia reperfusion injury and multisystem organ failure. Semin Pediatr Surg 13:11–17
Mallick IH, Yang W, Winslet MC, Seifalian AM (2004) Ischemia-reperfusion injury of the intestine and protective strategies against injury. Dig Dis Sci 49:1359–1377
Kalia N, Brown NJ, Wood RF, Hopkinson K, Fairburn B et al (2003) Effects of intestinal ischemia-reperfusion injury on rat peripheral blood neutrophil activation. Dig Dis Sci 48:1677–1684
Kozar RA, Holcomb JB, Hassoun HT, Macaitis J, DeSoignie R et al (2004) Superior mesenteric artery occlusion models shock-induced gut ischemia-reperfusion. J Surg Res 116:145–150
Stallion A, Kou TD, Latifi SQ, Miller KA, Dahms BB et al (2005) Ischemia/reperfusion: a clinically relevant model of intestinal injury yielding systemic inflammation. J Pediatr Surg 40:470–477
Carden DL, Granger DN (2000) Pathophysiology of ischaemia-reperfusion injury. J Pathol 190:255–266
Oldenburg WA, Lau LL, Rodenberg TJ, Edmonds HJ, Burger CD (2004) Acute mesenteric ischemia: a clinical review. Arch Intern Med 164:1054–1062
Collard CD, Gelman S (2001) Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesiology 94:1133–1138
Moore EE, Moore FA, Franciose RJ, Kim FJ, Biffl WL et al (1994) The postischemic gut serves as a priming bed for circulating neutrophils that provoke multiple organ failure. J Trauma 37:881–887
Bellingan GJ (2002) The pulmonary physician in critical care * 6: the pathogenesis of ALI/ARDS. Thorax 57:540–546
Souza DG, Vieira AT, Soares AC, Pinho V, Nicoli JR et al (2004) The essential role of the intestinal microbiota in facilitating acute inflammatory responses. J Immunol 173:4137–4146
de Arruda MJ, Poggetti RS, Fontes B, Younes RN, Souza AL Jr et al (2006) Intestinal ischemia/reperfusion induces bronchial hyperreactivity and increases serum TNF-alpha in rats. Clinics (Sao Paulo) 61:21–28
Mitsuoka H, Kistler EB, Schmid-Schonbein GW (2002) Protease inhibition in the intestinal lumen: attenuation of systemic inflammation and early indicators of multiple organ failure in shock. Shock 17:205–209
Wilmore DW, Smith RJ, O’Dwyer ST, Jacobs DO, Ziegler TR et al (1988) The gut: a central organ after surgical stress. Surgery 104:917–923
Mitsuoka H, Kistler EB, Schmid-Schonbein GW (2000) Generation of in vivo activating factors in the ischemic intestine by pancreatic enzymes. Proc Natl Acad Sci U S A 97:1772–1777
Yasuhara H (2005) Acute mesenteric ischemia: the challenge of gastroenterology. Surg Today 35:185–195
Granger DN, Korthuis RJ (1995) Physiologic mechanisms of postischemic tissue injury. Annu Rev Physiol 57:311–332
Ware LB, Matthay MA (2000) The acute respiratory distress syndrome. N Engl J Med 342:1334–1349
Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL (1994) Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 94:1621–1628
Koji T, Hishikawa Y, Ando H, Nakanishi Y, Kobayashi N (2001) Expression of Fas and Fas ligand in normal and ischemia-reperfusion testes: involvement of the Fas system in the induction of germ cell apoptosis in the damaged mouse testis. Biol Reprod 64:946–954
Nogae S, Miyazaki M, Kobayashi N, Saito T, Abe K et al (1998) Induction of apoptosis in ischemia-reperfusion model of mouse kidney: possible involvement of Fas. J Am Soc Nephrol 9:620–631
Fukuyama K, Iwakiri R, Noda T, Kojima M, Utsumi H et al (2001) Apoptosis induced by ischemia-reperfusion and fasting in gastric mucosa compared to small intestinal mucosa in rats. Dig Dis Sci 46:545–549
Haglund U (1994) Gut ischaemia. Gut 35:S73–S76
Genesca M, Sola A, Miquel R, Pi F, Xaus C et al (2002) Role of changes in tissular nucleotides on the development of apoptosis during ischemia/reperfusion in rat small bowel. Am J Pathol 161:1839–1847
Asano K, Miwa M, Miwa K, Hanayama R, Nagase H et al (2004) Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J Exp Med 200:459–467
Hart SP, Dransfield I, Rossi AG (2008) Phagocytosis of apoptotic cells. Methods 44:280–285
Wu R, Dong W, Wang Z, Jacob A, Cui T et al (2012) Enhancing apoptotic cell clearance mitigates bacterial translocation and promotes tissue repair after gut ischemia-reperfusion injury. Int J Mol Med 30:593–598
Borghi-Scoazec G, Scoazec JY, Durand F, Bernuau J, Belghiti J et al (1997) Apoptosis after ischemia-reperfusion in human liver allografts. Liver Transpl Surg 3:407–415
Contreras JL, Vilatoba M, Eckstein C, Bilbao G, Anthony Thompson J et al (2004) Caspase-8 and caspase-3 small interfering RNA decreases ischemia/reperfusion injury to the liver in mice. Surgery 136:390–400
Bell CW, Jiang W, Reich CF 3rd, Pisetsky DS (2006) The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 291:C1318–C1325
Silva MT, do Vale A, dos Santos NM (2008) Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications. Apoptosis 13:463–482
Atabai K, Jame S, Azhar N, Kuo A, Lam M et al (2009) Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J Clin Invest 119:3713–3722
Raymond A, Ensslin MA, Shur BD (2009) SED1/MFG-E8: a bi-motif protein that orchestrates diverse cellular interactions. J Cell Biochem 106:957–966
Finn WF (1981) Nephron heterogeneity in polyuric acute renal failure. J Lab Clin Med 98:21–29
Kubiak BD, Albert SP, Gatto LA, Vieau CJ, Roy SK et al (2011) A clinically applicable porcine model of septic and ischemia/reperfusion-induced shock and multiple organ injury. J Surg Res 166:e59–e69
Molls RR, Savransky V, Liu M, Bevans S, Mehta T et al (2006) Keratinocyte-derived chemokine is an early biomarker of ischemic acute kidney Injury. Am J Physiol Renal Physiol 290:F1187–F1193
Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K et al (2003) Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 14:2534–2543
Andreucci M, Michael A, Kramers C, Park KM, Chen A et al (2003) Renal ischemia/reperfusion and ATP depletion/repletion in LLC-PK(1) cells result in phosphorylation of FKHR and FKHRL1. Kidney Int 64:1189–1198
Bamberger MH, Qien L (1994) Molecular response to calcium channel blockers expressed during reperfusion following acute renal ischemia. Ann N Y Acad Sci 723:451–453
Chien CT, Chang TC, Tsai CY, Shyue SK, Lai MK (2005) Adenovirus-mediated bcl-2 gene transfer inhibits renal ischemia/reperfusion induced tubular oxidative stress and apoptosis. Am J Transplant 5:1194–1203
Qiao X, Chen X, Wu D, Ding R, Wang J et al (2005) Mitochondrial pathway is responsible for aging-related increase of tubular cell apoptosis in renal ischemia/reperfusion injury. J Gerontol A Biol Sci Med Sci 60:830–839
Padanilam BJ (2003) Cell death induced by acute renal injury: a perspective on the contributions of apoptosis and necrosis. Am J Physiol Renal Physiol 284:F608–F627
Lieberthal W, Koh JS, Levine JS (1998) Necrosis and apoptosis in acute renal failure. Semin Nephrol 18:505–518
Rana A, Sathyanarayana P, Lieberthal W (2001) Role of apoptosis of renal tubular cells in acute renal failure: therapeutic implications. Apoptosis 6:83–102
Sutton TA, Fisher CJ, Molitoris BA (2002) Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int 62:1539–1549
Kauvar DS, Lefering R, Wade CE (2006) Impact of hemorrhage on trauma outcome: an overview of epidemiology, clinical presentations, and therapeutic considerations. J Trauma 60:S3–S11
Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R et al (1995) Epidemiology of trauma deaths: a reassessment. J Trauma 38:185–193
Demetriades D, Murray J, Charalambides K, Alo K, Velmahos G et al (2004) Trauma fatalities: time and location of hospital deaths. J Am Coll Surg 198:20–26
Haas B, Nathens AB (2008) Pro/con debate: is the scoop and run approach the best approach to trauma services organization? Crit Care 12:224
Bahrami S, Zimmermann K, Szelenyi Z, Hamar J, Scheiflinger F et al (2006) Small-volume fluid resuscitation with hypertonic saline prevents inflammation but not mortality in a rat model of hemorrhagic shock. Shock 25:283–289
Cuschieri J, Gourlay D, Garcia I, Jelacic S, Maier RV (2002) Hypertonic preconditioning inhibits macrophage responsiveness to endotoxin. J Immunol 168:1389–1396
Gonzalez RJ, Moore EE, Ciesla DJ, Neto JR, Biffl WL et al (2002) Hyperosmolarity abrogates neutrophil cytotoxicity provoked by post-shock mesenteric lymph. Shock 18:29–32
Junger WG, Coimbra R, Liu FC, Herdon-Remelius C, Junger W et al (1997) Hypertonic saline resuscitation: a tool to modulate immune function in trauma patients? Shock 8:235–241
Murao Y, Hata M, Ohnishi K, Okuchi K, Nakajima Y et al (2003) Hypertonic saline resuscitation reduces apoptosis and tissue damage of the small intestine in a mouse model of hemorrhagic shock. Shock 20:23–28
Murao Y, Loomis W, Wolf P, Hoyt DB, Junger WG (2003) Effect of dose of hypertonic saline on its potential to prevent lung tissue damage in a mouse model of hemorrhagic shock. Shock 20:29–34
Pascual JL, Ferri LE, Seely AJ, Campisi G, Chaudhury P et al (2002) Hypertonic saline resuscitation of hemorrhagic shock diminishes neutrophil rolling and adherence to endothelium and reduces in vivo vascular leakage. Ann Surg 236:634–642
Rotstein OD (2000) Novel strategies for immunomodulation after trauma: revisiting hypertonic saline as a resuscitation strategy for hemorrhagic shock. J Trauma 49:580–583
Sheppard FR, Moore EE, McLaughlin N, Kelher M, Johnson JL et al (2005) Clinically relevant osmolar stress inhibits priming-induced PMN NADPH oxidase subunit translocation. J Trauma 58:752–757, discussion 757
Staudenmayer KL, Maier RV, Jelacic S, Bulger EM (2005) Hypertonic saline modulates innate immunity in a model of systemic inflammation. Shock 23:459–463
Alam HB (2011) Advances in resuscitation strategies. Int J Surg 9:5–12
Gonzalez EA, Moore FA, Holcomb JB, Miller CC, Kozar RA et al (2007) Fresh frozen plasma should be given earlier to patients requiring massive transfusion. J Trauma 62:112–119
Borgman MA, Spinella PC, Perkins JG, Grathwohl KW, Repine T et al (2007) The ratio of blood products transfused affects mortality in patients receiving massive transfusions at a combat support hospital. J Trauma 63:805–813
Silliman CC, Moore EE, Johnson JL, Gonzalez RJ, Biffl WL (2004) Transfusion of the injured patient: proceed with caution. Shock 21:291–299
Charles A, Shaikh AA, Walters M, Huehl S, Pomerantz R (2007) Blood transfusion is an independent predictor of mortality after blunt trauma. Am Surg 73:1–5
Offner PJ, Moore EE, Biffl WL, Johnson JL, Silliman CC (2002) Increased rate of infection associated with transfusion of old blood after severe injury. Arch Surg 137:711–716, discussion 716–717
Dunne JR, Malone DL, Tracy JK, Napolitano LM (2004) Allogenic blood transfusion in the first 24 hours after trauma is associated with increased systemic inflammatory response syndrome (SIRS) and death. Surg Infect (Larchmt) 5:395–404
Moore FA, Moore EE, Sauaia A (1997) Blood transfusion. An independent risk factor for postinjury multiple organ failure. Arch Surg 132:620–624, discussion 624–625
Yang FL, Subeq YM, Lee CJ, Lee RP, Peng TC et al (2011) Melatonin ameliorates hemorrhagic shock-induced organ damage in rats. J Surg Res 167:e315–e321
Noda T, Iwakiri R, Fujimoto K, Matsuo S, Aw TY (1998) Programmed cell death induced by ischemia-reperfusion in rat intestinal mucosa. Am J Physiol 274:G270–G276
Coopersmith CM, O’Donnell D, Gordon JI (1999) Bcl-2 inhibits ischemia-reperfusion-induced apoptosis in the intestinal epithelium of transgenic mice. Am J Physiol 276:G677–G686
Xu YX, Ayala A, Monfils B, Cioffi WG, Chaudry IH (1997) Mechanism of intestinal mucosal immune dysfunction following trauma-hemorrhage: increased apoptosis associated with elevated Fas expression in Peyer’s patches. J Surg Res 70:55–60
Hotchkiss RS, Schmieg RE Jr, Swanson PE, Freeman BD, Tinsley KW et al (2000) Rapid onset of intestinal epithelial and lymphocyte apoptotic cell death in patients with trauma and shock. Crit Care Med 28:3207–3217
Anne Morrison C, Moran A, Patel S, Vidaurre Mdel P, Carrick MM et al (2013) Increased apoptosis of peripheral blood neutrophils is associated with reduced incidence of infection in trauma patients with hemorrhagic shock. J Infect 66:87–94
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY et al (1998) Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101:890–898
Huynh ML, Fadok VA, Henson PM (2002) Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 109:41–50
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Jacob, A., Wang, P. (2014). Novel Therapeutic for Systemic Inflammation: Role of MFG-E8. In: Wang, P. (eds) MFG-E8 and Inflammation. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-8765-9_7
Download citation
DOI: https://doi.org/10.1007/978-94-017-8765-9_7
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-8764-2
Online ISBN: 978-94-017-8765-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)