
Abstract
It has been previously reported that miRNA regulations were involved in various biological processes. The deregulation activities of microRNA regulators potentially contribute to the pathopoiesis of various kinds of human cancers, and are candidate biomarkers for cancer diagnosis and prognosis. Until now, enormous studies have been conducted to explore potential miRNA biomarkers for different types of cancers. In this chapter, we will first provide a brief introduction about miRNAs biogenesis and their involvement in cancer pathopoiesis, and then reviewed the advances on current available miRNA profiling technologies. Then concise text will be exploited to describe the traditional experiment-dominate approaches for miRNA biomarker discovery. In the next part, intensive efforts are made on the review and summarization of miRNA–mRNA network based computational methods for the discovery of potential miRNA biomarkers. Afterwards, collect and list exsiting online databases relating to cancer miRNA biomarker discovery. Finally, we propose the perspective directions on this research area, and conclude the main context in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
MicroRNAs (miRNAs) are a class of small non-coding RNAs, approximately 22 nucleotides in length, which regulate gene expression at the post-transcriptional level through translation inhibition or mRNA cleavage (Bartel 2004). Since the discovery of the first miRNA (i.e., lin-4) in C. elegans (Lee et al. 1993; Wightman et al. 1993), enormous studies have been conducted to explore latent miRNA structures in a series of organisms, from viruses to advanced mammalians. Currently, within the most popular miRNA repository—miRBase (Release 20, June 2013) (Griffiths-Jones 2004), there have been 24,521 entries representing hairpin precursor miRNAs, expressing 30,424 mature miRNA products, among 206 distinct species. It is reported that most miRNAs are independently encoded in intergenic regions (Lee et al. 2004) or co-encoded within intron regions of other “host” protein-coding genes (Lin et al. 2008). Also, recent studies implicated the transfer RNAs (tRNAs) might be another origin for miRNA biogenesis (Schopman et al. 2010; Maute et al. 2013). The cardinal mechanism for the biological functions of miRNAs is to bind the 3’UTR (un-translated region) of their target genes through imperfect base pairing in animals (Reinhart et al. 2000), or perfect base pairing in plants (Dugas and Bartel 2004). Generally, the miRNA binding to its target genes will induce the mRNA degradation or protein translation inhibition, albeit several studies have shown its effect on the stabilization of target transcripts (Place et al. 2008).
Until now, there are more than 2,500 mature miRNAs identified in humans, which have potential to approximately regulate 33 % of human protein-coding genes (Lewis et al. 2005). It has been recently shown that miRNA regulations are involved in a wide variety of cellular processes, from cell proliferation, differentiation, development, to apoptosis (Ambros 2004; Bartel 2004). The alterations in miRNA expression have been associated with the pathogenesis and procession of various kinds of diseases, especially cancers (Jay et al. 2007). The abnormal miRNAs are reported to be capable as classifiers to distinguish tumour samples from Normal tissues (Raponi et al. 2009).
As implicated in the previous study (Bielekova and Martin 2004), some features were required to be as a disease biomarker: biological rational, clinical relevance, practicality and correlation with disease activity, etc. For miRNAs, they are involved in various biological processes and clinical related to the disease pathogenesis, as discussed in the above context. Besides, other advantageous features of miRNAs, such as manageable, durability and easily detected, make them more potential to be cancer biomarkers (Guerau-de-Arellano et al. 2012). In fact, enormous attempts have been made to explore candidate miRNA biomarkers in a series of cancers, such as breast cancer (Heneghan et al. 2010; Ramshankar and Krishnamurthy 2013), lung cancer (Gao et al. 2012), and gastric cancer (Li et al. 2012, 2013).
2 Advances of miRNA Expression Profiling Methods
The most straightforward approach to explore candidate cancer miRNA biomarkers should be the screening of abnormally expressed miRNAs between cancer tissues and normal tissues, via contemporary miRNA expression profiling detection platforms. Generally, there are two main experimental categories for these miRNA expression profiling techniques: high-throughput screening approaches and low-throughput detection methods. The first category consists of nucleic acid hybridization-based array technologies and cloning-sequencing based approaches, which can be used for simultaneous detection of many miRNAs in an individual experiment. The latter one includes small-scale detection methods, such as Northern Blot, Real time quantitative PCR (RT-qPCR), and in situ hybridization (ISH). The comparison information about these technologies is presented in Table 8.1.
2.1 High-Throughput Screening Approaches
Currently, microarray (also called Gene chip) technology may be the most widely used in the filed of transcript expression profiling detection. The implementation of this method is based on the hybridization of slide-localized miRNA-specific probes (Babak et al. 2004). MiRNAs in the sample are first fluorescent dye-labeled, and then hybridized on slide array with glass-printed probes. After eluting unstable connections, the miRNA abundance is measured according its fluorescent luminance. The main advantage of this approach is parallelized detection of hundreds of miRNAs in an individual experiment.
Another large-scale miRNA expression profiling method is Beadarray technology. Unlike the classical microarray approach, it uses magnetic microspheres tagged with unique DNA sequence to identify the bead. This bead can specifically bind to a chimeric probe, which is utilized to recruit a specific miRNA into a complex. The fluorescent labeling of the complex is implemented with the biotin on the probe. Therefore, the abundance of miRNA expression can be quantified according to the amount of fluorescence on the microspheres through flow instruments.
The aforementioned array-based technologies can merely exploited for the expression profiling of known miRNAs. Deep sequencing techniques are novel miRNA profiling approaches that could detect the expression of all the miRNAs expressed in the target sample, even for miRNAs that are never previously reported. The detailed sequencing procedures may be diverse for different platforms. Generally, the first step for all these techniques is the generation of a miRNA library. During this process, miRNAs are ligated to 5′ and 3′ adaptors for reverse transcription and PCR amplification to generate this library. After that, the miRNAs in the library are further simultaneously sequenced and eventually abundance quantified. Due to their higher specificity and accuracy, the deep sequencing technologies are very promising to take over the dominating position of microarray technique in the area of transcript expression profiling.
2.2 Low-Throughput Detection Methods
Even with low flux, low-throughput miRNA expression profiling methods are considered to be more reliable than high-throughput techniques. This is a trade-off problem. More specifically, northern blot method is declaimed as the “gold standard” for characterizing miRNA expression (Ahmed 2007), due to its high specificity. The method is more like a mini-version of microarray. The miRNA abundance in the sample is determined by the hybridization signal from the binding complex of query miRNA and pre-set probes. Except for its low throughput, another defect of this method is low sensitivity for low-abundance samples.
Different from northern blot technique, RT-qPCR is a relative high sensitive approach for miRNA expression characterization. The basic idea is to make miRNA molecules amplifiable, through adding adapters to their fragment ends. After that, the amount of miRNA in the sample is relatively quantified. The PCR amplification process makes it accessible for low-abundance specimen measurement. This technology can be used for quantification of both miRNA precursors and mature miRNAs.
Another small-scale miRNA profiling method is in situ hybridization, which was invented by American cell biologist Joseph Grafton Gall in 1969 (Gall and Pardue 1969). This technology can be used to measure and localize miRNAs within tissue sections and cells.
With the recent progress in miRNA expression profiling detection technologies, there have been growing intense interests in cancer miRNA biomarker discovery studies, as documented by the numbers of related published literatures from NCBI pubmed searching engine in the last 10 years (Fig. 8.1). In the following parts, much more context will be spent on the descriptive review on these cancer miRNA biomarker discovery studies.

Number of publications related to cancer miRNA biomarker discovery studies during the past decade. Bars represent the number of NCBI pubmed hits for query “(cancer[ti] OR carcinoma[ti] OR tumor[ti]) AND (miRNA*[ti] OR microRNA*[ti])” (Pane a) and “(cancer[ti] OR carcinoma[ti] OR tumor[ti]) AND (miRNA*[ti] OR microRNA*[ti]) AND (biomarker*[tiab] OR marker*[tiab])” (Pane b). The numbers for year 2013 are enumerated until 18th July
3 Traditional Approaches for Cancer miRNA Biomarker Discovery
The general procedures of traditional approaches for novel cancer miRNA biomarker discovery can be divided into three steps: (a) detection of differentially expressed miRNAs with high-throughput method (e.g., microarray) between cancer samples and control groups; (b) low-throughput technology (e.g., RT-qPCR) validation of outlier miRNAs detected above; (c) further confirmation of potential miRNA biomarkers on large-scale of case and control specimen via low-throughput experiments. The simplest bioinformatics tool for outlier miRNA identification in the first step is fold-change filtering (usually 2-fold). Following, many well-established statistical tools are employed for this issue, such as z-score, t-test, and Mann–Whitney test. Nevertheless, all these approaches do not take the heterogeneity of cancer samples into account.(MacDonald and Ghosh 2006) proposed a novel bioinformatics tool to infer chromosomal translocations only existing in the subset of disease samples . Afterwards, this concept was implemented to generate several other outlier gene detection algorithms (Tibshirani and Hastie 2007; Wu 2007; Lian 2008). These available methods could be applied for outlier miRNA screening from high-throughput experiments.
Now, let us exemplify the general routine of traditional approaches for cancer miRNA biomarker discovery. Through the aforementioned procedures, the study of (Li et al. 2012) revealed that miR-199a-3p in plasma as a potential diagnostic biomarker for gastric cancer. Another research group conducted genome-wide miRNA expression profiles detection followed with Real-Time quantitative RT-PCR (qRT-PCR) assays on gastric cancer samples and normal samples, and discovered three elevated expressed miRNA (miR-187(*), miR-371-5p and miR-378) in gastric cancer. Further validation study showed that miR-378 alone could produce 87.5 % sensitivity and 70.73 % specificity in discriminating gastric cancer patients from healthy controls, thus this miRNA was a potential diagnosis biomarker for gastric cancer.
4 The Reconstruction of miRNA–mRNA Network
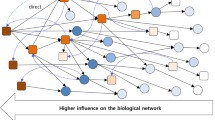
As the expression profiling data from high-throughput technologies (e.g., microarray) is always of high false positive rate, integrative analysis on high-throughput expression profiling data and miRNA–mRNA target network information may be a plausible approach for novel cancer miRNA biomarker discovery (Xu et al. 2011). The miRNA–mRNA network is more exactly a unidirectional graph, reflecting the regulation relationships from miRNAs to their target genes, as presented in Fig. 8.2. Due to the limit of current experimentally validated miRNA–mRNA target pairs (Sethupathy et al. 2006; Xiao et al. 2009; Hsu et al. 2011), the main resources for miRNA–mRNA network reconstruction are from computational prediction. Even with potential false positive or negative cases, many of the predicted miRNA–mRNA interactions are confirmed to be credible (Arora and Simpson 2008). Usually, the main resources for miRNA–mRNA network reconstruction are the common miRNA–mRNA target pairs shared by the prediction results from multiple computational approaches, such as PicTar (Krek et al. 2005), miRanda (Enright et al. 2003), and TargetScan (Lewis et al. 2003, 2005). Besides, the negative expression correlations between miRNAs and their putative targets, computed from matched miRNA and mRNA expression profiles can also be applied for the refinement of miRNA–mRNA network reconstruction (Tran et al. 2008; Zhang et al. 2013).

Schematic graph of miRNA–mRNA target network
5 Computational Based Approaches for Individual miRNA Biomarker Discovery
5.1 Based on Gene Expression Data and miRNA–mRNA Network Information
As the general miRNA functions are to regulate the gene expression at mRNA level, it is rational to infer miRNA deregulation from its target genes’ expression level changes. Indeed, a number of studies have reported the reverse correlations between expressions of miRNAs and their target genes (Krutzfeldt et al. 2005; Wang and Wang 2006). Base on this theory, (Cheng and Li 2008). proposed an algorithm to infer microRNA activities by combining gene expression data with miRNA–mRNA network information . The basic idea of this algorithm is to analyze the expression changes of target genes for miRNAs. The activity of a miRNA will be inferred to elevated, if the expressions of its target genes tend to be down-regulated, and vice versa. Applying this approach, the cancer miRNA expression patterns can be deduced according to the gene expression profiles between cancer and normal samples.
Similarly, another approach referred as Co-inertia analysis (CIA) was proposed for this issue. CIA is a multivariate coupling approach, which was initially introduced for ecological research (Doledec and Chessel 1994; Dray et al. 2003). It was used to explore the correlation of two sets of variables from two linked data tables. Stephen et al. (Madden et al. 2010) applied this method to detect miRNA activity in different biological conditions. In this case, the two linked tables were gene microarray expression data, and a miRNA frequency table on the same set of genes. The two linked tables were performed two simultaneous non-symmetric correspondence analyses (NSCs), which reduced each data table in a low dimensional space, by projecting each variable on to axes which best discriminate the coordinates of the projected points. Then these two reduced tables were linked to associate the miRNA activity with biological samples. This methodology can be used to identify miRNA deregulation patterns that distinguish disease and normal groups.
5.2 Based on miRNA and Gene Expression Profiles and miRNA–mRNA Network Information
As the miRNA–mRNA target relationships are presented a simplified network style, the topological features of this network may be helpful for the identification of candidate cancer miRNA biomarkers. (Xu et al. 2011). introduced an approach based on the miRNA-target dysregulated network (MTDN) to prioritize candidate disease miRNAs, and applied this method to predict novel miRNA biomarkers in prostate cancer. In this methodology, miRNA expression and mRNA expression data, and miRNA–mRNA interaction data were combined to construct MTDN in tumor and non-tumor conditions. Then a support vector machine (SVM) was trained with considering the expression fold change and network topological features of known prostate cancer miRNAs and non-prostate cancer miRNAs in MTDN. Finally, the novel prostate cancer miRNA biomarkers were prioritized with this SVM and in vitro experimentally validated in prostate cancer cell lines. This study also showed the function synergism of miRNAs that were involved in the specific disease or biological process.
In contrary to the functional cooperation, (Zhang et al. 2013). declaimed another bizarre characteristic of cancer miRNAs—strong independent regulation power, which denoted the number of exclusively regulated genes for an individual miRNA. This research group also proposed a novel pipeline to infer candidate cancer miRNA biomarkers. The negative correlations from paired miRNA and mRNA expression profiles, along with computational prediction miRNA-mRNA target pairs were combined to generate a reliable miRNA–mRNA network. This network was further reduced to a sub-network, which only consisted of miRNA nodes exhibiting deregulation patterns from the miRNA expression profiles. In this sub-network, the independent regulation power was calculated for each miRNA. Ultimately, miRNAs with significant great independent regulation power were predicted as potential cancer miRNA biomarkers. The afterwards in vitro experiment validation and systematic analysis confirmed the accuracy of this approach.
6 Computational Based Approaches for miRNA Network Biomarker Discovery
6.1 The Discovery of Cancer-Related miRNA–mRNA Regulatory Modules
The concept of miRNA regulatory modules (mRMs) was first proposed by Yoon and De Micheli (2005a, b) to indicate groups of miRNAs and target genes that were completely connected in the sub-groups and functionally corporate in specific biological processes. This notion was afterwards applied for cancer studies.
Through integrative analysis on matched miRNA expression and mRNA expression profiles, two cancer mRMs discovery algorithms were proposed based on fuzzy decision tree model (Bonnet et al. 2010) and correspondence latent dirichlet allocation (Liu et al. 2010). Afterwards, (Jayaswal et al. 2011) introduced a clustering method to infer miRNA regulatory modules involved in cancers, through deducing miRNA activities from microRNA gene expression data and computational miRNA–mRNA target information. Considering the miRNA function patterns, there should be negative expression correlations for an individual miRNA and its target genes. Based on this theory, Joung et al. and Tran et al. raised two approaches to discover functional mRMs by combining paired miRNA and mRNA expression data and miRNA–mRNA binding information, through Population-based probabilistic learning method (Joung et al. 2007) and rule induction method (Tran et al. 2008), respectively. Finally, a computational framework for the discovery of cancer related miRNA-gene modules was proposed by simultaneous integration of multiple types of genomic data, including matched miRNA and mRNA expression profile, computational miRNA–mRNA target information, and gene–gene interaction network data, which was generated by integrating protein–protein interaction data with DNA–protein interaction data (Zhang et al. 2011). The brief summary about the aforementioned approaches about cancer miRNA–mRNA regulatory modules discovery is presented in Table 8.2.
6.2 The Discovery of Cancer-Related MicroRNA Network Biomarkers
The regulations of miRNAs on mRNAs are only a miniature for the whole biological regulatory network. The incorporation of other information, such as transcriptional factor (TF) regulations may be used for better understanding of specific biological process. (Lu et al. 2011) designed a computational approach for identification of potential microRNA network biomarkers for the progression stages of gastric cancer . Within this approach, computational miRNA–mRNA target information and TF-miRNA regulation data were combined to generate a novel miRNA network for each individual miRNA. The significance of each miRNA network was evaluated according to its GSEA score, and miRNA networks with higher GSEA scores than pre-set threshold were declaimed as potential gastric cancer miRNA network biomarkers.
Except for miRNA regulations, the ultimate expression levels of miRNAs are determined by many factors, e.g., TF regulations. Through integrating analysis on miRNA–gene binding information and TF–gene binding information, Tran et al. introduced a novel way to discover miR–TF regulatory modules in human genome. In this study, many identified modules have been previously reported to be involved in cancer genesis and development (Tran et al. 2010).
7 Databases on Potential Cancer miRNA Biomarkers
With the data accumulation from cancer–miRNA association studies, there have been a couple of online databases that collected the cancer–miRNA associations via text-mining approaches on previous publications. The brief summary about these databases can be referred in Table 8.3.
MiR2Disease is a manually curated database collecting the miRNA deregulation patterns in various human diseases, including cancers (Jiang et al. 2009). It provides the detailed information about miRNA–disease relationships, experimentally validated miRNA targets, and corresponding literature references. Similarly, PhenomiR is another comprehensive repository of deregulation miRNA profiling data for different human diseases and biological processes (Ruepp et al. 2010, 2012). Based on self-defined text-mining rules, miRCancer (Xie et al. 2013) and dbDEMC (Yang et al. 2010) specially focus on the collection of cancer-related differentially expressed miRNAs information. More specifically, there have been also some established databases that merely provide miRNA expression profiling data on certain tumor type, such as S-MED (Sarver et al. 2010), CC-MED (Sarver et al. 2009), and others.
8 Future Directions
Although the current cancer miRNA studies have shed some light on our understanding of cancer genesis and development mechanisms, there is still a long way ahead in this new emerging research area. The heterogeneity of cancer is our main concern. Except the compensation of future more advanced miRNA expression detection technologies, another two research directions might also be potential solutions for this issue.
Recently, a new concept referred as personalized medicine (PM) has been proposed to indicate the customization of healthcare. The importance and urgency about PM has already been emphasized years ago (Long 2007). Therefore, the specific information about patients, such as race, genetic makeup, should also be integrated for the future discovery of cancer miRNA biomarkers.
Currently, network view shows that the disorder conditions (diseases, including cancers) may attribute to the deregulation of specific biological process, not simply the alteration of an individual biological molecule. Network biomarkers should be a better choice for cancer diagnosis. As reviewed above, there have been a couple of studies conducted for the identification of cancer miRNA network biomarkers. In the future, the integration of multi-layer information, such as genomic information, epigenomic information, and clinical information, is need for the discovery of miRNA network biomarkers.
9 Conclusions
This chapter summarizes current advances of miRNA expression detection technologies, the traditional approaches on cancer miRNA biomarker discovery based on these techniques. Computational-based methods on the identification of individual miRNA biomarker and miRNA network biomarker for cancer diagnosis and prognosis are also reviewed herein. Although studies of cancer miRNA biomarkers are still in their infancy, the evolving miRNA profiling measurement technologies, miRNA network information, and computational algorithms offer new insights on cancer mechanism investigation. We can expect the clinical application of miRNA biomarkers for the diagnosis, staging, and prognosis of cancers in the near future.
References
Ahmed FE. Role of miRNA in carcinogenesis and biomarker selection: a methodological view. Expert Rev Mol Diagn. 2007;7(5):569–603.
Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5.
Arora A, Simpson DA. Individual mRNA expression profiles reveal the effects of specific microRNAs. Genome Biol. 2008;9(5):R82.
Babak T, Zhang W, Morris Q, Blencowe BJ, Hughes TR. Probing microRNAs with microarrays: tissue specificity and functional inference. RNA. 2004;10(11):1813–9.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Bielekova B, Martin R. Development of biomarkers in multiple sclerosis. Brain J Neurol. 2004;127(Pt 7):1463–78.
Bonnet E, Tatari M, Joshi A, Michoel T, Marchal K, Berx G, Van de Peer Y. Module network inference from a cancer gene expression data set identifies microRNA regulated modules. PLoS One. 2010;5(4):e10162.
Cheng C, Li LM. Inferring microRNA activities by combining gene expression with microRNA target prediction. PLoS One. 2008;3(4):e1989.
Doledec S, Chessel D. Co-inertia analysis: an alternative method for studying species environment relationships. Freshw Biol. 1994;31(3):277–94.
Dray S, Chessel D, Thioulouse J. Co-inertia analysis and the linking of ecological data tables. Ecol. 2003;84(11):3078–89.
Dugas DV, Bartel B. MicroRNA regulation of gene expression in plants. Curr Opin Plant Biol. 2004;7(5):512–20.
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1.
Gall JG, Pardue ML. Formation and detection of RNA–DNA hybrid molecules in cytological preparations. Proc Natl Acad Sci USA. 1969;63(2):378–83.
Gao W, Lu X, Liu L, Xu J, Feng D, Shu Y. MiRNA-21: a biomarker predictive for platinum-based adjuvant chemotherapy response in patients with non-small cell lung cancer. Cancer Biol Ther. 2012;13(5):330–40.
Griffiths-Jones S. The microRNA registry. Nucleic acids Res. 2004;32(Database issue):D109–11.
Guerau-de-Arellano M, Alder H, Ozer HG, Lovett-Racke A, Racke MK. miRNA profiling for biomarker discovery in multiple sclerosis: from microarray to deep sequencing. J Neuroimmunol. 2012;248(1–2):32–9.
Heneghan HM, Miller N, Kelly R, Newell J, Kerin MJ. Systemic miRNA-195 differentiates breast cancer from other malignancies and is a potential biomarker for detecting noninvasive and early stage disease. Oncologist. 2010;15(7):673–82.
Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM et al. miRTarBase: a database curates experimentally validated microRNA–target interactions. Nucleic Acids Res. 2011;39(Database issue):D163–D169.
Jay C, Nemunaitis J, Chen P, Fulgham P, Tong AW. miRNA profiling for diagnosis and prognosis of human cancer. DNA Cell Biol. 2007;26(5):293–300.
Jayaswal V, Lutherborrow M, Ma DD, Yang YH. Identification of microRNA–mRNA modules using microarray data. BMC genomics. 2011;12:138.
Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37(Database issue): D98–104.
Joung JG, Hwang KB, Nam JW, Kim SJ, Zhang BT. Discovery of microRNA–mRNA modules via population-based probabilistic learning. Bioinformatics. 2007;23(9):1141–7.
Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495–500.
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–9.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54.
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23(20):4051–60.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20.
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115(7):787–98.
Li C, Li JF, Cai Q, Qiu QQ, Yan M, Liu BY, Zhu ZG. MiRNA-199a-3p in plasma as a potential diagnostic biomarker for gastric cancer. Annals Surg Oncol. 2012.
Li C, Li JF, Cai Q, Qiu QQ, Yan M, Liu BY, Zhu ZG. MiRNA-199a-3p: a potential circulating diagnostic biomarker for early gastric cancer. J Surg Oncol. 2013;108(2):89–92.
Lian H. MOST: detecting cancer differential gene expression. Biostatistics. 2008;9(3):411–8.
Lin SL, Kim H, Ying SY. Intron-mediated RNA interference and microRNA (miRNA). Front Biosci : J Virtual Libr. 2008;13:2216–30.
Liu B, Liu L, Tsykin A, Goodall GJ, Green JE, Zhu M, Kim CH, Li J. Identifying functional miRNA-mRNA regulatory modules with correspondence latent dirichlet allocation. Bioinformatics. 2010;26(24):3105–11.
Long M. Side effects of Tamiflu: clues from an Asian single nucleotide polymorphism. Cell Res. 2007;17(4):309–10.
Lu L, Li Y, Li S. Computational identification of potential microRNA network biomarkers for the progression stages of gastric cancer. Int J Data Min Bioinform. 2011;5(5):519–31.
MacDonald JW, Ghosh D. COPA–cancer outlier profile analysis. Bioinformatics. 2006;22(23):2950–1.
Madden SF, Carpenter SB, Jeffery IB, Bjorkbacka H, Fitzgerald KA, O’Neill LA, Higgins DG. Detecting microRNA activity from gene expression data. BMC Bioinform. 2010;11:257.
Maute RL, Schneider C, Sumazin P, Holmes A, Califano A, Basso K, Dalla-Favera R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc Natl Acad Sci USA. 2013;110(4):1404–9.
Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA. 2008;105(5):1608–13.
Ramshankar V, Krishnamurthy A. Lung cancer detection by screening—presenting circulating miRNAs as a promising next generation biomarker breakthrough. Asian Pac J Cancer Prev: APJCP. 2013;14(4):2167–72.
Raponi M, Dossey L, Jatkoe T, Wu X, Chen G, Fan H, Beer DG. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009;69(14):5776–83.
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403(6772):901–6.
Ruepp A, Kowarsch A, Schmidl D, Buggenthin F, Brauner B, Dunger I, Fobo G, Frishman G, Montrone C, Theis FJ. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010;11(1):R6.
Ruepp A, Kowarsch A, Theis F. PhenomiR: microRNAs in human diseases and biological processes. Methods Mol Biol. 2012;822:249–60.
Sarver AL, French AJ, Borralho PM, Thayanithy V, Oberg AL, Silverstein KA, Morlan BW, Riska SM, Boardman LA, Cunningham JM, et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer. 2009;9:401.
Sarver AL, Phalak R, Thayanithy V, Subramanian S. S-MED: sarcoma microRNA expression database. Lab Inv; J Tech Methods Pathol. 2010;90(5):753–61.
Schopman NC, Heynen S, Haasnoot J, Berkhout B. A miRNA–tRNA mix-up: tRNA origin of proposed miRNA. RNA Biol. 2010;7(5):573–6.
Sethupathy P, Corda B, Hatzigeorgiou AG. TarBase: a comprehensive database of experimentally supported animal microRNA targets. RNA. 2006;12(2):192–7.
Tibshirani R, Hastie T. Outlier sums for differential gene expression analysis. Biostatistics. 2007;8(1):2–8.
Tran DH, Satou K, Ho TB. Finding microRNA regulatory modules in human genome using rule induction. BMC Bioinform. 2008;9(Suppl 12):S5.
Tran DH, Satou K, Ho TB, Pham TH. Computational discovery of miR-TF regulatory modules in human genome. Bioinformation. 2010;4(8):371–7.
Wang X, Wang X. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res. 2006;34(5):1646–52.
Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75(5):855–62.
Wu B. Cancer outlier differential gene expression detection. Biostatistics. 2007;8(3):566–75.
Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37(Database issue):D105–110.
Xie B, Ding Q, Han H, Wu D. miRCancer: a microRNA-cancer association database constructed by text mining on literature. Bioinformatics. 2013;29(5):638–44.
Xu J, Li CX, Lv JY, Li YS, Xiao Y, Shao TT, Huo X, Li X, Zou Y, Han QL, et al. Prioritizing candidate disease miRNAs by topological features in the miRNA target-dysregulated network: case study of prostate cancer. Mol Cancer Ther. 2011;10(10):1857–66.
Yang Z, Ren F, Liu C, He S, Sun G, Gao Q, Yao L, Zhang Y, Miao R, Cao Y, et al. dbDEMC: a database of differentially expressed miRNAs in human cancers. BMC Genomics. 2010;11(Suppl 4):S5.
Yoon S, De Micheli G. Prediction and analysis of human microRNA regulatory modules. In: Conference proceedings: annual international conference of the IEEE Engineering in Medicine and Biology Society conference; 2005a, vol 5, p. 4799–802.
Yoon S, De Micheli G . Prediction of regulatory modules comprising microRNAs and target genes. Bioinformatics. 2005b;21 Suppl 2:ii93–100.
Zhang S, Li Q, Liu J, Zhou XJ. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules. Bioinformatics. 2011;27(13):i401–9.
Zhang WY, Zang J, Jing XH, Sun ZD, Yang DR, Guo F, Shen BR. Identification of candidate cancer miRNA biomarkers from miRNA regulatory network: with application to prostate cancer. RNA. 2013 (submitted).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Zhang, W., Shen, B. (2013). Identification of Cancer MicroRNA Biomarkers Based on miRNA–mRNA Network. In: Shen, B. (eds) Bioinformatics for Diagnosis, Prognosis and Treatment of Complex Diseases. Translational Bioinformatics, vol 4. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7975-4_8
Download citation
DOI: https://doi.org/10.1007/978-94-007-7975-4_8
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7974-7
Online ISBN: 978-94-007-7975-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)