
Abstract
Ehlers–Danlos syndrome (EDS) is a genetically and clinically heterogeneous group of connective tissue disorders that typically present with skin hyperextensibility, joint hypermobility, and tissue fragility. The major cause of EDS appears to be impaired biosynthesis and enzymatic modification of collagen. In this chapter, we discuss two types of EDS that are associated with proteoglycan abnormalities: the progeroid type of EDS and dermatan 4-O-sulfotransferase 1 (D4ST1)-deficient EDS. The progeroid type of EDS is caused by mutations in B4GALT7 or B3GALT6, both of which encode key enzymes that initiate glycosaminoglycan (GAG) synthesis. D4ST1-deficient EDS is caused by mutations in CHST14, which encodes an enzyme responsible for post-translational modification of GAG. The clinical and molecular characteristics of both types of EDS are described in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Ehlers–Danlos syndrome (EDS)
- Progeroid type
- B4GALT7 • B3GALT6
- Xylosylprotein beta 1,4-galactosyltransferase, polypeptide 7
- UDP-Gal:βGal β 1,3-galactosyltransferase polypeptide 6
- Dermatan 4-O-sulfotransferase 1 (D4ST1)-deficient EDS
- CHST14
10.1 Introduction
Ehlers–Danlos syndrome (EDS) is a heterogeneous connective tissue disorder that affects as many as 1 in 5,000 individuals. It is characterized by joint and skin laxity, and tissue fragility [44]. In a revised classification, Beighton et al. classified EDS into six major types and several minor types [2]. The major causes of EDS are thought to include abnormal collagen biosynthesis through dominant-negative effects, haploinsufficiency of mutant procollagen α-chains, or deficiencies in collagen processing enzymes [29]. Abnormal glycosaminoglycan (GAG) synthesis and incorrect post-translational modification of GAG in proteoglycans (PGs) were recently identified in the progeroid type of EDS (EDS, progeroid form; MIM#130070, MIM#615349) and dermatan 4-O-sulfotransferase 1 (D4ST1)-deficient EDS (EDS, musculocontractural type; MIM#601776), respectively. In this chapter, the clinical and molecular characteristics of both types of EDS are described.
10.2 Background
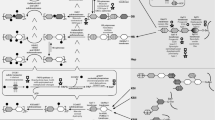
Glycosylation is the addition of a sugar chain (a glycan) to a protein (generating a glycoprotein) or lipid (generating a glycolipid). More than 40 human disorders are thought to be caused by abnormal glycosylation [15, 19]. PGs are composed of core proteins and one or more glycans with modifications. PGs are present in the extracellular matrix and have important diverse biological functions [5]. PG synthesis is initiated by the sequential addition of four monosaccharides (xylose [Xyl], two molecules of galactose [Gal] and glucuronic acid [GlcA]), known as a linker tetrasaccharide, to the serine residue of the core protein backbone (Fig. 10.1a). Additional sugar chains are extended from the linker tetrasaccharide by the addition of repeated disaccharides (usually consisting of 50–150 disaccharides in vivo). Afterwards, some sugars are modified by a series of epimerases (epimerization) and sulfotransferases (sulfation).

Proteoglycan biosynthesis and its defects in two types of EDS. (a) Normal state. The serine residue (Ser) of the core protein and the GAG chain are bound via a linker tetrasaccharide. In CS, the disaccharides are composed of N-acetylgalactosamine (GalNAc) [position A] and glucuronic acid [position B]. In DS, the disaccharides are composed of GalNAc [position A] and Iduronic acid (IdoA) [position B]. B4GALT7 and B3GALT6 add the first and second galactose (Gal) to the xylose of the linker tetrasaccharide (green arrows). D4ST1 then adds the active sulfate to the 4-O position of GalNAc (red arrows) on DS. (b, c) Progeroid type of EDS. The impaired B4GALT7 cannot elongate the glycan chain from the first galactose (b). The impaired B3GALT6 cannot add the second galactose and the following glycan chain (c). (d) D4ST1-deficient EDS. The impaired/inactive D4ST1 cannot add the sulfate to GalNAc. Gal galactose, GlcA glucuronic acid, S active sulfate, Ser serine, Xyl xylose
GAGs are long unbranched polysaccharides consisting of repeating disaccharide units. GAGs are highly negatively charged because of the acidic sugar residues and/or sulfation. Consequently, GAG can change its conformation, attract cations, and bind water. Hydrated GAG gels enable joints and tissues to absorb large pressure changes, providing tissue elasticity. Post-translational modifications such as epimerization, sulfation, and acetylation/deacetylation result in the formation of diverse motifs in the GAG chains, which can bind to a large variety of ligands. Therefore, GAG chains play important roles in regulating growth factor signaling, cell adhesion, proliferation, differentiation, and motility [3, 5, 45].
GAGs can be divided into two groups: (1) galactosaminoglycans such as chondroitin sulfate (CS) and dermatan sulfate (DS), and (2) glucosaminoglycans such as hyaluronic acid, keratan sulfate, heparan sulfate, and heparin [42]. Two types of glycosylation are known: O-glycosylation and N-glycosylation (Fig. 10.2a). Most GAGs (except for keratan sulfate and hyaluronic acid) are O-glycans that bind to the glycan via an oxygen molecule in the serine or threonine residue of the core protein (Fig. 10.2a). Notably, failure to add the first or second galactose residue of the tetrasaccharide results in the progeroid type of EDS (Fig. 10.1b, c).

Chemical structures of proteoglycan and disaccharides. (a) Chemical structure of O-linked and N-linked glycan. O-linked glycan can be linked via the O-element of serine or threonine. The diagram shows linking for serine. (b) Chemical structures of the disaccharide units of CS (left) and DS (right)
The CS and DS GAGs are produced via the same pathway (Fig. 10.3a). In this pathway, after the linker tetrasaccharide attaches to the serine residue of the core protein, GalNAc (N-acetyl galactosamine) transferase I elongates the glycan branch to create CS/DS. The enzyme C5-carboxy epimerase transforms glucuronic acid (GlcA) to iduronic acid (IdoA), which is specific for dermatan/DS (Fig. 10.3a). DS actually exists in a CS/DS hybrid state, containing GlcA–GalNAc and IdoA–GalNAc disaccharides (Figs. 10.2b and 10.3a) [12]. Dermatan 4-O-sulfotransferase 1(D4ST1) specifically transfers an active sulfate to the 4-O position on the GalNAc residue of dermatan. The transfer of the active sulfate is impaired in D4ST1-deficient EDS (Figs. 10.1d and 10.3b).

Effects of D4ST1 defects on the biosynthesis of CS and DS. (a) The starting structure is chondroitin with a repeating disaccharide consisting of GalNAc [position A] and GlcA [position B]. Sulfation by 6-O-GalNAc sulfotransferase and 4-O-GalNAc sulfotransferase creates CS from chondroitin. To produce DS, first, C5-carboxy epimerase replaces GlcA with IdoA [position C]. This process is bidirectional as indicated by the bi-directional arrows. Then, D4ST1 adds sulfates to dermatan creating DS and prevents back epimerization. DS is often detected as a CS/DS hybrid. (b) In D4ST1-deficient EDS, back epimerization from IdoA to GlcA occurs. Consequently, neither DS nor dermatan are detected in fibroblasts derived from patients. Gal galactose, GlcA glucuronic acid, S active sulfate, Ser serine, Xyl xylose
10.3 The Progeroid Type of EDS (type 1: MIM#130070, type2: MIM#615349)
Alternative Names (MIM#130070)
-
Xylosylprotein 4-β-galactosyltransferase deficiency
-
XGPT deficiency
-
Galactosyltransferase I deficiency
10.3.1 Clinical Manifestations
Hernandez et al. reported five unrelated males in 1979, 1981, and 1986 representing a distinct variant of EDS. These males presented with a progeroid facial appearance, mild intellectual disability, and multiple nevi, in addition to hyperextensibility and fragility of skin, a high propensity for bruising, and joint hypermobility (particularly of the digits) [16–18]. A wrinkled face, curly and fine hair, scant eyebrows/eyelashes, telecanthus, periodontitis, multiple caries, low set/prominent ears, pectus excavatum, winged scapulae, and pes planus were observed in all five patients. Cryptorchidism and inguinal hernia were also noticed in four of the patients. Interestingly, the occurrence of the disorder in all of these patients was sporadic and the ages of their fathers were relatively advanced (33–55 years old). These characteristics prompted Hernandez et al. to speculate that the syndrome is caused by a de novo mutation [16].
In 1987, Kresse et al. reported a Danish male patient who was born to non-consanguineous healthy parents [26]. This patient presented with the clinical features observed in the original five patients, as well as a triangular head with a tiny face, frontal bossing, mid-face hypoplasia, a broad nasal bridge, prominent deep-set eyes, a small mouth, dental anomalies, low-set ears, short stature, osteopenia of all bones, dysplasia of some bones, and hypotonia. In 2004, Faiyaz-Ul-Haque et al. reported two patients from a large consanguineous Qatari family. The clinical features of both Qatari patients and the Danish patient seemed to be different from those of the original five patients [14].
10.3.2 Genetic Information
10.3.2.1 B4GALT7
In 1999, two different research groups [1, 33] identified compound heterozygous mutations of gene for xylosylprotein beta 1,4-galactosyltransferase, polypeptide 7 (B4GALT7, NM_007255.2), c.557C>A (p.Ala186Asp) and c.617T>C (p.Leu206Pro) in the Danish patient reported by Kresse in 1987 [26]. The two Qatari patients from a large consanguineous family were analyzed in 2004 [14]. Based on the hypothesis of autosomal recessive inheritance, haplotype analysis using microsatellite markers for the limited candidate loci delineated a homozygous region from D5S469 and D5S2111, which harbors B4GALT7 [14]. A homozygous missense mutation (c.808C>T, p.Arg270Cys) in B4GALT7 was identified. Interestingly, the clinical phenotype of the Qatari patients was milder than that of the Danish one.
B4GALT7 was cloned by Okajima et al. [34]. The gene consists of six coding exons with a 948-bp open reading frame. This gene encodes xylosylprotein β-1,4-galactosyltransferase, polypeptide 7 (B4GALT7; aliases: galactosyltransferase I, XGPT1, and XGALT1), which is 327 amino acids long and its molecular weight is 37.4 kDa. B4GALT7 is a type II transmembrane protein localized in the Golgi apparatus, and is a key initiator of GAG synthesis as it attaches the first galactose of the linker tetrasaccharide of PGs (Fig. 10.1a, b).
10.3.2.2 B3GALT6
In 2013, Nakajima et al. have identified compound heterozygous mutations of B3GALT6 (NM_080605.3) in three patients with progeroid form of EDS [32]. This intronless gene has a 990-bp open reading frame and encodes UDP-Gal:βGal β 1,3-galactosyltransferase polypeptide 6 (alternatively galactosyltransferase -II: GalT-II), which is 329 amino acids long and its molecular weight is 37.1kDa. It is also the type II transmembrane protein localized in the Golgi apparatus, and it attaches the second galactose of the tetrasaccharide linker of PGs (Fig. 10.1a, c). So far, two missense (c.16C>T, p.Arg6Trp and c.925T>A, p.Ser309Thr), two frameshift deletions (c.353delA, p.Asp118Alafs*160 and c.588delG, p.Arg197Alafs*81) and one in-frame deletion (c.415_423del, p.Met139Ala141del) were reported in this type of EDS [32].
10.3.3 Biochemical Characteristics
10.3.3.1 B4GALT7
Kresse et al. reported that their patient’s fibroblasts produced only PG chain-free core proteins (molecular weight: 46 and 44 kDa) whereas control fibroblasts produced normal PG chains [26]. Additionally, the GAG-free core protein in that patient contained unsubstituted xylose residues (Fig. 10.1b).
Okajima et al. measured the enzyme activity of exogenously expressed proteins (wild type, p.Ala186Asp, p.Leu206Pro) in XGalT-1/B4GALT7-deficient CHO cells [33]. In total cell lysates, the enzyme activity of the p.Ala186Asp mutant was approximately 50 % lower than that of the wild-type protein, whereas the activity of the p.Leu206Pro mutant was almost undetectable. Interestingly, the wild-type and p.Ala186Asp proteins were localized in the Golgi apparatus whereas the p.Leu206Pro mutant existed in the cytoplasm. The α-helix disrupted by p.Leu206Pro may alter the protein’s conformation, thus impairing intracellular trafficking and enzyme activity [33].
B4GALT7 activity in fibroblasts from another patient with a homozygous mutation, c.808C>T (p. Arg270Cys), was also lower than that of controls [40]. The extracellular matrix around the B4GALT7Arg270Cys mutant fibroblasts was disorganized without banded fibrils. Furthermore, the proliferation of B4GALT7Arg270Cys fibroblasts was significantly reduced to 45 % of the level of control fibroblasts [40].
Bui et al. measured galactosyltransferase activity of B4GALT7 mutants expressed in CHO pgsB-618 cells using 4-methylumbelliferyl-β-D-xylopyranoside as acceptor substrate. The enzyme activities of the p.Arg270Cys, p.Ala186Asp, and p.Leu206Pro mutants were decreased to 60, 11, and 0 % (undetectable) of that of the wild-type enzyme [4]. It has been reported that the clinical features of patients with the homozygous p.Arg270Cys mutation appear to be milder than those of patients with compound heterozygous mutations, including p.Ala186Asp or p.Leu206Pro, supporting the different effects of these mutations.
10.3.3.2 B3GALT6
Nakajima et al. measured the galactosyltransferase activity of B3GALT6 in vitro using soluble-FLAG-tagged proteins for wild-type and mutant (p.Ser309Thr) which was observed common in two families and revealed the enzyme activity of the mutant protein was significantly decreased compared to the wild-type [32].
10.4 D4ST1-Deficient EDS (MIM#601776)
Alternative Names
-
Ehlers–Danlos syndrome, type VIB, formerly
-
Ehlers–Danlos syndrome, Kosho type
-
Ehlers–Danlos syndrome, musculocontractural type
-
Adducted thumbs, clubfoot, and progressive joints and skin laxity syndrome
-
Adducted thumb-clubfoot syndrome (ATCS)
-
Dündar syndrome
-
Arthrogryposis, distal, with peculiar faces and hydronephrosis
10.4.1 Clinical Manifestations
The kyphoscoliosis type of EDS (formerly known as, EDS type VI) is characterized by generalized joint laxity, severe muscular hypotonia and scoliosis at birth, scleral fragility, and rupture of the ocular globe [2]. This disorder is essentially caused by lysyl hydroxylase deficiency (EDS type VIA); other patients with similar clinical manifestations but without lysyl hydroxylase deficiency were classified as EDS type VIB.
In 2005, Kosho et al. reported two unrelated patients with fragile and hyperextensible skin, a high propensity for bruising, generalized joint laxity, kyphoscoliosis, and the major features of EDS VI, as well as a characteristic craniofacial appearance, and multiple congenital contractures [25]. Lysyl hydroxylase deficiency was excluded in these patients by analysis of the urinary deoxypyridinoline:pyridinoline ratio, and the patients were tentatively classified as EDS VIB. Kosho et al. subsequently reported on four additional unrelated patients and concluded that the patients represented a new type of EDS [23]. Notably, all six patients had homozygous or compound heterozygous mutations in CHST14 [31]. Loss-of-function mutations in CHST14 were independently found in 11 patients from four families with a rare arthrogryposis syndrome known as “adducted thumb-clubfoot syndrome (ATCS)” [9–11, 21, 43] and in three patients from two families who were originally classified as suffering from EDS VIB [27]. Malfait et al. suggested that these patients had the same disorder, which they termed “musculocontractural EDS” [27]. Shimizu et al. described the clinical characteristics of two additional patients together with a review of all of the patients reported at that time; their findings support the notion that the three independently identified conditions represent a single type of EDS [41]. Conversely, Janecke et al. claimed that the disorder should not be categorized as a type of EDS because of the presence of atypical clinical features, including facial dysmorphism, multiple congenital contractures, visceral anomalies, and impaired biosynthesis of DS as a cause of the disorder, and proposed the term DS-deficient adducted thumb-clubfoot syndrome [20]. In their response, Kosho et al. provided clinical and etiological evidence from which the disorder could be categorized as a type of EDS, because of the presence of all major features of EDS, including connective tissue fragility which required special and appropriate management of these patients. Decorin-mediated impaired assembly of collagen fibrils was the primary cause of progressive connective tissue fragility in this type [24]. Therefore, Kosho et al. proposed that the term D4ST1-deficient EDS (adducted thumb-clubfoot syndrome) was appropriate for this syndrome [24]. The current OMIM (http://www.ncbi.nlm.nih.gov/omim) registration of this disorder is EDS, musculocontractural type.
To date, descriptions of 26 patients (12 males, 14 females) from 17 families have been published [9–11, 21, 23, 25, 27, 30, 31, 41, 43, 46, 48, 49]. This syndrome is characterized by a unique set of clinical features consisting of progressive systemic manifestations, including tissue fragility (e.g., skin hyperextensibility and fragility, progressive spinal and foot deformities, and large subcutaneous hematomas) and various malformations (e.g., facial features, congenital eye/heart/gastrointestinal defects, congenital multiple contractures). We have summarized the main clinical features of this syndrome in each organ in Table 10.1
10.4.1.1 Craniofacial Features
The characteristic craniofacial features apparent at birth or during early infancy include a large fontanelle, hypertelorism, short and down slanting palpebral fissures, blue sclerae, a short nose with hypoplastic columella, low-set and rotated ears, a high or cleft palate, a long philtrum, a thin upper-lip vermilion, a small mouth, and micro-retrognathia (Fig. 10.4a, b). Slender and asymmetrical facial shapes with a protruding jaw are generally observed from school age onwards (Fig. 10.4c, d).

Clinical photographs of patients with D4ST1-deficient EDS. (a–d) Facial features of a patient at 23 days (a), 3 years (b), and 16 years (c, d) of age. (e, f) Images of the hand in a patient with an adducted thumb at 1 month of age (e) and cylindrical fingers at 19 years of age (f). (g–i) Images of the foot in a patient with bilateral clubfeet at 1 month of age (g, h) and progressive talipes deformities (planus and valgus) at 19 years of age (i). (j–m) Radiographs of a 16-year-old patient show diaphyseal narrowing of the phalanges and metacarpals (j, k) and kyphoscoliosis with tall vertebral bodies (l, m). (n, o) Cutaneous features of a 19-year-old patient with hyperextensibility (n), atrophic scars, and fistula formation (o). (p) A massive cranial subcutaneous hematoma in the head of a 6-year-old patient after falling onto the floor. (q) A subcutaneous hematoma in the leg of a 16-year-old patient (All figures were originally published in Kosho et al. [23] except Fig. 10.4p, which was published in Kosho et al. [25])
10.4.1.2 Skeletal Features
Congenital multiple contractures, particularly adduction-flexion contractures of the thumbs and talipes equinovarus, are the main skeletal features (Fig. 10.4e, g, h). Fingers with a “tapering”, “slender”, and “cylindrical” shape are also common (Fig. 10.4f). Aberrant finger movement was described in three patients. Four patients had tendon abnormalities, including anomalous insertion of the flexor muscles, which probably caused the congenital contractures. Spinal deformities (e.g., scoliosis and kyphoscoliosis) and talipes deformities (e.g., planus and valgus) (Fig. 10.4i) occurred and progressed during childhood. Marfanoid habitus, recurrent joint dislocations, and pectus deformities (e.g., flat and thin, excavatum, and carinatum) were also evident. Bone mineral density was decreased in five patients and normal in two. Urine concentrations of the N-telopeptide of collagen type I, an osteoclast marker, were increased in three patients, whereas serum bone-specific alkaline phosphatase concentrations, an osteoblast marker, were normal in three, suggesting that increased osteoclast activity but normal osteoblast activity could cause osteopenia or osteoporosis. Radiologically, diaphyseal narrowing of the phalanges and metacarpals was noted in six patients (Fig. 10.4j, k). Talipes valgus and planus or cavum, with diaphyseal narrowing of the phalanges and metatarsals, were noted in six patients. Tall vertebral bodies were noted in five patients (Fig. 10.4l, m).
10.4.1.3 Cutaneous Features
Cutaneous features were apparent in most patients, including hyperextensibility to redundancy (Fig. 10.4n), a high propensity for bruising, fragility leading to atrophic scars (Fig. 10.4o), acrogeria-like fine palmar creases or wrinkles, hyperalgesia to pressure, and recurrent subcutaneous infections with fistula formation. The palmar creases increased and became deeper with age.
10.4.1.4 Cardiovascular Features
Large subcutaneous hematomas were common, and frequently required intensive treatment, including hospital admission, blood transfusion, and surgical drainage (Fig. 10.4p, q). The lesions were thought to be caused by the rupture of a subcutaneous artery or vein. Bleeding time was prolonged in two patients (9 min and 11 min) and was normal in three. Intranasal administration of 1-desamino-8-D-arginine vasopressin prevented the development of large subcutaneous hematomas after trauma [49]. Four patients had congenital heart defects including an atrial septal defect in three, a patent ductus arteriosus in one, and coarctation of the aorta in one. Five patients had cardiac valve abnormalities including one who underwent surgery for infectious endocarditis, which was probably caused by aortic valve or mitral valve regurgitation.
10.4.1.5 Respiratory Features
Three adult patients developed pneumothorax or hemopneumothorax requiring chest tube drainage.
10.4.1.6 Gastrointestinal Features
Numerous gastrointestinal abnormalities were reported, including diverticular perforation in two adult patients, constipation in seven patients, abdominal pain in two patients, and other disorders in one patient (common mesentery, absence of the gastrocolic omentum with a spontaneous volvulus of small intestine, gastric ulcer, and malrotation with duodenal obstruction).
10.4.1.7 Genitourinary Features
Urological complications included nephrolithiasis or cystolithiasis in five patients, hydronephrosis in three, a dilated or atonic bladder with recurrent urinary tract infection in two, and a horseshoe kidney in one. Cryptorchidism was observed in eight male patients, including one who underwent orchiopexy because of hypogonadism in adulthood. Poor breast development was noted in five adolescent or adult patients. No pregnant females have been reported.
10.4.1.8 Ophthalmologic Features
Various ophthalmological complications have been reported, including strabismus in 12 patients, refractive errors in nine, glaucoma or elevated intraocular pressure in six, microcornea or microphthalmia in three, and retinal detachment in three.
10.4.1.9 Hearing Impairment
Six patients had hearing impairments, including for high-pitched sounds in three.
10.4.1.10 Growth
Patients showed mild prenatal growth retardation as the mean birth length was −0.5 standard deviations (SD), the mean birth weight was −0.6 SD, and the mean birth occipitofrontal circumference (OFC) was −0.2 SD. Postnatal growth was also mildly impaired, as the patients were generally slender with relative macrocephaly. The mean height was −0.9 SD, the mean weight was −1.5 SD, and the mean OFC was −0.2 SD.
10.4.1.11 Development and Neuromuscular Features
Gross motor developmental delay was observed in 14 patients, as the median age of independent walking was 2 years 1 month. Two patients, aged 15 years and 32 years, could not walk unassisted. An underlying myopathic process was observed in two patients. Mild intellectual disability was apparent in four patients. One patient had a global psychomotor delay at 1.5 years of age, but his intellectual quotient was approximately 90 at the age of 7 years 2 months. Brain imaging showed ventricular enlargement and/or asymmetry in seven patients, absence of the left septum pellucidum in one patient, and a short corpus callosum, mildly prominent Sylvian fissures, and periventricular nodular heterotopias. Two patients had spinal cord tethering.
10.4.2 Genetic Information
Autosomal recessive inheritance was considered based on the presence of this syndrome in consanguineous families [11, 27, 31]. Three independent groups have performed homozygosity mapping and/or linkage analysis and each showed that the gene carbohydrate (N-acetylgalactosamine 4-O) sulfotransferase 14 (CHST14, NM_130468.3) was responsible for this syndrome [11, 27, 31].
The CHST14 gene was first cloned by Evers et al. [13]. It contains one coding exon (1,131-bp open reading frame) and is localized at 15q15.1. This gene encodes D4ST1, a 376 amino acid type II transmembrane protein (molecular weight: 43 kDa), that is localized in the Golgi membrane. It transfers a sulfate group from 3′-phosphoadenosine 5′-phosphosulfate to position 4 of the GalNAc residues in dermatan to generate DS (Figs. 10.1a and 10.3a). Northern blotting revealed that CHST14 is mainly expressed in heart, placenta, liver, and pancreas, and is weakly expressed in lung, skeletal muscle, and kidney [13].
To date, 11 pathogenic mutations of CHST14 have been identified: p.Val49*, p.Lys69*, p.Arg135_Leu137delinsGlyThrGln, p.Phe209Ser, p.Arg213Pro, p.Lys226Alafs*16, p.Arg274Pro, p.Pro281Leu, p.Cys289Ser, p.Tyr293Cys, and p.Glu334Glyfs*107 [11, 27, 30, 31, 46, 48]. (p.Val48* was corrected to p.Val49*; Erratum in Am J Med Genet Part A 161A(2):403 (2013)) (p.Arg135Gly and p.Leu137Gln were originally reported by Dündar et al., but lately registered as c.403_410delCGCACCCTinsGGCACCCA, p.Arg135_Leu137delinsGlyThrGln in The Human Gene Mutation Database: https://portal.biobase-international.com/hgmd/pro/genesearch.php). Because these are protein truncation mutations and missense mutations, it seems likely that the mutations cause a loss of function.
10.4.3 Biochemical Information
Dündar et al. reported that DS-derived IdoA-GalNAc(4S) disaccharide was undetectable in fibroblasts derived from a patient with a homozygous p.Arg213Pro mutation. They also reported that GlcA-GalNAc(4S) content was greatly increased in the fibroblast extract and the culture media obtained from cultures of fibroblasts derived from this patient as compared with control fibroblasts [11]. It was also found that the amount of nonsulfated disaccharides (GlcA-GalNAc and IdoA-GalNAc) was increased in the cell extract and its media from the patient’s fibroblast as compared with normal control fibroblasts. From these results, Dündar et al. proposed that epimerization of GlcA to IdoA by C5-carboxy epimerase is followed by sulfation of the C4 hydroxyl on the adjacent GalNAc residue by D4ST1. This process generates DS from dermatan and prevents back-epimerization from IdoA to GlcA [11, 28].
Miyake et al. measured the sulfotransferase activity of COS7 cells transfected with wild-type and mutant D4ST1 harboring the p.Lys69*, p.Pro281Leu, p.Cys289Ser, or p.Tyr293Cys mutations. The enzyme activity of the mutants was as low as that in mock transfected cells, suggesting that these missense mutations result in the loss of function [31]. The disaccharide composition of the decorin GAG chain isolated from the patient’s fibroblasts consisted only of CS, without DS, while the chains isolated from normal fibroblasts consisted of CS/DS hybrid chains [31]. Furthermore, the level of nonsulfated dermatan was negligible in the patient’s fibroblasts [31]. Thus, in this syndrome, the CS/DS chain is replaced with the CS chain, even though the core proteins are normal.
10.4.4 Pathology and Pathophysiology
Of the major DS proteoglycans in skin, decorin was a focus of research because it binds to collagen fibrils via its core protein and its GAG chains act as interfibrillar bridges [38, 39]. Three α collagen chains are self-assembled to generate tropocollagen, in the form of a triple helix. Tropocollagen then self-assembles to form collagen fibrils via decorin (Fig. 10.5a). Collagen fibrils are assembled into a collagen fiber, known as the collagen bundle, via the antiparallel complex of the CS/DS hybrid GAG chains of decorin, which acts like a bridge to provide a space between individual fibrils and tighten the collagen fiber (Fig. 10.5c, d).

Putative model of abnormal collagen bundle assembly in D4ST1-deficient EDS. (a) Tropocollagen directly binds to decorin and forms a collagen fibril. The blue lines represent the CS/DS hybrid chain. (b) Illustration of the sliding filament model showing reversible longitudinal slippage between the antiparallel GAG chains. The black lines represent unspecified GAGs. (c, d) In the normal state, the CS/DS chains can bend against the direction of mechanical compression and rebound to the original structure (c). Thus, the collagen bundles are refractory to compression stress (d). (e, f) In D4ST1-deficient EDS, the CS/DS chains are replaced with CS chains (red lines). These chains cannot resist mechanical compression, resulting in irreversible scattering of the collagen fibrils. (g) The size and shape of the collagen fibrils are highly variable in decorin-deficient mice
The GAGs span collagen fibrils in the extracellular matrix of skin and tendons, and the length of the GAG chain determines the width of the interfibrillar gap [35, 36]. Elasticity of the extracellular matrix is explained by the sliding filament model, which allows reversible longitudinal slippage between the antiparallel GAG chains (Fig. 10.5b) [39]. Because tissue stability and elasticity depend on the structure of the GAG bridges, irreversible damage can occur if the bridges are inelastic [39].
Decorin is composed of a horseshoe-shaped core protein (molecular weight: ~45 kDa) and a single CS/DS hybrid chain on the N-terminal side (Fig. 10.5a) [22, 47]. Weber et al. reported that the model structure of decorin consists of an arch in which the inner concave surface is formed from a curved β-sheet and the outer convex surface is formed from α-helices. They also proposed that one tropocollagen fiber lies within the decorin convex and another interacts with one arm of the arch [47]. The IdoA:GlcA ratio in DS ranges from ~10 to >90 % depending on the tissue type [39]. Importantly, L-IdoA residues in DS can easily undergo conformational changes, unlike GlcA in CS [6, 7]. Thus, the IdoA:GlcA ratio should be higher in more flexible tissues [39].
Light microscopic investigation of skin specimens from two patients showed that fine collagen fibers were predominant in the reticular to papillary dermis and the number of thick collagen bundles was markedly reduced [31]. Electron microscopic examination of the specimens showed that collagen fibrils were dispersed throughout the reticular dermis, whereas they were regularly and tightly assembled in control tissue. Surprisingly, each collagen fibril was smooth and round, with little variation in size or shape, similar to the fibril in the control tissue (Fig. 10.5d, f) [31]. The disaccharide composition of the decorin GAG chain from a patient’s fibroblasts only consisted of CS, without DS disaccharide, whereas control fibroblasts consisted of a CS/DS hybrid [31]. The transition of decorin from the CS/DS hybrid chain to a CS chain probably decreases the flexibility of the GAG chain. The sliding filament model proposes that mechanical compression might also work in the CS chain of D4ST1-deficient patients, but the inflexibility of the CS chain is unable to tolerate higher mechanical pressures or is too inelastic to maintain normal skin properties (Fig. 10.5e, f). This irreversible event could explain the progressive clinical course of this disease.
Interestingly, there were marked variations in the size and shape of dermal collagen fibrils in decorin-null mice (Fig. 10.5g) [8]. These findings suggest that the decorin core protein is important for collagen fibril formation, and that the CS/DS hybrid chain of decorin PG regulates the space between the collagen fibrils and form collagen bundles, as previously reported [37]. These findings suggest that the main pathological basis of this disorder could be insufficient assembly of collagen fibrils.
However, Dündar et al. reported that the light microscopic and electron microscopic findings of a patient’s skin were unchanged compared to the normal control [11]. Malfait et al. reported that, in their patient, most collagen bundles were small diameter in size, and some were composed of collagen fibrils of varying diameter that were separated by irregular interfibrillar spaces [27]. In addition, the fibroblasts exhibited an elongated and/or dilated endoplasmic reticulum. So far, definitive histopathologic characteristics have not been established, so further studies are strongly encouraged to determine the major histological characteristics and underlying pathophysiology of this disorder.
Abbreviations
- CHST14:
-
Carbohydrate (N-Acetylgalactosamine 4-O) Sulfotransferase 14
- D4ST1:
-
Dermatan 4-O-sulfotransferase 1
- EDS:
-
Ehlers–Danlos Syndrome
- GAG:
-
Glycosaminoglycan
- Gal:
-
Galactose
- GalNAc:
-
N-Acetylgalactosamine
- GlcA:
-
Glucuronic Acid
- IdoA:
-
Iduronic Acid
- PG:
-
Proteoglycan
- Xyl:
-
Xylose
References
Almeida R, Levery SB, Mandel U, Kresse H, Schwientek T, Bennett EP, Clausen H (1999) Cloning and expression of a proteoglycan UDP-galactose:beta-xylose beta1,4-galactosyltransferase I. A seventh member of the human beta4-galactosyltransferase gene family. J Biol Chem 274:26165–26171
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ (1998) Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet 77:31–37
Bishop JR, Schuksz M, Esko JD (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446:1030–1037
Bui C, Talhaoui I, Chabel M, Mulliert G, Coughtrie MW, Ouzzine M, Fournel-Gigleux S (2010) Molecular characterization of beta1,4-galactosyltransferase 7 genetic mutations linked to the progeroid form of Ehlers-Danlos syndrome (EDS). FEBS Lett 584:3962–3968
Bulow HE, Hobert O (2006) The molecular diversity of glycosaminoglycans shapes animal development. Ann Rev Cell Dev Biol 22:375–407
Casu B, Petitou M, Provasoli M, Sinay P (1988) Conformational flexibility: a new concept for explaining binding and biological properties of iduronic acid-containing glycosaminoglycans. Trends Biochem Sci 13:221–225
Catlow KR, Deakin JA, Wei Z, Delehedde M, Fernig DG, Gherardi E, Gallagher JT, Pavao MS, Lyon M (2008) Interactions of hepatocyte growth factor/scatter factor with various glycosaminoglycans reveal an important interplay between the presence of iduronate and sulfate density. J Biol Chem 283:5235–5248
Danielson KG, Baribault H, Holmes DF, Graham H, Kadler KE, Iozzo RV (1997) Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol 136:729–743
Dündar M, Demiryilmaz F, Demiryilmaz I, Kumandas S, Erkilic K, Kendirci M, Tuncel M, Ozyazgan I, Tolmie JL (1997) An autosomal recessive adducted thumb-club foot syndrome observed in Turkish cousins. Clin Genet 51:61–64
Dündar M, Kurtoglu S, Elmas B, Demiryilmaz F, Candemir Z, Ozkul Y, Durak AC (2001) A case with adducted thumb and club foot syndrome. Clin Dysmorphol 10:291–293
Dündar M, Müller T, Zhang Q, Pan J, Steinmann B, Vodopiutz J, Gruber R, Sonoda T, Krabichler B, Utermann G, others (2009) Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am J Hum Genet 85:873–882
Esko JD, Kimata K, Lindahl U (2009) Proteoglycans and sulfated glycosaminoglycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (eds) Essentials of glycobiology. Cold Spring Harbor, New York
Evers MR, Xia G, Kang HG, Schachner M, Baenziger JU (2001) Molecular cloning and characterization of a dermatan-specific N-acetylgalactosamine 4-O-sulfotransferase. J Biol Chem 276:36344–36353
Faiyaz-Ul-Haque M, Zaidi SH, Al-Ali M, Al-Mureikhi MS, Kennedy S, Al-Thani G, Tsui LC, Teebi AS (2004) A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danlos syndrome resembling the progeroid type. Am J Med Genet Part A 128A:39–45
Freeze HH (2006) Genetic defects in the human glycome. Nat Rev Genet 7:537–551
Hernandez A, Aguirre-Negrete MG, Gonzalez-Flores S, Reynoso-Luna MC, Fragoso R, Nazara Z, Tapia-Arizmendi G, Cantu JM (1986) Ehlers-Danlos features with progeroid facies and mild mental retardation. Further delineation of the syndrome. Clin Genet 30:456–461
Hernandez A, Aguirre-Negrete MG, Liparoli JC, Cantu JM (1981) Third case of a distinct variant of the Ehlers-Danlos Syndrome (EDS). Clin Genet 20:222–224
Hernandez A, Aguirre-Negrete MG, Ramirez-Soltero S, Gonzalez-Mendoza A, Martinez y Martinez R, Velazquez-Cabrera A, Cantu JM (1979) A distinct variant of the Ehlers-Danlos syndrome. Clin Genet 16:335–339
Jaeken J, Hennet T, Freeze HH, Matthijs G (2008) On the nomenclature of congenital disorders of glycosylation (CDG). J Inherit Metab Dis 31:669–672
Janecke AR, Baenziger JU, Müller T, Dündar M (2011) Loss of dermatan-4-sulfotransferase 1 (D4ST1/CHST14) function represents the first dermatan sulfate biosynthesis defect, “dermatan sulfate-deficient adducted thumb-clubfoot syndrome”. Hum Mutat 32:484–485
Janecke AR, Unsinn K, Kreczy A, Baldissera I, Gassner I, Neu N, Utermann G, Müller T (2001) Adducted thumb-club foot syndrome in sibs of a consanguineous Austrian family. J Med Genet 38:265–269
Kobe B, Deisenhofer J (1993) Crystal structure of porcine ribonuclease inhibitor, a protein with leucine-rich repeats. Nature 366:751–756
Kosho T, Miyake N, Hatamochi A, Takahashi J, Kato H, Miyahara T, Igawa Y, Yasui H, Ishida T, Ono K, others (2010) A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am J Med Genet Part A 152A:1333–1346
Kosho T, Miyake N, Mizumoto S, Hatamochi A, Fukushima Y, Yamada S, Sugahara K, Matsumoto N (2011) A response to: loss of dermatan-4-sulfotransferase 1 (D4ST1/CHST14) function represents the first dermatan sulfate biosynthesis defect, “dermatan sulfate-deficient Adducted Thumb-Clubfoot Syndrome”. Which name is appropriate, “Adducted Thumb-Clubfoot Syndrome” or “Ehlers-Danlos syndrome”? Hum Mutat 32:1507–1509
Kosho T, Takahashi J, Ohashi H, Nishimura G, Kato H, Fukushima Y (2005) Ehlers-Danlos syndrome type VIB with characteristic facies, decreased curvatures of the spinal column, and joint contractures in two unrelated girls. Am J Med Genet Part A 138A:282–287
Kresse H, Rosthoj S, Quentin E, Hollmann J, Glossl J, Okada S, Tonnesen T (1987) Glycosaminoglycan-free small proteoglycan core protein is secreted by fibroblasts from a patient with a syndrome resembling progeroid. Am J Hum Genet 41:436–453
Malfait F, Syx D, Vlummens P, Symoens S, Nampoothiri S, Hermanns-Le T, Van Laer L, De Paepe A (2010) Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum Mutat 31:1233–1239
Malmström A (1984) Biosynthesis of dermatan sulfate. II. Substrate specificity of the C-5 uronosyl epimerase. J Biol Chem 259:161–165
Mao JR, Bristow J (2001) The Ehlers-Danlos syndrome: on beyond collagens. J Clin Invest 107:1063–1069
Mendoza-Londono R, Chitayat D, Kahr WH, Hinek A, Blaser S, Dupuis L, Goh E, Badilla-Porras R, Howard A, Mittaz L, others (2012) Extracellular matrix and platelet function in patients with musculocontractural Ehlers-Danlos syndrome caused by mutations in the CHST14 gene. Am J Med Genet Part A 158A:1344–1354
Miyake N, Kosho T, Mizumoto S, Furuichi T, Hatamochi A, Nagashima Y, Arai E, Takahashi K, Kawamura R, Wakui K, others (2010) Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum Mutat 31:966–974
Nakajima M, Mizumoto S, Miyake N, Kogawa R, Iida A, Ito H, Kitoh H, Hirayama A, Mitsubuchi H, Miyazaki O, others (2013) Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am J Hum Genet 92:927–934
Okajima T, Fukumoto S, Furukawa K, Urano T (1999) Molecular basis for the progeroid variant of Ehlers-Danlos syndrome. Identification and characterization of two mutations in galactosyltransferase I gene. J Biol Chem 274:28841–28844
Okajima T, Yoshida K, Kondo T, Furukawa K (1999) Human homolog of Caenorhabditis elegans sqv-3 gene is galactosyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J Biol Chem 274:22915–22918
Scott JE (1988) Proteoglycan-fibrillar collagen interactions. Biochem J 252:313–323
Scott JE (1992) Morphometry of cupromeronic blue-stained proteoglycan molecules in animal corneas, versus that of purified proteoglycans stained in vitro, implies that tertiary structures contribute to corneal ultrastructure. J Anat 180(Pt 1):155–164
Scott JE (1995) Extracellular matrix, supramolecular organisation and shape. J Anat 187(Pt 2):259–269
Scott JE (1996) Proteodermatan and proteokeratan sulfate (decorin, lumican/fibromodulin) proteins are horseshoe shaped. Implications for their interactions with collagen. Biochemistry 35:8795–8799
Scott JE (2003) Elasticity in extracellular matrix ‘shape modules’ of tendon, cartilage, etc. A sliding proteoglycan-filament model. J Physiol 553:335–343
Seidler DG, Faiyaz-Ul-Haque M, Hansen U, Yip GW, Zaidi SH, Teebi AS, Kiesel L, Gotte M (2006) Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (beta4GalT-7). J Mol Med (Berl) 84:583–594
Shimizu K, Okamoto N, Miyake N, Taira K, Sato Y, Matsuda K, Akimaru N, Ohashi H, Wakui K, Fukushima Y, others (2011) Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: observation of two additional patients and comprehensive review of 20 reported patients. Am J Med Genet Part A 155A:1949–1958
Sisu E, Flangea C, Serb A, Zamfir AD (2011) Modern developments in mass spectrometry of chondroitin and dermatan sulfate glycosaminoglycans. Amino Acids 41:235–256
Sonoda T, Kouno K (2000) Two brothers with distal arthrogryposis, peculiar facial appearance, cleft palate, short stature, hydronephrosis, retentio testis, and normal intelligence: a new type of distal arthrogryposis? Am J Med Genet 91:280–285
Steinmann B, Royce PM, Superti-Furga A (2002) The Ehlers -Danlos syndrome. In: Royce PM and Steinmann B (eds) Connective tissue and its heritable disorders. (2nd edition) Wiley-Liss, Inc., New York
Sugahara K, Mikami T, Uyama T, Mizuguchi S, Nomura K, Kitagawa H (2003) Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol 13:612–620
Voermans NC, Kempers M, Lammens M, van Alfen N, Janssen MC, Bonnemann C, van Engelen BG, Hamel BC (2012) Myopathy in a 20-year-old female patient with D4ST-1 deficient Ehlers-Danlos syndrome due to a homozygous CHST14 mutation. Am J Med Genet Part A 158A:850–855
Weber IT, Harrison RW, Iozzo RV (1996) Model structure of decorin and implications for collagen fibrillogenesis. J Biol Chem 271:31767–31770
Winters KA, Jiang Z, Xu W, Li S, Ammous Z, Jayakar P, Wierenga KJ (2012) Re-assigned diagnosis of D4ST1-deficient Ehlers-Danlos syndrome (adducted thumb-clubfoot syndrome) after initial diagnosis of Marden-Walker syndrome. Am J Med Genet A 158A:2935–2940
Yasui H, Adachi Y, Minami T, Ishida T, Kato Y, Imai K (2003) Combination therapy of DDAVP and conjugated estrogens for a recurrent large subcutaneous hematoma in Ehlers-Danlos syndrome. Am J Hematol 72:71–72
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Miyake, N., Kosho, T., Matsumoto, N. (2014). Ehlers–Danlos Syndrome Associated with Glycosaminoglycan Abnormalities. In: Halper, J. (eds) Progress in Heritable Soft Connective Tissue Diseases. Advances in Experimental Medicine and Biology, vol 802. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7893-1_10
Download citation
DOI: https://doi.org/10.1007/978-94-007-7893-1_10
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7892-4
Online ISBN: 978-94-007-7893-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)